DOI:
10.1039/D1MA00453K
(Paper)
Mater. Adv., 2022,
3, 1617-1628
Oxygen vacancies enable excellent electrochemical kinetics of carbon coated mesoporous SnO2 nanoparticles in lithium ion batteries†
Received
21st May 2021
, Accepted 31st October 2021
First published on 14th December 2021
Abstract
Herein, we report the synthesis, characterization and electrochemical performance of carbon coated mesoporous SnO2 nanoparticles (NPs) prepared by adopting a simple hydrothermal process. BET analysis shows that the formed SnO2 nanoparticles are porous with a high surface area which accommodates volume expansion. Morphological and Raman studies reveal that the carbon coating on SnO2 by a non-conventional carbon coating (NCC) process resulted in the formation of a homogenous carbon layer with a thickness of around 4–10 nm with predominant graphitic nature. The influence of the thermal treatment of SnO2 in air and carbon coating, which decide the oxygen vacancies, particle size, electrical conductivity and electrochemical performance, has been systematically studied. Electrochemical studies reveal that carbon coated (by the NCC process) H-SnO2 (calcined at 400 °C) with an average particle size and carbon coating thickness of 3 nm and 66 nm, respectively, delivered a discharge capacity of 440 mA h g−1 at a 1C rate with excellent electrochemical stability. Furthermore, it delivers capacities of 396 mA h g−1, 272 mA h g−1 and 129 mA h g−1 at 5C, 8C and 10C, respectively. The excellent electrochemical performance of carbon coated H-SnO2 by the NCC process can be attributed to the oxygen vacancies which can inhibit Sn coarsening and enhance electrical conductivity together with the carbon coating, high surface area, short Li+-ion diffusion length, and porous structure of SnO2 which buffers the volume expansion.
1. Introduction
Over the last three decades, lithium-ion batteries (LIBs) have been extensively utilized in portable electronic devices.1,2 As an important component of LIBs, the anode material plays a crucial role in determining the energy and power performance of the LIB device. However, for successful implementation of LIBs for hybrid electric vehicle (HEV) applications or electric vehicle (EV) applications, material level challenges such as (i) low ionic and electronic conductivity and (ii) large volume expansion (>300% for alloying reactions having high capacity) of anode materials upon cycling3,4 need to be addressed. In order to address these challenges, the development of an anode material with high capacity, long cycling stability and good rate capability by a facile synthesis route is essential. Commercial graphite has been mostly used as the anode material in LIBs due to its low and flat working potential, long cycle life and low cost.5 However, its application is limited due to its low power capability and the safety hazards associated with Li metal plating at higher current rates.6 Various anode materials like silicon,7 germanium,8 and metal-oxides like Fe2O3,9 Fe3O4,10 Co3O4,11 and SnO212 have been studied previously as alternative anode materials in place of graphite in LIBs. Among these anode materials, SnO2 is of particular interest, as it is cost-effective, is abundantly available, has a low discharge potential (0.5 V) and delivers a high capacity of 785 mA h g−1 (for 4 Li-ions) and 1494 mA h g−1 (for 8 Li-ions), which are higher in comparison with graphite.5,13–15 However, the issues associated with SnO2, such as large volume expansion/contraction problems (∼300%) during lithiation/de-lithiation, low intrinsic electrical conductivity as well as aggregation of Sn nanoparticles during cycling, hamper the realization of superior electrochemical performance.16 In order to overcome these issues, various strategies such as carbon coating, nanosizing and forming composites with TiO2, graphene or CNT and also the use of some hybrid anodes with excellent electrochemical performance as next generation electrode materials for LIBs have been studied.17–19 However, in spite of extensive efforts to improve the electrochemical performance of anode materials, the methodology developed for the preparation of these composites involves tedious and multi-step processes, making it less suitable for commercial application. Recently, generation of point defects such as oxygen vacancies in metal oxide based electrode materials has attracted special interest in tuning the electrochemical performance of lithium ion batteries.20 Based on previous reports, oxygen vacancies in metal oxides can act as electron donors and thereby increase carrier concentration.20 Recently, Dong and Li et al. reported black and yellow colored tin oxide nanoparticles with numerous oxygen vacancies which greatly enhanced the electrical conductivity and improved the rate capability.21,22 Very recently, Hu et al. reported that the sandwich-structured NiTi/SnO2/NiTi hybrid anode could successfully control the volume expansion problem and control Sn aggregation during repeated lithiation/delithiation and eventually result in improving the lithium storage capacity and stability of LIBs during cycling.23 None of the developed strategies address the issues of SnO2 and they only focus either on the synthesis of nanostructured SnO2 with some defects or on using some carbon allotropes to improve the electrical conductivity and volume expansion, but not simultaneously. In order to corroborate these issues, in the present study a unique hybrid strategy has been adopted, in which the performance of SnO2 is expected to be improved by combining the abovementioned strategies, i.e., (i) synthesis of mesoporous nanostructured SnO2 by a hydrothermal method using a surfactant assisted process and (ii) carbon coating on mesoporous nanostructured SnO2 by conventional carbon coating (CC) and non-conventional carbon coating (NCC) processes using sucrose as the carbon precursor. The resulting polysaccharide coated SnO2 was dried and finally carbonized to obtain uniform carbon coated SnO2. The Li-ion diffusion length can be reduced significantly using nanostructured SnO2 and the uniform carbon coating ensures higher electronic conductivity.24,25 It is interesting to note that the developed carbon coating process on mesoporous SnO2 results in a promising electrochemical performance, i.e., high capacity at higher current rates with good cyclic performance compared to SnO2 synthesized without a surfactant as well as carbon coating by a conventional carbon coating process.
2. Experimental section
Synthesis of mesoporous SnO2 and carbon coating by a dehydration assisted polymerization (DAP) process
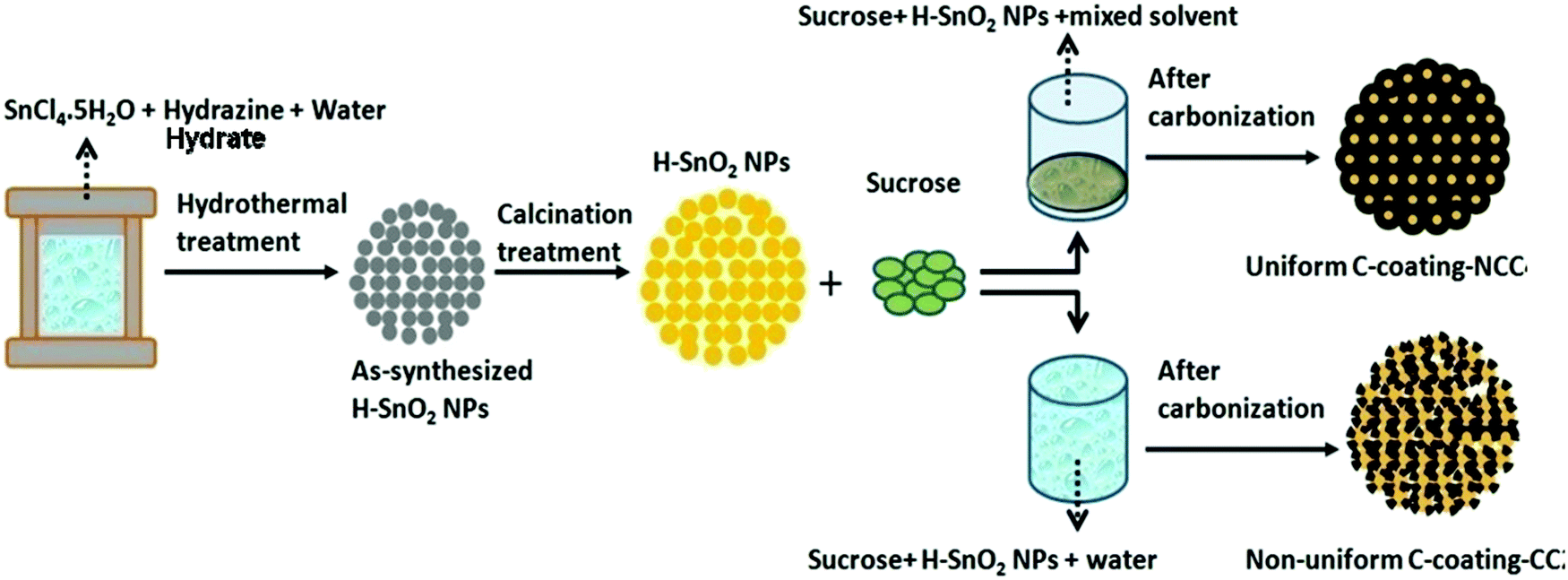
SnO2 nanoparticles were prepared by a surfactant assisted hydrothermal method using SnCl5·5H2O and hydrazine hydrate as the Sn precursor and surfactant, respectively. In a typical synthesis, 3.5 g SnCl5·5H2O and 1.2 g hydrazine hydrate were dissolved in distilled water (180 mL) under stirring using a magnetic stirrer for 30 min, which resulted in a semi-transparent colourless solution. The solution was then transferred to a Teflon-lined autoclave and heated at 160 °C for 5 h before it was cooled in air. The products were washed with DI water, dried in an oven at 100 °C for 6 h and finally calcined at different temperatures, namely, 300 °C, 400 °C and 450 °C, to obtain crystalline SnO2 particles. The SnO2 samples calcined at 300 °C appear white in colour, whereas the samples calcined at 400 °C and 450 °C appear yellow in colour. All the samples are labelled as H-SnO2. Similarly, for comparison, pure SnO2 nanoparticles synthesized without using a surfactant were calcined under similar experimental conditions and were labelled as pure SnO2. Pure SnO2 and H-SnO2 nanoparticles were then carbon-coated using a dehydration assisted polymerization (DAP) process, i.e., the non-conventional carbon coating (NCC) process, where four different carbon weight percentages with the sucrose to water ratio maintained at 1.216, 1.736, 2.439 and 3.472, respectively, were used, and accordingly the samples were labelled as NCC-1 to NCC-4. For comparison, carbon coating was also carried out on SnO2 using a conventional carbothermal process where carbon coating was carried out using sucrose without the addition of a dehydrating agent and its was labelled as H-SnO2-CC. Carbon coated SnO2 powders were then calcined in an argon atmosphere at 500 °C for 5 h to obtain a thin and uniform carbon layer around the particles. The entire process including synthesis of nanoparticles and subsequent carbon coating by conventional and non-conventional processes to yield thin, uniform and ordered carbon coated H-SnO2 nanoparticles as the final product is schematically shown in Fig. 1.
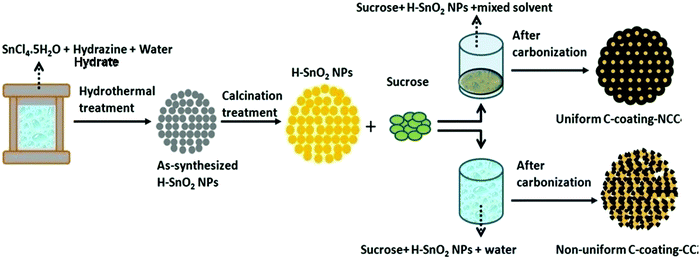 |
| | Fig. 1 Schematics of the synthesis of H-SnO2 and subsequent carbon coating by conventional and non-conventional carbon coating processes. | |
Materials characterization
The X-ray diffraction (XRD) measurement (Bruker D8 Advance X-ray diffractometer with Cu-Kα radiation, λ = 1.5406 Å) was carried out in the range of 2θ = 10°–80° to characterize the structural properties of the sample. The surface area, pore volume and pore diameter were measured using a BET surface area analyzer (Micromeritics ASAP 2020, V3.00 H). Thermogravimetric (TG) (model: STA 449 F3 Jupiter NETZSCH, Germany) analysis was carried out to measure the carbon content in carbon coated SnO2/H-SnO2 under an oxygen atmosphere in the temperature range of 0–800 °C. Raman analysis was performed in the range of 1000 to 2000 cm−1 at room temperature to know the nature of carbon coating using a Raman spectrometer (model: Horiba Jobin Yvon LabRAM HR-800) in which an argon ion laser was used as the source at an excitation wavelength of 514 nm. The optical properties of the SnO2 materials were measured using a spectrophotometer (Varian Cary 5000, UV-vis NIR). The morphology of all samples before and after carbon coating were measured using a field emission scanning electron microscope (FE-SEM, S-4300-SE/N microscope, Hitachi, Japan). Before FE-SEM analysis, a thin gold layer was coated on all materials by a sputtering process to make them conductive. The structural properties and carbon coating thickness on the surface of SnO2 were measured using a high-resolution transmission electron microscope (HR-TEM, TECNAI 200 kV, FEI, The Netherlands). The oxidation state, elemental composition and oxygen vacancies of the material were measured by X-ray photoelectron spectroscopy (XPS) analysis using an ESCA-Omicron XPS system with Mg-Kα as the excitation source.
Electrochemical characterization
The electrochemical performances of pure SnO2/H-SnO2 with and without carbon coating were evaluated by assembling half-cells in a Swagelok type cell in a glove box filled with pure argon gas. The working (SnO2) electrode was fabricated by mixing 80 wt% active material (SnO2), 10 wt% carbon black, and 10 wt% poly(vinylidene difluoride) (PVDF) in an appropriate amount of solvent, N-methyl-2-pyrrolidone (NMP). The resulting slurry was coated on copper foil, dried at 60 °C for 12 h and then kept in a vacuum oven for 6 h at 120 °C. Li foil was used as both the counter and reference electrodes. 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC; 1:1, wt%) was used as the electrolyte and a microporous membrane (Celgard 2400) was used as the separator. The prepared cells were aged for 12 h before the electrochemical measurement. The galvanostatic charge–discharge analysis was performed using an Arbin Instruments battery tester (model: BT2000) at various current densities in the range of 0.1–10C in the voltage window of 0.02 to 2 V at room temperature. AC-impedance analysis was carried out using an impedance/gain phase analyzer (Solartron-1260) in the frequency range of 100 kHz to 0.01 Hz with an AC signal amplitude of 5 mV. The electrochemical capacity was calculated based on the SnO2 mass used in the electrode.
:DMC; 1:1, wt%) was used as the electrolyte and a microporous membrane (Celgard 2400) was used as the separator. The prepared cells were aged for 12 h before the electrochemical measurement. The galvanostatic charge–discharge analysis was performed using an Arbin Instruments battery tester (model: BT2000) at various current densities in the range of 0.1–10C in the voltage window of 0.02 to 2 V at room temperature. AC-impedance analysis was carried out using an impedance/gain phase analyzer (Solartron-1260) in the frequency range of 100 kHz to 0.01 Hz with an AC signal amplitude of 5 mV. The electrochemical capacity was calculated based on the SnO2 mass used in the electrode.
3. Results and discussion
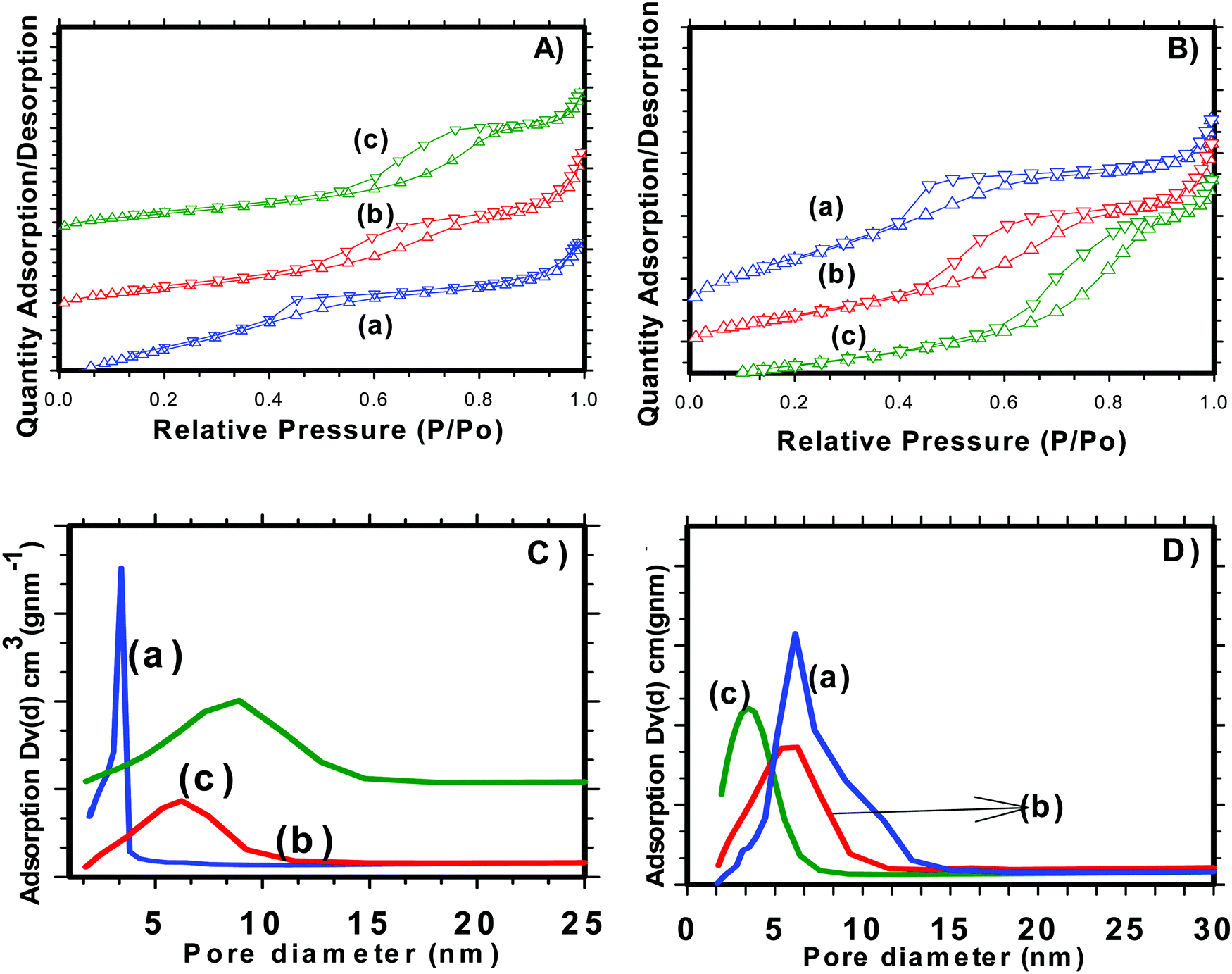
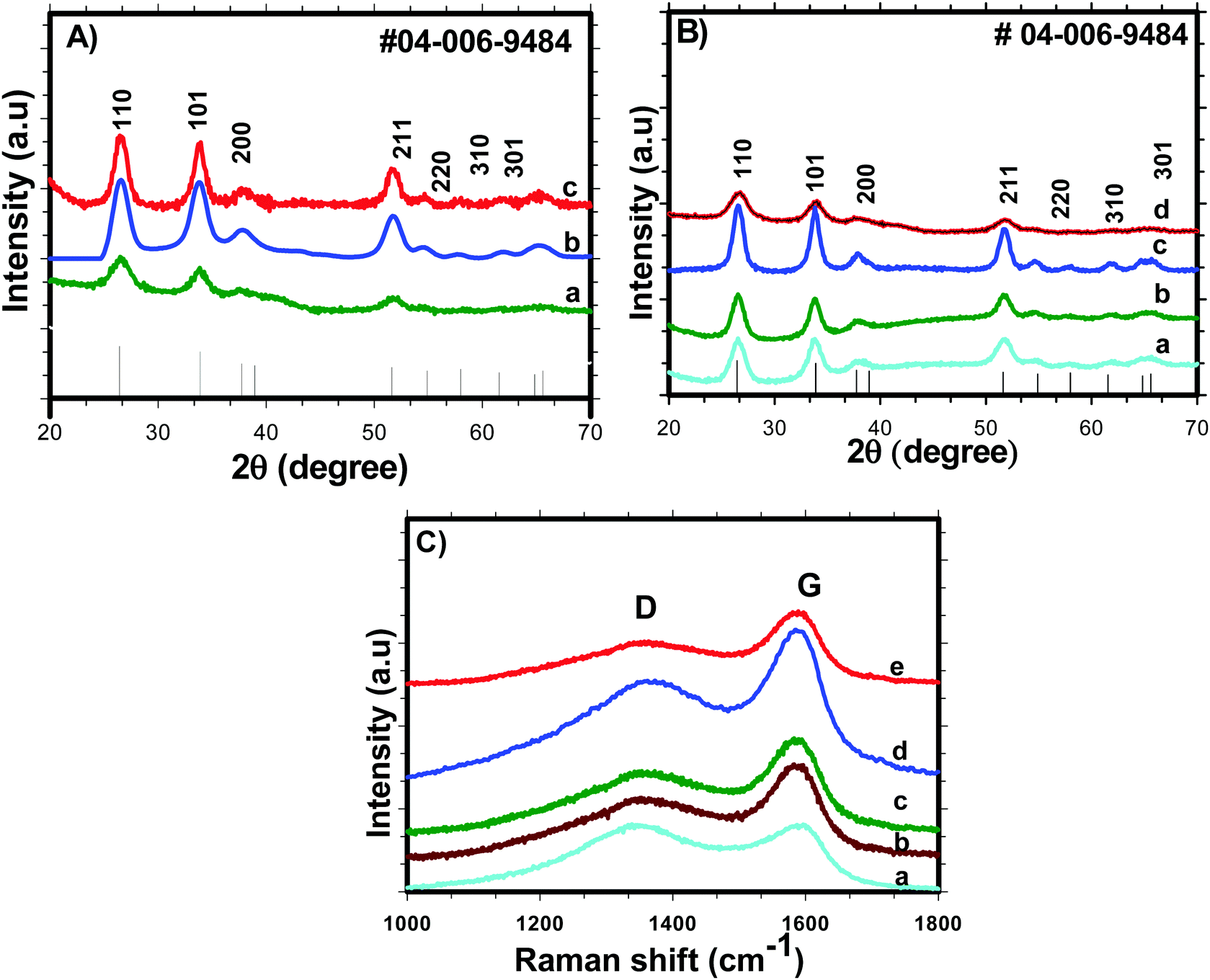
Nitrogen adsorption/desorption isotherms were used to examine various textural parameters like specific surface area, pore volume and pore size distribution of SnO2 nanoparticles. The N2 adsorption–desorption isotherm curves of H-SnO2 and SnO2 nanoparticles are shown in Fig. 2(A) and (B) and their respective pore size distributions are shown in Fig. 2C and D, respectively. As illustrated in Fig. 2A and B, both SnO2 and H-SnO2 exhibit type IV adsorption isotherms with a H2 type hysteresis loop in the relative pressure region of 0.4–0.9 p/po, which indicates the presence of mesopores.22 The adsorption/desorption isotherm of H-SnO2 showing a type IV isotherm with a hysteresis loop above 0.6 p/p0 reveals the presence of a mesoporous structure. The BET surface area decreases from 128 to 111.8 m2 g−1 and from 96 to 45.7 m2 g−1 for H-SnO2 and SnO2 nanoparticles, respectively, when the calcination temperature increases from 300 to 450 °C. The pore diameters of H-SnO2 and pure SnO2 nanoparticles are 5.2, 6.2 and 8.8 nm and 3.5, 5.8 and 6.1 nm for samples calcined at 300, 400 and 450 °C, respectively. The above results clearly show that with the increase in calcination temperature the surface area decreases and the pore diameter increases, which is attributed to the increase in the size of particles at higher calcination temperatures. Since porosity and surface area are high for H-SnO2, it is highly beneficial to accommodate the volume expansion occurring during electrochemical cycling and to increase the lithium ion active sites.24,25 Considering the above advantages of H-SnO2 over pure SnO2, the former has been chosen for further studies in terms of carbon coating, characterization and electrochemical performance evaluation. Fig. 3A shows the XRD pattern of H-SnO2, which was calcined at different temperatures, namely, 300, 400 and 450 °C, respectively. It can be seen that H-SnO2 calcined at different temperatures showed a single phase belonging to the tetragonal phase of SnO2 (ICDD # 04-006-9484; space group, P42/mnm; lattice parameters, a = 5.747 Å and c = 3.185 Å) without any impurities. The crystallite size of H-SnO2 nanoparticles was calculated using Scherrer's formula,26 and it was found that with increasing calcination temperature the crystallite size increases and the calculated crystallite sizes for different SnO2 particles are shown in Table S1 (ESI†). Fig. 3B(a)–(d) show the X-ray diffraction patterns of carbon coated H-SnO2 samples by non-conventional process under the aforementioned conditions for NCC-1 to NCC-4. It is observed that all samples exhibited a single phase SnO2 structure indicating that carbon coating had no significant influence on the structure or crystallinity of the samples. The crystallite sizes of all the samples calculated using Scherrer's equation26 are listed in Table S1 (ESI†). The existence of intrinsic structural defects and its influence on the structural disorder upon an increase in temperature are discussed based on the results of UV-Vis, DRS, and XPS analyses. Fig. 3C shows the Raman spectra of H-SnO2 carbon-coated using non-conventional (NCC-1–NCC-4) and conventional coating processes. As shown in Fig. 3C the spectrum is deconvoluted into two Gaussian peaks such as the G-band at 1585 cm−1 and the D-band at 1355 cm−1 corresponding to ordered sp2 graphitic carbon and sp3 disordered carbon, respectively.27–29 The G band is assigned to the second-order scattering of the E2g mode observed from the vibration of in-plane sp2 carbon, while the D band is related to the A1g phonon of the sp3 carbon atoms of disordered graphite.28 Although all carbon coated H-SnO2 samples showed the presence of D and G bands, the quality of carbon coating was monitored by calculating the intensity ratio of D and G bands, i.e. (ID/IG), which is used to evaluate the ordered and disordered nature of carbon coating quantitatively. It is reported that a carbon with a low ID/IG ratio leads to the formation of more graphitic (sp2) structured carbon in comparison to disordered (sp3) carbon. The sp2 nature helps in improving the electronic conductivity of electrode materials which is advantageous for energy storage applications.30 The calculated ID/IG ratios for NCC-1, NCC-2, NCC-3 and NCC-4 are 1, 0.66, 0.69 and 0.58, respectively, implying that the ID/IG ratio decreases with an increase in the sucrose to water ratio of 1.216, 1.736, 2.439 and 3.472, respectively. Particularly, when the sucrose to water ratio (NCC-4) increases to 3.472, the ID/IG ratio decreases to 0.58, indicating the presence of a high concentration of ordered sp2 carbon in carbon coated (NCC-4) SnO2 particles. For comparison, Raman analysis was also carried out for H-SnO2, which was carbon-coated by a conventional carbon coating process and the spectrum is shown in Fig. 3C(e). The spectrum shows the presence of D and G bands and the calculated ID/IG ratio is 0.68, which shows that most of the carbon has disordered nature. It is interesting to note that the ID/IG ratio of carbon coated by the non-conventional carbon coating process (NCC-4) is less than that of carbon coated by the conventional carbon coating process (CC), indicating that the dehydrating agent used in the former leads to the formation of a graphitic carbon layer with a smaller carbon content (<10 wt %), whereas a low degree of graphitization is observed for the latter with a high carbon content (weight of 50% carbon taken during the synthesis). Based on the Raman spectral analysis, it is concluded that, among all other conditions and conventional carbon coating techniques, H-SnO2 carbon coated under NCC-4 conditions resulted in a graphitic carbon rich layer around SnO2 nanoparticles which is expected to improve the electronic conductivity and to buffer the volume expansion.11,30,31 The morphologies of the H-SnO2 nanoparticles calcined at different temperatures and carbon coated H-SnO2 nanoparticles are shown in Fig. 4. The FE-SEM images of pure H-SnO2 nanoparticles are shown in Fig. 4(A)–(C). The FE-SEM images show the presence of porous spherical SnO2 particles with agglomeration. It is observed that the increase of calcination temperature (T = 300°–450 °C) has effectively resulted in particle size increase. Further, the bulk composition of H-SnO2 nanoparticles was found to be 1.76 [see Fig. S1, ESI†]. The porous structure could be beneficial for SnO2 as it accommodates volume expansion during Li+ ion insertion/extraction and is expected to improve the electrochemical performance in terms of specific capacity and cyclic stability.32
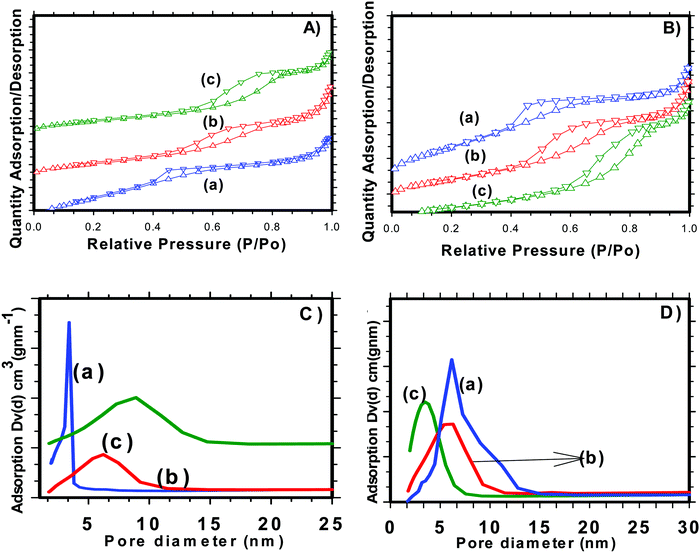 |
| | Fig. 2 N2 adsorption–desorption isotherms of (A) H-SnO2 and (B) SnO2, and pore size distribution of (C) H-SnO2 and (D) SnO2 calcined at (a) 300 °C, (b) 400 °C and (c) 450 °C. | |
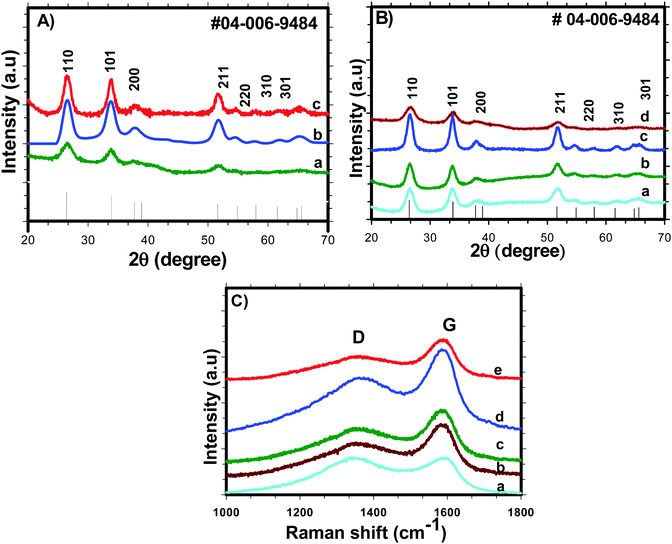 |
| | Fig. 3 XRD patterns of (A) H-SnO2 calcined at different temperatures: (a) 300 °C, (b) 400 °C and (c) 450 °C; (B) XRD patterns of H-SnO2 (calcined at 400 °C) carbon-coated by a non-conventional carbon coating process under NCC-1 to NCC-4 conditions; and (C) Raman spectra of non-conventional carbon coated (NCC-1–NCC-4) (a–d) and (e) conventional carbon (CC) coated H-SnO2. | |
 |
| | Fig. 4 FE-SEM images of H-SnO2 calcined at different temperatures: (A) 300 °C, (B) 400 °C and (C) 450 °C; (D) low and (E) high magnification TEM images of non-conventional carbon coated H-SnO2 (NCC-4); the inset shows the particle size distribution histogram; (F) the corresponding SAED pattern. | |
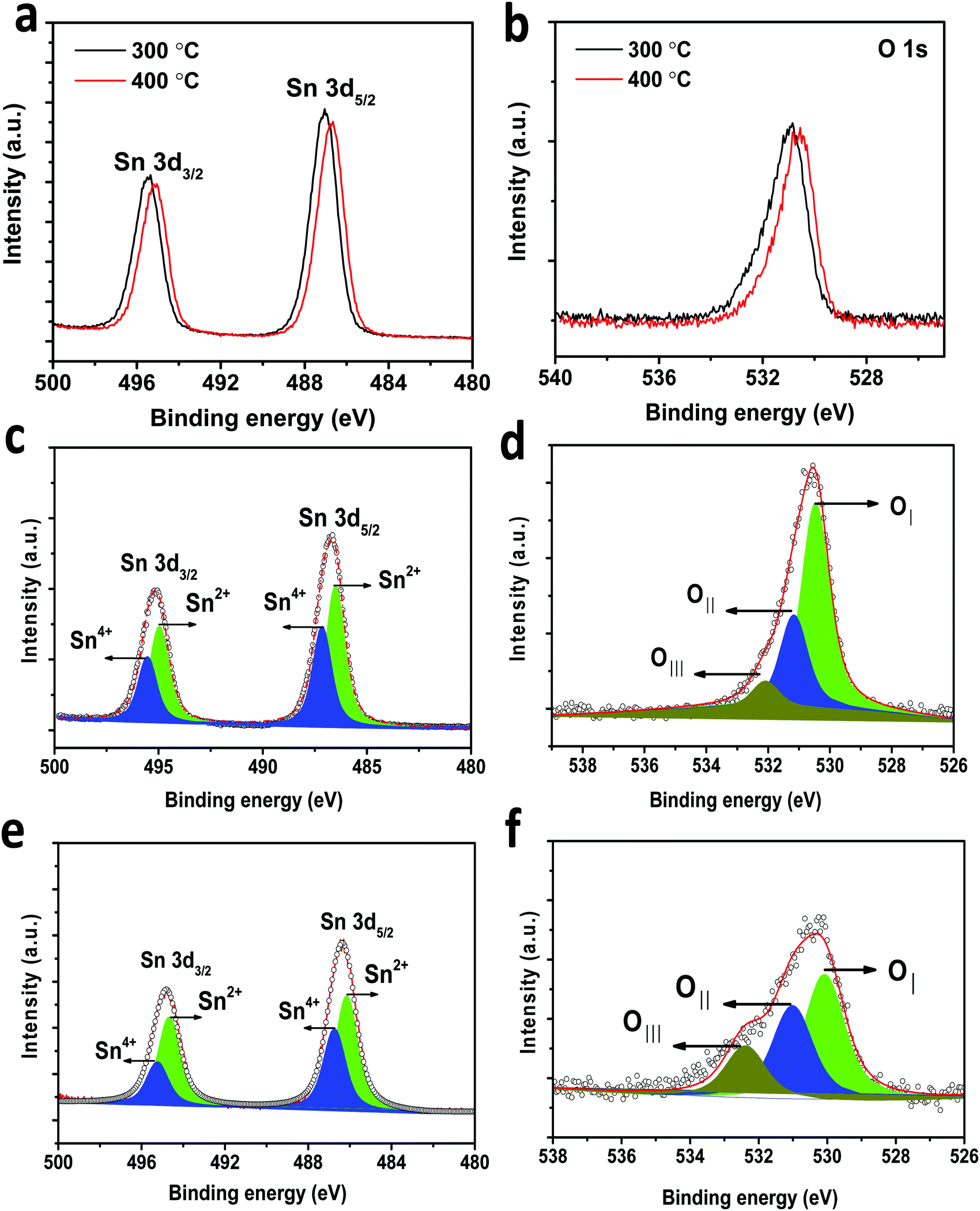
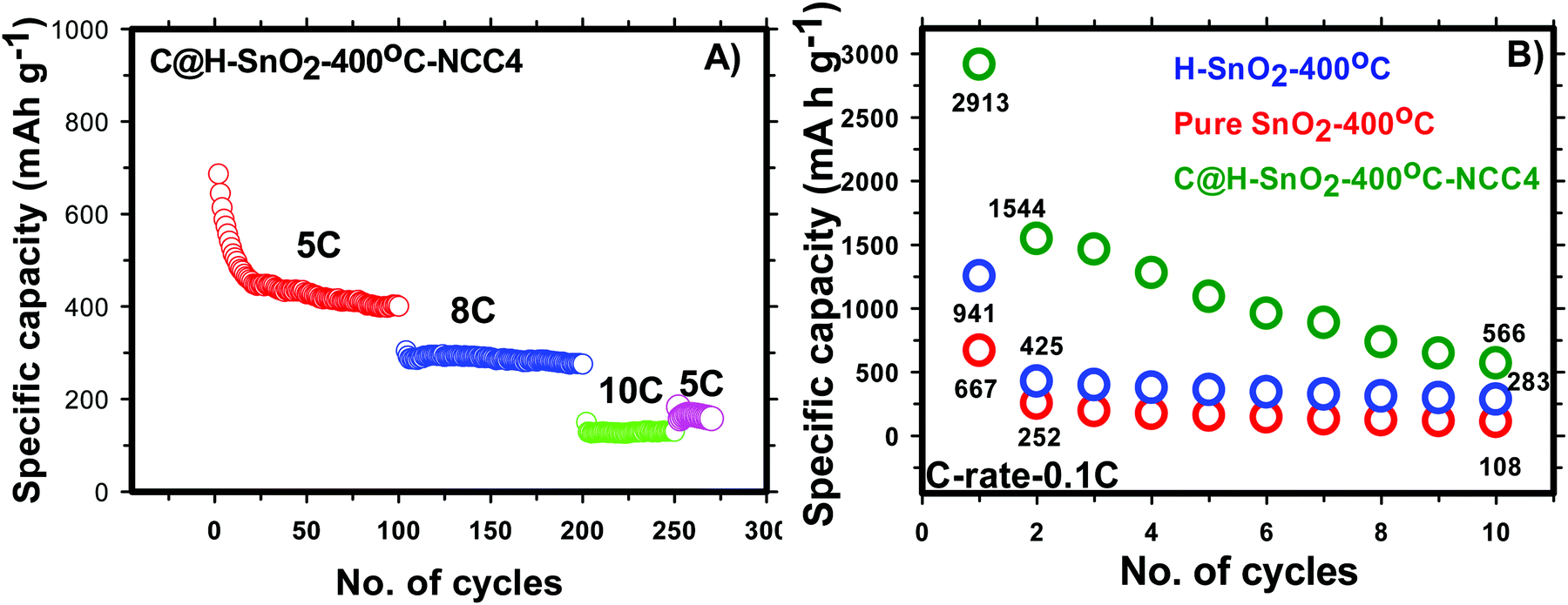
In-depth morphology and particle size distribution were analysed by HR-TEM analysis and the images are shown in Fig. 4(D) and (E). It is reported that, in addition to the graphitic nature of the carbon layer, uniform distribution of the carbon layer equally influences the electronic conductivity of Li-ion battery electrode materials. Fig. 4D presents the low magnification image showing nearly spherical SnO2 particles that are uniformly coated by a thin layer of carbon. The high magnification TEM image of carbon coated H-SnO2 shown in Fig. 4E confirms the existence of carbon coating with a thickness of ∼3–8 nm, which encapsulates H-SnO2 nanoparticles. This carbon coating layer on H-SnO2 not only increases the electronic conductivity of SnO2 but also endows it with the ability to withstand large volume expansion occurring during the Li+ insertion/extraction process.10,33 Further, there is the existence of lattice fringes with spacing values of 0.335 nm and 0.239 nm as shown in the high resolution TEM image in Fig. 4E, which match the d-spacing values of the (110) and (200) planes of SnO2, respectively, revealing the presence of tetragonal SnO2.34 Further, Fig. 4F displays the selected-area electron diffraction (SAED) ring pattern with diffused spots indexed to the (110), (101), (200), and (211) planes of H-SnO2,35 confirming that carbon coated H-SnO2 nanostructures exhibit a tetragonal phase with a polycrystalline structure, which is consistent with the XRD patterns shown in Fig. 3A and B. The crystal planes and crystallite size obtained from the HR-TEM analysis are consistent with the XRD results as discussed in earlier sections. Further, UV-Vis DRS absorption spectroscopy was also carried out to study the oxygen defects from the as-synthesized and calcined H-SnO2 nanoparticles shown in Fig. S2 (ESI†). The as-synthesized H-SnO2 nanoparticles showed an absorption onset around 400 nm which reveals the typical band to band transition characteristics of pure SnO2 materials, whereas the H-SnO2 nanoparticles calcined at different temperatures exhibited a shift towards the visible-light region, and its visible-light absorption increases with the increase in calcination temperature, attributed to the presence of oxygen vacancies (Vo) as reported previously.36–38 Hence, the effect of oxygen vacancies could successfully influence both the structural and optical properties of SnO2 shown in Table S1 (ESI†). The amount of carbon in carbon coated H-SnO2 nanoparticles was analyzed by thermogravimetric analysis and the results are shown in Fig. S3(A)–(D) (ESI†). As shown in Fig. S3 (ESI†), two weight losses can be observed in the TGA profile, where the initial weight loss occurs at a temperature less than 200 °C, which is due to the decomposition of water molecules, and the other weight loss observed between 350 and 550 °C corresponds to the oxidation of carbon. The carbon contents calculated from the weight loss in the temperature range of 350–550 °C {Fig. S3(A)–(D), ESI†} due to oxidation of carbon are 3.5, 5.7, 7 and 5.8 wt% for NCC-1, NCC-2, NCC-3 and NCC-4, respectively. In order to analyze the presence of tin and carbon and their oxidation states in carbon coated H-SnO2 nanoparticles, X-ray photoelectron spectroscopy (XPS) analysis was conducted. The binding energy of the C 1s peak at 284.5 eV is used as the standard reference point for binding energy correction. The high resolution wide scan and narrow spectra of Sn-3d and O-1s of SnO2 calcined at 300 °C and 400 °C are shown in Fig. 5a and b, respectively. The peaks of Sn-3d and O-1s for SnO2 calcined at 400 °C shifted towards lower binding energies compared with SnO2 calcined at 300 °C, attributed to the formation of a lower oxidation state of Sn on their surface. After careful deconvolution of the XPS spectra of Sn-3d (Fig. 5c), the presence of two peaks corresponding to different oxidation states of SnO2, namely, Sn4+ and Sn2+, is observed. The presence of the high intensity Sn2+ peak in addition to the Sn4+ peak is mainly due to the partial oxidation of SnO2 by thermal treatment in air.36 The presence of Sn2+ represents the formation of numerous oxygen vacancies, which can greatly enhance the electronic conductivity of the SnO2 material.37 In general, the difference in binding energies (BEs) observed between Sn-3d5/2 and Sn-3d3/2 peaks is 8.42 eV, which is a typical characteristic of SnO2. In the present study, the difference in binding energies (ΔBE) obtained for pure and carbon coated H-SnO2 NCC-4, i.e., ΔBE, is in the range of 8.013 to 8.422 eV (see Table S2, ESI†) which is consistent with the standard ΔBE value (∼ 8.42 eV) of Sn2+/4+ of SnO2.39,40 Also, the binding energies of Sn-3d5/2 and Sn-3d3/2 peaks are consistent with the binding energies of standard SnO2 reported previously,39 confirming that Sn exists in +2 and +4 oxidation states. Fig. 5d displays the deconvoluted high-resolution O-1s spectrum of H-SnO2-400 °C nanoparticles. The peak OI at a lower binding energy (530.5 eV) is indexed to O2− of H-SnO2, attributed to the coordination of oxygen bound to tin atoms, which is in good agreement with earlier reported values. The higher binding energy peaks such as OII (531.2 eV) and OIII (532.08 eV) were assigned to chemisorbed oxygen which arises due to the adsorption of surface hydroxyl groups, corresponding to O–H bonds or carbonyl groups. Overall, it is worth revealing that peaks at higher binding energy values (531.2–532.08 eV) correspond to oxygen deficiencies,41,42 which give rise to non-stoichiometric defects in SnO2 NPs. Further, the Sn 3d and O 1s spectra of carbon coated H-SnO2 (400 °C) under NCC-4 conditions also show similar results to those observed for H-SnO2 NPs without carbon coating (Fig. 5e and f). The electrochemical performances of H-SnO2 and carbon coated H-SnO2 nanoparticles were investigated in a half-cell in the potential window ranging from 0.02 to 2 V to study the influence of porous oxygen deficient carbon coated H-SnO2 in improving the electrochemical performance of SnO2 nanoparticles. From the Raman analysis, NCC-4 was considered as the optimum carbon coating and hence the H-SnO2 samples calcined at different temperatures were carbon-coated under NCC-4 conditions and the electrochemical performances of the corresponding carbon coated H-SnO2 were systematically evaluated to find the optimum annealing temperature of SnO2 nanoparticles. The capacities delivered by pure SnO2 and H-SnO2 as shown in Fig. S4 and S5 (ESI†) clearly indicate the influence of the surfactant. SnO2 synthesized using hydrazine hydrate (surfactant) (Fig. S5, ESI†) delivered a high discharge capacity compared to pure SnO2 (Fig. S4, ESI†) at the 1st, 2nd and 10th cycles for a 0.1C rate, respectively. This comparison shows that SnO2 synthesized using hydrazine hydrate having a smaller particle size and increased porosity displayed improved electrochemical performance. However the capacity delivered was poor and therefore carbon coating was found to increase the capacity by improving the electronic conductivity and buffering volume expansion. Fig. S6 (ESI†) shows the electrochemical performance of H-SnO2 carbon-coated by the non-conventional carbon coating process under NCC-4 conditions. Fig. S6A (ESI†) shows the charge–discharge profile of carbon coated H-SnO2 calcined at 300 °C, 400 °C and 450 °C and tested at a 0.1C rate. It delivered 2nd cycle discharge capacities of 845, 1544 and 941 mA h g−1 for carbon coated H-SnO2 calcined at 300, 400 and 450 °C, respectively. For a 1C rate, it delivered discharge capacities of 773, 812 and 659 mA h g−1 for H-SnO2 calcined at 300 °C, 400 °C and 450 °C, respectively (Fig. S6B, ESI†). The loss in capacity is due to the irreversible loss caused by SEI layer formation and the irreversible change of SnO2 to Sn.43 As shown in Fig. S6C (ESI†), carbon coated H-SnO2-NCC-4 delivered capacities of 275, 230 and 208 mA h g−1, respectively,
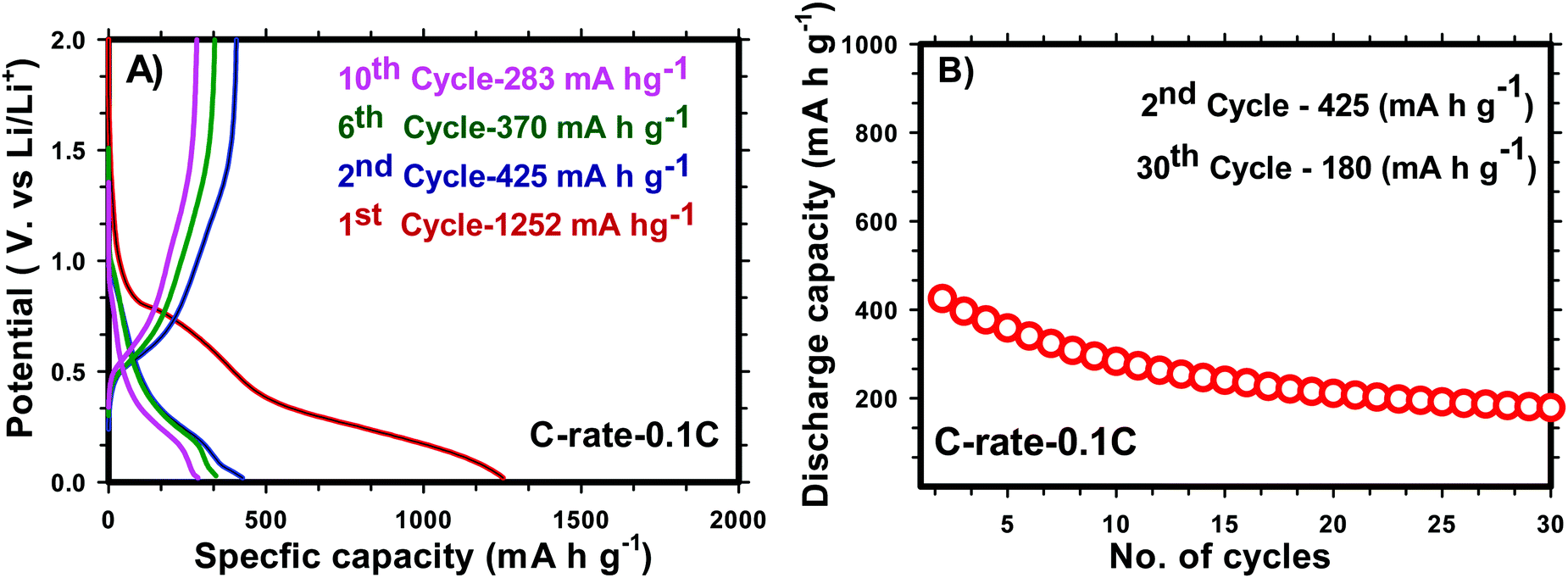
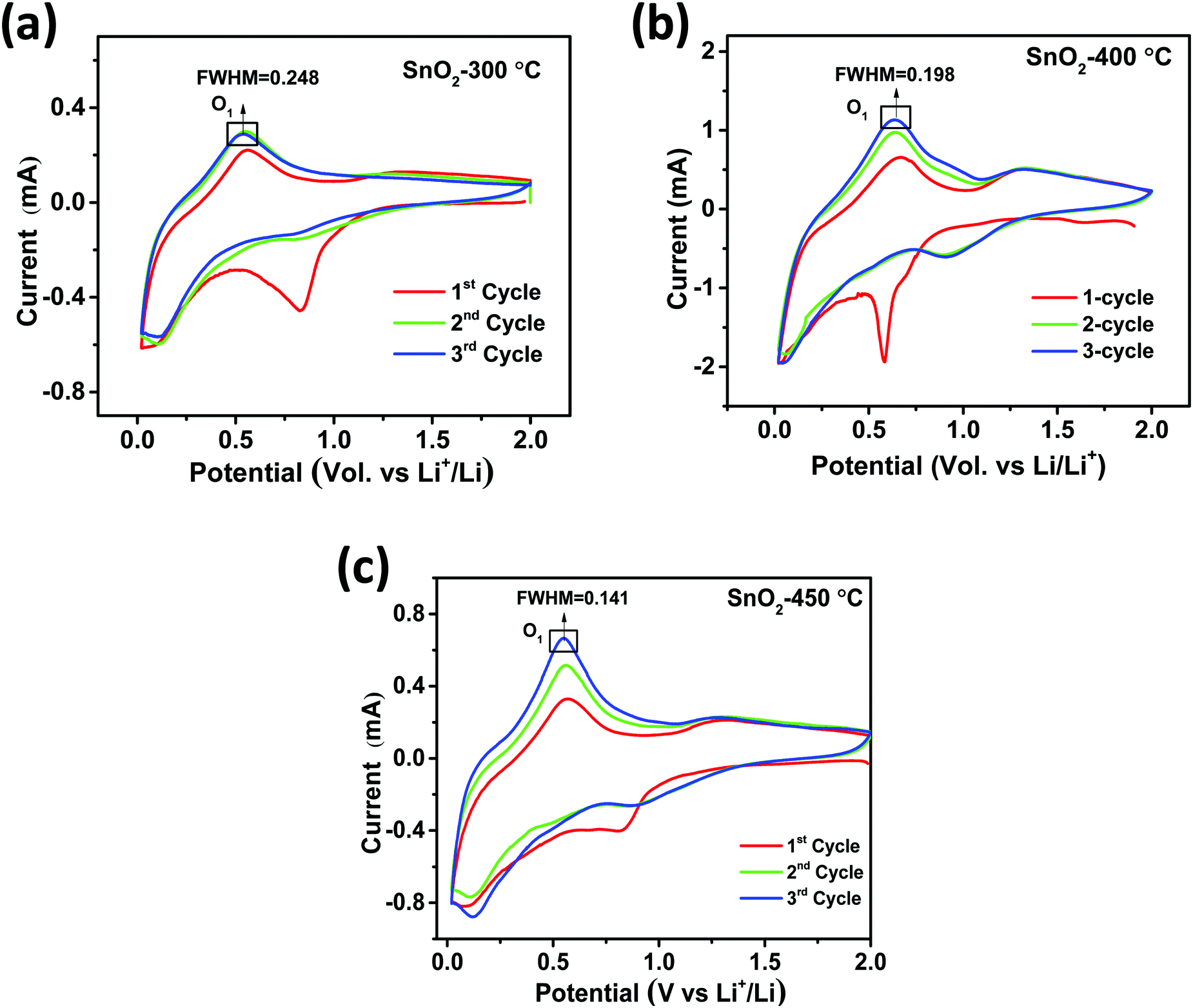
for H-SnO2 calcined at 300, 400 and 450 °C, respectively, at a 1C rate after 10 cycles. The H-SnO2 calcined at 400 °C showed better electrochemical performance and higher capacity retention due to a smaller particle size and optimum textural parameters making lithium ion diffusion faster during the charge–discharge process. As carbon coated H-SnO2-400 °C (by NCC-4) showed better electrochemical performance, rate capability studies were carried out to examine the improvement in electrochemical performance. Fig. 6A depicts the results of rate capability studies carried out from 5C to 10C and it can be seen that carbon coated H-SnO2 (by NCC-4) delivered capacities of 454, 292, and 129 mA h g−1 at 5C, 8C and 10C, respectively. Fig. 6B shows a comparison of the discharge capacities of pure SnO2, H-SnO2 and carbon coated H-SnO2-400 °C-NCC-4 tested at a 0.1C rate. Interestingly, uniform carbon coated H-SnO2 (by NCC-4) with oxygen defects showed high areal capacity of 2–4 mA h cm−2, which could meet the requirements for industrial applications [see Fig. S7, ESI†]. The results clearly show the influence of the porous structure and carbon coating in improving the electrochemical performance of SnO2 nanoparticles. Comparative studies were carried out for H-SnO2 nanoparticles carbon-coated by the conventional coating process (CC) in the absence of a dehydrating agent and it is seen that H-SnO2 calcined at 400 °C and carbon-coated by the conventional method delivered capacities of 1252, 425, 370 and 283 mA h g−1 at 0.1C for the 1st, 2nd, 6th and 10th cycles, respectively, as shown in Fig. 7, indicating that the capacity is low compared to that of H-SnO2 carbon-coated by NCC-4 which delivers 2913, 1544 and 566 mA h g−1 at 0.1C for the 1st, 2nd and 10th cycles (Fig. 6B), respectively. Further, the cyclic stability data (Fig. 7B) reveal that the capacity of carbon coated H-SnO2 by CC is very low compared with carbon coated H-SnO2 by NCC-4 (Fig. 6B). The electrochemical performance of carbon coated SnO2 prepared by the non-conventional process exhibits a high specific capacity and good cyclic stability in comparison with SnO2, H-SnO2 and H-SnO2 carbon-coated by the CC process. The poor capacity of H-SnO2 carbon coated by the CC process is due to improper carbon coverage and higher carbon coating thickness which hinder lithium ion diffusion. Further, the absence of a dehydrating agent also reduces proper catenation of the carbon precursor, leading to inhomogeneous carbon layer encapsulation on SnO2 nanoparticles, and therefore the capacity and stability fade when the electrochemical cycle number increases. SnO2 without carbon coating can be poorly conducting in the cases with and without hydrazine hydrate. Further, cyclic voltammetry (CV) was carried out for H-SnO2 calcined at 300, 400 and 450 °C in order to understand the electrochemical characteristics of SnO2 nanoparticles and the results are shown in Fig. 8a–c. Based on the literature, the cathodic peak appearing at 0.6–0.75 V corresponds to the reduction of SnO2−x to metallic Sn and the formation of a solid electrolyte interphase (SEI) film.44 The peak located at 0.1 V was assigned to the alloying reaction between Sn and Li. The anodic peak at 0.55 V represents the de-alloying reaction of LixSn and the broad peak at 1.3 V is assigned to the reversible reaction between Sn and Li2O.45,46 It is clear from Fig. 8a–c that the oxidation peaks of H-SnO2 synthesized at 400 °C and 450 °C with oxygen vacancies became more intense than those associated with H-SnO2-300 °C. The effect of oxygen vacancies on the kinetic behaviour of lithium ion storage was further examined by comparing the FWHM values of the oxidation (O1s) peaks of all H-SnO2 samples, H-SnO2-300, 400 and 450 °C. It is noticed that the FWHM values of oxygen vacancy rich H-SnO2-400 and 450 °C are smaller and the peaks are highly intense compared to H-SnO2-300 °C, which proves the presence of a high concentration of oxygen vacancies which accelerates the electrode reaction as well as enhances the electronic conductivity of the SnO2 electrode during the charge–discharge process.37,39 Further, the above CV scans are in good agreement with the voltage profiles, revealing the charge and discharge process (see Fig. S5 and S6, ESI†).
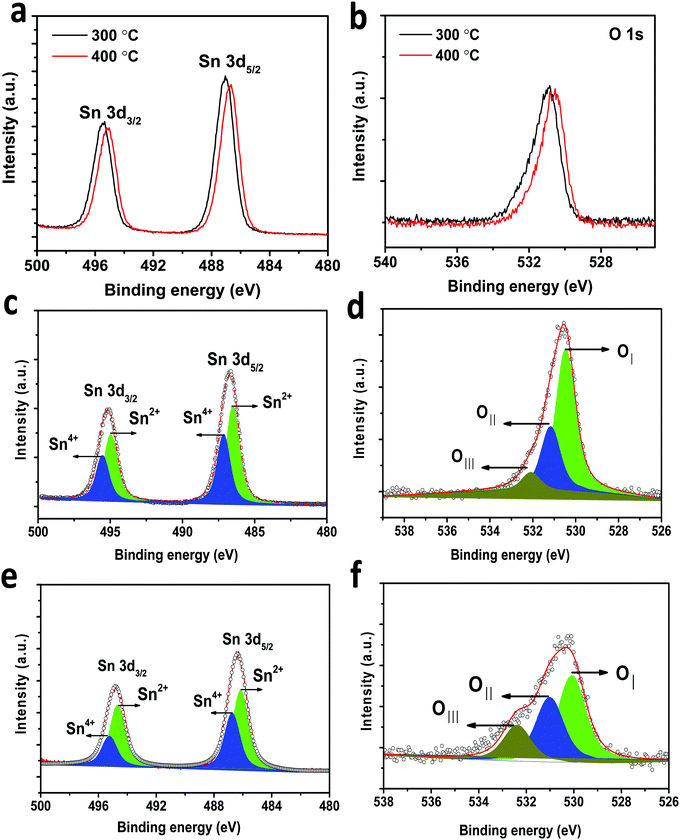 |
| | Fig. 5 XPS spectra for (a) Sn 3d of H-SnO2-300 °C and 400 °C, (b) O 1s of H-SnO2- 300 °C and 400 °C; (c and d) deconvoluted Sn 3d and O 1s spectra of H-SnO2-400 °C; and (e and f) deconvoluted Sn 3d and O 1s spectra of carbon coated H-SnO2-400 °C (NCC-4). | |
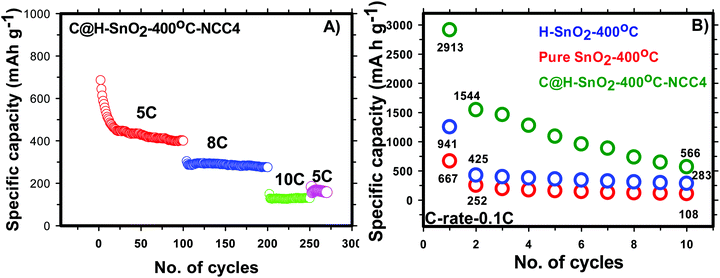 |
| | Fig. 6 (A) Rate capability of carbon coated H-SnO2-400 °C-NCC-4 and (B) comparison of the discharge capacities of SnO2, H-SnO2 and carbon coated H-SnO2-400 °C-NCC-4 tested at a 0.1 C rate. | |
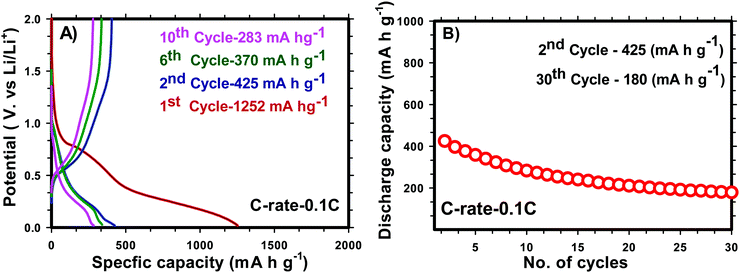 |
| | Fig. 7 Charge–discharge profile (A) and cyclic stability (B) (0.1 C rate) of carbon coated H-SnO2 by a CC process. | |
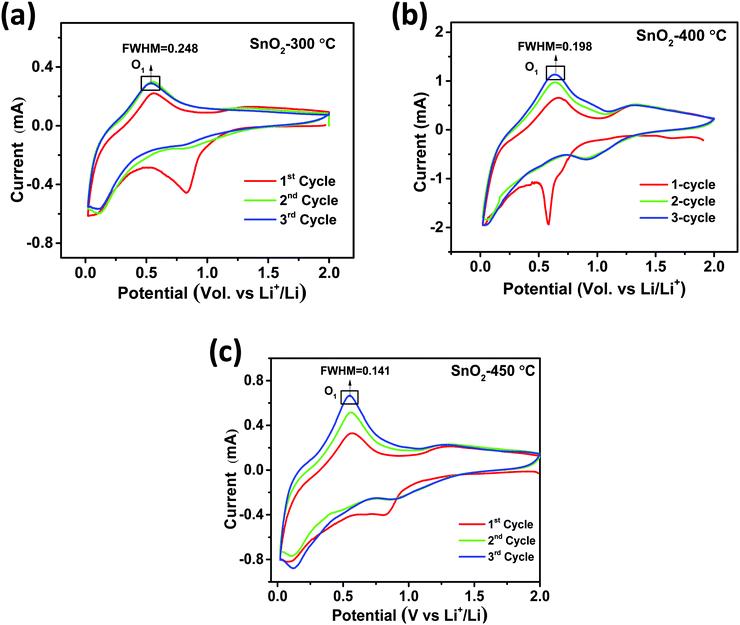 |
| | Fig. 8 Cyclic voltammetry of H-SnO2 calcined at (a) 300 °C, (b) 400 °C and (c) 450 °C. | |
The phase deviation of carbon coated H-SnO2 with and without oxygen vacancies during electrochemical cycling (i.e., lithiation/de-lithiation) was further investigated by ex situ XRD analysis and the results are shown in Fig. S8a and b (ESI†). Interestingly, the intense diffraction peaks of (110) and (101) planes without any diffraction peak of Sn appeared in the first cycle of (lithiation/de-lithiation stage) carbon coated H-SnO2-400 °C compared to H-SnO2-300 °C (Fig. S8a and b, ESI†). In contrast, after 50 cycles, the Li22Sn5 phase is formed together with a strong diffraction peak of Sn metal at the full lithiation stage (Fig. S9, ESI†), which resulted from the stress developed during lithiation induced coarsening of the Sn phase.47 Interestingly, oxygen vacancies control coarsening of Sn in the Sn/Li2O reaction, and thereby it maintains conversion reactions in the lithiated state. Thus suppression of Sn coarsening by oxygen vacancies can be responsible for the fast inter-diffusion kinetics in Sn/Li2O interfaces, resulting in good reversibility of SnO2 in the de-lithiated state, and enable excellent stability during cycling.47 Hence the abundance of oxygen vacancies in carbon coated H-SnO2 nanoparticles resulted in fast electrochemical kinetics, maintaining excellent reversibility and cyclability of the SnO2 electrode, which has been already discussed in the battery performance test above.
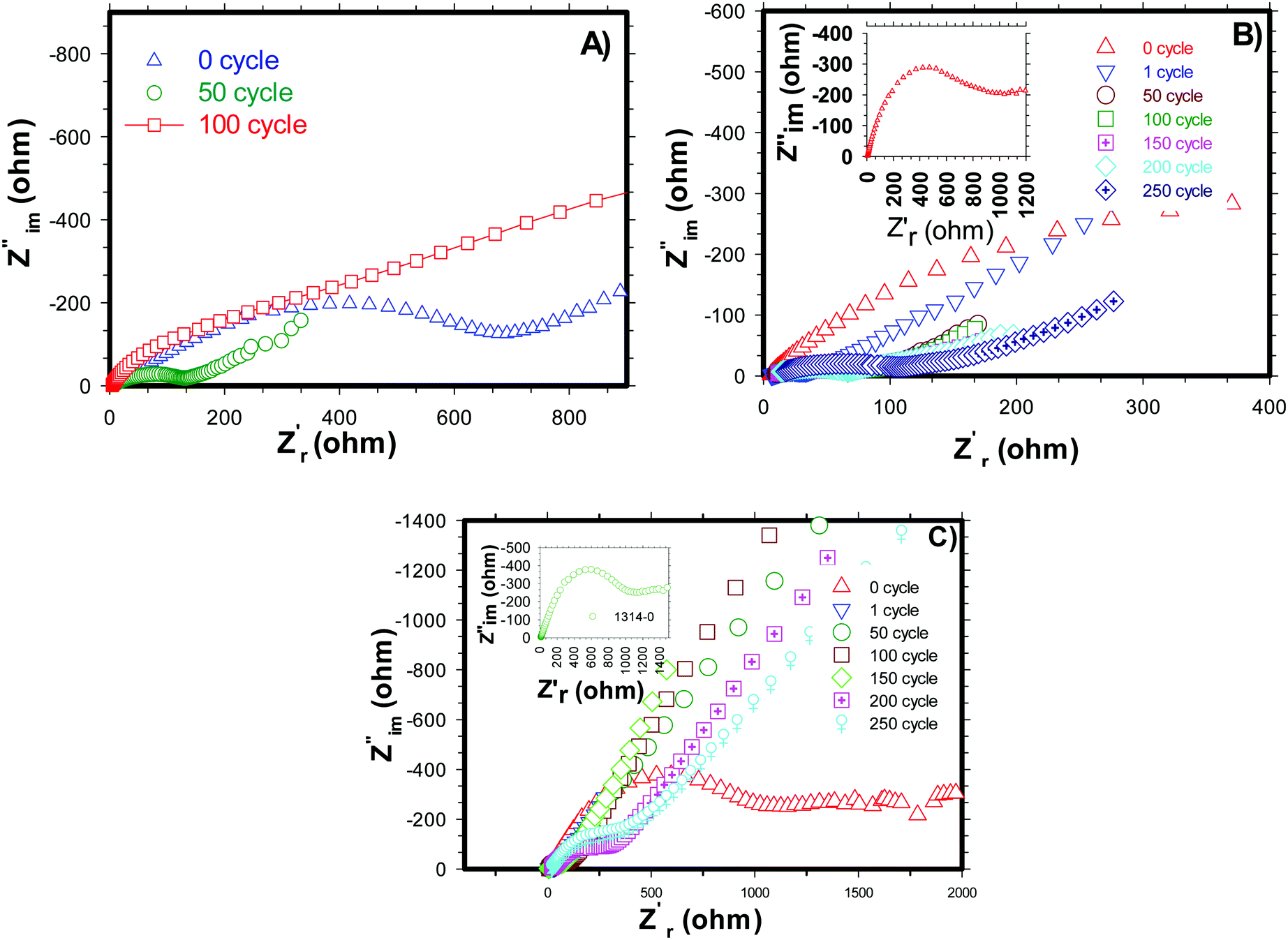
Electrochemical impedance spectroscopy (EIS) analysis was carried out for pure and carbon coated (by NCC-4 and CC) H-SnO2 nanoparticles after 0, 50, 100, 150, and 200 cycles and the results are shown in Fig. 9A–C. From the simulation of the equivalent circuit, it is noticed that the charge transfer resistance (Rct) values of H-SnO2 were found to be 867 Ω, 148 Ω and 640.5 Ω after 0, 50, and 100 cycles, respectively (see Fig. 9A), whereas, as expected, the charge transfer resistance (Rct) values of carbon coated H-SnO2 nanoparticles under NCC-4 conditions were found to display a decreasing trend: 990 Ω, 101.7 Ω, 97.6 Ω and 57.7 Ω after 0, 50, 100 and 150 cycles, respectively (see Fig. 9B). Finally, the decline in charge transfer resistance with the increase in cycle number is due to the greatly enhanced electronic contact achieved in the nanostructure, thereby improving the stable cyclic performance. But, after 200 cycles, the charge transfer resistance (Rct) was found to be 75.2 Ω, which is responsible for capacity fading. The increased resistance after 200 cycles may be due to the gradual increase in the thickness of the SEI layer formed on H-SnO2 grains, which acts as a potential barrier, leading to the blockage of charge carriers, responsible for the increase in the resistivity of the material. The low resistance obtained under NCC-4 conditions is due to the uniform graphitic carbon coating on H-SnO2 nanoparticles as confirmed by FE-SEM, HR-TEM and Raman analysis. In the present study, another interesting point noticed was that, in addition to uniform carbon coating, surface defects like oxygen vacancies were also involved in reducing the resistivity of the carbon coated H-SnO2 anode material, which in turn improves the electronic conductivity of the H-SnO2-NCC-4 anode material.37,48 The overall observation supports the prediction that both uniform carbon coating and the presence of oxygen vacancies (VO) play an important role in improving the electronic conductivity of the H-SnO2-NCC-4 anode material compared to pure H-SnO2 nanoparticles. Furthermore, the resistance of H-SnO2 carbon-coated by the conventional carbon coating process was measured and is shown in Fig. 9C. The charge transfer resistance (Rct) shows an increasing trend from the 50th to the 200th cycle, causing fast capacity fading as discussed earlier, which is due to the amorphous form of SnO2 as proved by XRD patterns and non-uniform carbon coating confirmed by FE-SEM images. Overall, as expected from AC impedance measurement results, the H-SnO2 anode carbon-coated under NCC-4 conditions resulted in decreasing charge transfer resistance compared to pure H-SnO2 and H-SnO2 carbon-coated under CC conditions. Hence the EIS results reveal that the uniform and graphitic nature of carbon in carbon coated H-SnO2 not only improves fast reaction kinetics, but also enhances ionic conductivity as well as electronic conductivity, thus showing excellent electrochemical performance.
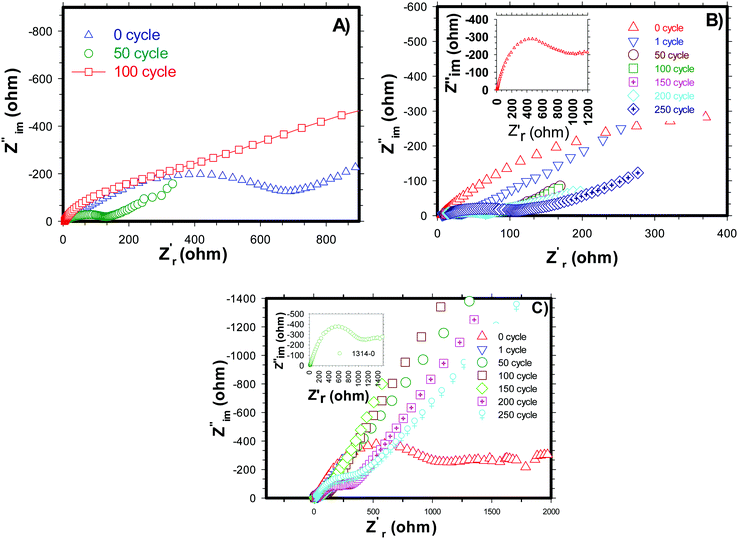 |
| | Fig. 9 Impedance spectra of (A) H-SnO2, (B) non-conventional carbon coated (NCC-4) H-SnO2 and (C) conventional carbon coated (CC) H-SnO2. | |
4. Conclusions
In summary, porous H-SnO2 nanoparticles with excellent electrochemical properties were synthesized via a surfactant assisted hydrothermal method. In the present study, a unique synthesis technique was successfully developed using a hydrazine hydrate assisted surfactant method to create a mesoporous structure along with a novel carbon coating process to improve the electronic conductivity and to buffer volume expansion. The effects of calcination temperature and carbon coating conditions were studied in depth and it was observed that H-SnO2 nanoparticles obtained after calcination at 400 °C represent an anode material with desirable electrochemical properties. It was also observed that carbon coating carried out using the NCC-4 process resulted in a graphitic and homogenous carbon layer around H-SnO2 nanoparticles in comparison with H-SnO2 nanoparticles carbon-coated using the CC process. This shows the influence of the dehydrating agent in creating catenation of carbon atoms and in forming uniform carbon coating around the active material. The carbon coated H-SnO2-NCC-4- 400 °C delivered capacities of 454, 292 and 129 mA h g−1 at 5C, 8C and 10C, respectively, with excellent stability up to 250 cycles. The improved electrochemical performance can be attributed to (a) the mesoporosity which can improve the surface area resulting in improved reaction sites, (b) uniform and graphitic carbon coating leading to improved electronic conductivity, and (c) oxygen vacancies which maintain the reversibility of SnO2 while inhibiting Sn coarsening during cycling. These unique properties effectively improve electronic conductivity, leading to excellent lithiation/de-lithiation, buffering volume expansion and excellent electrochemical stability. The synthesis process developed in the present study was accompanied by a unique non-conventional carbon coating technique which efficiently improved the electrochemical properties of the SnO2 anode by overcoming the major existing drawbacks of the SnO2 anode material. The porous structure accompanied by uniform graphitic carbon layer encapsulation of SnO2 nanoparticles not only improved the electronic conductivity and buffered the volume expansion, but also improved the rate capability and cyclic stability. With all the unique properties of the material developed in the present study, it may be considered as a promising next generation high-capacity anode electrode, especially at higher current rates, for LIBs in EV or HEV applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to Dr G. Sundararajan, former director, and Dr G. Padmanabham, director, ARCI for their consistent support and encouragement in carrying out this work. The authors highly acknowledge the financial support from ARCI-Technical Research Centre {Ref. No. AI/1/65/ARCI/2014 (c)} funded by the Department of Science and Technology (DST), Government of India.
References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scorasti, J.-M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–375 CrossRef PubMed.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- D. Larcher, S. Beattie, M. Morcrette, K. Edström, J. C. Jumas and J. M. Tarascon, J. Mater. Chem., 2007, 17, 3759–3772 RSC.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395–1397 CrossRef CAS.
- M. Yoshio, S. Kugino and N. Dimov, J. Power Sources, 2006, 153, 375–379 CrossRef CAS.
- D. Vrankovic, M. Graczyk-Zajac, C. Kalcher, J. Rohrer, M. Becker, C. Stabler, G. Trykowski, K. Albe and R. Riedel, ACS Nano, 2017, 11, 11409–11416 CrossRef CAS PubMed.
- A. M. Chockla, K. C. Klavetter, C. B. Mullins and B. A. Korgel, ACS Appl. Mater. Interfaces, 2012, 4, 4658–4664 CrossRef CAS PubMed.
- M. V. Reddy, T. Yu, C. H. Sow, Z. X. Shen, C. T. Lim, G. V. S. Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792–2799 CrossRef CAS.
- Y. Jiang, Z.-J. Jiang, L. Yang, S. Cheng and M. Liu, J. Mater. Chem. A, 2015, 3, 11847–11856 RSC.
- Y. Yao, J. Zhang, T. Huang, H. Mao and A. Yu, Int. J. Electrochem. Sci., 2013, 8, 3302–3309 CAS.
- H. X. Zhang, C. Feng, Y. C. Zhai, K. L. Jiang, Q. Q. Li and S. S. Fan, Adv. Mater., 2009, 21, 2299–2304 CrossRef CAS.
- J. Fan, T. Wang, C. Yu, B. Tu, Z. Jiang and D. Zhao, Adv. Mater., 2004, 16, 1432–1436 CrossRef CAS.
- X. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef CAS PubMed.
- J. Mei, T. Liao and Z. Sun, J. Energy Chem., 2018, 27, 117–127 CrossRef.
- Z. Chen, M. Zhou, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2012, 2, 95–102 CrossRef CAS.
- M. Han, Y. Mu and J. Yu, Mater. Adv., 2020, 1, 421–429 RSC.
- I. Elizabeth, R. B. Mathur, P. H. Maheshwari, B. P. Singh and S. Gopukumar, Electrochim. Acta, 2015, 176, 735–742 CrossRef CAS.
- L. Pan, Y. Zhang, F. Lu, Y. Du, Z. Lu, Y. Yang, T. Ye, Q. Liang, Y. Bando and X. Wang, Energy Storage Mater., 2019, 19, 39–47 CrossRef.
- H. S. Kim, J. B. Cook, H. Lin, J. S. Ko, S. H. Tolbert, V. Ozolins and B. Dunn, Nat. Mater., 2016, 16, 454–460 CrossRef PubMed.
- W. Dong, J. Xu, C. Wang, Y. Lu, X. Liu, X. Wang, X. Yuan, Z. Wang, T. Lin, M. Sui, I. W. Chen and F. Huang, Adv. Mater., 2017, 29, 1700136 CrossRef PubMed.
- N. Li, K. Du, G. Liu, Y. Xie, G. Zhou, J. Zhu, F. Li and H. M. Cheng, J. Mater. Chem. A, 2013, 1, 1536–1539 RSC.
- R. Hu, Y. Ouyang, D. Chen, H. Wang, Y. Chen, M. Zhu and M. Liu, Acta Mater., 2016, 109, 248–258 CrossRef CAS.
- X. Yu, S. Yang, B. Zhang, D. Shao, X. Dong, Y. Fang, Z. Li and H. Wang, J. Mater. Chem., 2011, 21, 12295–12302 RSC.
- J. S. Chen, Y. L. Cheah, Y. T. Chen, N. Jayaprakash, S. Madhavi, Y. H. Yang and X. W. Lou, J. Phys. Chem. C, 2009, 113, 20504–20508 CrossRef CAS.
- P. M. Pratheeksha, E. H. Mohan, B. V. Sarada, M. Ramakrishna, K. Hembram, P. V. V. Srinivas, P. J. Daniel, T. N. Rao and S. Anandan, Phys. Chem. Chem. Phys., 2017, 8, 175–188 RSC.
- P. Wu, N. Du, H. Zhang, C. Zhai and D. Yang, ACS Appl. Mater. Interfaces, 2011, 3, 1946–1952 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- Q. Gao, F. Qu, W. Zheng and H. Lin, J. Porous Mater., 2013, 20, 983–988 CrossRef CAS.
- E. Bekyarova, S. Sarkar, F. Wang, M. E. Itkis, I. Kalinina, X. Tian and R. C. Haddon, Acc. Chem. Res., 2013, 46, 65–76 CrossRef CAS PubMed.
- S. Nam, S. Kim, S. Wi, H. Choi, S. Byun, S.-M. Choi, S.-I. Yoo, K. T. Lee and B. Park, J. Power Sources, 2012, 211, 154–160 CrossRef CAS.
- M. Winter and J. O. Besenhard, Electrochim. Acta, 1999, 45, 31–50 CrossRef CAS.
- T. Moon, C. Kim, S.-T. Hwang and B. Park, Electrochem. Solid-State Lett., 2006, 9, A408–A411 CrossRef CAS.
- B. Nandan, B. Venugopal, S. Amirthapandian, B. Panigrahi and P. Thangadurai, J. Nanopart. Res., 2013, 15, 1–11 CrossRef.
- K. Kravchyk, L. Protesescu, M. I. Bodnarchuk, F. Krumeich, M. Yarema, M. Walter, C. Guntlin and M. V. Kovalenko, J. Am. Chem. Soc., 2013, 135, 4199–4202 CrossRef CAS PubMed.
- R. Hu, Y. Ouyang, T. Liang, X. Tang, B. Yuan, J. Liu, L. Zhang, L. Yang and M. Zhu, Energy Environ. Sci., 2017, 10, 2017–2029 RSC.
- C. J. Kevane, Phys. Rev., 1964, 133, A1431–A1436 CrossRef.
- Y. Wang, F. Su, J. Y. Lee and X. S. Zhao, Chem. Mater., 2006, 18, 1347–1353 CrossRef CAS.
- A.
K. Singh, A. Janotti, M. Scheffler and C. G. Van De Walle, Phys. Rev. Lett., 2008, 101, 1–4 Search PubMed.
- B. Venugopal, B. Nandan, A. Ayyachamy, V. Balaji, S. Amirthapandian, B. K. Panigrahi and P. Thangadurai, RSC Adv., 2014, 4, 6141–6150 RSC.
- Y. Zou and Y. Wang, ACS Nano, 2011, 5, 8108–8114 CrossRef CAS PubMed.
- J. Liang, Z. Cai, Y. Tian, L. Li, J. Geng and L. Guo, ACS Appl. Mater. Interfaces, 2013, 5, 12148–12155 CrossRef CAS PubMed.
- B. Huang, X. Li, Y. Pei, S. Li, X. Cao, R. C. Massé and G. Cao, Small, 2016, 12, 1945–1955 CrossRef CAS PubMed.
- S. Shi, T. Deng, M. Zhang and G. Yang, Electrochim. Acta, 2017, 246, 1104–1111 CrossRef CAS.
- M. Gao, X. Chen, H. Pan, L. Xiang, F. Wu and Y. Liu, Electrochim. Acta, 2010, 55, 9067–9074 CrossRef CAS.
- S. Li, W. Xie, S. Wang, X. Jiang, S. Peng and D. He, J. Mater. Chem. A, 2014, 2, 17139–17145 RSC.
- R. Hu, Y. Ouyang, D. Chen, H. Wang, Y. Chen, M. Zhu and M. Liu, Acta Mater., 2016, 109, 248–258 CrossRef CAS.
- N. Wu, W. Du, X. Gao, L. Zhao, G. Liu, X. Liu, H. Wu and Y.-B. He, Nanoscale, 2018, 10, 11460–11466 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00453k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Parakandy Muzhikara
Pratheeksha‡
ac,
Khasim Saheb
Bayikadi
d,
Pavan Srinivas
Veluri
a,
Mantripragada Rama
Krishna
e,
Bulusu Venkata
Sarada
f,
Tata Narasinga
Rao
a,
Paul Joseph
Daniel
c and
Srinivasan
Anandan
*a
*ab,
Parakandy Muzhikara
Pratheeksha‡
ac,
Khasim Saheb
Bayikadi
d,
Pavan Srinivas
Veluri
a,
Mantripragada Rama
Krishna
e,
Bulusu Venkata
Sarada
f,
Tata Narasinga
Rao
a,
Paul Joseph
Daniel
c and
Srinivasan
Anandan
*a