
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electroreduction of carbon dioxide to formate using highly efficient bimetallic Sn–Pd aerogels†
Maryam
Abdinejad‡
 *a,
Mozhgan Khorasani
Motlagh‡
b,
Meissam
Noroozifar
b and
Heinz Bernhard
Kraatz
*bc
*a,
Mozhgan Khorasani
Motlagh‡
b,
Meissam
Noroozifar
b and
Heinz Bernhard
Kraatz
*bc
aDepartment of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: M.Abdinejad@tudelft.nl
bDepartment of Physical and Environmental Sciences, University of Toronto Scarborough, 1265 Military Trail, Toronto, ON M1C 1A4, Canada. E-mail: bernie.kraatz@utoronto.ca
cDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON M5S 3H6, Canada
First published on 9th December 2021
Abstract
Electrochemical reduction of carbon dioxide (CO2) to valuable materials is a promising approach to suppress atmospheric CO2 levels. In order to bring this strategy to a commercial scale, the design of efficient, cost-effective, and robust catalysts is essential. Current advances in CO2 conversion technology use bimetallic components that enhance electrocatalysis via the introduction of binding site diversity. In this work, Sn–Pd bimetallic aerogels supported by carbon nanotubes (Sn–Pd/CNT) demonstrate selective electroreduction of CO2 to formate in ambient conditions. Amino substituents were introduced as an additional CO2 capture site (Sn–Pd/CNT–NH2), further enhancing the electrocatalytic activity and resulting in 91% formate selectively and a current density of −39 mA cm−2 at −0.4 V vs. RHE. The results demonstrate the potential of alloying Sn with other earth-abundant metals to promote the electrochemical conversion of CO2 to value-added materials. We believe this study provides valuable insights into the intricate relationship of bimetallic aerogels and shows the potential of the –NH2 group as a facilitator for CO2 capture and conversion that will inspire new forays into the development of competitive catalytic systems.
Introduction
Increasing carbon dioxide (CO2) levels in the atmosphere are considered as a major contributor to climate change.1 Electrocatalytic carbon dioxide reduction reactions (CO2RRs) to value added materials such as formic acid (HCOOH), carbon monoxide (CO), methane (CH4), methanol (CH3OH), and ethanol (C2H5OH), have drawn significant attention to mitigate climate change.2–8 Formate, in particular, is valued as an ideal candidate for hydrogen storage and fuel cell processes.9,10 However, due to the complexity of its multi-electron reaction pathway and competition with hydrogen evolution reactions (HERs), the reaction is sluggish and hindered by a large activation overpotential.11There has been considerable progress on improving electrocatalytic activity for CO2 reduction by introducing binding site diversity.12 Although various metals, such as Pd,13 Pb,14 Bi,15,16 Sn,17–19 Ag,20 and In21 themselves demonstrate high selectivity for formate production, recent reports show that bimetallic alloys of these metals can increase catalytic activity even further.22,23 Additionally, Sn-based24–27 and Pd-based28,29 bimetallic catalysts are superior in several aspects where monometallic catalysts are lacking: in reducing large overpotentials and improving surface stability towards CO2RR.30–34
In addition to tailoring the elemental composition affect catalytic performance, increasing the catalytically active surface area is another strategy that can be used to enhance current density.35,36 To this end, geometric factors such as metal dispersion, surface morphology, and porosity play a key role.37 Aerogels stand out among similar porous structural materials in facilitating electron transport and optimizing catalytically active surface area.38–45 Owing to their porous and three-dimensional (3D) structure, they can dramatically enhance mass transport and facilitate CO2RR.46–48 Liu et al.49 have reported a Sn nanoparticle–modified 3D carbon nanotube (CNT) aerogel whose unique porous, hierarchical structure was instrumental in increasing current density and effectively enhancing the selectivity of formate production. These studies stress the import of morphology control on the performance of Sn-based catalysts for the CO2RR.50
Knowing that Pd surface is poisoned by CO,51 in this project, we took an advantage of Sn surface to enhance the adsorption of CO2, but weakened the CO binding on Pd, and have designed a series of bimetallic Sn–Pd aerogels, to improve their stability and overall catalytic performance through the synergy between Pd and Sn surface for electroreduction of CO2. Aerogels of these bimetallic systems were employed to maximize the surface area of the catalytic layer through the creation of a highly porous nanostructure.45,48 Building on previous works, carbon nanotubes (CNTs) were incorporated throughout the aerogel to enhance the electron conductivity of the structure (Sn–Pd/CNT).52 Amine groups have been shown to have a high capability of capturing CO2,53–55 and are integrated into our design to serve as an additional CO2 capture site and localized proton source (Sn–Pd/CNT–NH2). Using this design, we could successfully reduce CO2 to formate with 91% selectivity and current density of −39 mA cm−2 at −0.4 V vs. RHE. Comparative studies of these bimetallic systems with their monometallic Sn and Pd aerogels counterparts will be discussed in this work.
Experimental
A series of 3D Pd, Sn–Pd, Sn–Pd–CNT, and Sn–Pd–CNT–NH2 aerogels were synthesized through a one-pot and surfactant-free technique by reducing H2PdCl4 and SnCl2 with glyoxylic acid using lyophilization (ESI† Part 3).48 The spongy, porous morphology characteristic of aerogels was visualized using Scanning Electron Microscopy (SEM) (Fig. 1a, and Fig. S1, ESI†). The uniform distribution of atoms within the aerogel's structure affirms a homogeneous distribution of corresponding Pd, Sn, N, and C components (Fig. 1b–e). Transmission electron microscopy (TEM) images highlight the structural presence of CNTs in the case of the Sn–Pd/CNT–NH2 aerogel (Fig. S2, ESI†). | ||
| Fig. 1 (a) Scanning Electron Microscopy (SEM) of Sn–Pd–CNT–NH2 aerogel with a scale bar of 10 μm. (b–e) Atom distribution of Sn, Pd, N, and C, respectively. | ||
X-Ray photoelectron spectroscopy (XPS) gives insight into the chemical environments of the bimetallic material, where the two characteristic peaks with the binding energy of 335 eV and 340 eV correspond to Pd 3d3/2 and Pd 3d5/2 and peaks at 486.1 and 492.5 eV are attributed to Sn 3d5/2 and Sn 3d3/2, respectively (Fig. 2a, b, and Fig. S3–S9, ESI†). In the presence of CNTs, a slight upward shift in the binding energy could be indicative of a highly dispersive and synergetic relationship between the two materials.56,57 The Sn–Pd/CNT–NH2 aerogel, was characterized further by X-ray diffraction (XRD) (Fig. S10, ESI†). Peaks seen at 40.1°, 46.6°, 68.1°, and 82.1° are characteristic diffraction peaks of Pd crystals; whereas, peaks at 26.5° and 34.4° belong to CNT58 and Sn which confirm the successful formation of the aerogel. These results are in agreement with previous reports.25,59,60
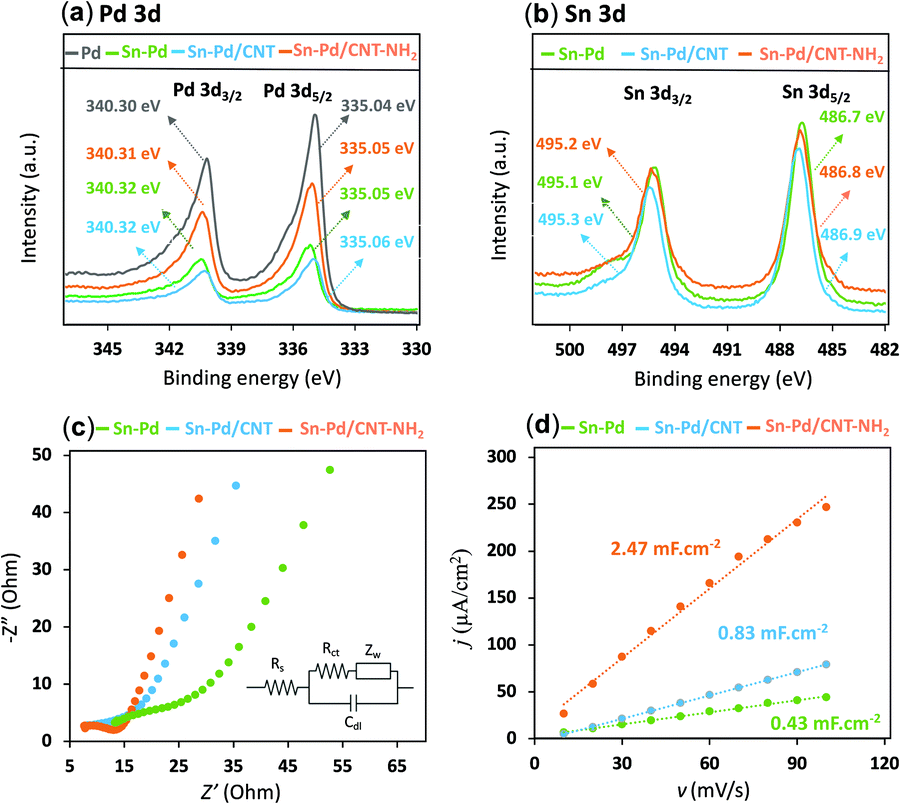 | ||
| Fig. 2 X-ray photoelectron spectroscopy (XPS) spectra survey comparison of: (a) Pd 3d spectra of Pd, Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 aerogels; (b) Sn 3d spectra of Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 aerogels. (c) Electrochemical impedance spectroscopy (EIS) of Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 in saturated solution with CO2 and 0.1 M KHCO3 with amplitude of 5 mV in a frequency range from 10 kHz to 0.1 Hz. (d) Current density difference between cathodic and anodic sweeps at −0.44 V vs. scan rates in 2.5 mM [Fe(CN)6]3−/4− and 0.2 M KNO3. | ||
The nature of the charge-transfer resistance in the presence and absence of CNTs was investigated using electrochemical impedance spectroscopy (EIS) (Fig. 2c). The semicircular arcs at the high-frequency regions of the Nyquist plots are characteristic of a capacitance double layer between the electrolyte and the electrode interface; which reflects the charge transfer resistance of an electrochemical reaction (Rct).61 The values of Rct from equivalent circuit fitting for Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 are an estimated 23.1, 17.9, and 8.7 Ω, respectively. The lower Rct in Sn–Pd/CNT and Sn–Pd/CNT–NH2 compared to Sn–Pd underscore the significant improvement to charge transfer and electrical conductivity the CNT and CNT–NH2 offer.62,63
The electronic structure of Sn–Pd-based catalysts has been reported previously. The bimetallic Pd–Sn alloys have a unique electronic structure similar to that of noble metals that forms when rehybridization of the Sn sp-orbitals and Pd d-orbitals occurs.64–66 In the case of Sn–Pd/CNT and Sn–Pd/CNT–NH2, the π-orbitals of CNTs can weakly bond with Sn–Pd through hybridization between the carbon π-orbitals and the high energy d-orbitals of the metals;67,68 strengthening the electronic structure and thereby facilitating charge transfer. To further examine the charge transfer of the synthesized aerogels, the standard exchange current density (i0) was calculated using the following equation (eqn (1)):69
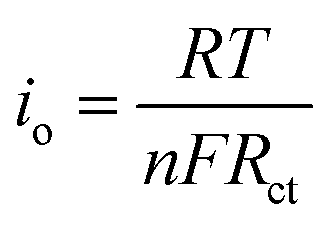 | (1) |
To shed light on the influence of CNTs and –NH2 on surface area activity, it is important to quantify the electrochemically active surface area (ECSA) of the modified electrode surfaces as described in ESI,† Part 4 (Fig. S11, ESI†).71 The Cdl was estimated by plotting the j (μA cm−2) at −0.44 V vs. Ag/AgCl against the scan rate (mV s−1) and it is twice the slopes of linear fit (Fig. 2d). The roughness factor (Rf) of the electrodes can be calculated using eqn (2):72,73
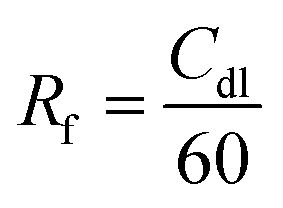 | (2) |
| ECSA = Rf × Selectrode | (3) |
Combining with the CVs results in Fig. S11 (ESI†) and actual catalyst load on glassy carbon electrode, the ECSA for Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 was calculated as 0.25, 0.49, and 1.46 cm2, respectively. The higher ECSA for the Sn–Pd/CNT–NH2 indicates its larger catalytic activity compared to Sn–Pd and Sn–Pd/CNT. The higher ESCA of Sn–Pd/CNT compared to Sn–Pd could be due to the greater charge transfer that CNT facilitates between the aerogel and the electrode surface.74–77 Further improvement to the ESCA is seen in the case of Sn–Pd/CNT–NH2, which can be due to enhanced electron–proton transfer of the −NH2 groups.57
Results and discussion
To evaluate the electroactivity of the synthesized complexes, electrochemical reduction of CO2 was studied with linear sweep voltammetry (LSV) in an aqueous 0.1 M KHCO3 solution. A two-compartment electrochemical H-cell comprised of a three-electrode system with immobilized aerogels on a glassy carbon working electrode (GCE), platinum counter electrode, and a silver/silver chloride (Ag/AgCl) reference electrode.78 To prepare the working electrodes, 10 μmol of the synthesized Pd–Sn aerogels were immobilized onto a 0.3 cm diameter GCE with a catalyst loading concentration of 1.57 × 10−7 mol cm−2 relative to the geometrical electrode area (0.071 cm2).55 To incorporate the CNTs, Sn–Pd and CNTs were dissolved in a DMF solution and sonicated for 20 minutes to form a homogeneous suspension of Sn–Pd/CNT and Sn–Pd/CNT–NH2.48Fig. 3a compares the LSV of bare CNT and CNT–NH2 with the immobilized aerogels Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 onto GCE in 0.1 M KHCO3 in the presence of CO2. Exposure of the aerogels to CO2 resulted in a dramatic increase to the current density beginning at −0.67, −0.42, and −0.38, V vs. RHE for compounds Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2, respectively, which could be due to either electroreduction of CO2 or HER (Fig. S12, ESI†). Comparing pristine CNT with CNT–NH2 in Fig. 3a finds a higher current density and lower potential in the presence of –NH2. This observation aligns with what has been reported previously.54 Achieving the highest catalytic activity with the lowest overpotential in the case of Sn–Pd/CNT–NH2 further highlights the to the role of –NH2 in capturing CO2.
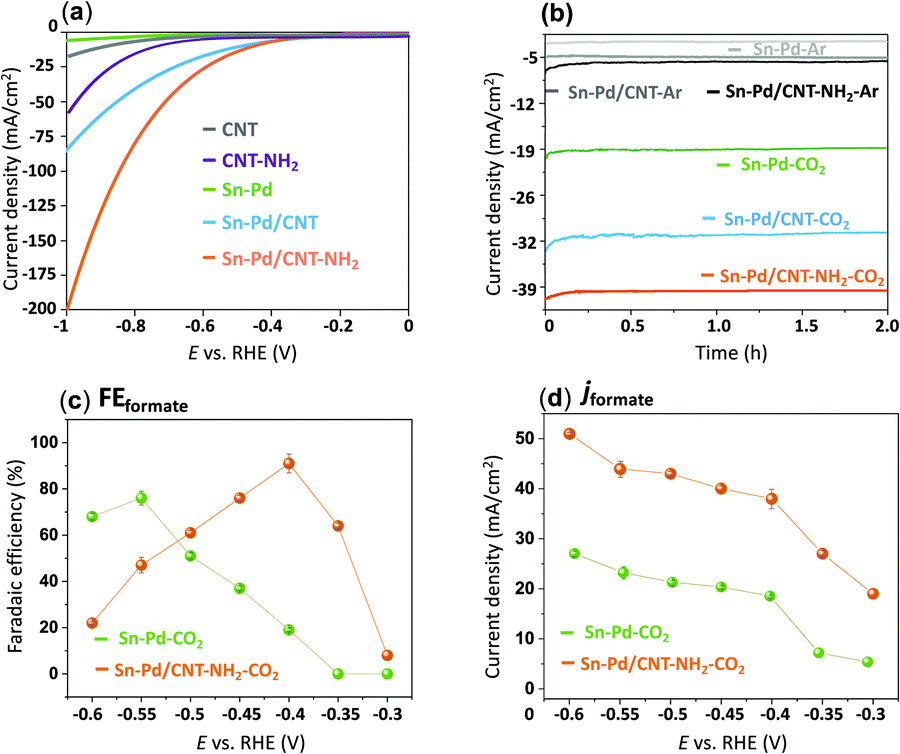 | ||
| Fig. 3 (a) Linear sweep voltammetry (LSV) comparison of bare CNT, bare CNT–NH2, Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 in 0.1 M KHCO3; (b) chronoamperometry comparison of Sn–Pd, Sn–Pd/CNT, Sn–Pd/CNT–NH2 in 0.1 M KHCO3 at −0.4 V vs. RHE. (c) Faradaic efficiency (FEformate), and (d) current density (jformate) comparison of Sn–Pd, and Sn–Pd/CNT–NH2 at −0.30, −0.35, −0.4, −0.45, −0.5, −0.55 and −0.6 V vs. RHE in 0.1 M KHCO3. | ||
The proficiency of the bimetallic Sn–Pds towards CO2 electroreduction was evaluated under chronoamperometric conditions. The experiments were conducted at −0.3, −0.35, −0.4, −0.45, −0.5, −0.55 and −0.6 V vs. RHE in 0.1 M KHCO3 for 2 h under sealed conditions with vigorous magnetic stirring. Formate and H2 were detected as the sole products of the electrochemical CO2RR after gas chromatography (GC) sampling of the reaction headspace and 1H NMR and HPLC analysis of the liquid solution. As shown in Fig. S14–S16 (ESI†), the product distribution is strongly dependent on the applied potential. A greater decrease in FEformate was seen at higher overpotentials in competition with an uptick in HERs.54
The optimal overpotential for Sn–Pd was found to be −0.55 V vs. RHE, and much lower optimal potential of −0.4 V vs. RHE was observed in the case of Sn–Pd–CNT and Sn–Pd/CNT–NH2 (Fig. 3b). This could be due to the support of the CNT scaffold, and –NH2 group which works to both improve conductivity and CO2 capture throughout the catalytic layer. Introducing amino groups, such as in Pd/CNT–NH2, served dual purposes as a localized proton source and as an additional CO2 capture site, facilitate even higher catalytic activity and selectivity. To confirm these phenomena and important role of CNT and –NH2 in the CO2RR, the FEformate and jformate of Sn–Pd with Sn–Pd/CNT–NH2 was compared in Fig. 3c and d at the potentials of −0.3 V to −0.6 vs. RHE in 0.1 M KHCO3. It clearly indicates both product selectivity and catalytic activity have been enhanced ∼2-folds at the optimal potential of −0.4 V vs. RHE in the case of Sn–Pd/CNT–NH2.
Fig. 4a demonstrates the current densities and selectivities of both Sn–Pd/CNT (FEformate: 83%, jformate: −29.6 mA cm−2), and Sn–Pd/CNT–NH2 (FEformate: 91%, jformate: −39.1 mA cm−2) at −0.4 V vs. RHE were significantly higher than that of Sn–Pd (FEformate: 19%, jformate: −18.2 mA cm−2). The long-term, operational stability of Sn–Pd/CNT–NH2 was monitored continuously for 4 h in KHCO3 at −0.4 V vs. RHE (Fig. 4b). The current density (−39 mA cm−2) was maintained for the entire duration and FEformate remained constant over 90%. No significant decrease in current density and product selectivity was observed during this period SEM (Fig. 4c), atomic force microscopy (AFM) (Fig. 4d), and XPS (Fig. S17, ESI†) found no significant alterations in surface morphology before and after long-term electrolysis. This phenomenon was further examined and discussed using FTIR and described in detail in the ESI† (Fig. S18).
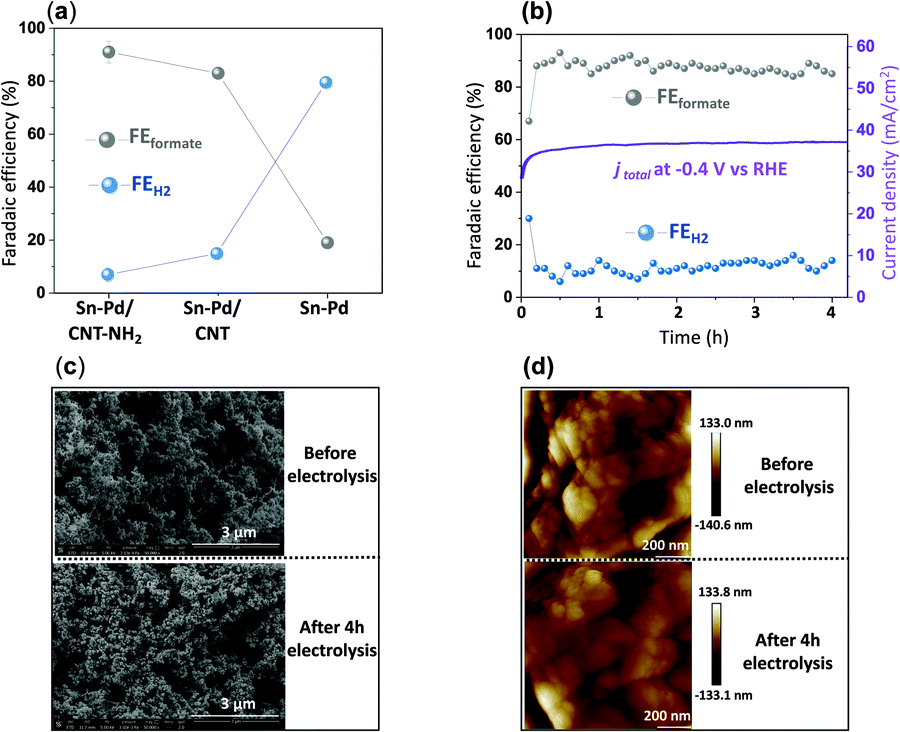 | ||
| Fig. 4 (a) Faradaic efficiency (FE) and current density (j) comparison of Sn–Pd, Sn–Pd/CNT, and Sn–Pd/CNT–NH2 at −0.4 V vs. RHE in 0.1 M KHCO3; (b) long-term stability studies of Sn–Pd/CNT–NH2 for CO2 electroreduction operated in 0.1 M KHCO3 at −0.4 V vs. RHE for 4 h; (c) SEM, and (d) AFM comparison of Sn–Pd/CNT–NH2 before and after 4 hours electrochemical CO2RR. | ||
The ability of the Sn–Pd/CNT–NH2 aerogel to capture and adsorb CO2 was measured at varying temperatures based on the weight change of the aerogel before and after the introduction of CO2.79 Sn–Pd/CNT–NH2 (12 mg) was heated to 110 °C in a sealed chamber under N2 with a flow rate of 40 sccm to remove any air or existing moisture. After applying CO2, the weight change of the sample during adsorption and desorption process was calculated every 2 min at 25 °C under CO2 with a flow rate of 40 sccm. The CO2 uptake per mole of Sn–Pd/CNT–NH2 slowly increased over time, with a maximum uptake of 6.89 mmol g−1 over 48 minutes (Fig. S19a, ESI†). Next, the adsorption of the aerogel was measured at several temperatures under CO2 atmosphere (Fig. S19b, ESI†). CO2 uptake improved with increasing temperatures, reaching a maximum of 7.82 mmol g−1 at 75 °C. The stability of the aerogel was examined over several adsorption/desorption cycles at 75 °C and no dramatic change in the level of adsorption was observed.
To obtain insight on the CO2 electroreduction reaction mechanism, Tafel slopes were calculated and performed in Fig. S20 (ESI†). The slopes were measured to be 97, 84, and 82 mV dec−1, for Sn–Pd, Sn–Pd/CNT and Sn–Pd/CNT–NH2, respectively. Achieving smaller Tafel slope values in the case of Sn–Pd/CNT–NH2, suggest that less driving force (overpotential) is required to improve the reaction rate,80,81 which is also in agreement with previous reports on the bimetallic Sn–Pd alloy which shown to be highly dependent on the catalysts’ surface electronic properties.82 The achieved Tafel values are close to the theoretical value for a rapid one-electron transfer step followed by a rate-determining step (RDS).25 Reducing CO2 to formate at such low overpotential,25,83 suggest that *OCHO is the key intermediate for the CO2RR electroreduction.84 The Sn's ideal *OCHO binding energy supports this phenomenon. These results are also consistent with previous reports on observing *OCHO on Sn-based electrodes using spectroscopic techniques.85,86
In summary, using porous, bimetallic Sn–Pd-based aerogels paired with CNT–NH2, we could significantly increase their catalytic efficiency and product selectivity compared to their monometallic analogues, highlighting the importance of synergistic binding site diversity between Sn and Pd (Table S1, ESI†). To the best of our knowledge, this is one of the lowest overpotentials that has been reported for bimetallic compounds for CO2RR applications (Table S2, ESI†).30,34,82,87–98 The essential role of the –NH2 group in acting as an additional CO2 capture site and as an extra proton source played a significant role in increasing the catalytic activity and selectivity of this system.99,100 The remarkable catalytic performance of the Sn–Pd aerogels reported herein make it a promising candidate for CO2RR on a large scale.
Conclusions
In conclusion, we have synthesized and characterized a new class of Pd–Sn aerogels outfitted with CNTs and –NH2 groups. In this work, the introduction of binding site diversity increased the current density ∼36-fold higher than its monometallic constituents. The exceptional catalytic performance achieved with Sn–Pd/CNT–NH2 demonstrates high selectivity (FEformate: 91%), current density (jformate: −39.1 mA cm−2), and stability at low overpotential of 0.4 V. The potential for bimetallic tunability and the unique 3D structure of aerogels facilitated greater CO2 capture and conversion and makes them an attractive class of electrocatalysts for electroreduction of CO2 to value-added materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC-DG-2016-06122), and the University of Toronto. We gratefully acknowledge Dr Tom Burdyny and Marcel Bus at the Delft University of Technology for their support on this work.Notes and references
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. B. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS PubMed.
- M. Usman, M. Humayun, M. D. Garba, L. Ullah, Z. Zeb, A. Helal, M. H. Suliman, B. Y. Alfaifi, N. Iqbal, M. Abdinejad, A. A. Tahir and H. Ullah, Nanomater., 2021, 11 Search PubMed.
- J. He, C. Wu, Y. Li and C. Li, J. Mater. Chem. A, 2021, 9, 19508–19533 RSC.
- M. Abdinejad, C. Dao, B. Deng, F. Dinic, O. Voznyy, X. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 9549–9557 CrossRef CAS.
- M. Abdinejad, M. N. Hossain and H.-B. Kraatz, RSC Adv., 2020, 10, 38013–38023 RSC.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- D. Lei, Q. Qing, W. Pei, Z. Xiaojing, H. Chengyi, L. Pengxin, Q. Ruixuan, C. Mei, O. Daohui, X. Chaofa, M. Shiguang, W. Binghui, F. Gang, Z. Peng and Z. Nanfeng, Sci. Adv., 2021, 3, e1701069 Search PubMed.
- M. Abdinejad, L. F. B. Wilm, F. Dielmann and H. B. Kraatz, ACS Sustainable Chem. Eng., 2021, 9, 521–530 CrossRef CAS.
- S. A. Al-Tamreh, M. H. Ibrahim, M. H. El-Naas, J. Vaes, D. Pant, A. Benamor and A. Amhamed, ChemElectroChem, 2021, 8, 3207–3220 CrossRef CAS.
- Y. Ling, Q. Ma, Y. Yu and B. Zhang, Trans. Tianjin Univ., 2021, 27, 180–200 CrossRef CAS.
- P. Ding, H. Zhao, T. Li, Y. Luo, G. Fan, G. Chen, S. Gao, X. Shi, S. Lu and X. Sun, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- B. Jiang, X.-G. Zhang, K. Jiang, D.-Y. Wu and W.-B. Cai, J. Am. Chem. Soc., 2018, 140, 2880–2889 CrossRef CAS PubMed.
- N. Zouaoui, B. D. Ossonon, M. Fan, D. Mayilukila, S. Garbarino, G. de Silveira, G. A. Botton, D. Guay and A. C. Tavares, J. Mater. Chem. A, 2019, 7, 11272–11281 RSC.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2020, 10, 743–750 CrossRef CAS.
- S. Liu, X. F. Lu, J. Xiao, X. Wang and X. W. (David) Lou, Angew. Chem., Int. Ed., 2019, 58, 13828–13833 CrossRef CAS PubMed.
- L. Lu, X. Sun, J. Ma, Q. Zhu, C. Wu, D. Yang and B. Han, Sci. China: Chem., 2018, 61, 228–235 CrossRef CAS.
- S. Liu, J. Xiao, X. F. Lu, J. Wang, X. Wang and X. W. (David) Lou, Angew. Chem., Int. Ed., 2019, 58, 8499–8503 CrossRef CAS PubMed.
- Yiliguma, Z. Wang, C. Yang, A. Guan, L. Shang, A. M. Al-Enizi, L. Zhang and G. Zheng, J. Mater. Chem. A, 2018, 6, 20121–20127 RSC.
- M. Abdinejad, I. Santos da Silva and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 9791–9797 RSC.
- W. Luo, W. Xie, M. Li, J. Zhang and A. Züttel, J. Mater. Chem. A, 2019, 7, 4505–4515 RSC.
- W. Zhu, B. M. Tackett, J. G. Chen and F. Jiao, Top. Curr. Chem., 2018, 376, 41 CrossRef PubMed.
- L. Xie, J. Liang, C. Priest, T. Wang, D. Ding, G. Wu and Q. Li, Chem. Commun., 2021, 57, 1839–1854 RSC.
- X. An, S. Li, A. Yoshida, Z. Wang, X. Hao, A. Abudula and G. Guan, ACS Sustainable Chem. Eng., 2019, 7, 9360–9368 CrossRef CAS.
- S. Zhao, S. Li, T. Guo, S. Zhang, J. Wang, Y. Wu and Y. Chen, Nano-Micro Lett., 2019, 11, 62 CrossRef PubMed.
- F. Cheng, X. Zhang, K. Mu, X. Ma, M. Jiao, Z. Wang, P. Limpachanangkul, B. Chalermsinsuwan, Y. Gao, Y. Li, Z. Chen and L. Liu, Energy Technol., 2021, 9, 2000799 CrossRef CAS.
- Y. Wei, J. Liu, F. Cheng and J. Chen, J. Mater. Chem. A, 2019, 7, 19651–19656 RSC.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C. T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283–286 CrossRef CAS.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS PubMed.
- C. H. Lee and M. W. Kanan, ACS Catal., 2015, 5, 465–469 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- X. Zheng, Y. Ji, J. Tang, J. Wang, B. Liu, H.-G. Steinrück, K. Lim, Y. Li, M. F. Toney, K. Chan and Y. Cui, Nat. Catal., 2019, 2, 55–61 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- N. Chaoui, M. Trunk, R. Dawson, J. Schmidt and A. Thomas, Chem. Soc. Rev., 2017, 46, 3302–3321 RSC.
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
- H. Ge, Z. Gu, P. Han, H. Shen, A. M. Al-Enizi, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2018, 531, 564–569 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4266 CrossRef CAS PubMed.
- R. Du, W. Jin, H. Wu, R. Hübner, L. Zhou, G. Xue, Y. Hu and A. Eychmüller, J. Mater. Chem. A, 2021, 9, 17189–17197 RSC.
- C. Ziegler, A. Wolf, W. Liu, A.-K. Herrmann, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2017, 56, 13200–13221 CrossRef CAS PubMed.
- X. Jiang, R. Du, R. Hübner, Y. Hu and A. Eychmüller, Matter, 2021, 4, 54–94 CrossRef CAS.
- R. Du, X. Jin, R. Hübner, X. Fan, Y. Hu and A. Eychmüller, Adv. Energy Mater., 2020, 10, 1901945 CrossRef CAS.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed.
- M. Wang, B. Zhang, J. Ding, N. Xu, M. T. Bernards, Y. He and Y. Shi, ACS Sustainable Chem. Eng., 2020, 8, 4983–4994 CrossRef CAS.
- M. Abdinejad, C. Ferrag, M. N. Hossain, M. Noroozifar, K. Kerman and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 12870–12877 RSC.
- Z. Chen, S. Yao and L. Liu, J. Mater. Chem. A, 2017, 5, 24651–24656 RSC.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W.-B. Cai, X. Chen, R. Si, F. Yang, S. Miao, J. Wang, G. Wang and X. Bao, Nano Res., 2017, 10, 2181–2191 CrossRef CAS.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X. A. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- M. Abdinejad, Z. Mirza, X. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- M. Abdinejad, C. Dao, X. Zhang and H. B. Kraatz, J. Energy Chem., 2021, 58, 162–169 CrossRef.
- D. Wu, X. Chen, S. Lu, Y. Liang, F. Xu and R. Fu, Microporous Mesoporous Mater., 2010, 131, 261–264 CrossRef CAS.
- N. Roy, A. Ejaz and S. Jeon, Appl. Surf. Sci., 2021, 151973 Search PubMed.
- M. Terrones, W. K. Hsu, A. Schilder, H. Terrones, N. Grobert, J. P. Hare, Y. Q. Zhu, M. Schwoerer, K. Prassides, H. W. Kroto and D. R. M. Walton, Appl. Phys. A: Mater. Sci. Process., 1998, 66, 307–317 CrossRef CAS.
- X. Liu, X. Li, D. Wang, R. Yu, Y. Cui, Q. Peng and Y. Li, Chem. Commun., 2012, 48, 1683–1685 RSC.
- A. M. Sheikh, E. L. Silva, L. Moares, L. M. Antonini, M. Y. Abellah and C. F. Malfatti, Am. J. Min. Metall., 2014, 2, 64–69 Search PubMed.
- A. Eshghi and M. kheirmand, Int. J. Hydrogen Energy, 2017, 42, 15064–15072 CrossRef CAS.
- S.-Y. Yang, K.-H. Chang, H.-W. Tien, Y.-F. Lee, S.-M. Li, Y.-S. Wang, J.-Y. Wang, C.-C. M. Ma and C.-C. Hu, J. Mater. Chem., 2011, 21, 2374–2380 RSC.
- Y. Simsek, L. Ozyuzer, A. T. Seyhan, M. Tanoglu and K. Schulte, J. Mater. Sci., 2007, 42, 9689–9695 CrossRef CAS.
- N. Tsud, T. Skála, F. Šutara, K. Veltruská, V. Dudr, S. Fabík, L. Sedláček, V. Cháb, K. C. Prince and V. Matolín, Surf. Sci., 2005, 595, 138–150 CrossRef CAS.
- A. F. Lee, C. J. Baddeley, M. S. Tikhov and R. M. Lambert, Surf. Sci., 1997, 373, 195–209 CrossRef CAS.
- C. Tayran and M. Çakmak, Eur. Phys. J. B, 2019, 92, 240 CrossRef CAS.
- S. M. Kozlov, F. Viñes and A. Görling, J. Phys. Chem. C, 2012, 116, 7360–7366 CrossRef CAS.
- F. Daneshvar, H. Chen, K. Noh and H.-J. Sue, Nanoscale Adv., 2021, 3, 942–962 RSC.
- E. P. Randviir, Electrochim. Acta, 2018, 286, 179–186 CrossRef CAS.
- M. Zareie Yazdan-Abad, M. Noroozifar, A. R. Modaresi Alam and H. Saravani, J. Mater. Chem. A, 2017, 5, 10244–10249 RSC.
- W. Deng, L. Zhang, L. Li, S. Chen, C. Hu, Z.-J. Zhao, T. Wang and J. Gong, J. Am. Chem. Soc., 2019, 141, 2911–2915 CrossRef CAS PubMed.
- S. Levine and A. L. Smith, Discuss. Faraday Soc., 1971, 52, 290–301 RSC.
- Y. Cheng, P. Hou, H. Pan, H. Shi and P. Kang, Appl. Catal., B, 2020, 272, 118954 CrossRef CAS.
- V. Yarlagadda, G. Lin, P. Y. Chong and T. Van Nguyen, J. Electrochem. Soc., 2015, 163, A5134–A5143 CrossRef.
- M. E. Birch, T. A. Ruda-Eberenz, M. Chai, R. Andrews and R. L. Hatfield, Ann. Occup. Hyg., 2013, 57, 1148–1166 CAS.
- Y. He, H. Zhu, Q. Liu, J. Liu, H. Zhang, R. Li, Z. Li and J. Wang, RSC Adv., 2016, 6, 70999–71005 RSC.
- V. Bogdanovskaya, I. Vernigor, M. Radina, V. Andreev, O. Korchagin and V. Novikov, Catalysis, 2020, 10 Search PubMed.
- M. Abdinejad, C. Dao, B. Deng, M. E. Sweeney, F. Dielmann, X. Zhang and H. B. Kraatz, ChemistrySelect, 2020, 5, 979–984 CrossRef CAS.
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 12219–12223 CrossRef CAS PubMed.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- J. S. Yoo, R. Christensen, T. Vegge, J. K. Nørskov and F. Studt, ChemSusChem, 2016, 9, 358–363 CrossRef CAS PubMed.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- A. Dutta, A. Kuzume, M. Rahaman, S. Vesztergom and P. Broekmann, ACS Catal., 2015, 5, 7498–7502 CrossRef CAS.
- D. H. Won, C. H. Choi, J. Chung, M. W. Chung, E.-H. Kim and S. I. Woo, ChemSusChem, 2015, 8, 3092–3098 CrossRef CAS PubMed.
- X. Zheng, P. De Luna, F. P. García de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler, Y. Cui, X. Du, P. Yang and E. H. Sargent, Joule, 2017, 1, 794–805 CrossRef CAS.
- L. Fan, Z. Xia, M. Xu, Y. Lu and Z. Li, Adv. Funct. Mater., 2018, 28, 1706289 CrossRef.
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., Int. Ed., 2017, 56, 3645–3649 CrossRef CAS PubMed.
- J. Wu, F. G. Risalvato, S. Ma and X.-D. Zhou, J. Mater. Chem. A, 2014, 2, 1647–1651 RSC.
- W. J. Dong, C. J. Yoo and J.-L. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 43575–43582 CrossRef CAS PubMed.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- V. S. K. Yadav, Y. Noh, H. Han and W. B. Kim, Catal. Today, 2018, 303, 276–281 CrossRef CAS.
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef.
- J. He, K. E. Dettelbach, A. Huang and C. P. Berlinguette, Angew. Chem., Int. Ed., 2017, 56, 16579–16582 CrossRef CAS PubMed.
- S. Y. Choi, S. K. Jeong, H. J. Kim, I.-H. Baek and K. T. Park, ACS Sustainable Chem. Eng., 2016, 4, 1311–1318 CrossRef CAS.
- A. Zhang, R. He, H. Li, Y. Chen, T. Kong, K. Li, H. Ju, J. Zhu, W. Zhu and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 10954–10958 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, G. Passard, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2013, 135, 9023–9031 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01057c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |