
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–gelation property relationships of phenolic glycosides of pentose sugars: pH dependent controlled release of curcumin†
Navendu P.
Pathak
a,
Arunava
Sengupta
b and
Somnath
Yadav
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology (ISM), Dhanbad, 826004, Jharkhand, India. E-mail: somnath@iitism.ac.in
bDepartment of Chemistry, Techno India University, Kolkata, 700091, West Bengal, India
First published on 18th March 2022
Abstract
Herein, we explored a library of structurally simple phenolic glycopyranosides of pentose sugars as organogelators, with the objective of finding a correlation between the sugar configuration and gelation ability. Of the twenty eight compounds investigated, only four–two each from the β-arabino- and α-xylo-series – were found to be capable of acting as organogelators. Thorough characterization of supramolecular gels using techniques such as FESEM, AFM, rheology, FTIR spectroscopy and WXRD led to the identification of one arabinoside derivative as being able to form the most mechanically robust organogel at loadings of about 0.2–0.3% (w/v). The mechanism of self-assembly involved mainly H-bonding interactions as evidenced from FTIR spectroscopy, WXRD and theoretical studies, although some evidence of π–π stacking interactions was also observed. This gelator was then used to prepare a curcumin entrapped organogel in mustard oil that was explored for the pH dependent controlled release of a drug model – curcumin.
Introduction
The design and synthesis of low molecular weight gelators capable of forming soft materials through self-assembly have gained attention over the past two decades.1,2 A lot of results in this direction have been obtained from the studies on the structure–property relationships at the molecular level for understanding the parameters that lead to self-assembly.3,4 Among the various categories of molecules that have been used for the synthesis of low molecular weight gelators, carbohydrates are unique as biocompatible and easily available precursors for the design and synthesis of supramolecular gelators.5,6 This stems from the fact that multiple hydroxyl groups provide the opportunity for self-assembly through hydrogen bonding interactions. Together with this, a balance of hydrophobicity can be easily provided by a plethora of protecting group manipulations using the well-established chemistry of carbohydrate synthesis. This strategy has been successfully implemented in the reports of a variety of carbohydrate based hydrogelators as well as organogelators.3,7 In designing carbohydrate based gelators, several aspects have to be taken into account such as the sugar configuration and the anomeric configuration.8,9 Such configuration dependent gelation abilities have been investigated and reported by Shinkai and co-workers for hexose sugars.3,7–10 They synthesized and investigated the gelation property–structure relationship of both the anomers of 4,6-benzylidene derivatives of the methyl glycosides of some hexose sugars. The authors found that the gelation abilities varied with the monosaccharide sugar configuration among the hexoses as well as the anomeric configuration of a particular sugar. These conclusions contributed to the design of gelators using particular hexose sugars as can be seen in the reports of several 4,6-arylidine derivatives of glucose for diverse applications.11–19 However, such fundamental investigations into the structure–property relationships of other classes of sugars are severely limited. We had previously reported a simple phenolic glycoside of arabinose as an organogelator20 as part of our ongoing work on the design of arabinose based gelators.21–23 To gain a better understanding of the structure–gelation property relationship of these classes of monosaccharides, we have investigated and herein report the same for the phenolic glycosides of pentose sugars (Fig. 1). Furthermore, we have used the most effective organogelator for the controlled release of curcumin from biocompatible and edible vegetable oil at different pH values. | ||
| Fig. 1 (a) The library of phenolic glycosides of pentose sugars reported in this study. (b) Structures of the organogelators. | ||
Results and discussion
Synthesis of the library of phenolic glycosides of pentose sugars
A library of phenolic glycosides of the aldopentose sugars (D-arabinose, D-xylose, D-lyxose and D-ribose) were synthesized as depicted in Scheme 1 from tetra-O-acetylpyranose precursors. For the β-isomers 2a–d, 3a–d and 4a–d, the tetra-O-acetylpyranose precursors were treated with phosphorus tribromide to yield the corresponding anomeric bromides. This was then followed by treatment of the anomeric bromide with the corresponding sodium phenolate obtained by the reaction of phenol with NaH. Finally, global deprotection of the acetyl groups was carried out using a catalytic amount of sodium methoxide (NaOMe) in methanol to afford the desired β-isomers of the phenyl glycosides 2–4 in 23–50% yields over three steps. However, this method did not produce the desired product with ribose. The α-isomers of the phenyl glycosides were synthesized by treatment of the tetra-O-acetylpyranose precursors with the corresponding phenol in the presence of a catalytic amount of triflic acid, followed by global de-acetylation as above to afford the desired α-isomers of the phenyl glycosides 2′–4′ in 41–88% yields over two steps. However, this method did not produce the desired α-product with ribose, probably as it isomerized to the β-isomer 5 in solution. All the products were characterized in detail using spectroscopic techniques such as FTIR spectroscopy, 1H- and 13C-NMR spectroscopy, HRMS and specific rotation (see the ESI† for detailed characterization data). | ||
| Scheme 1 Synthesis of phenolic glycosides of pentose sugars – arabinose, xylose, lyxose and ribose. Reagents and conditions: (i) PBr3, AcOH, RT, 4 h, (ii) NaH, ArOH, DMF, 6 h, (iii) NaOMe (cat.), MeOH, RT, 1 h, and (iv) ArOH, DCM, TfOH (cat.), 0–25 °C, 6 h. | ||
Gelation properties
Gelation tests for all the synthesized compounds were carried out in a variety of solvents using the standard vial inversion method. For this purpose, a weighed amount of phenyl glycoside was taken in a 5 mL vial and dissolved in a particular solvent by mild heating. Then, the vial was allowed to stand at room temperature after which it was inverted to check for gelation. From the experiments, it was found that in addition to the previously reported compound, 1-(2-methoxyphenoxy)-β-D-arabinopyranoside (2b),20 the β-arabinopyranoside, 1-(4-methoxyphenoxy)-β-D-arabinopyranoside (2d) and α-xylopyranosides, 1-(phenoxy)-α-D-xylopyranoside (3′a) and 1-(4-methoxyphenoxy)-α-D-xylopyranoside (3′d) were effective gelators for common hydrocarbon based organic solvents (Table 1). Additionally, compound 2b was able to gel amyl alcohol. An exhaustive list of the solvents tested for gelation with all the synthesized compounds is presented in the ESI† (Tables S1 and S2). In general, for compound 2b, the minimum gelator concentration (MGC) typically was 0.2% (w/v), while, for 2d, the MGCs typically ranged from 0.3% to 0.5% (w/v) for the solvents it was able to gel. For compound 3′a, the MGCs ranged from 0.5% to 0.8% (w/v) and, for 3′d, the MGCs ranged from 0.3% to 0.5% (w/v). Additionally, the gelation of the compounds was also tested for different edible and cosmetic vegetable oils such as mustard oil, coconut oil and olive oil. It was found that 2b was able to form gels in mustard oil (MGC = 1% w/v) and coconut oil (MGC = 0.3% w/v), while 2d, 3′a and 3′d formed gels only with coconut oil.| Solvent | 2b (MGCb, Tgc) | 2d (MGCb, Tgc) | 3′a (MGCb, Tgc) | 3′d (MGCb, Tgc) |
|---|---|---|---|---|
| a G = gel; PG = partial gel; S = solution; PS = partially soluble. b MGC = minumum gelation concentration % (w/v). c T g = gelation temperature. | ||||
| Benzene | G, (0.2%, 58–59 °C) | G, (0.3%, 52–53 °C) | G, (0.7%, 54–55 °C) | G, (0.3%, 48–49 °C) |
| Toluene | G, (0.2%, 61–62 °C) | G, (0.3%, 60–61 °C) | G, (0.5%, 57–58 °C) | G, (0.5%, 58–59 °C) |
| o-Xylene | G, (0.2%, 67–68 °C) | G, (0.5%, 60–61 °C) | G, (0.8%, 58–59 °C) | G, (0.3%, 56–57 °C) |
| m-Xylene | G, (0.2%, 68–69 °C) | G, (0.5%, 65–66 °C) | G, (0.8%, 63–64 °C) | G, (0.3%, 60–61 °C) |
| p-Xylene | G, (0.2%, 70–71 °C) | G, (0.5%, 59–60 °C) | G, (0.8%, 56–57 °C) | G, (0.3%, 59–60 °C) |
| Chlorobenzene | G, (0.2%, 64–65 °C) | G, (0.5%, 58–59 °C) | G, (0.5%, 55–56 °C) | G, (0.5%, 63–64 °C) |
| Amyl alcohol | G (1%, 52–53 °C) | S | S | S |
| n-Butyl alcohol | S | S | S | S |
| Methanol | S | S | S | S |
| Ethanol | S | S | S | S |
| Water | S | S | S | S |
| DMF | S | S | S | S |
| DMSO | S | S | S | S |
| Coconut oil | G (0.3%, 61–62 °C) | G (0.5%, 58–59 °C) | G (0.6%, 54–55 °C) | G (0.5%, 60–61 °C) |
| Mustard oil | G (1%, 55–56 °C) | PG | PG | PG |
| Olive oil | PG | S | S | S |
From these results, it can be concluded within certain limitations that among the phenolic glycosides of the pentose sugars in the pyranose form only β-arabinosides and α-xylosides were able to form gels. Again the gelation abilities were also dependent on the substituents on the phenols. While the xyloside of simple phenol was able to form weak gels, stronger gels in the arabino- and xylo-series were formed by the ortho- and para-methoxy derivatives.
Microstructural characterization of the gels
To gain insight into the microstructure of organogels, field emission scanning electron micrographs (FESEM) of the corresponding p-xylene xerogels of the gelators were also obtained. From the micrographs, it was observed that 2b self-assembled through a fibrillar network (Fig. 2a) with well defined thin fibres. The gelator 2d also self-assembled through fibres, but the fibres were thicker (Fig. 2b). In contrast, organogelators 3′a and 3′d self-assembled in sheet like manners (Fig. 2c and d). The gels were also characterized through atomic force microscopy studies which revealed that the morphology of the gel microstructures correalted well with those observed from the FESEM experiments. | ||
| Fig. 2 (a) FESEM image of the p-xylene xerogel of 2b. (b) FESEM image of the p-xylene xerogel of 2d. (c) FESEM image of the p-xylene xerogel of 3′a. (d) FESEM image of the p-xylene xerogel of 3′d. (e) 2D AFM image of the p-xylene xerogel of 2b. (f) 3D AFM image of the p-xylene xerogel of 2b. (g) 2D AFM image of the p-xylene xerogel of 2d. (h) 3D AFM image of the p-xylene xerogel of 2d. (i) 2D AFM image of the p-xylene xerogel of 3′a. (j) 3D AFM image of the p-xylene xerogel of 3′a. (k) 2D AFM image of the p-xylene xerogel of 3′d. (l) 3D AFM image of the p-xylene xerogel of 3′d. | ||
Mechanical properties of the gels
To compare the mechanical properties of different gels, rheological experiments were carried out at 1% (w/v) concentration of the gelators in the same solvent – p-xylene. For each of the gels of 2b, 2d, 3′a and 3′d in p-xylene, two separate experiments were carried out viz. (i) dynamic strain sweep (DSS) experiments and (ii) dynamic frequency sweep (DFS) experiments (Fig. S5–S8, ESI†). The DSS experiments at a constant frequency quantify the limiting strain (depicted by γ) beyond which the gel transitions from a visco-elastic solid like material to a fluid like material, providing the opportunity to compare the mechanical strength of the gels from different gelators in an identical solvent. From a comparison of the values of γ (Table 2) for each of the gels from gelators 2b, 2d, 3′a and 3′d in p-xylene, it can be clearly seen that 2b (γ = 0.2822%) formed the mechanically most robust gel as compared to 2d (γ = 0.0713%), 3′a (γ = 0.0445%), and 3′d (γ = 0.0245%). The above conclusions were also reinforced by the DFS experiments, the graphs from which represent the linear visco-elastic region at which the gels are stable. From the DFS experiments, the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ values of the gels could also be calculated. The tanδ value reflects the ratio of the loss modulus (G′′) to the storage modulus (G′) and the lower the value, the stronger the gel. As can be seen from Table 2, the gel from 2b possessed the lowest tanδ value among all the gels. These also correlate well with the morphology of the gels, where it was observed that 2b self-assembled through thinner entangled fibres compared to 2d which formed thicker and less entangled fibres or 3′a and 3′d which self-assembled through sheet like structures.
δ values of the gels could also be calculated. The tanδ value reflects the ratio of the loss modulus (G′′) to the storage modulus (G′) and the lower the value, the stronger the gel. As can be seen from Table 2, the gel from 2b possessed the lowest tanδ value among all the gels. These also correlate well with the morphology of the gels, where it was observed that 2b self-assembled through thinner entangled fibres compared to 2d which formed thicker and less entangled fibres or 3′a and 3′d which self-assembled through sheet like structures.
Experimental investigations into the self-assembly mechanism
To understand the mechanism of self-assembly during gelation, the organogels from 2b, 2d, 3′a and 3′d were also characterized by FTIR analysis in their crystalline and gel forms (Fig. S9, ESI†). Comparison of the IR spectra of organogelators 2b, 2d, 3′a and 3′d in their solid crystalline forms with those of their corresponding gels showed a considerable shift to a higher wave number, indicating O–H bonds and confirming that H-bonding plays a very important role in the self assembly of gelators. The shift to the higher wave number in the gel state compared to that in the solid crystalline state can be explained by the fact that the packing in the crystalline state is much more compact due to stronger H-bonding than that in the gel state.Additionally, wide angle X-ray diffraction (WXRD) experiments were carried out for the xerogels of 2b, 2d, 3′a and 3′d (Fig. S10, ESI†). The diffraction patterns of the p-xylene xerogels showed periodical peaks indicating lamellar organization. The xerogel of 2b obtained in this case for p-xylene showed much more crystalline phases and less amorphous phases than the benzene xergel reported previously.20 For the p-xylene xerogel of 2b, sharp peaks at 2θ values of 20.1° and 21.5° corresponding to d spacing values of 4.4 Å and 4.1 Å, respectively, indicated H-bonding interactions.23,24 Additionally, for the p-xylene xerogel of 2b, a sharp reflection peak was obtained at a 2θ value of 23.6° corresponding to a d spacing value of 3.8 Å, suggesting π–π stracking during the self-assembly.25 Similarly, the p-xylene xerogels of the compounds 2d, 3′a and 3′d also showed peaks that could be interpreted as contributers of H-bonding interactions during the self assembly. Thus, for 2d, sharp peaks at 2θ values of 19.1° and 21.3° corresponding to d spacing values of 4.6 Å and 4.2 Å, respectively, were observed; for the xerogel of 3′a, peaks at 2θ values of 18.5°, 19.3°, and 20.3° corresponding to d spacing values of 4.7 Å, 4.6 Å and 4.3 Å, respectively, were observed and for the xerogel of 3′d, peaks at 2θ values of 18.1° and 20.2° corresponding to d spacing values of 4.8 Å and 4.3 Å, respectively, were observed. Additional peaks for the p-xylene xerogels of 2d, 3′a and 3′d at 2θ values of 23°, 23.5° and 24.2°, respectively, were also observed with the corressponding d values of 3.9 Å, 3.7 Å and 3.6 Å, indicating the involvement of π–π stacking during the self-assembly.
Theoretical studies of the self-assembly process
To clearly understand the mechanism of self-assembly during gelation and to gather information on the particular reason behind the aggregation pattern in the gel state, theoretical studies were performed on 2b, 2d, 3′a and 3′d. Density functional theory (DFT) calculations were performed using the Gaussian 09 program.26 Optimisations were carried out with a hybrid exchange–correlation functional B3LYP.27–29 The triple-ζ quality basis set (TZVP)30 was used for oxygen, while the SVP basis set31 was used for carbon and hydrogen. Corresponding figures were made using the Chemcraft32 visualization program.FTIR analysis and WXRD experiments suggested that the H-bonding interaction is the main reason behind self-assembly in 2b, 2d, 3′a and 3′d. However, π–π stacking is also possible. To pinpoint the reason behind the aggregation, we performed geometry optimisation of dimers with both H-bonding and π–π stacking interactions. All these optimisations are carried out without any symmetry constraint to obtain the minimum energy configuration which is confirmed by vibrational frequency computation. The absence of the imaginary mode of vibration has been ascertained. The optimisations of all four monomers were performed using all the possible initial guess structures. For 2b, two possibilities have been examined. Both the initial guesses have converted to the most stable geometry (ΔE = 166 kcal mol−1) (Fig. 3a). The optimised geometries of the other 3 monomers were also optimized similarly (Fig. S13–S15, ESI†).
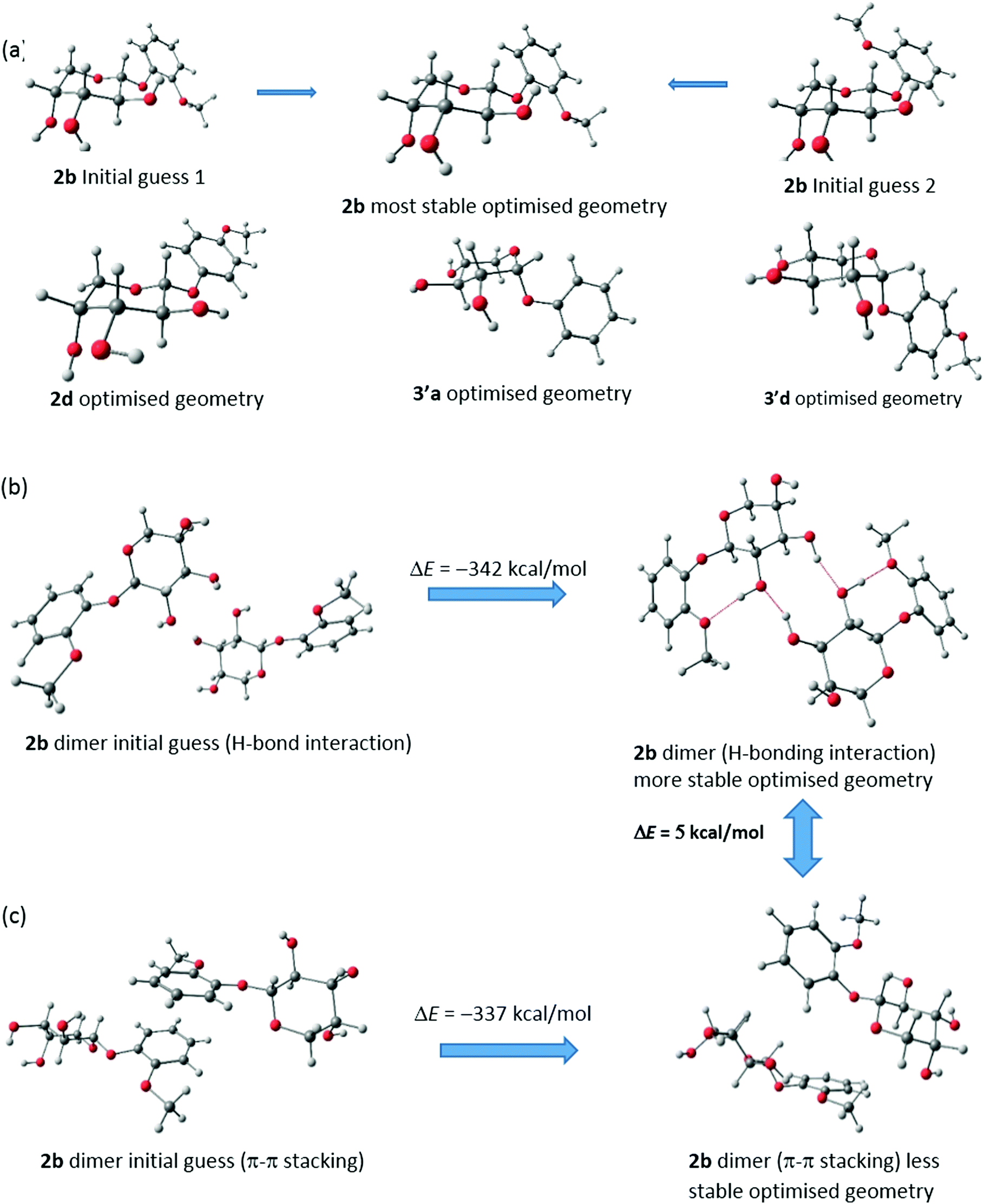 | ||
| Fig. 3 (a) Optimized geometries of (a) monomers of 2b, 2d, 3′a and 3′d. Optimized geometries of two differently oriented dimers of 2b by (b) H-bonding interactions (c) π–π stacking interactions. | ||
The general assumptions for the gel formation of these types of aromatic compounds are association with π–π stacking interactions as well as H-bonding interactions. We have taken both possibilities in mind and taken two differently oriented geometries of dimers. The first one deals with only π–π stacking whereas the second type includes the possibilities of H-bonding. Comparing the energy values of the optimised geometries, we found that the H-bonding interactions are more likely in the cases of 2b, 2d, 3′a and 3′d. Dimerization occurs predominantly through H-bonding interactions.
The optimisation of dimers of two different types shows that H-bonding interactions in 2b are favourable over π–π stacking by 5 kcal mol−1. In favour of this claim, we have presented the starting and optimised geometries of these two dimers and compared their energies (Fig. 3b and c). However, for both cases, final optimised geometries are heavily stabilised with respect to the initial guess structure and thus it is possible to have a smaller contribution of π–π stacking in the self-assembly process for gel formation which is also backed by our experimental findings with WXRD. Similar dimerization possibilities have also been studied for 2d, 3′a and 3′d and are shown in Fig. S13–S15 (ESI†). Similar trends have been reported for these three cases. Distortions in the π–π stacking interactions throughout the optimisation process have been observed for all three compounds. Also, it is observed that, for 2b, the H-bonding interactions are much stronger than for the other three compounds (2d, 3′a and 3′d) as additional H-bonds can be found. Further studies are being carried out considering the H-bonding interactions and forming three-dimensional structured aggregates with four monomers. The entrapment of curcumin is also being investigated using DFT studies with these initial findings.
Entrapment of curcumin in oragnogels and characterization of the curcumin-entrapped gel
Organogels have been explored as media for the controlled release of drugs for some time.33–36 This application can be attributed to the fact that as smart materials organogels are responsive to various stimuli which can be used to trigger the release of the entrapped drugs. Such applications are mainly restricted to organogels of amino acids37–40 and peptides,41 phospholipids,42 fatty acids43 and their derivatives,44 steroids45 and alkylamides.46 In contrast, carbohydrate based organogelators have rarely been explored for controlled release studies. Due to the importance of this area of research and the biocompatibility and eco-friendly nature of our organogels, we explored the controlled release of curcumin as a model drug, from the organogel of 2b. Since the organic solvents that were gelled by 2b are not biocompatible and toxic, we selected mustard oil as the medium for the curcumin release studies. The entrapment of curcumin into the 2b gel was carried out by simply heating a solution of the former in mustard oil with the latter and then allowing the mixture to stand at ambient temperature till the gel formation was observed by the inversion test. From a series of experiments, it was determined that a maximum of 0.5% (w/v) of curcumin could be entrapped in the mustard oil gel and loading of curcumin beyond this resulted in the formation of a very unstable gel.To gain insight into the effect of curcumin on the mechanical properties and stability of the mustard oil gels of 2b, the native mustard oil gel and the mustard oil gel loaded with curcumin were subjected to rheological experiments (Fig. S11 and S12, ESI†). For this purpose, the results obtained from the DSS and DFS experiments of the mustard oil gel of 2b at 1.5% (w/v) and the 1.5% curcumin loaded gels of 2b with a gelator concentration of 1.5% (Table 3) were compared (Table 3). It was found that the value of γ for the curcumin loaded gel was slightly higher than that for the native gel. Also, the tanδ value of the former was slightly lower than that of the latter, which indicated that the incorporation of curcumin into the gel led to a mechanically more robust gel. This could be due to the additional H-bonding interactions provided by the phenolic groups of curcumin and additional π–π interactions provided by the phenyl rings of curcumin during self-assembly, leading to more robust cross-linking. Again, the gelation of curcumin in the presence of sugars has also been reported to be induced which may also contribute to the enhanced mechanical properties observed in our case.47
Curcumin release with pH as stimulus
Due to the precedence of the use of pH as a stimulus to control the release of drugs from gels,48 we explored the controlled release of curcumin from the curcumin entrapped gel using the former as a trigger. The curcumin release experiments were carried out from the curcumin entrapped gel at different pH values using phosphate buffer solution (PBS) at 25 °C. The gelator–curcumin ratio was kept constant in all cases, wherein 2b (20 mg) was mixed with curcumin (2.0 mg) and mustard oil (2 mL) in a 15 mL sample vial. Then the curcumin entrapped gel (2b + Cur) was prepared by warming the mixture to 50 °C to dissolve the solids after which it was left to stand at room temperature for the gel to form. Then 4 mL of phosphate buffer solution was added on top of the gel so that the curcumin could be released from the gel to the phosphate buffer (Fig. 4). | ||
| Fig. 4 Images of the curcumin release experiments: (a) gels of 2b + Cur in MO; (b) gels of 2b + Cur in MO and 4 mL phosphate buffer solution at pH 5.8; (c) after 12 h and (d) after 30 h; (e) biphasic mixture of 2b + Cur in MO and phosphate buffer, after 30 h (f) and after 40 h release (g). The mixture after 49 h shows the complete degradation of the gel and no discernible separation of the oil and PBS layers. | ||
The release of curcumin from the gel medium to the phosphate buffer medium was monitored through UV-vis spectroscopy of the phosphate buffer at fixed intervals (Fig. 5a and Fig. S17–S20, ESI†). The graph for the comparative release of curcumin from the 2b + Cur gel at different pH values is plotted in Fig. 5b. The data for the 100% release of curcumin are listed in Table 4. At a pH of 8, the curcumin release was very fast and within 5 hours 100% of the curcumin was released into the buffer medium. The release time of curcumin from the gel was found to increase with the decreasing pH of the buffer medium. Thus, for 100% release of the curcumin, the times taken were about 18 h at pH 7.6, about 27 h at pH 7.4, 33 h at pH 7 and about 49 h at pH 5.8. This led us to conclude that the gels were suitable for the slow release of curcumin through transdermal applications which are associated with a pH of 5–6. The release profiles of the curcumin from the gels were also seen to be strongly correlated with the gel degradation (Fig. 4). Again the degradation of the gel structure did not affect the chemical structure of the gelator at all the pH values studied, as the gelator could be re-isolated from the mixture after the study. Therefore, it can be concluded that the disruption of the self-assembly resulting in the degradation of the gel was related to the pH due to the decrease of the H-bonding necessary for the self-assembly, especially under the basic pH. From control studies, it was also observed that the gels were less stable in basic medium (96 h) than in acidic medium (>150 h). This was probably due to the disruption of the H-bonding responsible for self-assembly by strongly basic ions which themselves preferred to form H-bonds with the H-bond donor sites in the gelator. Again, curcumin is more soluble in a basic pH solution than in an acidic pH solution, and this could also contribute to its faster release in the basic pH solution compared to the acidic pH solution. Another factor that could contribute to the slower release in the acidic pH solution is the fact that the degree of protonation of curcumin or the gelator is much higher than in the basic pH solution. The increased protonation could also lead to a comparatively slower degradation of the gel at these pH values compared to the basic pH value.
 | ||
| Fig. 5 (a) UV-Visible spectra of the release of curcumin from 2b + Cur at pH 5.8 in phosphate buffer solution. (b) Comparative release profiles of curcumin at different pH values. | ||
|
2b + Cur ratio (4:1, 10 mg for each experiment), volume of mustard oil (1 mL) |
|||||
|---|---|---|---|---|---|
| pH 5.8 | pH 7.0 | pH 7.4 | pH 7.6 | pH 8.0 | |
| Curcumin release time | 49 h | 33 h | 27 h | 18 h | 5 h |
Kinetics of the release of curcumin
The kinetic release profile plays a vital role in understanding the formulation concerning the release rates and to find out whether the drug release occurs in an understandable pattern. To gain an insight into the kinetics of the curcumin release from the gel at pH 5.8, the release data were analysed with hypothetical drug release kinetic approaches such as the zero-order kinetic plot and first-order kinetics plot (Fig. S21a and S22, ESI†).49 The approximation accuracy of individual models was evaluated in terms of the correlation coefficient (R2) value. In the case of zero-order kinetics (Fig. S21, ESI†), the data obtained were plotted as the cumulative drug release vs. time; the R2 value of the release was found to be 0.8594 which is less than the R2 value of 0.9486 obtained from the plot of the first-order kinetics (Fig. S22, ESI†). This could possibly be indicative of the release of the curcumin from the gel by first order kinetics with respect to the former. However, from a closer inspection of the data for the release via zero-order kinetics, it can be observed that the initial release up to nearly 20 h was associated with a very low release, which increased sharply subsequently. Therefore, assuming that the release occurred in two stages, a linear fit of the zero-order kinetics data from 20 h onwards was made (Fig. S21b, ESI†). This was associated with a correlation factor of 0.9879, which is higher than that of the linear fit from the first-order kinetics. Therefore, the release can also be explained by the zero-order kinetics model if we ignore the initial 20 h of the release during which time the actual release was very low.50 Such a release may be associated with the stability of the gel with respect to time. Initially, the gel was stable and the release was very slow. Subsequently, with the degradation of the gel, the release accelerated. The disintegration of the gel starts to operate at a similar time interval and then is rapidly accelerated resulting in an increase of the regression coefficient. Notably, during the drug release experiment, the degradation and erosion of the gel phase could be visually detected (Fig. 4). The degradation of the gel structure was also detected through the absorption studies where the presence of gelator in the release medium could be detected through UV-vis spectra, thus confirming that the erosion of the gel was a major contributor towards the release of curcumin. Therefore, a complex mechanism of the curcumin release involving the degradation of the gel in multiple steps involving diffusion, the erosion of the gel and the formation of oil in PBS emulsions/micelles seems to be operative. Initially, the release may be through a diffusion mechanism and as with time the gel degraded, the release became faster with the concomitant formation of oil encapsulated emulsions/micelles, as is evident from Fig. 4g where no discernible boundary was visible between the oil and PBS layers.Conclusions
In this work, we have explored the gelation ability of a library of phenolic glycosides of pentose sugars, wherein it was found that β-arabinoses (2b and 2d) and α-xylosides (3′a and 3′d) were able to act as organogelators. Again with respect to the substituents on the phenols in the arabino-series, it was seen that the ortho-methoxy substituted phenolic arabinoside 2b formed a mechanically more robust gel than the para-methoxy substituted phenolic arabinoside 2d. Similarly, in the xylo series, the unsubstituted phenolic xyloside 3′a formed a mechanically more robust gel than the para-methoxy substituted phenolic xyloside 3′d. All the gels were characterized through a variety of techniques and it was observed that the mechanical strength of the gels correlated with the morphology of the gel structure. The self-assembly of the gelators occurred mostly through H-bonding forces although some evidence of π–π stacking was also observed. This was evident from experimental studies such as FTIR spectroscopy and WXRD and was reinforced through theoretical modelling studies. The mechanical strength of the organogels from the ortho-methoxyphenol β-glycoside of arabinose 2b was found to be greater than those for the para-derivative 2d or the corresponding xyloside gels from 3′a and 3′d. This observation correlates well with our previous studies where we have reported several structurally simple β-arabinoside derivatives as LMOGs at very low concentrations. Organogelator 2b was also able to gel mustard oil, a feature that was used to entrap curcumin in a gel matrix for potential application as a controlled release medium. Entrapment of curcumin in the 2b-mustard organogel led to an increase in the robustness of the gel, possibly through the contribution of the phenolic hydroxyls of curcumin leading to greater cross-linking of the self-assembled 2b fibres. The release of curcumin was explored in media with different pH values and it was found that the slowest release occurred at pH 5.8, implying that 2b-curcumin could be potentially used for transdermal applications. The curcumin release was associated with the degradation of the gel medium and followed first order kinetics with respect to time.Author contributions
Navendu P. Pathak carried out the experimental work and wrote the preliminary draft of the manuscript. Arunava Sengupta was responsible for the theoretical modelling studies. Somnath Yadav was responsible for the conceptualization of the work, generating funds, the overall supervision of the project and writing the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Council for Scientific and Industrial research (CSIR), India [grant number 02(0362)/19/EMR-II], for financial support.Notes and references
- N. Zweep and J. H. van Esch, The design of molecular gelators. In Functional molecular gels, ed. B. Escuder and J. F. Miravet, Royal Society of Chemistry: UK, 2013; Vol 1, pp 1–3 Search PubMed.
- P. Dastidar, Gels, 2019, 5, 15 CrossRef CAS PubMed.
- O. Gronwald and S. Shinkai, Chem. – Eur. J., 2001, 7, 4328–4334 CrossRef CAS.
- S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596–5637 RSC.
- J. Morris, J. Bietsch, K. Bashaw and G. Wang, Gels, 2021, 7, 24 CrossRef CAS PubMed.
- N. Basu, A. Chakraborty and R. Ghosh, Gels, 2018, 4, 52 CrossRef PubMed.
- O. Gronwald, K. Sakurai, R. Luboradzki, T. Kimura and S. Shinkai, Carbohydr. Res., 2001, 331, 307–318 CrossRef CAS PubMed.
- K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai and D. Reinhoudt, Chem. – Eur. J., 1999, 5, 2722–2729 CrossRef CAS.
- K. Yoza, Y. Ono, K. Yoshihara, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai and D. N. Reinhoudt, Chem. Commun., 1998, 907–908 RSC.
- R. Luboradzki, O. Gronwald, M. Ikeda, S. Shinkai and D. N. Reinhoudt, Tetrahedron, 2000, 56, 9595–9599 CrossRef CAS.
- D. Wang, A. Chen, J. Morris and G. Wang, RSC Adv., 2020, 10, 40068–40083 RSC.
- J. Morris, P. Kozlowski and G. Wang, Langmuir, 2019, 35, 14639–14650 CrossRef CAS PubMed.
- A. Chen, D. Wang, L. P. Samankumara and G. Wang, Synthesis, 2019, 2897–2908 CAS.
- A. Chen, L. P. Samankumara, C. Garcia, K. Bashaw and G. Wang, New J. Chem., 2019, 43, 7950–7961 RSC.
- A. Chen, I. S. Okafor, C. Garcia and G. Wang, Carbohydr. Res., 2018, 461, 60–75 CrossRef CAS PubMed.
- A. Chen, S. B. Adhikari, K. Mays and G. Wang, Langmuir, 2017, 33, 8076–8089 CrossRef CAS PubMed.
- G. Wang, N. Goyal, H. P. R. Mangunuru, H. Yang, S. Cheuk and P. V. N. Reddy, J. Org. Chem., 2015, 80, 733–743 CrossRef CAS PubMed.
- N. Goyal, H. P. R. Mangunuru, B. Parikh, S. Shrestha and G. Wang, Beilstein J. Org. Chem., 2014, 10, 3111–3121 CrossRef PubMed.
- A. Chen, D. Wang, J. Bietsch and G. Wang, Org. Biomol. Chem., 2019, 17, 6043–6056 RSC.
- N. P. Pathak, Rajkamal and S. Yadav, Chem. Commun., 2020, 56, 2999–3002 RSC.
- Rajkamal, D. Chatterjee, A. Paul, S. Banerjee and S. Yadav, Chem. Commun., 2014, 50, 12131–12134 RSC.
- Rajkamal, N. P. Pathak, T. Halder, S. Dhara and S. Yadav, Chem. – Eur. J., 2017, 23, 11323–11329 CrossRef CAS PubMed.
- Rajkamal, N. P. Pathak, D. Chatterjee, A. Paul and S. Yadav, RSC Adv., 2016, 6, 92225–92234 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- D. K. Maiti and A. Banerjee, Chem. – Asian J., 2013, 8, 113–120 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. B. Izmaylov, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, Jr.J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785–789 CrossRef PubMed.
- P. J. Stevens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- http://www.chemcraftprog.com/ .
- J. Bietsch, M. Olson and G. Wang, Gels, 2021, 7, 134 CrossRef CAS PubMed.
- C. L. Esposito, P. Kirilov and V. G. Roullin, J. Controlled Release, 2018, 271, 1–20 CrossRef CAS PubMed.
- J. Mayr, C. Saldias and D. D. Diaz, Chem. Soc. Rev., 2018, 47, 1484–1515 RSC.
- S. S. Sagiri, B. Behera, R. R. Rafanan, C. Bhattacharya, K. Pal, I. Banerjee and D. Rousseau, Soft Mater., 2014, 12, 47–72 CrossRef CAS.
- M. Khuphe, B. Mukonoweshuro, A. Kazlauciunas and P. D. Thornton, Soft Matter, 2015, 11, 9160–9167 RSC.
- S. Kaplan, M. Colak, H. Hosgoren and N. Pirinccioglu, ACS Omega, 2019, 4, 12342–12356 CrossRef CAS PubMed.
- B. Hu, W. Sun, H. Li, H. Sui and S. Li, Int. J. Pharm., 2018, 547, 637–647 CrossRef CAS PubMed.
- B. Hu, H. Yan, Y. Sun, X. Chen, Y. Sun, S. Li, Y. Jing and H. Li, Artif. Cells, Nanomed., Biotechnol., 2020, 48, 266–275 CrossRef CAS PubMed.
- W. Gao, Y. Liang, X. Peng, Y. Hu, L. Zhang, H. Wu and B. He, Biomaterials, 2016, 105, 1–11 CrossRef CAS PubMed.
- G. Bayer, S. Grasselli, A. Malchiodi and I. S. Bayer, Colloids Surf., A, 2021, 619, 126537 CrossRef CAS.
- E. Yadav, A. K. Khatana, S. Sebastian and M. K. Gupta, New J. Chem., 2021, 45, 415–422 RSC.
- B. Martin, F. Brouillet, S. Franceschi and E. Perez, AAPS PharmSciTech, 2017, 18, 1261–1269 CrossRef CAS PubMed.
- A. K. Bandela, V. K. Hinge, D. S. Yarramala and C. P. Rao, ACS Appl. Mater. Interfaces, 2015, 7, 11555–11566 CrossRef CAS PubMed.
- S. Uzan, D. Barıs, M. Colak, H. Aydın and H. Hosgoren, Tetrahedron, 2016, 72, 7517–7525 CrossRef CAS.
- S. Wong, J. Zhao, C. Cao, C. K. Wong, R. P. Kuchel, S. De Luca, J. M. Hook, C. J. Garvey, S. Smith, J. Ho and M. H. Stenzel, Nat. Commun., 2019, 10, 582 CrossRef CAS PubMed.
- R. Pan, G. Liu, Y. Zeng, X. He, Z. Ma, Y. Wei, S. Chen, L. Yang and L. Tao, Polym. Chem., 2021, 12, 2457–2463 RSC.
- R. Gouda, H. Baishya and Z. Qing, J. Dev. Drugs, 2017, 6, 2–10 Search PubMed.
- P. J. Haddow, M. A. da Silva, D. B. Kaldybekov, C. A. Driess, E. Hoffman, V. Hutter, V. V. Khutoryanskiy, S. B. Kirton, N. Mahmoudi, W. J. McAuley and M. T. Cook, Macromol. Biosci., 2021, 2100432 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/d1ma00907a |
| This journal is © The Royal Society of Chemistry 2022 |