
A multi-functional microfluidic device compatible with widefield and light sheet microscopy†
Regan P.
Moore
 a,
Ellen C.
O'Shaughnessy
b,
Yu
Shi
a,
Ana T.
Nogueira
b,
Katelyn M.
Heath
a,
Klaus M.
Hahn
b and
Wesley R.
Legant
*ab
a,
Ellen C.
O'Shaughnessy
b,
Yu
Shi
a,
Ana T.
Nogueira
b,
Katelyn M.
Heath
a,
Klaus M.
Hahn
b and
Wesley R.
Legant
*ab
aJoint Biomedical Engineering Department, University of North Carolina at Chapel Hill, North Carolina State University, Chapel Hill, NC 27599, USA. E-mail: legantw@email.unc.edu
bPharmacology Department, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
First published on 22nd November 2021
Abstract
We present a microfluidic device compatible with high resolution light sheet and super-resolution microscopy. The device is a 150 μm thick chamber with a transparent fluorinated ethylene propylene (FEP) cover that has a similar refractive index (1.34) to water (1.33), making it compatible with top-down imaging used in light sheet microscopy. We provide a detailed fabrication protocol and characterize the optical performance of the device. We demonstrate that the device supports long-term imaging of cell growth and differentiation as well as the rapid addition and removal of reagents while simultaneously maintaining sterile culture conditions by physically isolating the sample from the dipping lenses used for imaging. Finally, we demonstrate that the device can be used for super-resolution imaging using lattice light sheet structured illumination microscopy (LLS-SIM) and DNA PAINT. We anticipate that FEP-based microfluidics, as shown here, will be broadly useful to researchers using light sheet microscopy due to the ability to switch reagents, image weakly adherent cells, maintain sterility, and physically isolate the specimen from the optics of the instruments.
Introduction
Light sheet microscopy (LSM), reduces photobleaching and phototoxicity and is able to image cells for longer durations and at higher speeds than confocal or widefield instruments.1 The most common variants of light sheet microscopy for high resolution, sub-cellular imaging are top-down systems that utilize optics which are immersed into the same solution as the specimen, two of which are lattice light sheet microscopy (LLSM)2 and dual-view plane illumination microscopy (diSPIM).3 These instruments utilize two independent dipping lenses, one for excitation and one for detection, positioned above the specimen. LLSM uses structured optical lattices to illuminate the sample, improving axial resolution while maintaining the rapid imaging speed and reduced phototoxicity inherent to SPIM techniques.2 diSPIM typically utilizes a Gaussian (unstructured) light sheet but makes use of both objectives for excitation and detection sequentially. Combining the views from the two objectives results in isotropic volumetric resolution.3Both LLSM and diSPIM have been critical in studies observing embryogenesis,2–5 intracellular dynamics,2,4,6–10 multicellular dynamics4,11,12 and super-resolution microscopy.13,14 While the advantages of these systems are clear, difficulties in sample preparation have limited their use in many applications. In LLSM and diSPIM, samples are mounted on a coverslip and submerged in a bath together with the imaging optics. To position the sample within the working distance of the excitation and detection objectives, the specimen itself is oriented at an oblique angle relative to the optics (Fig. 1A and B). To acquire a three-dimensional volume, either the sample is scanned through the beam by a motorized stage, or the beam and detection objective are scanned together through the specimen.
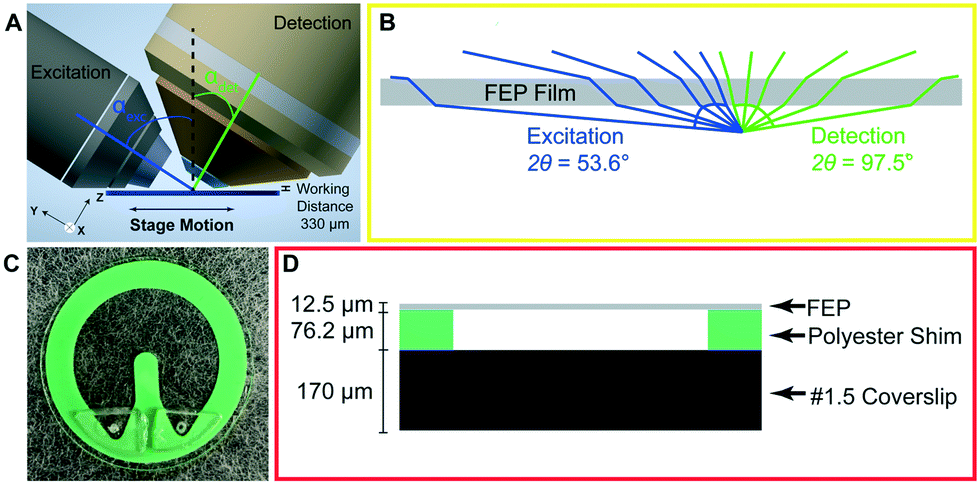 | ||
| Fig. 1 Microfluidic chip compatible with light sheet microscopy. A) Illustration demonstrating the position of the chip relative to the objectives for light sheet microscopy. The objectives are positioned above the sample with a working distance of 330 μm. The excitation objective (left) is 0.6 NA and is positioned at an αexc = 57.6°. The detection objective (right) is 1.0 NA and is positioned at an αdet = 32.4°. The sample is scanned left-to-right via a motorized stage. B) Refraction due to top FEP layer of chip. Light rays from the excitation objective (blue) have an angle of acceptance (2θ) of 53.6°. Light rays from the detection objective (green) have an angle of acceptance of 97.5°. C) Fully assembled microfluidic chip. D) Schematic cross-sectional view of chip. The chip consists of a top FEP layer from 12.5 μm stock. This is glued (blue) to the main body of the device which is 76.2 μm thick polyester shim stock. The polyester shim stock is glued to the base of the chip, a #1.5 (170 μm), 25 mm diameter coverslip. | ||
The physical and optical requirements of top-down light sheet microscopes generate constraints on sample preparation including: unimpeded optical access between the specimen and lenses, a sample bath of large enough volume to submerge both the sample and the objectives, and, in some cases, physical coupling between the moving coverslip and the specimen to achieve scanning. This precludes imaging of non-, or weakly-adherent cells without extracellular matrix (ECM), adhesives, or embedding media (i.e., low-melt agarose). Keeping samples viable long-term requires the imaging bath to be filled with media, often making it difficult to maintain sterility in an open chamber. In addition to the need for large amounts of costly reagents, the size of the imaging bath makes it difficult to rapidly perturb the specimen with small molecule inhibitors which must diffuse and re-equilibrate within the entire chamber.
Microfluidic devices could potentially be used to address these challenges; however, they must be designed to accommodate the unique optical requirements of light sheet microscopes. Many of these devices are constructed from a combination of glass and poly(dimethylsiloxane), PDMS, elastomer. However, the refractive index differences between these materials (1.515 for glass and 1.43 for PDMS) and the surrounding media (1.333 for water) causes shearing of both the illumination and detection wavefronts, which is exacerbated by the obliquely mounted LSM objectives (Fig. 2, S1 and S2†). Even with conventional imaging using an orthogonally mounted objective, refractive index differences between the device and the ideal immersion media of the lens could lead to substantial spherical aberration (Fig. S2†). Microfluidic devices, with index-matched materials, that are compatible with LSM and other modalities will not only increase the diversity of samples that can be imaged, including non-adherent cells, but would also increase the diversity of experiments that can be performed, including the application and washout of small molecule inhibitors.
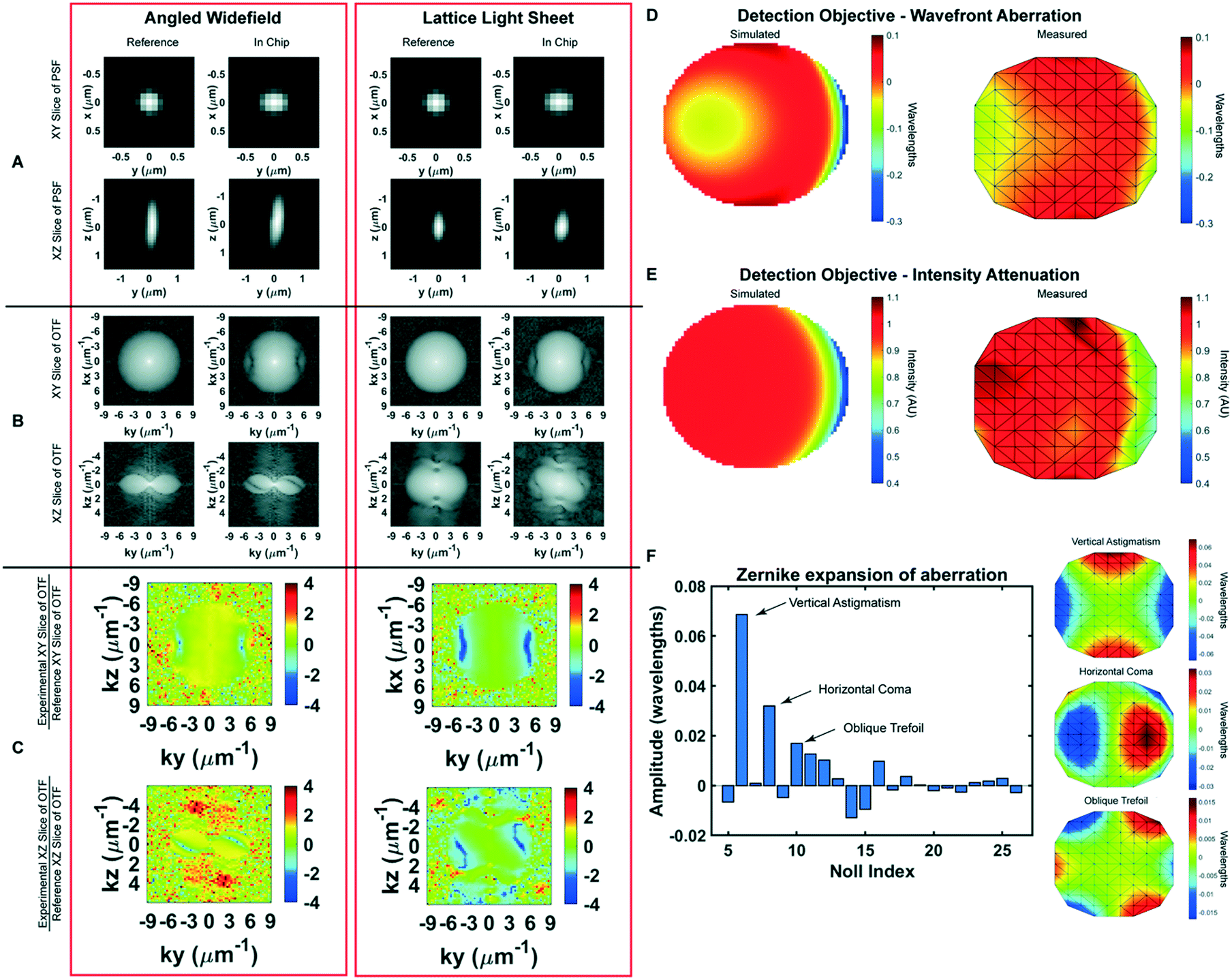 | ||
| Fig. 2 Optical characterization of microfluidic chip A) angled widefield and lattice light sheet point spread functions (PSFs) in the chip. The PSFs become wider in the Y-axis as light passes through the FEP film. B) Optical transfer functions (OTFs) in the chip. Aberration from the FEP film is particularly present at the high frequency components of the PSF. C) Logarithmically scaled heatmap of the ratio between the magnitude of the aberrated OTF to the magnitude of the reference OTF. Heatmaps of the ratio more clearly demonstrate the aberration generated by the FEP film. D) Wavefront abberation generated by the FEP film. Modeled aberration in wavelengths that the FEP introduces to the wavefront using the optics software Code V (left). Measured abberation of the wavefront generated by the FEP film using a Shack–Hartmann sensor (right). E) Intensity attenuation due to the presence of the FEP film. Relative intensity of light after passing through the film modeled in Code V (left). Measured intensity relative to expected intensity using a Shack–Hartmann sensor (right). F) Relative weight of measured abberation modes shown in Zernike polynomials. The FEP film generates aberrations, excluding tip, tilt and defocus, consisting mostly of vertical astigmatism (Noll index = 6), horizontal coma (Noll index = 8), and oblique trefoil (Noll index = 10) as shown in graphs at right. | ||
Two recent papers describe the fabrication of microfluidic devices compatible with LSM. Han et al. utilize a newly developed bio-compatible polymer, Bio-133, that has a refractive index very close to water and minimizes aberrations in both lightsheet and conventional imaging modalities.7 This polymer can be spin coated to generate the top layer of an imaging device or can be micropatterned using soft-lithography techniques for experiments requiring fluid exchange. This is an exciting development, although current demonstrations do have several drawbacks. In the previously fabricated devices, the polymer layer is relatively thick, 50 μm, consuming ∼24 percent of the 210 μm working distance underneath the 0.8 NA objectives often used in diSPIM.7 Furthermore, the proprietary polymer is costly, its composition is not available, and it can be purchased from only a single source. In another recent manuscript, Fan et al. present a microfluidic system to continuously flow suspended cells through the light sheet.15 A 3D volume can be reconstructed from these subsequent planes. While this device is ideal for high-throughput imaging, it is limited to imaging suspended cells for a limited duration due to flow.
To complement these developments, here we describe an alternative microfluidic chip that is compatible with both inverted and light sheet microscopy. Cells and media are contained in a thin region between a coverslip and an FEP cover, thus physically isolating the specimen from the optics while minimizing refractive index differences between the film and surrounding media. Within the device, both the internal fluid and specimen move together with the stage during imaging. Thus, we are no longer limited to strongly adherent cells or to embedding samples within adherent hydrogels to couple them to the stage motion. The chip allows us to introduce reagents in a small volume and diversifies the types of solution we can image in without exposing the objectives to these compounds. In summary, by coupling microfluidics with light sheet microscopy we are able to study a greater diversity of cells and cellular processes, rapidly perform transient perturbations, and perform cost-effective super-resolution imaging with only minor trade-offs in optical performance.
Results
Assembling and imaging within the microfluidic device
The microfluidic device is built around a 25 mm diameter coverslip, meets the optical constraints, and maximizes the working distance of top-down light-sheet microscopes. In our implementation of lattice light sheet microscopy, the objective lenses are situated above the sample and have a working distance of 330 μm between the physical edges of the lenses and the specimen focus (Fig. 1A). We note that this geometry would also be readily applied to existing diSPIM systems that have a working distance of 210 μm. The bottom layer of the device is a glass coverslip which is separated from the FEP top by a 76.2 μm polyester shim that can be laser cut into diverse shapes. Here we demonstrate a u-shaped chamber through which we can flow cells, media, and reagents, Fig. 1C and D. We attach the shim to the coverslip using an optically clear, UV-curable glue, Norland 81. To seal the device, we take stock 12.5 μm FEP, which is an inert and transparent film, stretch it as described in more detail below, and adhere it to the shim. We then glue acrylic ports on top of this film to facilitate loading the device. Together, this makes an imaging chamber that is reproducibly 140–160 μm thick and can be mounted not only on LLS microscopes but also on standard widefield imaging systems.To minimize aberration introduced from refractive index changes, we used a thin FEP film as the top layer of the device. FEP has a refractive index (RI) of 1.344, comparable to water (1.333), PBS (1.335) and cell culture media (DMEM, 1.337).16 We uniaxially stretched the FEP film to reduce the thickness and minimize aberrations. The thickness of the film can be reduced by up to 44% by uniaxial stretching the film to 200% of its original width (Fig. S1A†). However, we observed that this stretching may introduce micro-holes that decrease the integrity of the film over time. Thus, we stretched the film to 110% of its original length, which although it does not substantially decrease the film thickness, it provides sufficient tension within the film to prevent wrinkles during fabrication without introducing defects. Because of the oblique angle between the objectives and film, the refractive index mismatch between the film and surrounding solution generates aberrations in one lateral direction (Y), and in the axial direction (Z). The x-axis remains parallel to the film regardless of incident angle and there are no additional aberrations in this direction (Fig. 2A and B). To quantify this, we compared experimentally-measured point spread functions (PSFs) using both widefield and light sheet illumination (Fig. 2A–C and S1B, Table S1†). In absence of the film, we observed widefield detection PSFs with full width at half maximal (FWHM) intensities of 0.38 × 0.38 × 1.38 microns when measured at 560 nm excitation and 605 nm emission, in close agreement with vectorial electromagnetic diffraction theory intensities of 0.33 × 0.33 × 1.18 μm.17 When imaging through the film and the chip volume, these values increased along the y dimension due to propagation through the film to 0.46 microns, an increase of 21%. When we compare the PSFs of the bead in the chip to the bead on top of the chip we recover a Strehl ratio of 0.7097. Under lattice light sheet illumination, the lateral resolution was degraded similarly to widefield, while the axial resolution was maintained due to the optical sectioning provided by the light sheet. The FHWM measurements increased from 0.39 × 0.37 × 0.82 microns in absence of the film to 0.37 × 0.47 × 0.83 microns when imaging through the chip and resulted in a Strehl ratio of 0.7857. However, we note that FWHM measurements by themselves are very poor descriptions of overall resolution. For example, the film causes the PSF to be sheared about the x-axis, which is not revealed by the above measurements. Therefore, we sought to fully describe the loss in imaging performance by comparing the optical transfer functions for both reference and measurements within the microfluidic device. Here, the reduction in frequency-space support along the y-axis is apparent by the elliptical shape of the OTF in the XY plane and the more complex effect due to wavefront shearing and attenuation of the marginal rays can be seen by the two angled lines of reduced support at the periphery of the OTF in the YZ plane (Fig. 2B). Taking the ratio of the reference and the in-chip OTFs allows us to precisely quantify the filtering effect as a function of spatial frequency (Fig. 2C). Finally, following a procedure similar to that of previous studies,18 we also quantified both wavefront aberration and intensity attenuation due to the microfluidic chip using optical simulations and additionally by direct experimental measurement of the pupil wavefront using a Shack–Hartmann (SH) wavefront sensor. We simulated the optical configuration of the FEP film and the 1.0 NA detection objective at a 32.4° angle (αdet) and perpendicular to the film using Code V, a commercially available optical simulation suite (Fig. 2D and E and S2†). In parallel, we performed the same measurements experimentally by illuminating a single bead on the glass surface within the microfluidic chip, measuring the emission wavefront on the SH sensor, and comparing to beads on top of the FEP film. We observed good agreement between simulations and experimental measurements, revealing a peak-to-peak wavefront error across the 1.0 NA detection pupil of less than λ/10 for all but the most marginal rays (Fig. 2D and E). We note that although the entire emission pupil illuminates the SH sensor, because of the microlens size and pitch, we are not able to experimentally measure the aberration at the very edge of the pupil. For this reason, our experimental measurements are limited to roughly 90% of the pupil diameter. Further, for these measurements, both the chip and imaging chamber were filled with phosphate buffered saline. In this case, we did not observe significant differences when imaging on the glass coverslip vs. on the underside of the film (data not shown). However, the performance will degrade substantially if the solutions in the imaging chamber and the chip are not index matched. Zernike decomposition of the aberration revealed the primary contributions, neglecting tip, tilt, and defocus, to be from vertical astigmatism, horizontal coma, and oblique trefoil terms (Fig. 2F). While most of the pupil did not experience substantial intensity attenuation, there was a maximum of 60% reduction in transmission for the most oblique rays (Fig. 1B and 2E). As expected, the wavefront aberration increased with increasing FEP film thickness (Fig. S1C, Table S1†); however, we observed only a modest increase in attenuation with film thickness (Fig. S1D†), suggesting that most of the loss occurred due to Fresnel reflections at the dielectric interfaces rather than absorption within the film. Finally, the wavefront aberration and intensity attenuation from the thin FEP film are less than what would occur with a #1.5 glass coverslip top (Fig. S2E and F†).
Together, our results demonstrate that while FEP does not exactly match the RI of the imaging solution, when a 12.5 μm thick film is used and stretched to provide tension, the aberration due to the microfluidic device is within λ/10 over most of the excitation and detection pupils, comparable to the range of most commercial optical components. Below, we demonstrate that this device is biologically compatible and supports both high, and super-resolution 4D light sheet imaging.
Sterile culture of cells and long-term cell growth
The microfluidic device can be sealed off from the external environment, allowing it to be taken on and off the microscope and remain sterile. It is also biocompatible and non-toxic to cells. This allows us to repeatedly observe processes that occur over multiple days, including long-term cell growth. To demonstrate this, we grew U2OS cells for 3 days, imaged them every 24 hours, and quantified cell proliferation on an inverted widefield microscope. Gas exchange across the FEP membrane will only occur if the top portion of the chip is exposed to air and not when the chip is submerged and the membrane partitions two liquid interfaces. Thus, samples in the chip can be maintained in normal bicarbonate buffered cell culture media within a CO2 controlled incubator, but when submerged for imaging, pH must be controlled with CO2 dependent buffers. The smaller volume in the chip leads to decreased nutrient availability and increased waste accumulation as previously described.19 We therefore tested two frequencies of media changes to determine the relationship between nutrient availability and growth rate in the devices. As a control we plated U2OS cells in a glass bottom 6-well plate and imaged and exchanged media every 24 hours (Fig. 3A). In both the control dish and microfluidic chips, cells continued to divide throughout the multiday time course and reached confluence after 72 hours (Fig. 3A–C). Fitting the data to an exponential growth curve allowed us to estimate the doubling times as 1.61 ± 0.52 days for the control, 1.95 ± 0.14 days for the 12 h media change and 2.29 ± 0.27 days for the 24 h media change (Fig. 3D). In all cases, cells continued to divide and maintain normal morphology. We anticipate that more frequent media changes or continuous perfusion of the chip may increase the rate of growth closer to the control.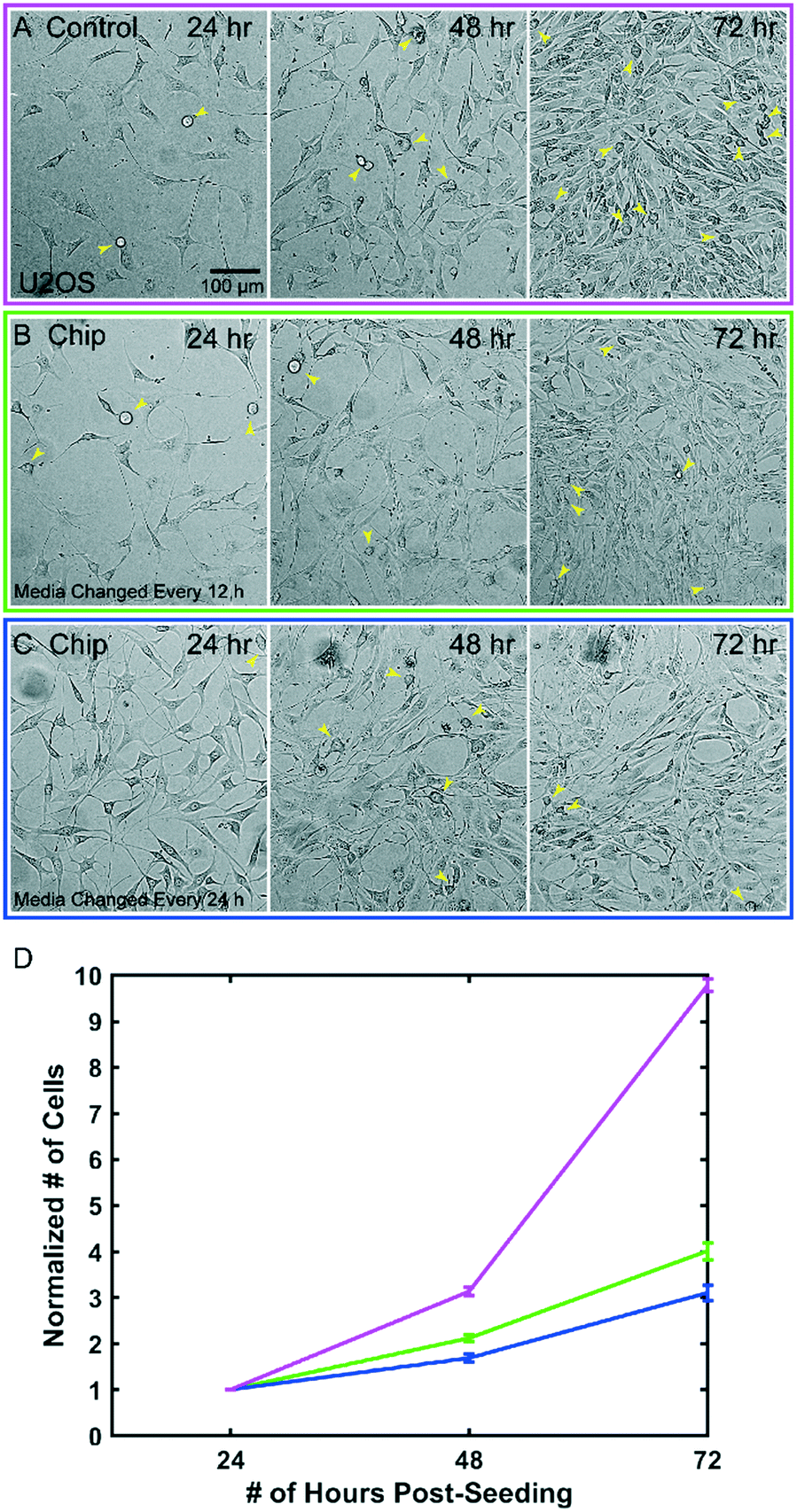 | ||
| Fig. 3 Cells are viable and proliferate within the microfluidic chip. Representative images from a single field of view comparing: A) control cells plated in a glass bottom six-well plate, B) cells plated in the chip with media changes performed every 12 h, and C) cells plated in the chip with media changes performed every 24 h D) quantification of cell growth in each condition for n = 3 chips per condition, 4 ROIs per chip, 4 ROIs for controls, error bars are SEM. Yellow arrows indicate dividing cells in each image. | ||
Imaging primary, weakly-adherent cells
To demonstrate the diversity of cell types and cellular processes that we can image with the chip, we observed megakaryocytes (MKs) undergoing the final stage of their differentiation process, termed megakaryopoiesis. This is a dynamic process in which cells span large 3D volumes and undergo constant, rapid change. MKs are large, polyploid hematopoietic cells that reside primarily in the bone marrow, where they produce platelets. They undergo a series of dramatic morphological changes to transition from dense spherical cells to widely spread cells with many branching, thin protrusions, in a process called proplatelet formation (PPF).20 Because megakaryocytes are very weakly adherent, previous light sheet imaging of these cells has required embedding the cells under a drop of low-melt agarose which causes additional aberrations.4 We labelled the demarcation membrane system (DMS), a unique feature of mature MKs, and imaged healthy cells at all stages of PPF (Fig. 4A–C, Movie S1†). Because the chip couples the movement of the cell suspension to the scanning stage, we were able to scan these weakly adherent cells through the lightsheet and acquire high resolution 3D images. Repeating these scans over 10 hours allowed us to observe the entirety of PPF and track individual extensions over time. The spatiotemporal resolution afforded by LLSM allowed us to clearly track protrusion growth during PPF (Fig. 4D, Movie S2†) and resolve the convolutions of the DMS (slices, Fig. 4A–C). We anticipate that use of these microfluidic devices will greatly aid in further understanding this dynamic process.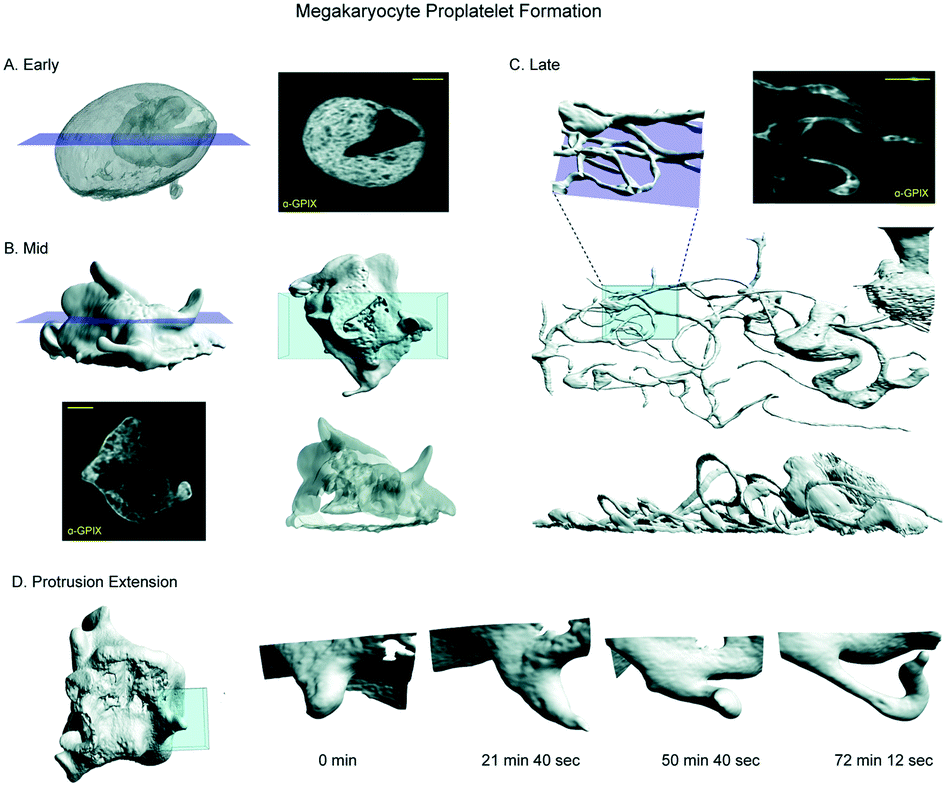 | ||
Fig. 4 Imaging weakly adherent cells within the microfluidic chip. Megakaryocytes are weakly adherent hematopoietic cells that undergo a dynamic process called proplatelet formation as part of a larger differentiation called megakaryopoiesis. A) Cells in early proplatelet formation are mostly round and are characterized by extensive membrane convolutions shown by labeling with GPIX antibody. Purple plane indicates position of slice view (left). B) In mid-proplatelet formation, the cells begin to form protrusions. A volume rendering of a region (blue, top right) demonstrates the complex membrane folds (bottom right). Purple plane indicates position of slice view (bottom, left) C) by late proplatelet formation, the protrusions have grown from small buds into long, thin, branching extensions as shown in the volume rendering (blue, middle). A slice (purple plane) through this region shows branching and bending of the extension (top, right). The protrusions are extensive and take up a larger area than the cell body as shown in the volume rendering (bottom). D) Protrusion growth during mid-proplatelet formation demonstrating the transition from a small bud, to a longer, thin extension. Time in min![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :s. All scale bars are 10 μm. :s. All scale bars are 10 μm. | ||
Applying small molecule inhibitors and long-term imaging within the microfluidic device
Without the use of microfluidics, the application and washout of small molecule inhibitors is challenging due to the time needed to thoroughly mix or replace the large volume (10–40 mL depending on LSM implementation) of the imaging chamber, and the amount of costly reagents needed for this volume. The 40 μL volume of the chip is less than 0.1% of the total volume of the bath that the objectives are immersed in. To demonstrate rapid perturbation of cellular response within the microfluidic chip, we imaged U2OS cells stably expressing Lifeact::RFP every 2 min for 40 min prior to the addition of the myosin II ATPase inhibitor, blebbistatin (Fig. 5A, Movie S3†). After 20 timepoints, we exchanged the media in the chip to media containing 50 μM blebbistatin (frames with red border, Fig. 5A). Within minutes, we observed the dissolution of stress fibers (arrowheads, Fig. 5A, yellow inset). With the loss of contractility, the cells shifted from a polarized, spindle shape, to a less defined shape with extensive lamellipodia and dramatic ruffling at the leading edge (Fig. 5A). After 90 min, we replaced the media containing blebbistatin with fresh, prewarmed growth media (frames with green border, Fig. 5A). Stress fibers rapidly reappeared and cells recovered their initial phenotype within minutes (arrowheads, Fig. 5A, magenta inset). All media exchanges were completed on the microscope and imaging was restarted without perturbing saved imaging locations. We note that PDMS based microfluidic devices can absorb hydrophobic reagents and thus make drug dosing and washing challenging.21 In contrast, FEP based microfluidic chips are resistant to drug absorption, enabling rapid washout of reagents.22,23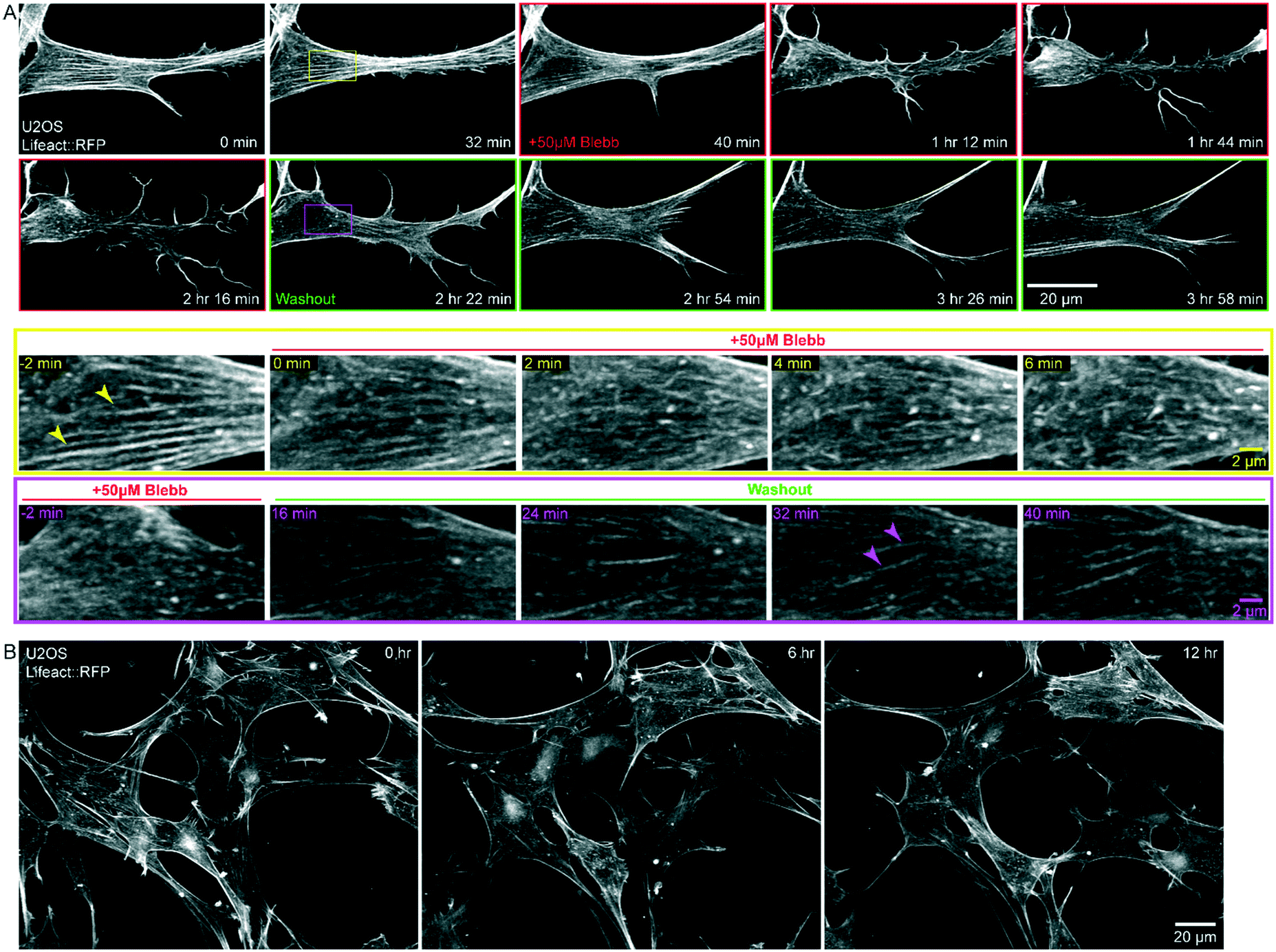 | ||
| Fig. 5 Drug treatment/washout and long-term imaging within the microfluidic chip. U2OS cells stably expressing Lifeact::RFP were grown in the microfluidic chip. A) Application of 50 μm blebbistatin causes significant morphological changes which can be reversed upon washout. Yellow inset demonstrates the fading of stress fibers (arrowheads) upon the application of blebbistatin. Magenta inset shows that upon washout the stress fibers reform in the same ROI, although this occurs on a slightly longer timescale. B) Continuous light sheet imaging of cells within the microfluidic chip over 12 hours. Cells remained viable with normal morphology throughout the time course. | ||
Long-term imaging of biological specimens requires careful control of environmental conditions as well as sample sterility. However, this can be challenging in conventional light sheet microscopes where the immersion optics are in contact with the same media as the sample. To circumvent these challenges, we plated cells in media within the microfluidic chip, sterilely sealed it, and filled the microscope sample chamber with 1X PBS. We imaged U2OS cells stably expressing Lifeact::RFP every 5 min for 12 hours. Cells remained healthy as demonstrated by a normal spread area and actin dynamics (Fig. 5B, Movie S4†). This demonstrates that the imaging optics can be physically isolated from the specimen and growth media, without introducing additional aberrations, as long as the refractive index of the solution filling the sample bath is similar to that of the media within the chip.
Super-resolution light sheet imaging in a microfluidic chip
Finally, we demonstrate that the microfluidic chip is compatible with both live- and fixed-cell super-resolution microscopy. By coupling LLSM with structured illumination microscopy (SIM) we previously demonstrated an improvement of 1.3–1.5× beyond the diffraction limit in living specimens.2 To demonstrate that this modality is compatible with the microfluidic device, we imaged U2OS cells stably expressing Lifeact::RFP and acquired 3D LLS-SIM volumes of these cells every 6 min for 2 hours, clearly seeing protrusion dynamics at the leading edge of the cell (Fig. 6A, Movie S5†). When performing this experiment, we observed that the FEP film, similar to other thin polymer films, acts as an optical retarder to modulate the polarization of the transmitted light. While this does not detrimentally affect the randomly polarized emission light, SIM illumination relies on high-contrast modulation of the excitation pattern which is maximized when the excitation polarization is orientated perpendicular to the direction of pattern modulation. To achieve this at the specimen, we oriented the FEP film to have its fast axis (direction of film extrusion and additional stretch) to be perpendicular to the polarization of the illumination light (Fig. S3†). In this case, we were able to maximize pattern contrast and see a clear resolution improvement using LLS-SIM over, non-SIM, Wiener deconvolved LLS (compare arrowheads, Fig. 6B). It is important to note that the birefringence that interferes with the modulation of the SIM illumination pattern will also interfere with other imaging modalities that depend on polarized light, including differential interference contrast (DIC).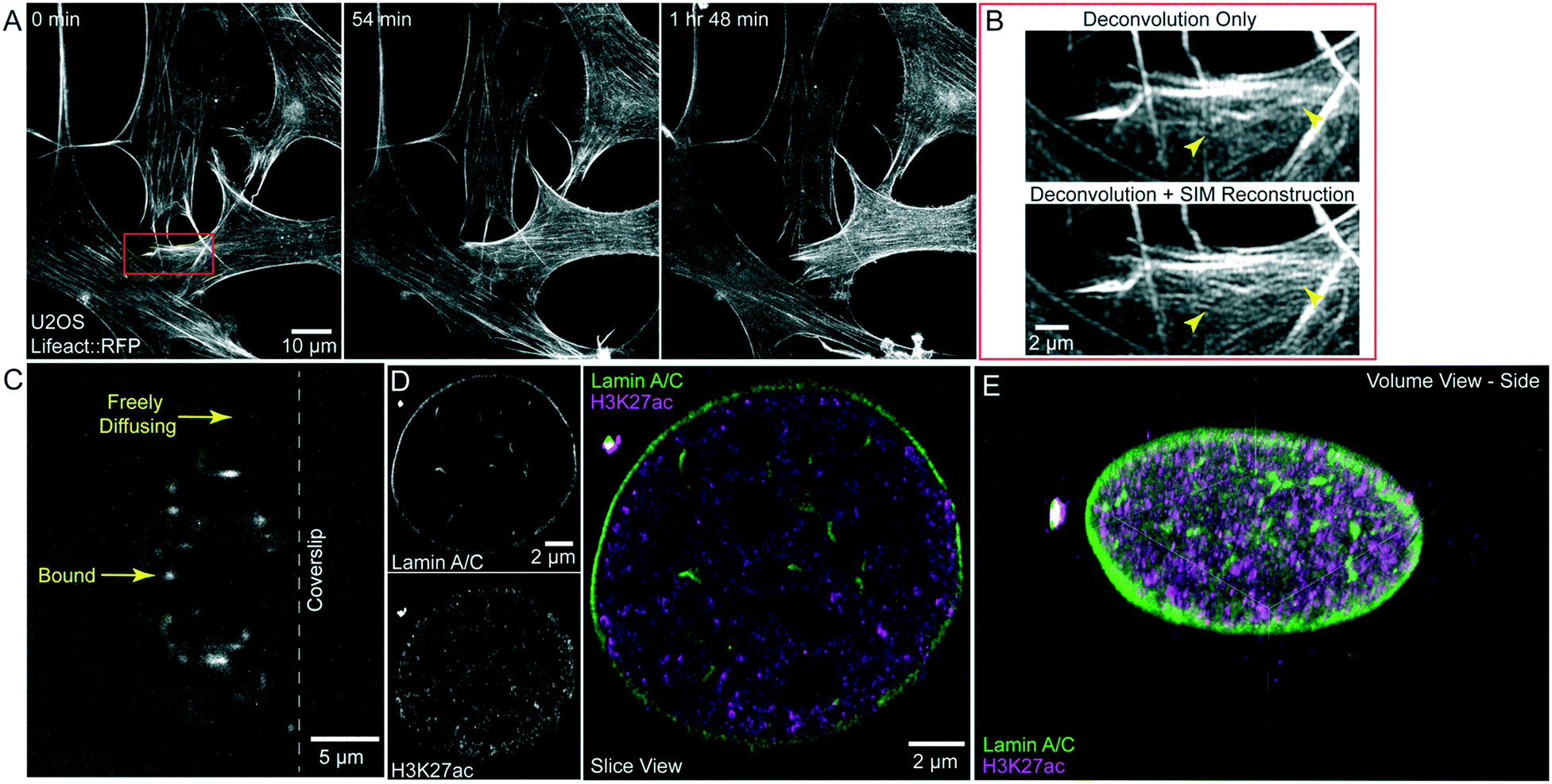 | ||
| Fig. 6 Super-resolution imaging within the microfluidic chip. A) U2OS cells stably expressing Lifeact::RFP were imaged using lattice light sheet coupled with structured illumination (LLS-SIM). Red box indicates ROI in panel B. B) ROI demonstrating the difference in images when using Wiener deconvolution compared to SIM reconstruction. The improvement in resolution clearly allows the delineation of separate filaments, compare arrowheads. C) Raw data of single molecule imaging demonstrating the higher intensity of bound imaging strands compared to freely diffusing probes. D) Single slice views of DNA PAINT data set of lamin A/C alone (top, left), H3K27 acetylation alone (bottom, left), and merged channels (right). E) Volume view of complete nucleus showing all localized molecules against lamin A/C (green) and H3K27ac (magenta). Bounding box dimensions are 14.4 μm × 15.3 μm × 5.22 μm. | ||
The small volume of the device is also useful for minimizing the amount of costly DNA oligo imaging strands used in DNA PAINT localization microscopy, a fixed-cell implementation of super-resolution microscopy.24 Fluorescent DNA oligos are diluted, freely diffuse and are stabilized when they bind a complementary strand tethered to a secondary antibody or nanobody.25 The freely diffusing population of imaging strands provides a pool of fluorescent signal that can replenish the strands that have been stabilized, imaged and bleached. The transient interaction provides increased contrast (compare arrows, Fig. 6C), and by acquiring many images we can generate a high-density localization microscopy dataset of the underlying structure. To demonstrate that the device can be used in this application, we plated and fixed unlabelled U2OS cells in a microfluidic chip that had previously been coated with fluorescent nanodiamonds.26 These nanodiamonds act as fiducials for image registration to account for drift during the imaging process and to account for sample shifts during probe exchange. We performed indirect immunofluorescence with primary antibodies against lamin A/C and the active histone post-translational modification, H3K27-acetylation, and secondary antibodies coupled to orthogonal DNA docking strands. Fluorescence imager strands were added sequentially to the device without removing the sample from the microscope. We acquired thousands of volumes over multiple days and reconstructed the localizations using the SMAP software.27 In total, we obtained ∼19 million and 15 million localizations acquired over 30000 and 20000 sequential 3D images of the specimen for the lamin A/C and H3K27ac datasets respectively. The average localization error from the two datasets combined was 20 nm in the X- and Y-axis and 50 nm in the Z-axis. The reconstructed image clearly shows the lamins localized to the nuclear envelop as well as detailed nuclear architecture including nuclear tunnels (Fig. 6D and E, Movie S6†). We can also see clusters of active chromatin, denoted by the signal from H3K27ac (Fig. 6D and E). As the DNA PAINT probes are costly, being able to utilize a very small volume at the desired concentration is advantageous for these experiments. For example, we purchased 300 μL of imaging strand which at the concentration used in the above experiment is enough for 10 experiments at ∼$100/experiment if diluted in the whole imaging bath. By using the microfluidic chip, we can complete 10000 experiments at 10¢/experiment.
Discussion
We have presented a microfluidic device that is compatible with both inverted microscopes and top-down light sheet microscopes. The device is reproducible and is assembled with readily available materials. We demonstrate that the device can be used to sterile culture cells for multiple days (Fig. 3) and does not impede the rapid and gentle imaging inherent to LLSM or its super-resolution variants (Fig. 4–6). With both simulations and experimental measurements, we demonstrate that the aberration introduced by thin FEP films, even when used at the oblique angles and high numerical aperture lenses associated with LLSM, is less than λ/10 over most of the excitation and detection pupils. This is comparable to the specifications of commercial grade optics used on many of these systems.The same advantages that we show here for DNA PAINT, namely the ability to flow in small volumes of imaging reagents, would also be beneficial for the future studies on spatial transcriptomics.28,29 The robustness of the device for multiday imaging and the ability to rapidly exchange probes makes this an attractive device for highly multiplexed data sets.
Finally, we note that in our current work, we have not utilized any optical correction to compensate for aberrations from imaging through the FEP film. This demonstrates the wide applicability to existing top-down light sheet microscopes without any additional modifications, however we anticipate that instruments with adaptive optical correction could further compensate for the small residual aberration.4,18 However, the attenuation of the most marginal rays in the pupil may still cause a degradation from ideal performance. We believe that the optical characterization, fabrication protocol, and imaging demonstrations presented here will enable future devices based on similar principles and will greatly increase the diversity of sample types and experimental conditions that can be observed via light sheet microscopy.
Materials and methods
Fabrication of devices
We fabricate the microfluidic chips using a glass coverslip base, a chamber pattern of polyester shim stock, and an FEP top (Fig. 1D and S4†). We use #1.5 glass coverslips cleaned in 1 M KOH under sonication for 30 min. We then rinse with distilled water three times, sonicate in distilled water for 30 min, rinse with distilled water three more times, and store in distilled water. We use 0.003′′ polyester shim stock (Argus) for the devices. Other shim stock thickness can be chosen to vary the height of the chamber to match the working distance of the objective and the final chip dimensions. We use a Universal PLS 6.150D laser cutter to cut the shim stock. For this specific model, we used the following settings: 5% power, 40% speed, 1000 ppi, although these will need to be optimized based on shim thickness and laser cutter age/model. We attach the patterned shims to the cleaned coverslips using Norland 81 Optical Adhesive. The coverslip/shim is secured between two microscope slides and secured with binder clips and then placed under a UV LED (M365LP1-C1, Thorlabs) at a distance of approximately 3 in from the light source. Devices were cured for 5 min under an illumination power of 35 mW. After the glue has cured, we wash the coverslip and shim in 1 M KOH, shim side up, for 1 min in a sonicator. We rinse the chips extensively with dH2O and blow dry with compressed N2. Chips can be stored at this point in the process indefinitely.We prepare the top layer of 0.5 mil FEP (C.S. Hyde) by cutting it into 10′′ × 3′′ strips, washing it for 1 min in 70% ethanol and then for 1 min in distilled water. We attach the film to an 8′′ wide vice using tape and stretch it uniaxially to 110% of its original width (Fig. S4†). We secure the stretched FEP to an acrylic frame using double-sided tape and cut away excess FEP. To activate the FEP to allow it to stick with the optical glue we plasma treat it with a Harrick Plasma PDC-32G cleaner on ‘High’ for 3 min. We apply Norland 81 optical adhesive dropwise to the shim and then place the frame with the FEP over the coverslips and sandwich between two glass slides with binder clips. We then place this whole assembly under the UV light and cure at 35 mW for five minutes. As a final hardening step for the glue, we sandwich the frame/FEP/coverslip between two pieces of acrylic, secure with binder clips, and place in a 50 °C oven for 12 h. The next day we cut the FEP to release the assembled device with a scalpel.
Finally, we attach 1/32′′ thick acrylic ports to allow sample loading. Ports were cut on the same laser cutter referenced above at the following settings: 100% power, 15% speed, 500 ppi. We activate the FEP in the plasma cleaner for 3 min before applying glue to the acrylic top. We aspirate glue out of the port holes prior to curing with the UV light for 1 min. Any glue that remains in the holes is removed gently using a 0.55 mm drill bit. The final device is then cured at 50 °C for 12 hours to strengthen the bond between the FEP and ports.
Measurement of optical aberrations
To measure the optical aberration introduced by the microfluidic chip, we compared 3D images of 100 nm diameter fluorescent beads (Fluospheres, Thermo F8801) within the device to those adhered to the outer surface of the FEP film. We constructed microfluidic chips using FEP films of three different thickness: 0.5 mil, 1 mil, and 2 mil corresponding to 12.5, 25, and 50 μm thick respectively. To measure the thickness of the stretched FEP film, we subtracted the z-position of a bead on the top of the film and a bead adhered to the bottom surface of the film, inside of the chip and measured several regions per chip spaced ∼1 mm apart. To measure aberrations, for each chip, we imaged 3D volumes of isolated beads at three separate fields of view and computed FWHM values by least square fitting of 1D Gaussian function to line cuts through the center of the bead along each dimension. OTF comparisons were generated by first registering and averaging the three beads from each condition in real space and then Fourier transforming the image. We measured pupil functions using a Shack–Hartmann wavefront sensor by replacing the tube lens before the camera with a microlens array (Edmund Optics 64-479). The sensor was calibrated by measuring the focus spot displacement for each lenslet in response to preconfigured Zernike mode aberrations applied to a deformable mirror (Alpao DM-69) located at a conjugate rear-pupil image plane. We determined aberrations by imaging isolated fluorescent beads on top of and within the microfluidic chip and computing the SH-lenslet spot displacement. To compensate for variation in the 3D location of each bead, we expanded the observed foci displacement between the reference beads and those within the chip with a Zernike basis (first 26 terms under Noll indexing) corresponding to the calibrated images Shack–Hartmann images and then discarded Zernike terms for tip, tilt, and defocus. The remaining displacement fields were converted to pupil wavefront measurements as described in Liu et al. 2018.4 Transmission attenuation across the pupil due to the FEP film was measured by comparing the total integrated intensity of each lenslet focus relative to those from control beads on top of the film.Simulation of optical aberrations
To simulate optical aberrations when imaging through the FEP film, we used the optical design package Code V (Synopsys). We created a series of geometric models for the FEP film surrounded by PBS corresponding to the angled detection objective, an upright detection objective, and the angled excitation objective. For the angled detection objective, we positioned the film at a 32.4° angle (αdet) to the chief ray and propagated rays covering an angular acceptance of 97.5°, matching the orientation and NA of the lens. For the upright detection objective, we positioned the film parallel to the chief ray and propagated rays covering an angular acceptance of 97.5°. For the excitation objective, we positioned the film at a 57.6° angle (αexc) to the chief ray and propagated rays over an angular acceptance of 53.6°, matching the orientation and NA of the excitation lens. The refractive index of FEP film in the model was 1.345 and of glass was 1.52. The whole system was modelled to be submerged in 1X PBS with a refractive index of 1.335. The spatially dependent aberration across the input pupil was determined using the Pupil Map function in Code V with the tip, tilt, piston, and defocus removed. The spatially dependent attenuation was simulated also using the Pupil Map function, after activating polarized ray tracing and normalized to the maximum ray intensity. For convenience we have included the Code V models for each simulation in the ESI.†Cell culture
U2OS and HeLa cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (VWR) and 1% penicillin/streptomycin (Gibco).Megakaryocytes (MKs) were isolated from murine fetal livers as previously described Shivdasani & Schulze, 2005.30 In brief, fetal livers from E15.5 embryos were homogenized to a single cell suspension and cultured in DMEM (Corning) supplemented with 10% FBS (Gemini), 2 mM Glutamax (Gibco), 1% penicillin/streptomycin (Gibco) and mTPO-conditioned media for 4–6 days. Mature MKs were isolated by a 3%/1.5% BSA (Roche) density gradient.
Widefield imaging for long-term growth
To image the microfluidic device on a widefield microscope we secured the device to the bottom of a glass bottom 6-well plate using PDMS and let it cure at 50 °C for 3 hours. We then coated the internal chamber of the devices with 10 μg mL−1 of human plasma fibronectin (Millipore) at 4 °C overnight. Control glass-bottom six-well plates were also coated with fibronectin. The next day the plates were UV sterilized for 20 min prior to plating cells. We plated ∼20000 cells per device. Cells were plated and maintained post-plating in DMEM (Gibco) supplemented with 10% FBS (VWR), 1% penicillin/streptomycin (Gibco), and 10 mM HEPES (Gibco). Samples were then kept at 37 °C with 5% injected CO2. To allow gas exchange, the chips were not immersed during this step. The HEPES provided additional buffering for the brief (<15 min) amount of time that cells were being imaged daily. To accommodate for the decreased nutrient availability and increased waste accumulation as previously described in microfluidic devices,19 cells required more frequent media changes than conventional cell culture dishes. We tested media changes every 24 hours and every 12 hours and compared growth rate. Cells proliferated more with media changes every 12 hours, (Fig. 3D). Widefield imaging was completed on a Nikon Ti2 Eclipse microscope using a CFI Plan Apochromat 10× objective using brightfield illumination.
LLS imaging
All light sheet imaging was performed at 37 °C on a modified version of the instrument described in Chen et al., 2014.2 Key modifications relevant to this work are the use of a 0.6 NA excitation lens (Thorlabs, TL20X-MPL) and a 1.0 NA detection lens (Zeiss, Objective W “Plan-Apochromat” 20x/1.0, model # 421452-9800) which allowed an effective working distance of 330 μm below the objective surfaces. Detailed imaging conditions for each dataset are described in Table S2.†Imaging megakaryocytes
Polyploid MKs were isolated by a BSA density gradient and incubated with 2 mg mL−1 α-GPIX-Alexa647 antibody for 15 min at 37 °C. Cells were washed with imaging media and resuspended for loading in microfluidic chips. Chips were coated with 100 mg mL−1 fibrinogen (Enzyme Research Laboratories) for 45 min at 37 °C and washed twice with DPBS (Corning) and perfused with Ham's F12 imaging media (Caisson) supplemented with 2% FBS (Gemini) and 15 mM HEPES (Gibco). Cells were plated in prepared chips and incubated at 37 °C, 10% CO2 for 4 hours before imaging. Ports were sealed with bone wax (VWR) and chips were mounted for imaging in Ham's F12 imaging media supplemented with 2% FBS (Gemini) and 15 mM HEPES (Gibco).Imaging fluorescent U2OS cells
Microfluidic devices were coated in 10 μg mL−1 fibronectin at 4 °C overnight. They were then UV sterilized for 20 min prior to plating cells. Cells were plated and maintained in Leibovitz's media (Gibco) supplemented with 10% FBS (VWR) and 1% penicillin/streptomycin (Gibco). The plates were stored at 37 °C without injected CO2. The media in each device and control wells was replaced every 12 hours. Blebbistatin wash-in: prewarmed 50 μM blebbistatin in Leibovitz's media was added to the device after 40 min of imaging. We continued to image cells for 90 min. We then washed out the blebbistatin using prewarmed Leibovitz's media and imaged to observe the recovery of the cells. For long-term imaging, we filled the sample chamber with 1X PBS and sealed the ports of the device using bone wax. For SIM imaging, cells were cultured as above. All images were reconstructed using custom written Matlab code based on the procedure outlined in Gustafsson et al., 2008.31 For comparing the resolution improvement in the SIM image to Wiener deconvolution, the 5 SIM images taken at different lateral phases were summed together and standard Wiener deconvolution was applied using an identical NSR filtering parameter to that applied in the SIM reconstruction.DNA PAINT imaging
Microfluidic chips were coated with 100 nm carboxylated, fluorescent nanodiamonds (NDNV-100 nm-high, Adamás). These served as fiducials for image registration as they are stable and do not photobleach. To make a solution of nanodiamonds we vortexed the stock solution (1 mg mL−1) and made a dilution of 125 ng mL−1 in dH2O. We vortexed this dilution for 1 min, sonicated for 10 min, and vortexed for 1 min. Prior to incubating with nanodiamonds, we first coated the inside of the chip with 0.2% poly-L lysine (Sigma) for 20 minutes, rinsed with diH20 and then dried. We then filled the chip with 200 μL of the solution, allowing the excess solution to come out the opposite port. This generates the most even distribution of fiducials in the chip. We incubated the chips at room temperature for 20 min and then flushed with dH2O, aspirated out remaining liquid and allowed to air dry.We sterilized the coated chips prior to plating cells for at least 20 min with UV light. We then plated ∼35–40000 cells in the chip in DMEM supplemented with 10% FBS (VWR) and 1% penicillin/streptomycin (Gibco) covered the chip with 2 mL of excess media and incubated overnight to allow cells to attach. We left the ports open to allow us to fix the cells and the excess media allowed some nutrient exchange and prevented the cells from drying out. We fixed the cells ∼24 hours after plating using 4% PFA in 1X PBS for 20 min. After fixation, we flushed the chips with 1X PBS quickly three times and then added fresh 1X PBS for 5 min. We then flowed in 200 μL of 0.2% Triton X in 1X PBS to wash out the PBS and make sure that the chip was filled with permeabilization solution. Cells were incubated in this solution at room temperature for 20 min. We washed the chips with 1X PBS as previously described and then added 10% normal goat serum (Thermo) to block non-specific binding and incubated overnight at 4 °C. We then diluted the primary antibodies against lamin A/C (Santa Cruz, sc-376248) and H3K27ac (Abcam, EP16602) at a concentration of 1:200 and 1:100, respectively, in 10% normal goat serum and added 200 μL of this solution to each chip. The chips were incubated in primary antibody solution overnight at 4 °C. The next day, we washed the chips by flushing in 20 μL of 1X washing buffer (Massive Photonics) and let it sit for 5 min. We diluted the secondary antibodies (Massive Photonics, Massive-AB 1-Plex) conjugated to the complimentary DNA oligo (Massive Photonics, ATTO 655) at a concentration of 1:200 and incubated in a humid chamber at room temperature for 60 min. We flushed in 200 μL of 1X washing buffer and let sit for 5 min and repeated this three times. The imaging strands were diluted in imaging buffer at a concentration of 0.75 nM (imager 2, complimentary to anti-rabbit secondary) and 0.2 nM (imager 1, complimentary to anti-mouse secondary) and added to the chips sequentially.
We used SMAP to localize the molecules in all imaging frames using a calibrated astigmatic PSF.27 The calibrated point spread function is generated using the “calibrate3DsplinePSF” module in SMAP. We then applied a difference of Gaussian filter (DoG) with an amplitude of 1.2 and a dynamic threshold with an amplitude of 1.2 and used 3D spline fitting based on the calibrated PSF to fit the centroid of all single molecules. After localizing all molecules in all imaging frames, we then corrected for the drift between frames using the nanodiamond fiducials, which were detected by localizations that persist across more than 10 imaging frames. Fiducials were later discarded from the molecules used for final image reconstruction. The corrected localizations with lateral precision worse than 100 nm, axial precision worse than 200 nm or intensity less than 1000 counts were filtered out to remove spurious localizations. Localizations persistent in consecutive frames with a displacement less than 50 nm are grouped as the same molecule and the average coordinates are used in the final reconstruction. Finally, for image rendering, all localizations are binned into 30 nm by 30 nm squares, with intensity representing the number of localizations in each bin.
Author contributions
Conceived project: Kl. M. H., W. R. L. Designed experiments: R. P. M., E. C. O., Y. S., Kl. M. H., W. R. L. Provided biological advice: R. P. M., E. C. O., A. T. N., W. R. L. Created new reagents: R. P. M. Performed experiments: R. P. M., E. C. O., Y. S., A. T. N., Ka. M. H., W. R. L. Wrote paper with input from all authors: R. P. M., W. R. L. Supervised research: Kl. M. H., W. R. L. Directed research: W. R. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank David Adalsteinsson for assistance with the ImageTank software. We would like to thank Synopsys for academic access to the Code V software and Weimin Shi for his technical assistance with the optical simulations. This work was funded in part by grants from the National Institutes of Health (1R01HL133668 and 1DP2GM136653). WRL acknowledges additional support from the Searle Scholars program, the Beckman Young Investigator Program, and the Packard Fellowship for Science and Engineering.Notes and references
- J. M. Girkin and M. T. Carvalho, J. Opt., 2018, 20, 053002 CrossRef.
- B.-C. Chen, W. R. Legant, K. Wang, L. Shao, D. E. Milkie, M. W. Davidson, C. Janetopoulos, X. S. Wu, J. A. Hammer, Z. Liu, B. P. English, Y. Mimori-Kiyosue, D. P. Romero, A. T. Ritter, J. Lippincott-Schwartz, L. Fritz-Laylin, R. D. Mullins, D. M. Mitchell, J. N. Bembenek, A.-C. Reymann, R. Böhme, S. W. Grill, J. T. Wang, G. Seydoux, U. S. Tulu, D. P. Kiehart and E. Betzig, Science, 2014, 346, 1257998-1-12, DOI:10.1126/science.1257998.
- A. Kumar, Y. Wu, R. Christensen, P. Chandris, W. Gandler, E. McCreedy, A. Bokinsky, D. A. Colón-Ramos, Z. Bao, M. McAuliffe, G. Rondeau and H. Shroff, Nat. Protoc., 2014, 9, 2555–2573 CrossRef CAS PubMed.
- T.-L. Liu, S. Upadhyayula, D. E. Milkie, V. Singh, K. Wang, I. A. Swinburne, K. R. Mosaliganti, Z. M. Collins, T. W. Hiscock, J. Shea, A. Q. Kohrman, T. N. Medwig, D. Dambournet, R. Forster, B. Cunniff, Y. Ruan, H. Yashiro, S. Scholpp, E. M. Meyerowitz, D. Hockemeyer, D. G. Drubin, B. L. Martin, D. Q. Matus, M. Koyama, S. G. Megason, T. Kirchhausen and E. Betzig, Science, 2018, 360, eaaq1392, DOI:10.1126/science.aaq1392.
- M. Roh-Johnson, G. Shemer, C. D. Higgins, J. H. McClellan, A. D. Werts, U. S. Tulu, L. Gao, E. Betzig, D. P. Kiehart and B. Goldstein, Science, 2012, 335, 1232–1235 CrossRef CAS.
- F. Aguet, S. Upadhyayula, R. Gaudin, Y. Chou, E. Cocucci, K. He, B.-C. Chen, K. Mosaliganti, M. Pasham, W. Skillern, W. R. Legant, T.-L. Liu, G. Findlay, E. Marino, G. Danuser, S. Megason, E. Betzig and T. Kirchhausen, Mol. Biol. Cell, 2016, 27, 3418–3435, DOI:10.1091/mbc.E16-03-0164.
- X. Han, Y. Su, H. White, K. M. O'Neill, N. Y. Morgan, R. Christensen, D. Potarazu, H. D. Vishwasrao, S. Xu, Y. Sun, S. Huang, M. W. Moyle, Q. Dai, Y. Pommier, E. Giniger, D. R. Albrecht, R. Probst and H. Shroff, Lab Chip, 2021, 21, 1549–1562, 10.1039/D0LC01233E.
- Z. Lavagnino and D. W. Piston, Microsc. Microanal., 2017, 23, 2322–2323 CrossRef.
- A. M. Valm, S. Cohen, W. R. Legant, J. Melunis, U. Hershberg, E. Wait, A. R. Cohen, M. W. Davidson, E. Betzig and J. Lippincott-Schwartz, Nature, 2017, 546, 162–167 CrossRef CAS.
- N. Yamashita, M. Morita, W. R. Legant, B.-C. Chen, E. Betzig, H. Yokota and Y. Mimori-Kiyosue, J. Biomed. Opt., 2015, 20, 101206 CrossRef.
- B. Eismann, T. G. Krieger, J. Beneke, R. Bulkescher, L. Adam, H. Erfle, C. Herrmann, R. Eils and C. Conrad, J. Cell Sci., 2020, 133, jcs.245043-1-11, DOI:10.1242/jcs.245043.
- B. Qin, C. Fei, A. A. Bridges, A. A. Mashruwala, H. A. Stone, N. S. Wingreen and B. L. Bassler, Science, 2020, 369, 71–77 CrossRef CAS.
- W. R. Legant, L. Shao, J. B. Grimm, T. A. Brown, D. E. Milkie, B. B. Avants, L. D. Lavis and E. Betzig, Nat. Methods, 2016, 13, 359–365 CrossRef PubMed.
- F. Wäldchen, J. Schlegel, R. Götz, M. Luciano, M. Schnermann, S. Doose and M. Sauer, Nat. Commun., 2020, 11, 887 CrossRef PubMed.
- Y.-J. Fan, H.-Y. Hsieh, S.-F. Tsai, C.-H. Wu, C.-M. Lee, Y.-T. Liu, C.-H. Lu, S.-W. Chang and B.-C. Chen, Lab Chip, 2021, 21, 344–354 RSC.
- V. T. Hoang, G. Stępniewski, K. H. Czarnecka, R. Kasztelanic, V. C. Long, K. D. Xuan, L. Shao, M. Śmietana and R. Buczyński, Appl. Sci., 2019, 9, 1145 CrossRef CAS.
- B. M. Hanser, M. G. L. Gustafsson, D. A. Agard and J. W. Sedat, Opt. Lett., 2003, 28, 801–803 CrossRef.
- R. Mcgorty, D. Xie and B. Huang, Opt. Express, 2017, 25, 17798–17810 CrossRef PubMed.
- A. L. Paguirigan and D. J. Beebe, Integr. Biol., 2009, 1, 182–195 CrossRef CAS.
- K. R. Machlus, J. N. Thon and J. E. Italiano, Br. J. Haematol., 2014, 165, 227–236 CrossRef.
- B. J. van Meer, H. de Vries, K. S. A. Firth, J. van Weerd, L. G. J. Tertoolen, H. B. J. Karperien, P. Jonkheijm, C. Denning, A. P. IJzerman and C. L. Mummery, Biochem. Biophys. Res. Commun., 2017, 482, 323–328 CrossRef CAS PubMed.
- K. Ren, W. Dai, J. Zhou, J. Su and H. Wu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8162–8166 CrossRef CAS.
- E. Sano, C. Mori, N. Matsuoka, Y. Ozaki, K. Yagi, A. Wada, K. Tashima, S. Yamasaki, K. Tanabe, K. Yano and Y. Torisawa, Micromachines, 2019, 10, 793–816, DOI:10.3390/mi10110793.
- R. Jungmann, C. Steinhauer, M. Scheible, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Nano Lett., 2010, 10, 4756–4761 CrossRef CAS PubMed.
- F. Schueder, J. Lara-Gutiérrez, B. J. Beliveau, S. K. Saka, H. M. Sasaki, J. B. Woehrstein, M. T. Strauss, H. Grabmayr, P. Yin and R. Jungmann, Nat. Commun., 2017, 8, 2090 CrossRef.
- N. Nunn, N. Prabhakar, P. Reineck, V. Magidson, E. Kamiya, W. F. Heinz, M. D. Torelli, J. Rosenholm, A. Zaitsev and O. Shenderova, Nanoscale, 2019, 11, 11584–11595 RSC.
- J. Ries, Nat. Methods, 2020, 17, 870–872 CrossRef CAS PubMed.
- E. Lubeck, A. F. Coskun, T. Zhiyentayev, M. Ahmad and L. Cai, Nat. Methods, 2014, 11, 360–361 CrossRef CAS.
- P. L. Ståhl, F. Salmén, S. Vickovic, A. Lundmark, J. F. Navarro, J. Magnusson, S. Giacomello, M. Asp, J. O. Westholm, M. Huss, A. Mollbrink, S. Linnarsson, S. Codeluppi, Å. Borg, F. Pontén, P. I. Costea, P. Sahlén, J. Mulder, O. Bergmann, J. Lundeberg and J. Frisén, Science, 2016, 353, 78–82 CrossRef.
- R. A. Shivdasani and H. Schulze, Curr. Protoc. Immunol., 2005, 67, 22F.6.1–22F.6.13 Search PubMed.
- M. G. L. Gustafsson, L. Shao, P. M. Carlton, C. J. R. Wang, I. N. Golubovskaya, W. Z. Cande, D. A. Agard and J. W. Sedat, Biophys. J., 2008, 94, 4957–4970 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00600b |
| This journal is © The Royal Society of Chemistry 2022 |