DOI:
10.1039/D2GC03858G
(Paper)
Green Chem., 2023,
25, 348-356
A stable and recyclable Z-scheme g-C3N4/rGO/BiVO4 heterojunction photocatalyst for site-selective C-3 formylation of indoles with methanol as a formyl source under visible light†
Received
14th October 2022
, Accepted 30th November 2022
First published on 1st December 2022
Abstract
A stable and recyclable Z-scheme g-C3N4/rGO/BiVO4 heterojunction photocatalyst was constructed by a facile two-step hydrothermal procedure. The photocatalyst was used for the synthesis of C-3 formylated indoles from indoles with methanol as an atom-economical formyl source for the first time, and 30% g-C3N4/rGO/BiVO4 exhibited superior photocatalytic efficiency. Quenching experiments showed that the photogenerated electrons and holes were the main photoactive species to achieve the reaction. This formylation reaction has the advantages of exceptional functional group tolerance, reusable photocatalysts, and mild reaction conditions. Gram scale synthesis indicated the future prospects of this strategy for real applications.
Introduction
Indole derivatives are widespread in natural products, drugs and pesticide molecules. Their complex chemical structures and significant pharmacological activities promoted these derivatives to become attractive targets for organic chemists in recent decades.1 C-3 formylated indoles are pivotal intermediates for the synthesis of indole derivatives.2 In addition, the formyl group at C-3 of indole is also a directing group (DG) for C–H bond activation at the C-2,3 C-4,4 or C-5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 position. Therefore, tremendous efforts have been devoted by chemists for the preparation of C-3 formylated indoles. The classical strategy was electrophilic formylation of indoles, including the Vilsmeier–Haack6 and Reimer–Tiemann7 reactions (Scheme 1a). However, a stoichiometric amount of POCl3 or NaOH was used in these methods. To overcome these shortcomings, various alternative formylating reagents were developed for the C-3 formylation of indoles under photochemical, electrochemical and thermochemical conditions. The first type was amines, such as N,N-dimethylaminoethanol,8N-methylaniline,9N,N-dimethylaniline,10 tetramethylethylenediamine (TMEDA),11 hexamethylenetetramine (HMTA)12 and trimethylamine13 (Scheme 1b). The second type was carbonyl compounds, for instance, glyoxylic acid,14 HCOOH,15 HCHO (aq.)16 and N-formylpyrrolidine17 (Scheme 1c). In addition, DMSO,18 PhSCF2H,19 and DMF20 were also used as formylating reagents (Scheme 1d). Despite the significant progress achieved in recent years, most approaches required poor atom-economical formylating reagents, unrecyclable homogeneous transition metal catalysts, external oxidants, and a high reaction temperature, which greatly reduced its economy and practicality. Therefore, finding mild and cost-effective methods for C-3 formylation of indoles is still highly desirable.
5 position. Therefore, tremendous efforts have been devoted by chemists for the preparation of C-3 formylated indoles. The classical strategy was electrophilic formylation of indoles, including the Vilsmeier–Haack6 and Reimer–Tiemann7 reactions (Scheme 1a). However, a stoichiometric amount of POCl3 or NaOH was used in these methods. To overcome these shortcomings, various alternative formylating reagents were developed for the C-3 formylation of indoles under photochemical, electrochemical and thermochemical conditions. The first type was amines, such as N,N-dimethylaminoethanol,8N-methylaniline,9N,N-dimethylaniline,10 tetramethylethylenediamine (TMEDA),11 hexamethylenetetramine (HMTA)12 and trimethylamine13 (Scheme 1b). The second type was carbonyl compounds, for instance, glyoxylic acid,14 HCOOH,15 HCHO (aq.)16 and N-formylpyrrolidine17 (Scheme 1c). In addition, DMSO,18 PhSCF2H,19 and DMF20 were also used as formylating reagents (Scheme 1d). Despite the significant progress achieved in recent years, most approaches required poor atom-economical formylating reagents, unrecyclable homogeneous transition metal catalysts, external oxidants, and a high reaction temperature, which greatly reduced its economy and practicality. Therefore, finding mild and cost-effective methods for C-3 formylation of indoles is still highly desirable.
 |
| | Scheme 1 The protocols for the C-3 formylated indoles with different formyl sources. | |
Methanol can serve as a readily available and atom-economical bulk raw material for various chemical conversions.21 The selective transformation of methanol into value-added chemicals under mild conditions still remains a huge challenge, because it is limited by low selectivity. Even if the conversion can be achieved, it is mostly under harsh conditions. For example, methanol as a C1 source was used for the N-formylation of amines, but the transformation required a transition metal complex as the catalyst, such as a [Ru] complex,22 a [Mn] complex,23 an [Fe] complex,24 and [Cu]/TEMPO.25 The C-2 formylation of quinolines and quinoxalines with methanol was reported by using the catalytic system of TBHP/Fe(acac)3/AcOH.26 Despite significant advances, the fly in the ointment was that these formylation reactions with methanol entirely required heating conditions and unrenewable transition metal catalysts or transition metal catalysts combined with oxidants in a homogeneous catalytic system. It is worth mentioning that all of the formylation reactions of amines involved the conversion of methanol into formaldehyde. Delightfully, recent studies indicated that dehydrogenation of methanol to formaldehyde could also be achieved in heterogeneous photocatalytic systems under mild conditions.27 These heterogeneous photocatalysts could be easily isolated and reused compared with homogeneous catalysts.28 Moreover, the photoexcited electrons and holes (charge carriers) could bypass the external redox reagent to complete the chemical conversion. From the above studies, we conceived an idea that heterogeneous semiconductors may be used as photocatalysts for the formylation of indoles with cheap and easily available methanol under mild conditions.
Generally, few semiconductors simultaneously have high valence band (denoted as CB) and conduction band (denoted as CB) potentials. If such a semiconductor exists, its optical absorption range will be narrow. In addition, the single semiconductor tends to undergo fast recombination of photoexcited charge carriers, which greatly reduces the photochemical conversion efficiency and hinders its practical applications.29 Constructing an artificial Z-scheme heterojunction by coupling two semiconductors for efficient charge carrier separation is a hopeful way to overcome these disadvantages.30 Classically, a Z-scheme heterojunction system contains a reduction semiconductor with a negative conduction band potential and an oxidation semiconductor with a positive valence band potential. Polymeric photocatalysts are cost-effective semi-conductive solid materials derived from polymerizing organic substances,31 such as g-C3N4 (denoted as CN). CN with a moderate band gap (Eg) of approximately 2.7 eV and high chemical and thermal stability is drawing the attention of chemists and materialists in various photocatalytic applications.32 It is a reduction semiconductor due to a relatively negative CB potential of about −1.2 eV vs. NHE.30 Bismuth vanadate (BiVO4 (denoted as BVO)) is a promising oxidation semiconductor due to its narrow Eg of approximately 2.4 eV and a relatively positive VB potential of about 2.8 eV vs. NHE.33 Theoretically, the band gap structure of CN can perfectly match that of BVO to form a Z-scheme heterojunction. As a result, the Z-scheme CN/BVO heterojunction photocatalyst has been constructed for the photodegradation of rhodamine B (RhB)34 and tetracycline hydrochloride (TC).35 Furthermore, according to previous reports,36 reduced graphene oxide (denoted as rGO) exhibited high conductivity, outstanding mechanical properties and a large surface area, and could be used as an electron transport medium between the two semiconductors in a Z-scheme system to improve the electron transport rate and photoreaction efficiency. Hence, the introduction of rGO into the Z-scheme CN/BVO heterojunction to enhance the efficiency of the reaction is desired. Until now, there was only a case that the Z-scheme CN/rGO/BVO heterojunction was used for the photodegradation of TC.37
Taking into account the above aspects, and as part of our continuing interest in photocatalysis,38 herein a new cost-effective, mild approach was developed for the site-selective C-3 formylation of indoles with methanol catalyzed by the Z-scheme CN/rGO/BVO ternary heterojunction photocatalyst under irradiation of a 10 W blue LED at room temperature (Scheme 1e).
Results and discussion
The 30% CN/rGO/BVO (30 wt% of CN/rGO in BVO) ternary composite was prepared through a modified hydrothermal procedure37 (Part 2.3, ESI†). According to the analogous procedure of the 30% CN/rGO/BVO composite, 10 wt% of CN/rGO in BVO and 50 wt% of CN/rGO in BVO composites with different weights of CN/rGO (33.4 mg/1.7 mg and 166.6 mg/8.3 mg) were obtained and denoted as 10% CN/rGO/BVO and 50% CN/rGO/BVO, respectively.
The XRD analyses of the as-prepared specimens are shown in Fig. 1. The characteristic peak at 10.9° could be attributed to the (002) reflection of pure graphene oxide (denoted as GO) sheets. The broad peak at 24.7° could belong to the (002) reflection of pure rGO sheets.39 Two peaks of CN appeared at 13.2° and 27.5°, which were attributed to the interlayer and interplanar packing of aromatic systems from the crystal planes (100) and (002),40 respectively. All of the peaks of BVO could belong to monoclinic BVO (JCPDS 83-1699). The diffraction peak of rGO in CN/rGO, rGO/BVO binary and CN/rGO/BVO ternary composites was not observed obviously; a possible reason could be the low content or diffraction intensity of rGO. To confirm the existence of rGO in the ternary composite, the Raman spectrum of 30% CN/rGO/BVO was tested (Fig. S3†). The D band and G band of rGO were observed at 1353 and 1602 cm−1, respectively. Furthermore, the diffraction peaks attributed to BVO could be found in CN/BVO, rGO/BVO binary and CN/rGO/BVO ternary composites.
 |
| | Fig. 1 XRD patterns for the as-synthesized specimens. | |
The complete morphological properties of the as-obtained specimens can be seen in the TEM and SEM images (Fig. 2). Fig. 2a shows the wrinkled structure of CN. The TEM image of the BVO particles with a smooth surface and large size and a cross-linked structure is shown in Fig. 2b. Fig. 2c shows a typical feature of the layered structure, which was ascribed to the rGO sample. The TEM images in Fig. 2d–f show that CN, rGO and BVO coexisted in the 30% CN/rGO/BVO composite. This result confirmed that the CN/rGO/BVO ternary hybrid was fabricated. The clear lattice fringes with a spacing of 0.43 and 0.29 nm were observed in the HRTEM image of the 30% CN/rGO/BVO composite (Fig. 2g), which was attributed to the (011) and (004) crystal planes of monoclinic BVO. Additionally, the SEM images of 30% CN/rGO/BVO showed that the regular surface of BVO became slightly blurred by hybridizing with CN/rGO (Fig. 2h and i). The SEM and corresponding elemental mapping images of 30% CN/rGO/BVO with the well-distributed C, N, Bi, V and O elements clearly illustrated the simultaneous hybridization of CN, rGO and BVO (Fig. 2j–o).
 |
| | Fig. 2 TEM images of (a) CN, (b) BVO, (c) rGO and (d–g) the 30% CN/rGO/BVO composite. SEM and relevant elemental mapping images (h–o) of the 30% CN/rGO/BVO composite. | |
The surface chemical status of the 30% CN/rGO/BVO composite was investigated by XPS. Fig. 3a shows that the 30% CN/rGO/BVO composite was composed of C, N, Bi, V and O. The three peaks of the C 1s spectrum appeared at 284.8, 285.7 and 288.6 eV (Fig. 3b), and originated from the C–C and C–N bonds of CN, and sp2-hybridized C in the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N bond, respectively.29b As shown in Fig. 3c, the three peaks of the N 1s spectrum at 399.2, 400.5 and 401.4 eV belonged to the CN–C, N–(C)3 and N–H groups, respectively.29b
C–N bond, respectively.29b As shown in Fig. 3c, the three peaks of the N 1s spectrum at 399.2, 400.5 and 401.4 eV belonged to the CN–C, N–(C)3 and N–H groups, respectively.29b
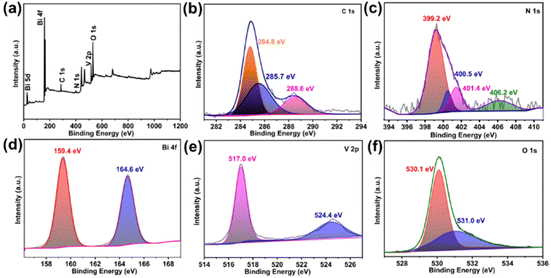 |
| | Fig. 3 XPS spectra of 30% CN/rGO/BVO: (a) survey, (b) C 1s, (c) N 1s, (d) Bi 4f, (e) V 2p and (f) O 1s. | |
Meanwhile, the weak peak at 406.2 eV could belong to the oxidized nitrogen N–‘O’.37 In Fig. 3d, two peaks can be seen at 159.4 eV and 164.6 eV, which were assigned to Bi 4f7/2 and Bi 4f5/2, respectively.35 The V 2p peaks could be deconvoluted to two peaks at 517.0 eV and 524.4 eV, and ascribed to V 2p3/2 and V 2p1/2, respectively (Fig. 3e).41 Furthermore, the two peaks at 530.1 and 531.0 eV were attributed to the O 1s spectrum in Fig. 3f.37 Obviously, these analyses of XRD, TEM, SEM and XPS proved that the as-obtained composite contained CN, rGO and BVO without any impurity.
Fig. 4a and Fig. S4a† show the UV-vis DRS spectra of the as-obtained specimens. The optical absorption edge of the 30% CN/rGO/BVO composite at 506 nm was the longest wavelength compared with the pure semiconductors, binary composites, and 10% and 50% CN/rGO/BVO ternary composites. The optical absorption properties of photocatalysts played a vital role in the analysis of the energy band structure and selection of the light source in photoreactions. The Eg could be calculated according to formula (1).42
where
α,
h,
ν,
A and
Eg denote the photo-absorption coefficient, Planck constant, incident photon frequency, a constant and the band gap, respectively. The values of
n for the indirect transition of CN and the direct transition of BVO were 4 and 1.
37 The
Eg of CN and BVO could be estimated to be 2.76 eV and 2.48 eV, respectively (Fig. S4b
†), and it was consistent with the previous report.
43 The
Eg of 30% CN/rGO/BVO was calculated to be 2.45 eV, originating from the absorption edge at 506 nm. The band edge positions of BVO and CN were calculated according to empirical formulas
(2) and
(3).
Herein,
EVB and
ECB are the edge potentials of VB and CB, respectively.
Ee is the energy of free electrons on the hydrogen scale (4.5 eV). The electronegativity
X values of CN and BVO are 4.66 and 6.04 eV, respectively.
44 Therefore, the
EVB and
ECB of CN were 1.54 and −1.22 eV, respectively. Similarly, the
EVB and
ECB of BVO were 2.78 and 0.30 eV, respectively. To further verify the valence band position, the valence band XPS of the as-prepared CN and BVO was performed. The
EVB of CN and BVO was 1.64 eV and 2.77 eV, respectively (Fig. S4c and d
†). These results were basically consistent with the above-mentioned calculations. The band edge positions of CN and BVO are shown in
Fig. 4b.
 |
| | Fig. 4 (a) UV-vis DRS of the as-synthesized different specimens. (b) The band edge positions of CN and BVO. | |
Photoluminescence (PL) spectra could be applied to show the recombination rate of the photoexcited charge carriers.45 In the PL spectra, the high fluorescence intensity implies that the charge carriers are easy to recombine, and that the photoexcited electrons are short-lived. In other words, a weak intensity of the luminous peak means that the photoexcited electrons have a long life.41 As shown in Fig. 5a and b, compared with pure BVO or CN, rGO modified BVO or CN exhibited a weaker luminous peak intensity, which originated from the fact that rGO could increase the electron transport capacity and retard charge carrier recombination. Obviously, the fluorescence intensity of the 30% CN/rGO/BVO ternary composite was the weakest compared with those of the 10% CN/rGO/BVO, 50% CN/rGO/BVO and other binary composites. It indicated that the 30% CN/rGO/BVO ternary composite had the lowest recombination rate of the charge carriers. Furthermore, photocurrent response measurements were performed to illustrate the separation efficiency of the charge carriers (Fig. 5c). The photocurrent of the 30% CN/rGO/BVO ternary composite was approximately one or two times than that of pure BVO or CN, respectively. This indicated that the construction of the ternary composite was efficient for the separation of the photoexcited charge carriers, and that the photoreactivity of 30% CN/rGO/BVO would be enhanced.
 |
| | Fig. 5 (a and b) PL spectra. (c) Photocurrent response plots of the as-synthesized samples. | |
We initiated our study by using the model reaction of 1-methyl-1H-indole (1a) with methanol in the presence of the 30% CN/rGO/BVO heterojunction photocatalyst at room temperature for 58 h under 10 W blue LED irradiation under a N2 atmosphere. Delightfully, the anticipated target product 2a was isolated in 72% yield (Table 1, entry 1). Next, single variable controlled experiments were performed, and the variables included a catalyst, an amine, an acid and an LED light source, respectively. These results indicated that the 30% CN/rGO/BVO photocatalyst and a 10 W blue LED were necessary conditions (Table 1, entries 2 and 6–9), and that the amine and the acid were also important additives (Table 1, entries 3–6). Thirdly, the yield of 2a was low under an air or O2 atmosphere (Table 1, entries 10 and 11). Due to the more positive potential of O2/O2˙− (ca. −0.33 eV vs. NHE)36b than the ECB (−1.22 eV vs. NHE) of CN, O2 could be reduced to O2˙−, which would consume the photoexcited electrons in the CB of CN. Meanwhile, protons continued to accumulate in the system, which was not conducive to the oxidative dehydrogenation of methanol, so the reaction could not achieve the desired effect under an air or O2 atmosphere. Subsequently, we systematically screened the types and doses of the catalyst (Table S1†), amine (Table S2†), and acid (Table S3†). When 10% CN/rGO/BVO, 30% CN/rGO/BVO and 50% CN/rGO/BVO were used with a dose of 5 mg, the yields of 2a were 65%, 72%, and 59%, respectively (Table S1,† entries 6–8). The 30% CN/rGO/BVO composite exhibited high catalytic activity. Subsequently, the catalytic performance of 30% CN/rGO/BVO with different doses was investigated (Table S1,† entries 9–13). When the dose was 8 mg, an optimal catalytic activity was observed. In addition, when we used tetrahydrofuran, ethanol or benzyl alcohol instead of methanol as the solvent, only a trace amount of 2a was obtained (Table 1, entries 12–14). According to previous works, the MeNHCy additive might be used as the source of the formylation reaction in the absence of methanol. Besides, no indole acetylation or benzoylation products were obtained when ethanol or benzyl alcohol was used as the reaction solvent, respectively. It indicated that the photocatalyst had good selectivity for methanol. Ultimately, the target product 2a was isolated with a yield of 82% under the optimal conditions: 1a (0.4 mmol), CH3NHCy (0.4 mmol), AcOH (0.8 mmol), and 30% CN/rGO/BVO (8 mg) in CH3OH (3 mL) under irradiation of a 10 W blue LED and a N2 atmosphere at room temperature for 58 h.
Table 1 Optimization of the reaction conditionsa
|

|
| Entry |
Variation from the reaction conditions |
Yieldb (%) |
|
Reaction conditions: 1a (0.4 mmol), MeNHCy (0.4 mmol), AcOH (0.8 mmol), 30% CN/rGO/BVO (5 mg), CH3OH (3 mL), 10 W blue LED, N2 balloon, room temperature, 58 h.
Isolated yields.
MeNHCy = N-methylcyclohexylamine.
n.r. = no reaction.
|
| 1 |
None |
72 |
| 2 |
Without 30% CN/rGO/BVO |
Trace |
| 3c |
Without MeNHCy |
16 |
| 4 |
Without AcOH |
Trace |
| 5 |
Without MeNHCy and AcOH |
Trace |
| 6 |
Without 30% CN/rGO/BVO, MeNHCy and AcOH |
Trace |
| 7d |
Without 10 W blue LED |
n.r. |
| 8 |
10 W purple LED instead of 10 W blue LED |
63 |
| 9 |
10 W white LED instead of 10 W blue LED |
24 |
| 10 |
Air instead of a N2 atmosphere |
29 |
| 11 |
O2 instead of a N2 atmosphere |
15 |
| 12 |
THF instead of CH3OH |
Trace |
| 13 |
EtOH instead of CH3OH |
Trace |
| 14 |
Benzyl alcohol instead of CH3OH |
Trace |
|
15
|
8 mg 30% CN/rGO/BVO
|
82
|
Having confirmed the optimal reaction conditions, we next sought to evaluate the scope of indoles with various functional groups (Scheme 2). The indoles or N-substituted indoles with either an electron-donating group (EDG) or an electron-withdrawing group (EWG) reacted successfully with methanol, leading to the target products. To our delight, different alkyl groups, for instance, methyl, ethyl, butyl and isopropyl groups were well suitable, and the products 2a and 2c–2e were realized in moderate to good yields (57–80%). However, the present protocol failed to realize the formylation of indoles when a strong EWG such as t-butyloxy carbonyl (Boc) was attached on the nitrogen atom. Presumably, this might have occurred since the electron density of the indole ring was decreased. A phenyl group blocked at the C-2 position of N-methylindole was also compatible; the product 2b was obtained in 70% yield. This result suggested that the influence of steric hindrance at the C-2 position was negligible. As far as the methyl group at the 4-, 5-, 6- or 7-position in the benzene ring of indoles, the transformation proceeded smoothly to produce the expected products (2j: 46%, 2o: 62%, 2r: 64%, 2w: 79%, 2z: 44% and 2ai: 65%). For the functionalized indoles with halogen (e.g., F, Cl, Br and I) in the benzene ring, the corresponding products were obtained smoothly in moderate to good yields. The yields of the formylated indoles with the same EDG or EWG at the C-7 position were superior to the yields of those at the C-4, C-5 and C-6 positions (e.g., 2tvs.2h; 2uvs.2l and 2q; 2aivs.2z; 2ahvs.2y, 2aj and 2aa). In particular, 4- or 7-cyano-1H-indole, 4-carboxylic acid methyl ester-1H-indole, 5-fluoro-6-bromo-1H-indole, and 7-azaindole could also be subjected to the optimal reaction conditions, and the corresponding products were afforded in moderate yields (2aa: 47%, 2ab: 68%, 2aj: 49%, 2ak: 69% and 2al: 43%). In general, the yields of N-methyl indoles with different substituents for C-3 formylation are higher than those of free NH-indoles.
 |
| | Scheme 2 Substrate scope of the C-3 formylation reaction of indoles. Reaction conditions: 1 (0.4 mmol), MeNHCy (0.4 mmol), AcOH (0.8 mmol), 30% CN/rGO/BVO (8 mg), CH3OH (3 mL), 10 W blue LED, N2 balloon, room temperature, 58 h. Isolated yields. | |
To showcase the applicability of the present method, a gram scale C-3 formylation reaction of indole was performed under modified conditions, and product 2a was isolated with a yield of 62% (Scheme 3a). In addition, we assessed the conversion of 2a into compounds 3 and 4 by the derivatization reaction (Scheme 3b). A condensation reaction occurred, in which 2a reacted with ethyl cyanoacetate, and compound 3 was obtained in 92% yield. Compound 4 was easily obtained in 91% yield by a one-step reaction of 2a, and 4 as a raw material could be further converted into 1-benzene acyl-2-(1-methylindol-3-yl)-benzimidazole derivatives, which are potential tubulin polymerization inhibitors.2a
 |
| | Scheme 3 The gram-scale reaction and the synthetic transformations of product 2a. | |
The reusability of 30% CN/rGO/BVO was investigated using the model reaction under the optimal conditions. After each cycle, the heterogeneous photocatalyst was separated by centrifugation, washed and dried and then reused in the successive C-3 formylation reactions (Part 5, ESI†). No apparent deactivation was observed during the five-run recycling and reuse processes of 30% CN/rGO/BVO (Fig. 6a). To further characterize the crystal structure stability, the XRD patterns of 30% CN/rGO/BVO before and after the fifth formylation reaction were inspected (Fig. 6b). It was clearly observed that their diffraction peaks were almost identical. The results indicated that 30% CN/rGO/BVO has good stability.
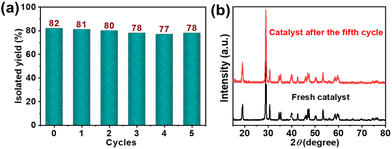 |
| | Fig. 6 (a) Reusability of the 30% CN/rGO/BVO photocatalyst. (b) XRD patterns of the 30% CN/rGO/BVO photocatalyst before and after the 5-cycle reaction. | |
Controlled experiments were performed to explore the tentative mechanism of the formylation reaction, as shown in Scheme 4. When 37% aqueous formaldehyde was used as the formyl source to react with 1a in THF, the desired product 2a was successfully obtained in 32% yield. It illustrated that formaldehyde might be an intermediate. An isotope labeling experiment with CD3OD instead of methanol was conducted, and the corresponding product 2am was verified by 1H NMR and HRMS. The 1H NMR results showed a 67% deuteration ratio of the formyl group in 2a (Scheme 5a). It proved that the deuterated formyl group was derived from deuterated methanol, and also revealed that the hydrogen of the formyl group in 2a originated from methanol. Next, another reaction intermediate species was also revealed by HRMS (Scheme 5b). When 1a reacted with CD3OD, intermediate IV′ was obtained. Analogously, intermediate IV was obtained using methanol instead of CD3OD. Under standard conditions (AcOH with a purity of 99.7% and dry methanol with a purity of 99.9%), 18O-2a and 16O-2a were obtained by using 10 equivalents of external H218O, and the ratio of 18O-2a to 16O-2a was 1.8 to 1 in HRMS (Scheme 5c). This result illustrated that the target product was obtained possibly through a hydrolysis process, and the oxygen of the formyl group stemmed from methanol under external water-free conditions. The effects of different quenchers were investigated to figure out the reactive species under the standard conditions (Table 2). The yield of 2a decreased significantly in the presence of CuCl2, AgNO3, KI, (NH4)2C2O4, or triethanolamine (TEOA) (Table 2, entries 1–5); these results showed that the electrons and holes as the main active species were involved in the reaction. When 2 equivalents of TEMPO or BHT were added, the yield of 2a decreased (Table 2, entries 6 and 7). Meanwhile, a BHT-trapped product was observed in HRMS (Fig. S10†). These analyses implied that a radical process might exist in the reaction. H2 was detected by GC under standard conditions (Scheme 6 and Fig. S11†). The above-mentioned ECB of BVO (0.30 eV vs. NHE) was more positive compared to the reduction potential of H+/H2 (0 eV), hence the electrons in the CB of BVO could not react with the protons to release H2. However, owing to the more negative ECB of CN (−1.22 eV vs. NHE), the protons could be reduced by the electrons in the CB of CN to produce H2. These results show that the electron transfer mode of the 30% CN/rGO/BVO heterojunction followed a Z-scheme rather than type II. In the absence of an amine and acid, the reaction of 5 and H2O led to the target product 2a in 89% yield under standard conditions (Scheme 7). It illustrated that 5 might be an intermediate substance in the conversion. According to Schemes 5b and 7, the conversion of 5 into 2a might have undergone the process of photoredox catalysis and hydrolysis.11a
 |
| | Scheme 4 HCHO (aq.) as a formyl source. | |
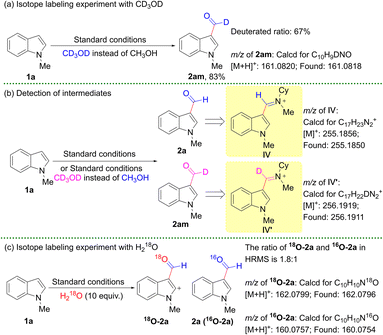 |
| | Scheme 5 Isotope labeling experiments. | |
 |
| | Scheme 6 H2 detection experiment. | |
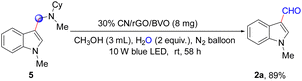 |
| | Scheme 7 Conversion of 5 into 2a. | |
Table 2 Quenching experimentsa
|

|
| Entry |
Quencher |
Yield of 2ab (%) |
Conclusion |
|
Reaction conditions: 1a (0.4 mmol), MeNHCy (0.4 mmol), AcOH (0.8 mmol), 30% CN/rGO/BVO (8 mg), CH3OH (3 mL), quencher (0.8 mmol), 10 W blue LED, N2 balloon, room temperature, 58 h.
Isolated yields.
|
| 1 |
CuCl2 |
Trace |
Electron |
| 2 |
AgNO3 |
Trace |
Electron |
| 3 |
KI |
7 |
Hole |
| 4 |
(NH4)2C2O4 |
17 |
Hole |
| 5 |
TEOA |
16 |
Hole |
| 6 |
TEMPO |
39 |
Radical |
| 7 |
BHT |
33 |
Radical |
On the basis of the above mechanistic investigations and previous reports,11a,46 a plausible mechanism was proposed, as depicted in Fig. 7. Initially, both BVO and CN could be excited by photons to generate electrons and holes, and the photogenerated electrons in the CB of BVO recombined with the holes in the VB of CN via a Z-scheme mode with the assistance of rGO. Simultaneously, electrons with reduction capacity in the CB of CN and holes with oxidation capacity in the VB of BVO were accumulated quickly. Subsequently, methanol was oxidized and dehydrogenated by the holes to form formaldehyde, and the protons were reduced by the electrons to produce H2. With the assistance of an acid, the dehydration reaction between formaldehyde and N-methylcyclohexylamine led to the formation of an iminium ion I. The in situ generated iminium ion I was nucleophilically attacked by the N-methylindole 1a to give the intermediate IIvia the well-known Mannich-type reaction. Next, III was obtained through the deprotonation of II. Afterwards, a photoredox process accompanied by deprotonation of III produced the iminium ion intermediate IV. The intermediate IV underwent hydrolysis to yield the desired 2a, and released the amine to accomplish the catalytic cycle.
 |
| | Fig. 7 A plausible mechanism. | |
Conclusions
In summary, we have successfully constructed a Z-scheme CN/rGO/BVO heterojunction, and developed a useful protocol for site-selective C-3 formylation of indoles with methanol as a formyl source catalyzed by a Z-scheme CN/rGO/BVO heterojunction photocatalyst with H2 evolution under visible light irradiation at room temperature. The photocatalytic effect of 30% CN/rGO/BVO was optimal in the formylation reaction. The recombination rate of the charge carriers for 30% CN/rGO/BVO was the lowest and the separation efficiency was the best, which were in accordance with the effect of photocatalysis. The Z-scheme mode of the heterojunction was confirmed by the rate of H2 evolution in the photocatalytic system. 37 examples of the site-selective C-3 formylated products were obtained from both indoles and N-substituted indoles. This formylation procedure has several advantages including good functional group compatibility, a stable and recyclable Z-scheme heterojunction photocatalyst, methanol as a cheap formyl source, H2 as a by-product, high atom economy, external H2O-free conditions (without external sacrificial reagents of electrons or holes), and mild reaction conditions. The gram-scale synthesis of 2a and the recyclability and reusability of the photocatalyst revealed great potential for practical applications.
Author contributions
Zhenkai Lei carried out most of the experiments and data analyses, and prepared the manuscript. Chenjiang Liu directed the project and supervised the whole experiment, and revised the manuscript. Fei Xue, Bin Wang, Shijie Wang, Yonghong Zhang, Yu Xia and Weiwei Jin provided experimental assistance. All authors discussed and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Program for Tianshan Innovative Research Team of Xinjiang Uygur Autonomous Region (2021D14011), the National Natural Science Foundation of China (21961037 and 21861036), the Key Program of Natural Science Foundation of Xinjiang Uygur Autonomous Region (2022D01D06), the Outstanding Youth Program Natural Science Foundation of Xinjiang Uygur Autonomous Region (2021D01E10), and the Graduate Innovation Project of Xinjiang Uygur Autonomous Region (XJ2021G036).
Notes and references
-
(a) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC
 ;
(b) X. Y. Liu and Y. Qin, Acc. Chem. Res., 2019, 52, 1877–1891 CrossRef CAS PubMed .
;
(b) X. Y. Liu and Y. Qin, Acc. Chem. Res., 2019, 52, 1877–1891 CrossRef CAS PubMed .
-
(a) Y. T. Wang, Y. J. Qin, N. Yang, Y. L. Zhang, C. H. Liu and H. L. Zhu, Eur. J. Med. Chem., 2015, 99, 125–137 CrossRef CAS ;
(b) V. Lakshmi Reddy, P. S. Prathima, V. J. Rao and R. Bikshapathi, New J. Chem., 2018, 42, 20152–20155 RSC ;
(c) C. Y. Wu, M. Hu, Y. Liu, R. J. Song, Y. Lei, B. X. Tang, R. J. Li and J. H. Li, Chem. Commun., 2012, 48, 3197–3199 RSC .
- V. Lanke and K. Ramaiah Prabhu, Org. Lett., 2013, 15, 6262–6265 CrossRef CAS PubMed .
- A. Singh, A. Dey, K. Pal, O. P. Dash and C. M. R. Volla, Org. Lett., 2022, 24, 1941–1946 CrossRef CAS PubMed .
- Z. Y. Wang, R. L. Guo, X. L. Zhang, M. Y. Wang, G. N. Chen and Y. Q. Wang, Org. Chem. Front., 2021, 8, 1844–1850 RSC .
- J. Zheng, L. J. Deng, M. F. Chen, X. Z. Xiao, S. W. Xiao, C. P. Guo, G. K. Xiao, L. L. Bai, W. C. Ye, D. M. Zhang and H. R. Chen, Eur. J. Med. Chem., 2013, 65, 158–167 CrossRef CAS .
- H. Wynberg, Chem. Rev., 1960, 60, 169–184 CrossRef CAS .
- M. Yan, R. C. Hider and Y. M. Ma, Org. Chem. Front., 2019, 6, 1168–1172 RSC .
- W. L. Wu and W. P. Su, J. Am. Chem. Soc., 2011, 133, 11924–11927 CrossRef .
- L. Sun, A. J. Yao, H. Y. Wang, L. M. Zhang, L. Z. Zeng, Z. L. Huang, M. Gao and A. W. Lei, Asian J. Org. Chem., 2018, 7, 1571–1574 CrossRef .
-
(a) X. Li, X. Y. Gu, Y. J. Li and P. X. Li, ACS Catal., 2014, 4, 1897–1900 CrossRef ;
(b) W. J. Zhang, J. T. Tang, W. G. Yu, Q. Huang, Y. Fu, G. C. Kuang, C. Y. Pan and G. P. Yu, ACS Catal., 2018, 8, 8084–8091 CrossRef .
- Q. D. Wang, J. M. Yang, D. Fang, J. M. Ren and B. B. Zeng, Tetrahedron Lett., 2017, 58, 2877–2880 CrossRef CAS .
- F. Ling, D. D. Cheng, T. Liu, L. Liu, Y. J. Li, J. Y. Li and W. H. Zhong, Green Chem., 2021, 23, 4107–4113 RSC .
-
(a) D. Z. Lin and J. M. Huang, Org. Lett., 2019, 21, 5862–5866 CrossRef CAS PubMed ;
(b) V. Dinesh and R. Nagarajan, J. Org. Chem., 2022, 87, 10359–10365 CrossRef CAS .
- S. Isamu, M. Mitsutomo, N. Masashi and M. Teruaki, Heterocycles, 1995, 40, 141–148 CrossRef .
- L. Q. Yang, Z. R. Liu, Y. J. Li, N. Lei, Y. L. Shen and K. Zheng, Org. Lett., 2019, 21, 7702–7707 CrossRef CAS PubMed .
- L. H. Li, Z. J. Niu and Y. M. Liang, Org. Biomol. Chem., 2018, 16, 7792–7796 RSC .
- H. Y. Fei, J. T. Yu, Y. Jiang, H. Guo and J. Cheng, Org. Biomol. Chem., 2013, 11, 7092–7095 RSC .
- N. M. Betterley, S. Kerdphon, S. Chaturonrutsamee, S. Kongsriprapan, P. Surawatanawong, D. Soorukram, M. Pohmakotr, P. G. Andersson, V. Reutrakul and C. Kuhakarn, Asian J. Org. Chem., 2018, 7, 1642–1647 CrossRef CAS .
- H. Y. Wang, X. G. Tong, Y. M. Huo, J. Y. Tang and C. F. Xia, Org. Chem. Front., 2020, 7, 4074–4079 RSC .
-
(a) K. Natte, H. Neumann, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2017, 56, 6384–6394 CrossRef ;
(b) B. B. Asare Bediako, Q. L. Qian, J. J. Zhang, Y. Wang, X. J. Shen, J. B. Shi, M. Cui, G. Y. Yang, Z. Wang, S. R. Tong and B. X. Han, Green Chem., 2019, 21, 4152–4158 RSC ;
(c) G. Choi and S. H. Hong, Angew. Chem., Int. Ed., 2018, 57, 6166–6170 CrossRef PubMed ;
(d) L. Y. Xie, Y. S. Liu, H. R. Ding, S. F. Gong, J. X. Tan, J. Y. He, Z. Cao and W. M. He, Chin. J. Catal., 2020, 41, 1168–1173 CrossRef .
- G. Choi and S. H. Hong, ACS Sustainable Chem. Eng., 2019, 7, 716–723 CrossRef CAS .
- S. Chakraborty, U. Gellrich, Y. Diskin-Posner, G. Leitus, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 4229–4233 CrossRef CAS .
- E. M. Lane, K. B. Uttley, N. Hazari and W. Bernskoetter, Organometallics, 2017, 36, 2020–2025 CrossRef CAS .
- M. C. Pichardo, G. Tavakoli, J. E. Armstrong, T. Wilczek, B. E. Thomas and M. H. G. Prechtl, ChemSusChem, 2020, 13, 882–887 CrossRef PubMed .
- Z. B. Xu and L. Z. Zhang, Org. Biomol. Chem., 2021, 19, 9476–9482 RSC .
-
(a) Z. Z. Li, A. Ivanenko, X. C. Meng and Z. S. Zhang, J. Hazard. Mater., 2019, 380, 120822 CrossRef PubMed ;
(b) N. Nalajala, K. N. Salgaonkar, I. Chauhan, S. P. Mekala and C. S. Gopinath, ACS Appl. Energy Mater., 2021, 4, 13347–13360 CrossRef .
- E. Koranteng, Y. Y. Liu, S. Y. Liu, Q. X. Wu, L. Q. Lu and W. J. Xiao, Chin. J. Catal., 2019, 40, 1841–1846 CrossRef .
-
(a) Z. Qin, W. J. Fang, J. Y. Liu, Z. D. Wei, Z. Jiang and W. F. Shangguan, Chin. J. Catal., 2018, 39, 472–478 CrossRef ;
(b) Y. X. Geng, D. Y. Chen, N. J. Li, Q. F. Xu, H. Li, J. H. He and J. M. Lu, Appl. Catal., B, 2021, 280, 119409 CrossRef .
- W. H. Zhang, A. R. Mohamed and W. J. Ong, Angew. Chem., Int. Ed., 2020, 59, 22894–22915 CrossRef PubMed .
- W. Zhang and R. Cao, Chem. Rev., 2022, 122, 5408–5410 CrossRef PubMed .
-
(a) U. Ghosh and A. Pal, J. Ind. Eng. Chem., 2019, 79, 383–408 CrossRef ;
(b) M. Jourshabani, B.-K. Lee and Z. Shariatinia, Appl. Catal., B, 2020, 276, 119157 CrossRef ;
(c) F. L. Zeng, H. L. Zhu, X. L. Chen, L. B. Qu and B. Yu, Green Chem., 2021, 23, 3677–3682 RSC ;
(d) L. Zhou, Z. Q. Zhang, M. M. Li, Q. Wang, J. N. Gao, K. B. Li and L. Lei, Green Chem., 2021, 23, 9617–9624 RSC ;
(e) B. Dam, A. K. Sahoo and B. K. Patel, Green Chem., 2022, 24, 7122–7130 RSC .
-
(a) L. del Olmo, M. Dommett, I. H. Oevreeide, A. Walsh, D. Di Tommaso and R. Crespo-Otero, J. Mater. Chem. A, 2018, 6, 24965–24970 RSC ;
(b) M. Ou, S. P. Wan, Q. Zhong, S. L. Zhang, Y. Song, L. N. Guo, W. Cai and Y. L. Xu, Appl. Catal., B, 2018, 221, 97–107 CrossRef ;
(c) L. C. Spangler, J. P. Cline, J. D. Sakizadeh, C. J. Kiely and S. McIntosh, Green Chem., 2019, 21, 4046–4054 RSC ;
(d) Q. X. Zhang, Z. Mao, K. X. Wang, N. T. S. Phan and F. Zhang, Green Chem., 2020, 22, 3239–3247 RSC .
- L. Li, M. Mao, X. J. She, J. J. Yi, M. Q. He, L. Pan, Z. G. Chen, H. Xu and H. M. Li, Mater. Chem. Phys., 2020, 241, 122308 CrossRef .
- C. X. Li, H. N. Che, C. B. Liu, G. B. Che, P. A. Charpentier, W. Z. Xu, X. Y. Wang and L. H. Liu, J. Taiwan Inst. Chem. Eng., 2019, 95, 669–681 CrossRef .
-
(a) P. Kumar, C. Joshi, A. Barras, B. Sieber, A. Addad, L. Boussekey, S. Szunerits, R. Boukherroub and S. L. Jain, Appl. Catal., B, 2017, 205, 654–665 CrossRef ;
(b) Y. Yang, J. J. Wu, T. T. Xiao, Z. Tang, J. Y. Shen, H. J. Li, Y. Zhou and Z. G. Zou, Appl. Catal., B, 2019, 255, 117771 CrossRef .
- D. L. Jiang, P. Xiao, L. Q. Shao, D. Li and M. Chen, Ind. Eng. Chem. Res., 2017, 56, 8823–8832 CrossRef .
-
(a) Z. R. Chen, W. W. Jin, Y. Xia, Y. H. Zhang, M. W. Xie, S. C. Ma and C. J. Liu, Org. Lett., 2020, 22, 8261–8266 CrossRef PubMed ;
(b) Z. R. Chen, F. Xue, T. X. Liu, B. Wang, Y. H. Zhang, W. W. Jin, Y. Xia and C. J. Liu, Green Chem., 2022, 24, 3250–3256 RSC ;
(c) Z. R. Chen, F. Xue, Y. H. Zhang, W. W. Jin, B. Wang, Y. Xia, M. W. Xie, A. Abdukader and C. J. Liu, Org. Lett., 2022, 24, 3149–3154 CrossRef PubMed ;
(d) T. Zhang, Y. H. Wang, B. Wang, W. W. Jin, Y. Xia, C. J. Liu and Y.
H. Zhang, Adv. Synth. Catal., 2022, 364, 1962–1968 CrossRef ;
(e) T. X. Liu, F. Xue, Z. R. Chen, Z. Cheng, W. Cao, B. Wang, W. W. Jin, Y. Xia, Y. H. Zhang and C. J. Liu, J. Catal., 2022, 414, 76–83 CrossRef .
- C. Q. Ma, K. Y. Yang, L. L. Wang and X. Wang, J. Appl. Biomater. Funct. Mater., 2017, 15, 1–6 Search PubMed .
- D. L. Jiang, J. J. Zhu, M. Chen and J. M. Xie, J. Colloid Interface Sci., 2014, 417, 115–120 CrossRef .
- F. Chen, Q. Yang, Y. L. Wang, J. W. Zhao, D. B. Wang, X. M. Li, Z. Guo, H. Wang, Y. C. Deng, C. G. Niu and G. M. Zeng, Appl. Catal., B, 2017, 205, 133–147 CrossRef .
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef .
-
(a) K. M. Ji, H. Arandiyan, P. Liu, L. Zhang, J. H. Han, Y. C. Xue, J. G. Hou and H. X. Dai, Nano Energy, 2016, 27, 515–525 CrossRef ;
(b) Z. Y. Zhang, D. L. Jiang, D. Li, M. Q. He and M. Chen, Appl. Catal., B, 2016, 183, 113–123 CrossRef ;
(c) F. Chen, Q. Yang, X. M. Li, G. M. Zeng, D. B. Wang, C. G. Niu, J. W. Zhao, H. X. An, T. Xie and Y. C. Deng, Appl. Catal., B, 2017, 200, 330–342 CrossRef ;
(d) S. Shanavas, S. Mohana Roopan, A. Priyadharsan, D. Devipriya, S. Jayapandi, R. Acevedo and P. M. Anbarasan, Appl. Catal., B, 2019, 255, 117758 CrossRef .
- S. Samanta, S. Khilari, D. Pradhan and R. Srivastava, ACS Sustainable Chem. Eng., 2017, 5, 2562–2577 CrossRef .
-
(a) Y. Y. Bu, Z. Y. Chen and C. J. Sun, Appl. Catal., B, 2015, 179, 363–371 CrossRef ;
(b) Y. C. Huang, W. J. Fan, B. Long, H. B. Li, F. Y. Zhao, Z. Liu, Y. X. Tong and H. B. Ji, Appl. Catal., B, 2016, 185, 68–76 CrossRef .
- C. M. Adolph, J. Werth, R. Selvaraj, E. C. Wegener and C. Uyeda, J. Org. Chem., 2017, 82, 5959–5965 CrossRef PubMed .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Shijie
Wang
a,
Yonghong
Zhang
a,
Shijie
Wang
a,
Yonghong
Zhang