
Poplar lignin structural changes during extraction in γ-valerolactone (GVL)†
Feng
Cheng
 ab,
Sarah
Liu
b,
Steven D.
Karlen
b,
Hoon
Kim
b,
Fachuang
Lu
b,
John
Ralph
bc,
Leida M.
Vázquez Ramos
ab,
George W.
Huber
*a and
James A.
Dumesic
ab
ab,
Sarah
Liu
b,
Steven D.
Karlen
b,
Hoon
Kim
b,
Fachuang
Lu
b,
John
Ralph
bc,
Leida M.
Vázquez Ramos
ab,
George W.
Huber
*a and
James A.
Dumesic
ab
aDepartment of Chemical and Biological Engineering, University of Wisconsin–Madison, Madison, WI 53726, USA. E-mail: gwhuber@wisc.edu
bDepartment of Energy, Great Lakes Bioenergy Research Center, University of Wisconsin–Madison, Madison, WI 53726, USA
cDepartment of Biochemistry, University of Wisconsin–Madison, Madison, WI 53726, USA
First published on 6th December 2022
Abstract
In this paper, we describe an approach for producing both high quality and high quantity of lignin through studying the structural change of lignin during treatment of poplar wood in γ-valerolactone (GVL) for a range of temperatures (from 80 to 120 °C) and reaction time at temperature (from 1 to 24 h). Various techniques, including nuclear magnetic resonance (NMR) spectroscopies (solution- and gel-state 1H–13C 2D HSQC and 31P) and gel-permeation chromatography (GPC) were applied to characterize the lignin structures. As the GVL-extracted lignin yield increases, the level of β-ether units decreases and the level of condensation products increases. The β-ether content, the aliphatic hydroxyl group content, and the molecular weight of the GVL-extracted lignin fractions were close to the poplar lignin from other preparation methods (e.g., enzyme lignin). A two-step hydrolytic process (120 °C, 2 × 15 min) gave a higher lignin yield (56.5% vs. 54.8%) with three times higher β-ether content (31.9% vs. 10.6%) than lignin extracted from a single-step process at 120 °C for 1 h. The results demonstrate that multiple-step cycling of cosolvent-assisted hydrolysis can help preserve more of the virgin ether-bond structures of GVL-extracted poplar lignin. Such a strategy can also be applied to a fully continuous-flow reactor system in future research to further improve both the productivity and quality of GVL-extracted lignin.
Introduction
As the second most abundant component in lignocellulosic biomass (up to one third of biomass weight), lignin cross-links to hemicelluloses to form a biopolymer network interface between amphiphilic cellulose microfibers and hydrophobic lignin.1 Lignin serves as a backbone to support biomass cells, prevent bacterial invasion, and promote water and nutrient transportation.2,3 With the presence of different types of chemical bonds, including van der Waals, hydrogen, and covalent bonds,4 lignin intertwines cellulose strands to generate a strong lignocellulosic matrix, leading to difficulties in fractionation and valorization of each primary biomass component. To improve upgrading efficiency of non-lignin components, lignin is usually removed first due to its high structural heterogeneity, recalcitrance, and inhibition to the enzymatic hydrolysis of polysaccharides.5–8 The impacts of the changes that occur in the lignin structure during biomass fractionation are poorly understood and vary with the deconstruction process.9 During fractionation, the β-aryl ether bonds can cleave, and new C–C bonds can form through multiple reaction pathways (e.g., aldol condensation, Michael addition, and electrophilic aromatic substitution),10–13 as lignin is removed efficiently from biomass at higher temperatures (above 170 °C) using delignification technologies, such as alkaline14 and sulfite pulping,15 ammonia fiber expansion, dilute acid,16 and hydrothermal pretreatments.17–19 The severe conditions of such techniques degrade the hemicellulose and lignin.20Lignin derives primarily from three monolignols, the hydroxycinnamyl alcohols p-coumaryl, coniferyl, and sinapyl alcohols that produce the p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units in the polymer. Apart from being depolymerized into low-value biofuels under severe and corrosive conditions, the existence of specific functional groups (multiple aliphatic and phenolic hydroxyl groups) enables polymerization of lignin into high-value vitrimer adhesives,21 polyesters,22,23 and polyurethane-based foam and cast films.24,25 Thereby, the demand for high-quality lignin products is growing, requiring an improvement in the fractionation of native-like lignin materials.13,26–29 Several lignin fractionation techniques have been explored including using organic solvents, ionic liquids,30 and acids.31 The organic solvent (Organosolv) fractionation methods (e.g., γ-valerolactone (GVL),32 tetrahydrofuran (THF),33 ethanol,34 and acetone) were found under acidic conditions to be the most effective for hydrolysis35–37 and saccharification38 of the polysaccharides, and in the separation and dissolution of lignin from the cell wall matrix,39–41 especially in hardwoods.34
GVL is a promising renewable solvent due to its high lignin solubility and lower partial pressure under reaction condition, GVL treatment of lignocellulosic biomass has been reported to produce higher-quality lignin by preserving more β-ether units and more aliphatic hydroxyl groups,42–45 as well as improving downstream hydrolysis and saccharification of cellulose46 by lowering the activation energy of glycosidic bond cleavage and increasing cellulose accessibility.37,47,48 GVL occurs naturally in fruit, and can be produced from catalytic hydrogenation of cellulose-derived levulinic acid and derivatives.49,50 GVL can be safely transported and stored by virtue of its high boiling point (205 °C),51 high flash point (81 °C), low vapor pressure (<0.1 kPa at 35 °C),52 and low melting point (−31 °C). Since 2014, various papers have been published to report methods of utilizing a combined solvent system including GVL and water with sulfuric acid to fractionate lignin, cellulose, and hemicelluloses.43,53 When heated to over 160 °C, most of the biomass can be dissolved in this mixed solvent system, which also promotes saccharification, and thereby high yields of soluble carbohydrates (70–90%).36,54 The GVL can be recycled, but first it must be separated from the aqueous carbohydrates fraction using liquid-state carbon dioxide, sodium chloride, organic solvents, or other techniques, then the GVL needs to be purified by removing the lignin and other co-products.46,47,55,56 In the last decade, GVL fractionation has been applied to a variety of biomass sources (e.g., bamboo, corn stover, eucalyptus wood, maple wood, poplar wood, and cotton stalk) with high yields of lignin (30–90%) under mild conditions (temperature 100–180 °C and reaction time 0.5–4 h).42,46,57–63
High quality lignin can be extracted at low temperature and long treatment times due to the high susceptibility of β-ether bonds for cleavage.64 However, high lignin yield requires effective dissociation of β-ether bonds within the lignin-cellulose matrix. Thus, further understanding the evolution of lignin structure during GVL pretreatment is needed to design suitable strategies for maximizing lignin production while simultaneously preserving the virgin structure of lignin. In the treatment, lignin structures are usually investigated through understanding their corresponding C–O and C–C interunit bonding patterns, molecular weight distribution, and the contents of hydroxyl groups.59,65
The β-O-4-ether bond is the predominant interunit linkage in lignin (up to 60%).66 Accordingly, the most influential factor during treatment is cleavage of β-O-4-ether bonds,59 which leads to lower quality of lignin-derived products.67,68 In contrast, the minor C–C bonds are found to be the most recalcitrant, and new C–C bonds are prone to being formed as the severe of the treatment increases,69 resulting in the loss of active oxygen-containing functional groups (e.g., aliphatic hydroxyl,59 aldehyde, ether, and methoxy groups).65
The molecular weight of lignin can play a vital role in product properties,70 and it is significantly impacted by the pretreatment conditions. In most cases, the molecular weight of lignin ranges from 0.4 to 16 kDa, with polydispersity of up to 3.38,60,71 In addition, the impact of operational conditions on the quantity of hydroxy groups (∼3.7 mmol g−1)38 remains unclear, yet they are essential for many strategies to synthesize polymers from lignin oligomers. Developing an understanding of the structural and chemical evolution of lignin during fractionation treatment will therefore be important to improve lignin quality and to better integrate lignin into downstream applications.
The objective of this paper is to understand how an effective process could be implemented where both high quality and yield of lignin can be reached. Specifically, the aim is to identify how the extraction process can be modified to produce lignin that retains the structure of the lignin in the poplar wood.
Experimental methods
Materials
Hybrid poplar line NM6 (Populus nigra × Populus maximowiczii) was obtained from the Great Lakes Bioenergy Research Center (GLBRC) in the form of debarked chips. The poplar was milled and sieved through a 5 mm screen prior to any analysis or reaction. GVL, sulfuric acid (2.5 M), and enzymes were purchased from Sigma-Aldrich. All chemicals were used as received without further purification.Experimental procedure of GVL pretreatment
GVL-assisted hydrolysis of poplar chips was conducted in a 450 mL Parr reactor equipped with a mechanical impeller. The reactor was loaded with 33.3 g poplar chips and 300 g of 85 mM sulfuric acid (H2SO4) in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (wt/wt) GVL:H2O to give a 10 wt% solids loading. The reaction was performed at a temperature range of 80–120 °C, with the autogenous pressure applied by liquid (6.9–28.8 psig). The reactor was heated up to reaction temperature at a rate of 4 °C min−1. Once the reaction was completely, the reactor was chilled down to room temperature within 10 min. After the GVL treatment reaction, the hydrolysate was filtered using a Whatman No. 1 filter (mesh size = 10 microns) with aid of house vacuum in the fumehood. The filter cake was rinsed with 100 g of pure GVL at 85 °C to transfer as much GVL-soluble lignin from the solid to the liquid phase as possible. The lignin-rich GVL phase was then diluted 10-fold by pouring it into a beaker containing 3.6-L deionized H2O, and the lignin precipitate was allowed to gradually settle to the bottom overnight. The lignin-rich GVL phase was then centrifuged at 2000 rpm for 4 min, and the lignin precipitate was transferred and collected in a beaker containing 300 mL fresh deionized H2O. To avoid powder aggregation, the collected lignin powder-containing solution was violently mixed until the solution was warmed up to 75 °C on a hot plate. The 75 °C lignin-containing solution was filtrated and the lignin filter cake was rinsed with 200 mL of 75 °C deionized H2O in triplicates to eliminate most of residual GVL from the lignin (targeting <0.3 wt%). Lastly, the GVL-extracted lignin was isolated by vacuum filtration using a Buchner funnel kit and dried in a vacuum oven at 50 °C overnight.
:1 (wt/wt) GVL:H2O to give a 10 wt% solids loading. The reaction was performed at a temperature range of 80–120 °C, with the autogenous pressure applied by liquid (6.9–28.8 psig). The reactor was heated up to reaction temperature at a rate of 4 °C min−1. Once the reaction was completely, the reactor was chilled down to room temperature within 10 min. After the GVL treatment reaction, the hydrolysate was filtered using a Whatman No. 1 filter (mesh size = 10 microns) with aid of house vacuum in the fumehood. The filter cake was rinsed with 100 g of pure GVL at 85 °C to transfer as much GVL-soluble lignin from the solid to the liquid phase as possible. The lignin-rich GVL phase was then diluted 10-fold by pouring it into a beaker containing 3.6-L deionized H2O, and the lignin precipitate was allowed to gradually settle to the bottom overnight. The lignin-rich GVL phase was then centrifuged at 2000 rpm for 4 min, and the lignin precipitate was transferred and collected in a beaker containing 300 mL fresh deionized H2O. To avoid powder aggregation, the collected lignin powder-containing solution was violently mixed until the solution was warmed up to 75 °C on a hot plate. The 75 °C lignin-containing solution was filtrated and the lignin filter cake was rinsed with 200 mL of 75 °C deionized H2O in triplicates to eliminate most of residual GVL from the lignin (targeting <0.3 wt%). Lastly, the GVL-extracted lignin was isolated by vacuum filtration using a Buchner funnel kit and dried in a vacuum oven at 50 °C overnight.
In a two-step GVL-assisted hydrolysis of poplar chips, the fresh poplar was first processed in the acidic GVL and H2O mixture solution at 90 °C for 1.5 h following the same protocol for the one-step treatment. After removal of the lignin-rich GVL solution, the biomass was treated a second time with fresh acidic GVL and H2O at 90 °C for 1.5 h. Collection of the second-stage lignin-rich GVL solution by filtration produced a second lignin sample from the process. These two lignin samples were characterized and compared with a single-step GVL hydrolysis produced at 90 °C and 3 h, as shown in Fig. S1.†
The estimated lignin yield is defined as the percentage of lignin that was successfully transferred from wood chips to GVL phase. The equation for calculating estimated lignin yield is shown below in eqn (1) where the content of lignin in poplar was determined using a facile spectroscopic method known as Cysteine–Assisted Sulfuric Acid (CASA) developed by Lu et al.,72 and the lignin content in the untreated poplar was measured to be 21.78 ± 0.38 wt% by CASA; for reference the Klason lignin content was measured to be 24%.73 All reactions were carried out at least twice. Some solvent was unavoidably lost during the reaction and post-treatment because it may be retained in the swollen wood chips.
 | (1) |
The reaction severity (Rt) is quantified according to eqn (2), modified from a previous study.74 The reaction severity was calculated by integrating the piecewise functions where ti and tf are the initial time and the final time of extraction at desired temperatures, respectively. Time t equals 0 is defined as the starting time for lignin extraction at the lowest temperature Tb (defined as a minimum temperature for lignin extraction, herein, it was assumed to be 75 °C according to the negligible reaction rate observed under such conditions). T(t) = kt + b, in which k represents the heating rate, and b is room temperature during the heating period. As the desired extraction temperature was achieved, T(t) = C, in which C is the final reaction temperature.
 | (2) |
Nuclear magnetic resonance (NMR) spectroscopic analysis of lignin structures
NMR spectra were acquired on a Bruker Biospin NEO 700 MHz spectrometer equipped with a 5 mm QCI 1H/31P/13C/15N cryoprobe with inverse geometry (proton coils closest to the sample), and spectral processing was performed using Bruker's Topspin 4.1.3 (Windows) software.The lignin in the original poplar chips and cellulose-rich solid residues obtained from GVL-assisted hydrolysis were extracted by the following procedure for gel-state NMR experiments. The original dried sample was ground in a Wiley mill and the particle size was reduced to 40 μm. The sample was then rinsed and sonicated using deionized H2O in triplicate, 80% (v/v) ethanol in triplicate, and then 100% acetone sequentially to remove volatile impurities. Once the sample was freeze-dried, it was ball-milled using a Fritsch planetary micro mill PULVERISETTE 7 premium line (Germany) fitted with 50 mL zirconia (ZrO2) grinding jars and 10 × 10 mm ball-bearings, set at 600 rpm. For the species of poplar, the samples were milled for 10 min in quintuplicate with a 5 min pause between each run to avoid excessive heating. In the next step, 5 wt% enzymes, cellulase (Trichoderma viride), and 45 mL sodium acetate buffer (pH = 5.0) were added into samples, mixed and shaken at 250 rpm at 35 °C for 3 days in duplicates to remove most of the cellulose and hemicelluloses from the sample. Lastly, after the enzyme-treated samples were rinsed with distillated water and dried, and premixed DMSO-d6/pyridine-d5 (4:1, vol/vol) was directly added into a 5 mm OD NMR tube for each sample to form a gel with the aid of sonication. More details of this procedure have been described in a published protocol.75,76
For solution-state NMR experiments, the central solvent peaks were used as the internal references (δC/δH: DMSO, 39.5/2.49 ppm). Standard Bruker implementations of the traditional suite of one-dimensional (1D) and two-dimensional (2D) [gradient-selected, 1H-detected; 1H–13C heteronuclear single-quantum coherence (HSQC)] NMR experiments were used for structural elucidation and assignment authentication. HSQC processing used typical matched Gaussian apodization in F2 (LB = −0.5 GB = 0.001) and squared cosine-bell apodization in F1.
NMR spectroscopic quantification of lignin inter-unit linkages
The relative contents of different lignin units, with their characteristic inter-unit linkages, were estimated using the integration of α-C/H correlation peaks verses the integration of aromatic-C/H correlation peaks in the HSQC spectra, according to the method of Li et al.77 as shown in eqn (3)–(6). In these equations, A represents the integration of α-C/H correlation peaks from β-ether (β-O-4) units; B represents the integration of α-C/H correlation peaks of phenylcoumaran (β-5) units; C represents the integration of α-C/H correlation peaks from the resinol (β–β) units; S2,6 represents the integration of the aromatic 2/6 H/C correlation peaks from syringyl units; S′2,6 represents the integration of 2/6 correlation peaks from benzyl-oxidized syringyl aromatic units; SCondensed represents the integration of the 2/6 peaks from the condensed syringyl units; G2 represents the integration of correlation from the aromatic 2-position of guaiacyl aromatic units. | (3) |
 | (4) |
 | (5) |
 | (6) |
Quantitative UV–visible (UV-Vis) spectrophotometric analysis of CASA lignin contents in poplar and cellulose-rich pulp
Prior to quantitative analysis, the original poplar and cellulose-rich pulps obtained from GVL-assisted hydrolysis were dried and ground in a Wiley Mill to decrease the particle size to 40 μm. Next, each ground sample was rinsed and sonicated using deionized H2O in triplicate, 80% (v/v) ethanol in triplicate, and 100% acetone sequentially. The sample was then freeze-dried and prepared for UV-Vis analysis on a Shimadzu UV-1800 UV spectrophotometer. 10 g L-cysteine was dissolved in 100 mL 72% sulfuric acid to prepare the stock solution. The solvent-extracted samples were dissolved in the cysteine stock solution with aid of stirring for 60 min at room temperature to ensure the complete dissolution of the sample. An appropriate amount of deionized H2O was used to dilute the prepared sample solution to tune the lignin concentration and to have a suitable UV-absorbance (<1.0) at 283 nm. The lignin content in the sample was estimated using the Beer–Lambert law and the sample mass (eqn (7)), in which ε is the UV-absorption coefficient of lignin at λ = 283 nm, with the unit of L g−1 cm−1; L is the length of light path (1 cm was used here); and C is the concentration of solubilized lignin. The detailed procedure for lignin estimation were described in the published CASA-lignin protocol.72 The measurements were carried out in triplicate.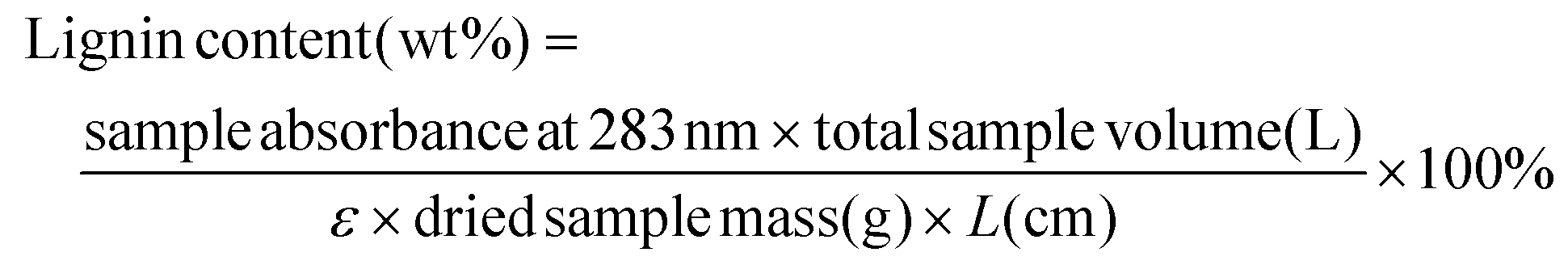 | (7) |
Gel-permeation chromatographic (GPC) analysis of the lignin molecular weight distribution
Analytical GPC was performed on a Shimadzu Prominence LC20 with a photodiode array detector (SPD-M20A). Separation was performed using five Tosoh TSKgel columns connected in series; α-guard column (6.0 mm ID × 30 cm L) into two sequential α-M columns (7.8 mm ID × 30 cm L × 30 μm particle size) and two α-2500 columns (7.8 mm ID × 30 cm L × 7 μm particle size) in a 50 °C oven, with 0.1 M lithium bromide (LiBr) in N,N-dimethylformamide (DMF) flowing at 0.3 mL min−1 for 200 min. The samples were prepared as 1 mg mL−1 solution of lignin in 0.1 M LiBr in DMF. The solutions were filtered through a 0.2 μm PTFE (polytetrafluoroethylene) membrane filter into 250 μL HPLC vials. The molecular weight distribution was calibrated at λ = 270 nm using PSS Polystyrene ReadyCal Standard Set M(p) 250–65000 Da (P/N 76552; Fluka, Sigma/Aldrich, St Louis, MO, USA) and acetovanillone (166 Da).
Quantitative 31P NMR analysis of hydroxy group content in lignin
The hydroxy group content in lignin was quantified using Quantitative 31P NMR analysis on a Bruker Avance 500 MHz spectrometer equipped with 5 mm BBFO probe. To prepare a sample for 31P NMR measurements, 30 mg of lignin sample was dissolved in an 0.5 mL solution with an anhydrous pyridine to chloroform-d ratio of 1.6:1 (vol/vol). The chromium(III) 2,4-pentanedionate and N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide were added into the NMR sample as relaxing reagent and internal standard compound, respectively. Lastly, 0.1 mL 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane was added into the NMR sample as phosphitylation reagent. The pulse program used here had inverse gated decoupling pulse (zgig) on the 31P nucleus. The acquisition time and relaxation delay were 0.8 s and 10 s, respectively. The number of scans was 128. The procedural details are described in the published protocol.78
Results and discussion
Lignin structural evolution during GVL treatment
GVL treatment reactions under different conditions (80–120 °C and 1–24 h) were carried out in duplicate. As shown in Fig. 1, the estimated lignin yield ranged from 10.2 wt% to 54.8 wt% within the GVL treatment conditions used. The estimated lignin yield increased with increasing temperature and/or reaction time, consistent with previous studies.59 However, further extension of reaction time from 12 to 24 h at 90 °C, as shown in Fig. S3† cause a decrease in the lignin yield. This is likely due to condensation reactions of the lignin forming GVL insoluble products. The β-ether unit content decreased from 69.0% to 10.6% as the estimated lignin yield increased. The percentage of the condensed structures (condensed content in Fig. 1) increased as the estimated lignin yield increased. More data obtained under various conditions can be found in Fig. S2 and S3.† Prior studies indicated that the condensation of lignin molecules may be attributed to the stability of native C–C bonds (e.g., β–β and β-5 bonds) as well as the formation of new bonds by electrophilic aromatic substitution by benzylic carbocation intermediates on the electron-rich aromatic rings.18,69,79 Benzylic carbocation intermediates are believed to be generated from dehydration of side-chain hydroxyl groups, and the electron-rich benzene ring is the resonance form of regular lignin units (e.g., guaiacyl unit).79 As shown in Table S1,† the relative content of β–β-coupled units slightly increases with reaction time. | ||
| Fig. 1 GVL-extracted lignin as a function of the lignin yield. Lignin yields were estimated on a basis of lignin quantity in the untreated poplar. All data points were obtained in duplicates using 90 wt% GVL and H2O mixture (9:1, wt/wt) and 100 mM H2SO4 at 80–120 °C and 1–24 h. The numerical values represent β-O-4 bond content. | ||
The molecular weight distribution of GVL lignin is shown in Fig. 2. The GVL lignin had a weight-average molecular weight of 1.96 kDa when produced at 80 °C and 1 h. The 1 kDa MW species, which represent lignin pentamers, was the largest peak under these conditions. Also, under this mild condition, the other smaller peaks at 0.4–0.5 kDa and 0.8 kDa represent lignin dimers and trimer fragments, respectively. As temperature and reaction time increased, these two peaks decreased, whereas a peak at 5 kDa increase. The 5 kDa peak had a degree of polymerization of about 25. The increase in the 5 kDa peak may be because the extraction of larger lignin polymers favors more severe conditions or because the lignin molecules extracted into the GVL liquid phase condensed more significantly under the more severe conditions.
 | ||
| Fig. 2 The molecular weight distribution of GVL lignin obtained under different pretreatment conditions. Both the values in the parentheses and the color darkness represent the condition severity, which was estimated using eqn (2) in the Experimental section. | ||
A slight decrease in weight average molecular weight was observed at 90 °C when the reaction time increased from 12 h to 24 h. This could be due to condensation of heavier lignin forming oligomers that are not soluble in the solvent.
The weight-average molecular weight (Mw) and PDI increased with temperature and time as shown in Table 1 and Fig. 3. The values of Mw, Mn and PDI are consistent with those of organosolv-isolated lignins reported previously.80 However, the number-average molecular weight (Mn) did not show a consistent change with temperature and time. The variance of PDI values under different conditions indicates that the molecular weights of GVL-lignin distribute less uniformly as the temperature becomes higher or reaction time becomes longer.
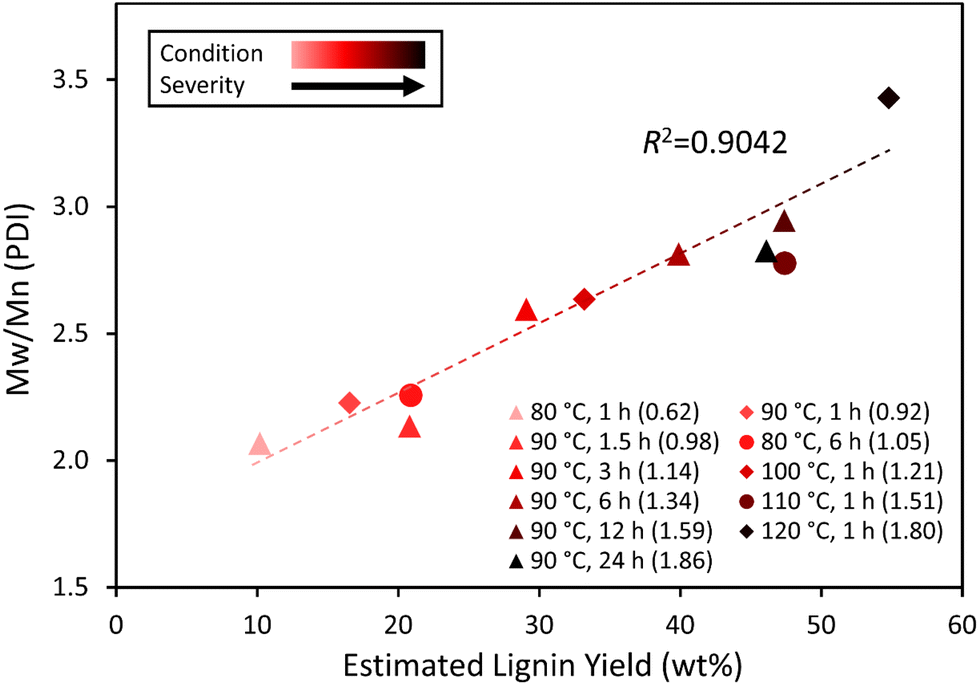 | ||
| Fig. 3 PDI of lignin as a function of the theoretical yield. Both the values in the parentheses and the color darkness represent the condition severity, which was estimated using eqn (2) shown in the experimental section. | ||
| GVL condition | M w (Da) | M n (Da) | M w/Mn (PDI) |
|---|---|---|---|
| 120 °C, 1 h | 2883 | 841 | 3.43 |
| 110 °C, 1 h | 2554 | 920 | 2.78 |
| 100 °C, 1 h | 2583 | 981 | 2.63 |
| 90 °C, 24 h | 2486 | 880 | 2.83 |
| 90 °C, 12 h | 2771 | 941 | 2.94 |
| 90 °C, 6 h | 2776 | 987 | 2.81 |
| 90 °C, 3 h | 2509 | 966 | 2.60 |
| 90 °C, 1.5 h | 1831 | 858 | 2.13 |
| 90 °C, 1 h | 2167 | 973 | 2.22 |
| 80 °C, 6 h | 2151 | 953 | 2.26 |
| 80 °C, 1 h | 1956 | 946 | 2.07 |
The 31P NMR result shows that the hydroxyl groups were clearly affected by temperature and reaction time, in particular, the aliphatic hydroxyl groups on the lignin side chain. Tables S2 and S3† show that the contents of aliphatic, p-hydroxyphenyl, and carboxylic acid hydroxyl groups generally decreased as the condition severity increased, which is consistent with a previous study,59 whereas the C5-substituted group exhibited the opposite trend. The aliphatic, p-hydroxyphenyl, and carboxylic acid hydroxyl groups are converted in the acidic GVL medium to form benzylic carbocation intermediates as precursors to condensed lignin. Tables S2 and S3† show that guaiacyl hydroxyl group seems to be slightly impacted by the reaction condition, perhaps due to slower reaction rate of nucleophilic guaiacyl nuclei during lignin condensation.69 The ratio of aliphatic to phenolic hydroxyl group was highly correlated with the condition severity and lignin yield (Fig. 4). The 2D HSQC NMR, GPC, and 31P NMR results showed that increasing the temperature and time of the reaction gave higher estimated lignin yields, but that the produced lignin was more condensed.
 | ||
| Fig. 4 Aliphatic to phenolic hydroxyl group molar ratio of GVL lignin as a function of estimated lignin yield. Phenolic hydroxyl groups contain C5-substituted, guaiacyl, and p-hydroxyphenyl hydroxyl groups. Both the values in the parentheses and the color darkness represent the condition severity, which was estimated using eqn (2) shown in the experimental section. | ||
Elucidating lignin structural evolution during GVL treatment
All results showed that the GVL lignins extracted under more severe conditions contained fewer β-O-4 bonds, greater Mw and PDI, fewer aliphatic –OH groups, and more phenolic –OH groups than those obtained under milder conditions. However, such evidence may not be sufficient to answer the questions: how does operating condition impact lignin structure evolution during lignin extraction from poplar in GVL? Does operating condition primarily change the structure of the lignin that has been extracted into the GVL liquid phase, leading to loss of β-O-4 bonds and aliphatic –OH groups and formation of condensed lignin molecules? Or does GVL selectively extract the fraction of lignin with different characteristic structures under corresponding operating conditions?A two-step 90 °C GVL hydrolysis was next carried out to understand the influence of operating conditions on lignin structure evolution during GVL treatment. The total lignin yield of the two-step hydrolysis was comparable to that of the single-step process (31.0 wt% vs. 29.1 wt%). Both the first and second lignin samples from the two-step hydrolysis contained higher β-O-4 bond content (51.2% and 50.8% vs. 43.8%) and similar aliphatic hydroxyl groups (3.64 mmol g−1 lignin and 3.60 mmol g−1 lignin vs. 3.58 mmol g−1 lignin) compared to the single-step treatment.
To investigate why the original lignin structure can be preserved better in the two-step hydrolysis procedure, the lignin in the untreated poplar feedstock and in the cellulose-rich solids obtained under the different heating temperatures (80–120 °C) and reaction times (1–24 h), were isolated by enzymatic digestion of the polysaccharides. The isolate their lignins were characterized by 2D HSQC NMR and GPC. Fig. 5 shows a comparison of the 2D HSQC NMR spectra between the lignin in the poplar cellulose-rich solids and the lignin recovered from the GVL extract produced at 90 °C and 1.5 h. It was observed that all characteristic peaks assigned to the original lignin structure, including β-aryl ether (β-O-4), phenylcoumaran (β-5), resinol (β–β), syringyl, guaiacyl, and p-hydroxybenzoate, remain almost unchanged between the lignin from poplar and that from cellulose-rich pulp, whereas these peaks are much weaker for the lignin from the GVL phase. An S′ peak (δC/δH: 106.0/6.49) assigned to condensed form of lignin was found in the GVL extracted lignin, and an extremely weak S′ peak can be found in the cellulose-lignin, whereas no such signal was found in the lignin from the original poplar. Fig. 5 shows that the lignin in cellulose-rich solids are structurally much resembles the original lignin in the poplar feedstock more closely than the lignin that was recovered from the GVL solution.
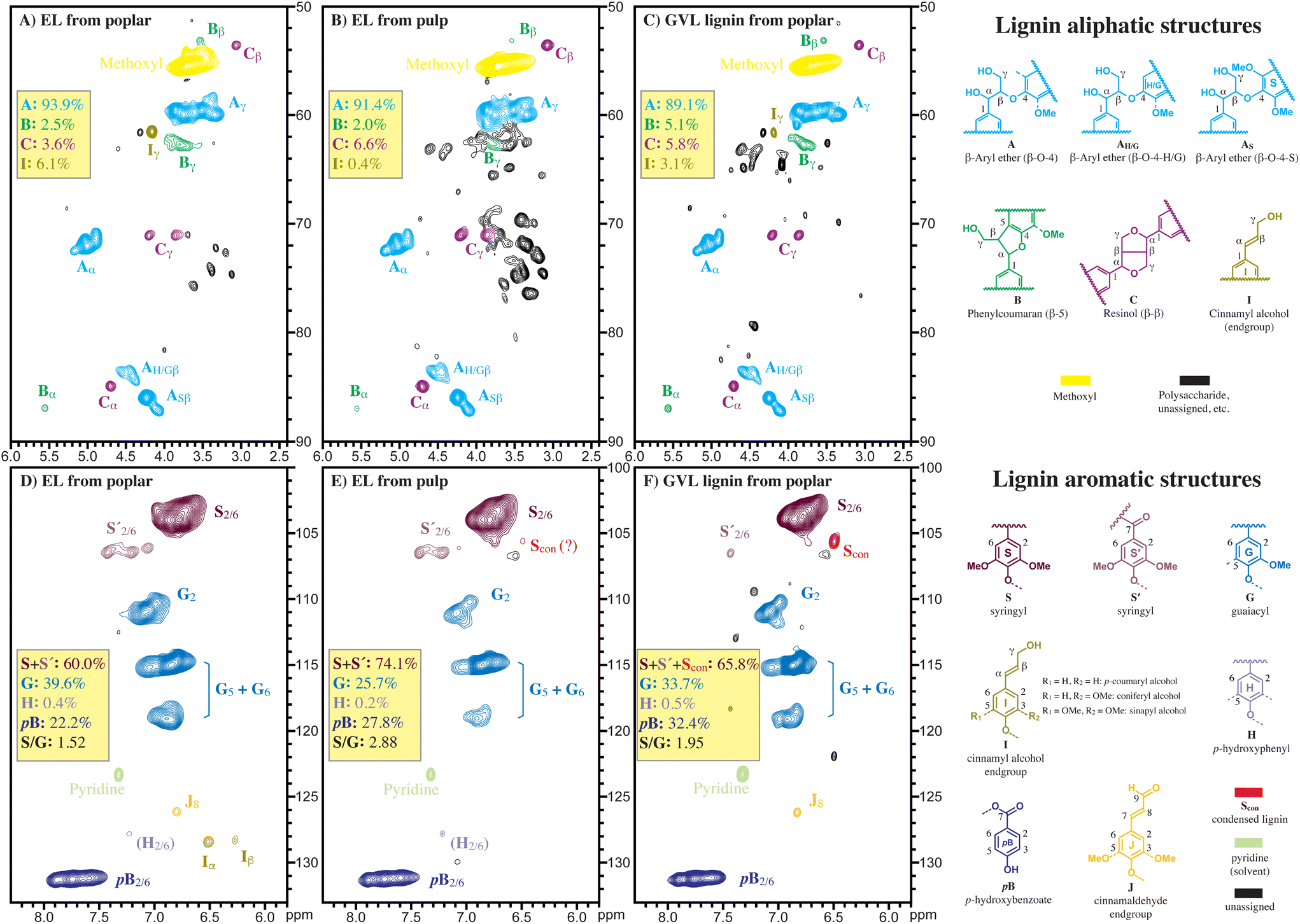 | ||
| Fig. 5 Partial 2D HSQC NMR spectra of (A) and (D) enzyme lignin isolated from poplar feedstock; (B) and (E) enzyme lignin isolated from cellulose-rich pulp; (C) and (F) GVL lignin extracted at 90 °C and 1.5 h. The top three spectra: lignin aliphatic region (δH 2.4–6.0 ppm, δC 50–90 ppm). The bottom three spectra: aromatic region (δH 5.8–8.3 ppm, δC 100–133 ppm). | ||
Fig. 6 shows the β-O-4 bond contents from the lignin in the cellulose-rich solids and the lignin recovered from the GVL liquid phase under different conditions. The β-ether content decreases with reaction time in both solid phase and liquid phase, suggesting that condensation reactions are occurring in both phases. However, the condensation reactions occur more rapidly in the liquid phase. As the temperature and reaction time increase, the β-ether content of the lignin in the solid phase is over three times higher than the GVL extracted lignin in liquid phase (Fig. 6(b)). This suggests that the linkages between lignin, cellulose and hemicellulose stabilize the lignin.4
 | ||
| Fig. 6 The β-O-4 bond content for lignin extracted into GVL liquid phase (blue curve) and that remained in cellulose-rich poplar solid phase (red curve) obtained in (a) different reaction times and (b) different temperatures. All samples were obtained using GVL and H2O mixture (9:1) and 100 mM H2SO4. | ||
Fig. S4† shows that the β-O-4 bond content of lignin in the different product phases during extraction. The lignin in the poplar feedstock has a β-O-4 bond content of 69.0%. After the first 1.5 h hydrolysis, 20.7 wt% linin was extracted into GVL liquid phase with a β-O-4 bond content of 51.2%, while 76 wt% of the lignin remained in poplar with a 63.9% β-O-4 bond content. 3.3 wt% lignin, including the portion soluble in GVL phase, the portion lost during operation, and the portion caused by analytic/measurement error, was not able to be recovered. After the second 1.5 h hydrolysis, another 10.2 wt% lignin was extracted with a β-O-4 bond content of 50.8%, while 55.3 wt% lignin remaining in the poplar still contains 56.4% β-O-4 bond content. The total lignin loss reached 13.8 wt%.
The GPC data show that the lignin in the poplar and cellulose-rich solid phases have similar peak molecular weight (Mw) of ∼5.7 kDa, regardless of operating conditions (as seen in Fig. 7). The lignin polymer remaining in the poplar solid phase consists of 25–30 phenolic monomer units. The lower Mw fraction of the lignin decreases as reaction time increases, as seen by the decreasing of the low Mw shoulder with an apparent Mw of ∼1–2 kDa. This change is particularly noticeable when the reaction time is extended from 12 to 24 hours. This result indicates that the lower MW lignin has a higher GVL solubility than the heavier MW lignin.
 | ||
| Fig. 7 The molecular weight distribution of lignin existed in the feedstock and cellulose-rich pulps obtained under different pretreatment conditions. | ||
GVL hydrolysis of the lignin powders (produced at 90 °C and 1.5 h) with different lignin concentrations was carried out at 90 °C for 1.5 h to understand the role of lignin concentration. Fig. 8 shows that the β-O-4 bond content of lignin in the GVL liquid phase decreases from 39.0% to 31.2% as the lignin concentration increases from 0.25 wt% to 4.18 wt%. In the two-step hydrolysis shown in Fig. S4,† the lignin concentration in the first 1.5 h period was 0.47 wt%, and that in the second 1.5 h period was 0.19 wt%. By comparison, the lignin concentration in the 3 h single-step hydrolysis reached 0.63 wt%. These results indicate that lignin condensation reactions occur in the liquid phase during the lignin extraction. An effective strategy of producing high-quality lignin should rapidly extract the lignin from the biomass and minimize the time that the lignin remains in the solution at reaction temperature (80–120 °C).
 | ||
| Fig. 8 The β-O-4 bond content for lignin dissolved in GVL H2O mixture phase. All samples were obtained using GVL and H2O mixture (9:1) and 100 mM H2SO4 at 90 °C and 1.5 h. The feedstock is the lignin obtained under 90 °C and 1.5 h GVL hydrolysis. | ||
Process improvement for both productivity and quality of lignin
Fig. 9 summarizes the impact of operating conditions on lignin yield and β-O-4 bond content of lignin, which have been comprehensively described in Fig. 1. For instance, the estimated lignin yield increased from 10.2 to 54.8 wt% with increased reaction temperature (from 80 to 120 °C) and constant reaction time at temperature. The estimated lignin yield also increased with increasing reaction time (from 1 to 12 h) for the same reaction temperature (90 °C). The β-O-4 bond content had the opposite trend, decreasing with increased temperature and decreasing with increased reaction time. Fig. 9 also shows that the two-step hydrolysis gave comparable lignin yield to a single-step hydrolysis of the same total extraction time at 90 °C (31.0 wt% vs. 29.1 wt%), but had a much higher β-ether content than the single-step hydrolysis (51.0% vs. 43.8%).Therefore, additional data points were added in Fig. 9, as compared to previous figures, to determine suitable operating condition used for the multiple-step hydrolysis process. | ||
| Fig. 9 The lignin theoretical yield obtained throughout 0.25–24 h at different temperatures. The values in the parentheses represent the β-O-4 bond content. | ||
Fig. 9 indicates that lignin can be extracted faster at higher temperatures. For example, it required 1 h to reach 10.2 wt% of lignin yield at 80 °C, 0.5 h to reach 21.7 wt% at 100 °C, and only 0.25 h to reach 46.9 wt% at 120 °C. In addition, the lignin yields at 80 °C for 6 h, 90 °C for 1.5 h, and 100 °C for 0.5 h were comparable (20.9 wt% vs. 20.8 wt% vs. 21.7 wt%). The lignin yields at 90 °C for 12 h, 110 °C for 1 h, and 120 °C for 0.25 h were also similar (47.4 wt% vs. 47.4 wt% vs. 46.9 wt%). With similar lignin yields, the conditions of 80 °C for 6 h, 90 °C for 1.5 h, and 100 °C for 0.5 h gave β-O-4 bond content of 49.7%, 51.2%, and 50.4%, respectively. The conditions of 90 °C for 12 h, 110 °C for 1 h, and 120 °C for 0.25 h gave lignin yields of 26.4%, 27.1%, and 32.6%, respectively. This behavior indicates that higher temperature and shorter time can achieve similar or even higher lignin yield and higher β-O-4 bond content as compared to lower temperature but longer reaction time. Therefore, 120 °C and 15 min was chosen as the operating condition used for the following multiple-step hydrolysis.
Considering the advantages of the two-step hydrolysis process, a four-step GVL lignin extraction at 120 °C and 15 min was carried out to further improve lignin yield and quality. In each step, the same volume of fresh GVL, H2O, and homogeneous sulfuric acid mixture solution was used to replace the former-steps liquid product containing extracted lignin. The fresh solution was mixed with the solid phase from the former step to extract more lignin. The lignin extracted in each step was quantified and characterized in terms of β-O-4 bond content. Fig. 10 shows that the estimated lignin yield generated in each of the four steps sequentially becomes smaller from dropping down from 46.9 wt% in the first step to 0.4 wt% in the fourth step. The β-O-4 bond content of lignin throughout these four steps decreased more gradually dropping from 32.6% in the first stet to 25.8% in the fourth step. Based on the observation from the former section, much shorter time is needed to extract comparable amount of lignin at higher temperature, as compared to that at lower temperature. Thus, the lignin yield from the first and second 15 min periods accounts for over 97% of total yield from all four steps. The estimated lignin yield from the first two steps was comparable to that from the single-step process at 120 °C and 1 h (56.5 ± 1.6 wt% vs. 54.8 ± 1.2 wt%).
 | ||
| Fig. 10 The estimated lignin yields and corresponding β-O-4 bond contents obtained from the four-step sequential GVL pretreatment at 120 °C for a total reaction time of 1 h. | ||
Further, in the multiple-step process, the β-O-4 content of lignin reached 31.9% in the first two steps, three times higher than the 10.6% observed in for the single-step process (as seen in Fig. 10), and this value is even higher than the β-O-4 bond content (23.7–24.6%) of commercial high-grade lignin (MetGen Co., Finland). This result is consistent with that observed in the two-step hydrolysis process. In the multiple-step process, as the main proportion of extracted lignin is moved out of reaction system at the early stage, the native structure of such lignin is more likely to be preserved due to shorter residence time, as compared to that of lignin from the single-step process. Therefore, based on above results, there is no need to run all four steps at 120 °C. Instead, the first two steps, which only consumed 0.5 h, have shown higher lignin yield and higher β-O-4 bond content than that from the single-step process at 120 °C for 1 h.
The strategy of multi-step GVL pretreatment might not be realistic in a series of batch reactors. However, this approach could be applied in a larger-scale continuous flow reactor by recycling part of the solid GVL-lignin stream back to the reactor. Thus, further research on designing a continuous reactor for GVL extraction and recycling a fraction of the solid biomass product is necessary to explore the feasibility of realistic GVL lignin extraction.
Conclusion
The results of 2D HSQC NMR, GPC, and 31P NMR measurements show that as the reaction severity increased, the β-O-4 content of GVL-extracted lignin and the aliphatic to phenolic hydroxyl group ratio decreased, while the amount of condensed lignin increases and the estimated lignin yield increased. Thus, the chemical structure of the lignin is directly related to the lignin yield. Another important result shows that the β-O-4 content of lignin in the solid poplar was 16 to 230% higher than the lignin in the GVL liquid phase under the same conditions. Additionally, lignin condensation increases as the lignin concentration in GVL liquid phase increased. This demonstrates that lignin undergoes degradation reactions in the liquid phase. All previous observations were confirmed by a two-step process, which gave both higher β-O-4 content and lignin yield than the single-step one. Lastly, based on the studies of the impact of operating condition on lignin extraction, it was found higher temperatures combined with shorter extraction times achieve similar lignin yields with a higher β-O-4 content, compared to lignins isolated at low extraction temperature for longer extraction times. A sequential multistep process was conducted at 120 °C and 15 min (for each step) gave 31.9% β-O-4 content, which was much greater than that (10.6%) obtained from the single-step process at 120 °C and 1 h. These results indicate that a fully continuous flow lignin extraction reactor system equipped with a simple product loop can improve both the productivity and quality of GVL-extracted lignin.Data and materials availability
All data are available in the main text or the ESI.†Author contributions
Feng Cheng contributed conceptualization, data curation, formal analysis, investigation, methodology, resources, validation, visualization, and writing; Sarah Liu and Fachuang Lu contributed methodology, resources, and data curation; Steven D. Karlen contributed data curation, formal analysis, and resources; Hoon Kim and John Ralph contributed NMR methodology, resources, and validation. Leida M. Vázquez Ramos contributed conceptualization. James A. Dumesic and George W. Huber contributed conceptualization, project administration, funding acquisition, supervision, and writing. All authors edited and approve the manuscript.Conflicts of interest
Authors declare that there is no conflict of interest.Acknowledgements
The authors thank Jason Coplien and David Alonso for valuable discussion on strategies for preserving lignin quality in GVL-treatment. This material is based upon work supported by the Great Lakes Bioenergy Research Center, U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018409.References
- L. Salmén and A.-M. Olsson, Interaction between hemicelluloses, lignin and cellulose: Structure-property relationships, J. Pulp Pap. Sci., 1998, 24, 99–103 Search PubMed.
- A. Ferrer, E. Quintana, I. Filpponen, I. Solala, T. Vidal, A. Rodríguez, J. Laine and O. J. Rojas, Effect of residual lignin and heteropolysaccharides in nanofibrillar cellulose and nanopaper from wood fibers, Cellulose, 2012, 19, 2179–2193 CrossRef CAS.
- J. H. Grabber, How do lignin composition, structure, and cross-linking affect degradability? A review of cell wall model studies, Crop Sci., 2005, 45, 820–831 CrossRef CAS.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, Characterization of lignin structures and lignin–carbohydrate complex (LCC) linkages by quantitative 13C and 2D HSQC NMR spectroscopy, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- J. V. Vermaas, L. Petridis, X. Qi, R. Schulz, B. Lindner and J. C. Smith, Mechanism of lignin inhibition of enzymatic biomass deconstruction, Biotechnol. Biofuels, 2015, 8, 1–16 CrossRef PubMed.
- F. Hu, S. Jung and A. Ragauskas, Pseudo-lignin formation and its impact on enzymatic hydrolysis, Bioresour. Technol., 2012, 117, 7–12 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Lignin structure and its engineering, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Biomass recalcitrance: engineering plants and enzymes for biofuels production, Science, 2007, 315, 804–807 CrossRef CAS PubMed.
- A. S. Patri, B. Mostofian, Y. Pu, N. Ciaffone, M. Soliman, M. D. Smith, R. Kumar, X. Cheng, C. E. Wyman and L. Tetard, A multifunctional cosolvent pair reveals molecular principles of biomass deconstruction, J. Am. Chem. Soc., 2019, 141, 12545–12557 CrossRef CAS PubMed.
- Y. Pu, F. Hu, F. Huang, B. H. Davison and A. J. Ragauskas, Assessing the molecular structure basis for biomass recalcitrance during dilute acid and hydrothermal pretreatments, Biotechnol. Biofuels, 2013, 6, 1–13 CrossRef PubMed.
- N. Li, Y. Li, C. G. Yoo, X. Yang, X. Lin, J. Ralph and X. Pan, An uncondensed lignin depolymerized in the solid state and isolated from lignocellulosic biomass: a mechanistic study, Green Chem., 2018, 20, 4224–4235 RSC.
- F. S. Chakar and A. J. Ragauskas, Review of current and future softwood kraft lignin process chemistry, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS.
- S. Constant, H. L. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. Van Es, R. J. Grisel, B. M. Weckhuysen, W. J. Huijgen and R. J. Gosselink, New insights into the structure and composition of technical lignins: a comparative characterisation study, Green Chem., 2016, 18, 2651–2665 RSC.
- Y. Zhao, Y. Wang, J. Zhu, A. Ragauskas and Y. Deng, Enhanced enzymatic hydrolysis of spruce by alkaline pretreatment at low temperature, Biotechnol. Bioeng., 2008, 99, 1320–1328 CrossRef CAS PubMed.
- J. Zhu, X. Pan, G. Wang and R. Gleisner, Sulfite pretreatment (SPORL) for robust enzymatic saccharification of spruce and red pine, Bioresour. Technol., 2009, 100, 2411–2418 CrossRef CAS PubMed.
- S. Tian, W. Zhu, R. Gleisner, X. Pan and J. Zhu, Comparisons of SPORL and dilute acid pretreatments for sugar and ethanol productions from aspen, Biotechnol. Prog., 2011, 27, 419–427 CrossRef CAS PubMed.
- J.-L. Wen, T.-Q. Yuan, S.-L. Sun, F. Xu and R.-C. Sun, Understanding the chemical transformations of lignin during ionic liquid pretreatment, Green Chem., 2014, 16, 181–190 RSC.
- P. K. Deralia, A. Jensen, C. Felby and L. G. Thygesen, Chemistry of lignin and hemicellulose structures interacts with hydrothermal pretreatment severity and affects cellulose conversion, Biotechnol. Prog., 2021, 37, e3189 CrossRef CAS PubMed.
- H. Alizadeh, F. Teymouri, T. I. Gilbert and B. E. Dale, Pretreatment of switchgrass by ammonia fiber explosion (AFEX), Appl. Biochem. Biotechnol., 2005, 124, 1133–1141 CrossRef PubMed.
- X. Ma, X. Yang, X. Zheng, L. Lin, L. Chen, L. Huang and S. Cao, Degradation and dissolution of hemicelluloses during bamboo hydrothermal pretreatment, Bioresour. Technol., 2014, 161, 215–220 CrossRef CAS PubMed.
- A. Moreno, M. Morsali and M. H. Sipponen, Catalyst-Free Synthesis of Lignin Vitrimers with Tunable Mechanical Properties: Circular Polymers and Recoverable Adhesives, ACS Appl. Mater. Interfaces, 2021, 13, 57952–57961 CrossRef CAS PubMed.
- C. Scarica, R. Suriano, M. Levi, S. Turri and G. Griffini, Lignin functionalized with succinic anhydride as building block for biobased thermosetting polyester coatings, ACS Sustainable Chem. Eng., 2018, 6, 3392–3401 CrossRef CAS.
- P. Chee, P. M. Yew, D. Kai and X. Loh, Reinforcement of aligned cellulose fibers by lignin-polyester copolymers, Mater. Today Chem., 2020, 18, 100358 CrossRef CAS.
- M. Zieglowski, S. Trosien, J. Rohrer, S. Mehlhase, S. Weber, K. Bartels, G. Siegert, T. Trellenkamp, K. Albe and M. Biesalski, Reactivity of isocyanate-functionalized lignins: A key factor for the preparation of lignin-based polyurethanes, Front. Chem., 2019, 7, 562 CrossRef CAS PubMed.
- T. Saito, J. H. Perkins, D. C. Jackson, N. E. Trammel, M. A. Hunt and A. K. Naskar, Development of lignin-based polyurethane thermoplastics, RSC Adv., 2013, 3, 21832–21840 RSC.
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, The catalytic valorization of lignin for the production of renewable chemicals, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Valorization of biomass: deriving more value from waste, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- N. A. Nguyen, S. H. Barnes, C. C. Bowland, K. M. Meek, K. C. Littrell, J. K. Keum and A. K. Naskar, A path for lignin valorization via additive manufacturing of high-performance sustainable composites with enhanced 3D printability, Sci. Adv., 2018, 4, eaat4967 CrossRef CAS PubMed.
- D. M. Alonso, S. H. Hakim, S. Zhou, W. Won, O. Hosseinaei, J. Tao, V. Garcia-Negron, A. H. Motagamwala, M. A. Mellmer and K. Huang, Increasing the revenue from lignocellulosic biomass: Maximizing feedstock utilization, Sci. Adv., 2017, 3, e1603301 CrossRef PubMed.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Ionic liquid pretreatment of lignocellulosic biomass with ionic liquid–water mixtures, Green Chem., 2011, 13, 2489–2499 RSC.
- D. Gao, C. Haarmeyer, V. Balan, T. A. Whitehead, B. E. Dale and S. P. Chundawat, Lignin triggers irreversible cellulase loss during pretreated lignocellulosic biomass saccharification, Biotechnol. Biofuels, 2014, 7, 1–13 CrossRef PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass, Green Chem., 2013, 15, 584–595 RSC.
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, THF co-solvent enhances hydrocarbon fuel precursor yields from lignocellulosic biomass, Green Chem., 2013, 15, 3140–3145 RSC.
- X. Pan, N. Gilkes, J. Kadla, K. Pye, S. Saka, D. Gregg, K. Ehara, D. Xie, D. Lam and J. Saddler, Bioconversion of hybrid poplar to ethanol and co–products using an organosolv fractionation process: Optimization of process yields, Biotechnol. Bioeng., 2006, 94, 851–861 CrossRef CAS PubMed.
- M. A. Mellmer, D. M. Alonso, J. S. Luterbacher, J. M. R. Gallo and J. A. Dumesic, Effects of γ-valerolactone in hydrolysis of lignocellulosic biomass to monosaccharides, Green Chem., 2014, 16, 4659–4662 RSC.
- B. Song, Y. Yu and H. Wu, Solvent effect of gamma-valerolactone (GVL) on cellulose and biomass hydrolysis in hot-compressed GVL/water mixtures, Fuel, 2018, 232, 317–322 CrossRef CAS.
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Solvent effects in acid–catalyzed biomass conversion reactions, Angew. Chem., Int. Ed., 2014, 53, 11872–11875 CrossRef CAS PubMed.
- H. Ji, C. Dong, G. Yang and Z. Pang, Valorization of lignocellulosic biomass toward multipurpose fractionation: furfural, phenolic compounds, and ethanol, ACS Sustainable Chem. Eng., 2018, 6, 15306–15315 CrossRef CAS.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. Doherty and I. M. O'Hara, Organosolv pretreatment of plant biomass for enhanced enzymatic saccharification, Green Chem., 2016, 18, 360–381 RSC.
- J. H. Lora and S. Aziz, Organosolv pulping: a versatile approach to wood refining, Tappi, 1985, 68, 94–97 CAS.
- W. Huijgen, A. Smit, P. De Wild and H. Den Uil, Fractionation of wheat straw by prehydrolysis, organosolv delignification and enzymatic hydrolysis for production of sugars and lignin, Bioresour. Technol., 2012, 114, 389–398 CrossRef CAS PubMed.
- H. Q. Lê, Y. Ma, M. Borrega and H. Sixta, Wood biorefinery based on γ-valerolactone/water fractionation, Green Chem., 2016, 18, 5466–5476 RSC.
- J. S. Luterbacher, A. Azarpira, A. H. Motagamwala, F. Lu, J. Ralph and J. A. Dumesic, Lignin monomer production integrated into the γ-valerolactone sugar platform, Energy Environ. Sci., 2015, 8, 2657–2663 RSC.
- X. Meng, S. Bhagia, Y. Wang, Y. Zhou, Y. Pu, J. R. Dunlap, L. Shuai, A. J. Ragauskas and C. G. Yoo, Effects of the advanced organosolv pretreatment strategies on structural properties of woody biomass, Ind. Crops Prod., 2020, 146, 112144 CrossRef CAS.
- B. Seemala, X. Meng, A. Parikh, N. Nagane, R. Kumar, C. E. Wyman, A. Ragauskas, P. Christopher and C. M. Cai, Hybrid catalytic biorefining of hardwood biomass to methylated furans and depolymerized technical lignin, ACS Sustainable Chem. Eng., 2018, 6, 10587–10594 CrossRef CAS.
- L. Shuai, Y. M. Questell-Santiago and J. S. Luterbacher, A mild biomass pretreatment using γ-valerolactone for concentrated sugar production, Green Chem., 2016, 18, 937–943 RSC.
- A. H. Motagamwala, W. Won, C. T. Maravelias and J. A. Dumesic, An engineered solvent system for sugar production from lignocellulosic biomass using biomass derived γ-valerolactone, Green Chem., 2016, 18, 5756–5763 RSC.
- M. J. Selig, S. Viamajala, S. R. Decker, M. P. Tucker, M. E. Himmel and T. B. Vinzant, Deposition of lignin droplets produced during dilute acid pretreatment of maize stems retards enzymatic hydrolysis of cellulose, Biotechnol. Prog., 2007, 23, 1333–1339 CrossRef CAS PubMed.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. Bruijnincx and B. M. Weckhuysen, High performing and stable supported nano-alloys for the catalytic hydrogenation of levulinic acid to γ-valerolactone, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- X. L. Du, Q. Y. Bi, Y. M. Liu, Y. Cao and K. N. Fan, Conversion of biomass–derived levulinate and formate esters into γ–valerolactone over supported gold catalysts, ChemSusChem, 2011, 4, 1838–1843 CrossRef CAS PubMed.
- Z. S. Baird, P. Uusi-Kyyny, J.-P. Pokki, E. Pedegert and V. Alopaeus, Vapor pressures, densities, and PC-SAFT parameters for 11 bio-compounds, Int. J. Thermophys., 2019, 40, 1–36 CrossRef.
- V. Pokorny, V. Stejfa, M. Fulem, C. Cervinka and K. Ruzicka, Vapor pressures and thermophysical properties of ethylene carbonate, propylene carbonate, γ-valerolactone, and γ-butyrolactone, J. Chem. Eng. Data, 2017, 62, 4174–4186 CrossRef CAS.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Nonenzymatic sugar production from biomass using biomass-derived γ-valerolactone, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- R. M. Trevorah, T. Huynh, T. Vancov and M. Z. Othman, Bioethanol potential of Eucalyptus obliqua sawdust using gamma-valerolactone fractionation, Bioresour. Technol., 2018, 250, 673–682 CrossRef CAS PubMed.
- H. Q. Lê, J.-P. Pokki, M. Borrega, P. Uusi-Kyyny, V. Alopaeus and H. Sixta, Chemical recovery of γ-valerolactone/water biorefinery, Ind. Eng. Chem. Res., 2018, 57, 15147–15158 CrossRef PubMed.
- W. Sun, R. Trevorah and M. Z. Othman, Fractionation of spent liquor from organosolv-pretreatment using lignin-incompatible extraction, Bioresour. Technol., 2018, 269, 255–261 CrossRef CAS PubMed.
- W. Fang and H. Sixta, Advanced Biorefinery based on the Fractionation of Biomass in γ–Valerolactone and Water, ChemSusChem, 2015, 8, 73–76 CrossRef CAS PubMed.
- S.-X. Li, M.-F. Li, P. Yu, Y.-M. Fan, J.-N. Shou and R.-C. Sun, Valorization of bamboo by γ-valerolactone/acid/water to produce digestible cellulose, degraded sugars and lignin, Bioresour. Technol., 2017, 230, 90–96 CrossRef CAS PubMed.
- Y.-J. Li, H.-Y. Li, X.-F. Cao, S.-N. Sun and R.-C. Sun, Understanding the distribution and structural feature of eucalyptus lignin isolated by γ-valerolactone/water/acid system, ACS Sustainable Chem. Eng., 2018, 6, 12124–12131 CrossRef CAS.
- M. A. Ahmed, J. H. Lee, A. A. Raja and J. W. Choi, Effects of gamma-valerolactone assisted fractionation of ball-milled pine wood on lignin extraction and its characterization as well as its corresponding cellulose digestion, Appl. Sci., 2020, 10, 1599 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz and J. A. Dumesic, Integrated conversion of hemicellulose and cellulose from lignocellulosic biomass, Energy Environ. Sci., 2013, 6, 76–80 RSC.
- R. M. Trevorah, T. Huynh, R. Brkljača and M. Z. Othman, Structural and Morphological Analysis of Cellulose Pulp Produced from the Fractionation of Eucalyptus obliqua Sawdust Using γ-Valerolactone, ACS Omega, 2021, 6, 4126–4136 CrossRef CAS PubMed.
- M. Wu, Z. Y. Yan, X. M. Zhang, F. Xu and R. C. Sun, Integration of mild acid hydrolysis in γ-valerolactone/water system for enhancement of enzymatic saccharification from cotton stalk, Bioresour. Technol., 2016, 200, 23–28 CrossRef CAS PubMed.
- L. Shuai and B. Saha, Towards high-yield lignin monomer production, Green Chem., 2017, 19, 3752–3758 RSC.
- S. Li, C. Zhao, F. Yue and F. Lu, Revealing structural modifications of lignin in acidic γ-Valerolactone-H2O pretreatment, Polymers, 2020, 12, 116 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph, B. Demedts, R. Vanholme and K. Morreel, Lignin biosynthesis and structure, Plant Physiol., 2010, 153, 895–905 CrossRef PubMed.
- R. Zhang, Q. Du, L. Wang, Z. Zheng, L. Guo, X. Zhang, X. Yang and H. Yu, Unlocking the response of lignin structure for improved carbon fiber production and mechanical strength, Green Chem., 2019, 21, 4981–4987 RSC.
- D. S. Zijlstra, A. de Santi, B. Oldenburger, J. de Vries, K. Barta and P. J. Deuss, Extraction of lignin with high β-O-4 content by mild ethanol extraction and its effect on the depolymerization yield, J. Visualized Exp., 2019, 143, 1–12 Search PubMed.
- K. Shimada, S. Hosoya and T. Ikeda, Condensation reactions of softwood and hardwood lignin model compounds under organic acid cooking conditions, J. Wood Chem. Technol., 1997, 17, 57–72 CrossRef CAS.
- T. Saito, R. H. Brown, M. A. Hunt, D. L. Pickel, J. M. Pickel, J. M. Messman, F. S. Baker, M. Keller and A. K. Naskar, Turning renewable resources into value-added polymer: development of lignin-based thermoplastic, Green Chem., 2012, 14, 3295–3303 RSC.
- X. Jiang, R. H. Narron, Q. Han, S. Park, H. M. Chang and H. Jameel, Tracing Sweetgum Lignin's Molecular Properties through Biorefinery Processing, ChemSusChem, 2020, 13, 4613–4623 CrossRef CAS PubMed.
- F. Lu, C. Wang, M. Chen, F. Yue and J. Ralph, A facile spectroscopic method for measuring lignin content in lignocellulosic biomass, Green Chem., 2021, 23, 5106–5112 RSC.
- J. M. Perez, C. Sener, S. Misra, G. E. Umana, J. Coplien, D. Haak, Y. Li, C. T. Maravelias, S. D. Karlen and J. Ralph, Integrating lignin depolymerization with microbial funneling processes using agronomically relevant feedstocks, Green Chem., 2022, 24, 2795–2811 RSC.
- R. P. Overend and E. Chornet, Fractionation of lignocellulosics by steam-aqueous pretreatments, Philos. Trans. R. Soc., A, 1987, 321, 523–536 CrossRef CAS.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Whole plant cell wall characterization using solution-state 2D NMR, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS PubMed.
- H. Kim and J. Ralph, Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d6/pyridine-d5, Org. Biomol. Chem., 2010, 8, 576–591 RSC.
- Y. Li, B. Demir, L. M. V. Ramos, M. Chen, J. A. Dumesic and J. Ralph, Kinetic and mechanistic insights into hydrogenolysis of lignin to monomers in a continuous flow reactor, Green Chem., 2019, 21, 3561–3572 RSC.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Determination of hydroxyl groups in biorefinery resources via quantitative 31P NMR spectroscopy, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Characterization and analysis of the molecular weight of lignin for biorefining studies, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03446h |
| This journal is © The Royal Society of Chemistry 2023 |