
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable polymer and coating system based on renewable raw materials†
Johannes G. H.
Hermens
 a,
Thomas
Freese
a,
Georgios
Alachouzos
a,
Mathieu L.
Lepage
a,
Keimpe J.
van den Berg
b,
Niels
Elders
b and
Ben L.
Feringa
*a
a,
Thomas
Freese
a,
Georgios
Alachouzos
a,
Mathieu L.
Lepage
a,
Keimpe J.
van den Berg
b,
Niels
Elders
b and
Ben L.
Feringa
*a
aStratingh Institute for Chemistry, Advanced Research Center Chemical Building Blocks Consortium (ARC CBBC), University of Groningen, Nijenborgh 7, 9747 AG, Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bDepartment Resin Technology, AkzoNobel Car Refinishes BV, 2171 AJ, Sassenheim, The Netherlands
First published on 24th November 2022
Abstract
Paints and coatings are widely used in modern society and their current production is mainly dependent on the petrochemical industry. The establishment of processes using sustainable alternative monomers based on biorenewable resources, using exclusively biobased reagents and green synthetic transformations are highly warranted for a more sustainable future. Herein, we report on a sustainable polymer and coating system based on the monomer methoxybutenolide, a biobased acrylate alternative. Methoxybutenolide and the comonomer dodecyl vinyl ether are synthesized from biobased platform chemicals using the environmentally benign synthetic transformations photooxygenation and vinylation. For the photooxygenation, a biobased photosensitizer was developed showing high quantum yields. The monomers were copolymerized using biomass derived (photo)initiators to yield fully biobased polymers and coatings with properties comparable to acrylate based coatings.
Introduction
Over the past decades, increased efforts have been taken to advance chemical science to a sustainable future.1,2 Technologies revolved around biobased chemicals and synthesis methods adherent to the Twelve Principles of Green Chemistry have been developed to be less dependent on petrochemical based materials and processes.3–5 In particular, polymers and coatings,6 which are used for preservation, protection and decoration of virtually all man-made materials used in society, are frequently based on olefin-derived acrylate monomers, whose multi-million ton scale production is heavily dependent on fossil fuels.7 These facts, paired with the undesirable emission of COx and other greenhouse gases in the conversion of finite fossil fuel feedstocks into molecules of higher application, highly warrant the development of sustainable alternatives and greener synthetic procedures.8,9 The coating industry successfully implemented the latter by the invention of new technologies such as acrylic dispersions in emulsion polymerization,10,11 solvent-free radiation-cured acrylics12 and acrylic powder coatings13 as green alternatives to solvent-borne coatings. Besides the sustainable aspect of less environmental pollution, these innovations significantly contributed to reduced risk of health hazards and improved industrial occupational safety, strengthening the Responsible Care initiative.14Now, development of alternative sustainable processes is generally led by strategies such as opening new biobased synthetic routes to commodity and specialty chemicals or through establishing novel biobased building blocks for the replacement of these chemicals.15 Despite significant progress, major challenges pertain to the development of completely sustainable processes.6 An integrated approach considering the use of biorenewable resources, biobased reagents, sustainable catalysis and green solvents is highly desired. The inherent higher complexity of bio renewable resources allows for a magnitude of different pathways to unique platform chemicals with different functional groups, whereas the development of biobased alternatives might lead to unprecedented reactivity.16 The exploration and progress in the field of green chemistry towards the utilization of biomass-derived building blocks to replace the non-sustainable acrylate monomers has been successfully attained. Recently, advances were made towards the development of acrylate alternatives, and the free radical polymerization thereof, derived from the platform chemicals levoglucosenone,17 tulipalin A,18 itaconic acid,19–21 itaconic anhydride,22 muconic acid,23,24 and levulinic acid.25 Implementing these acrylate alternatives results in promising polymer properties such as higher glass transition temperatures, higher stability and post-polymerization functionalization.
Previously we have shown the development of an attractive acrylate alternative based on a butenolide motif, derived from the platform chemical furfural.26 Furfural is converted quantitatively into the non-substituted acrylic acid alternative hydroxybutenolide via a [4 + 2] cycloaddition with singlet oxygen (1O2), which is generated via photosensitization. Hydroxybutenolide was readily converted into a set of various alkoxybutenolides (linear and branched) by heating it in the presence of an appropriate alcohol. The alkoxybutenolides were used in combination with comonomers as vinyl esters and in particular vinyl ethers to yield promising polymeric materials. High reaction rates were found for the combination of butenolides and vinyl ethers; hence a divinyl ether was chosen as a suitable crosslinker for the formation of coatings. UV-curing of the butenolide divinyl ether mixture led to excellent hard coatings with tunable material properties depending on the substituents at the butenolide, showing their effectiveness as acrylate replacements and high versatility for coatings.
Despite the merits of the various reported systems, the resulting biobased polymers often do not consist of 100% biobased carbon content due to the use of petrochemically derived comonomers, reagents for derivatization or additives.
In the design reported herein, we take an integrated approach, using renewable resources, sustainable transformations, solvents and reagents, to develop methodology for an integrated biobased polymer and coating system containing a significant amount of biobased carbon. All components used, including catalysts and polymerization initiators, originate from platform chemicals that are readily derived from lignocellulosic biomass. We report on the monomers methoxybutenolide and dodecyl vinyl ether, obtained from the biomass derived chemicals furfural27 and dodecanol,28 made in a sustainable fashion through catalytic photooxygenation and vinylation using calcium carbide, respectively. Building on the already sustainable photooxidation29 of furfural to hydroxybutenolide, the photosensitizer was replaced by a biobased, blue-light activated counterpart derived from the top value added platform chemicals levulinic acid and hydroxymethylfurfural (HMF).30 The free radical polymerization was then carried out using the biobased radical initiator ascaridole (derived from natural oil terpinene via photooxygenation),31 in combination with the sustainable solvent gamma-valerolactone (GVL, derived from levulinic acid),32 to ultimately yield fully biobased polymers with high conversions. Finally, using the same approach for dodecyl vinyl ether, a biobased crosslinker was synthesized from 1,4-butanediol.33 Together with a vanillin-derived UV photoinitiator, methoxybutenolide and the biobased crosslinker were cured, which resulted in a hard, transparent and fully biobased coating. Overall, the strategy reported herein demonstrates the viability of a polymerization platform where all components are derived from sustainable sources (Scheme 1).
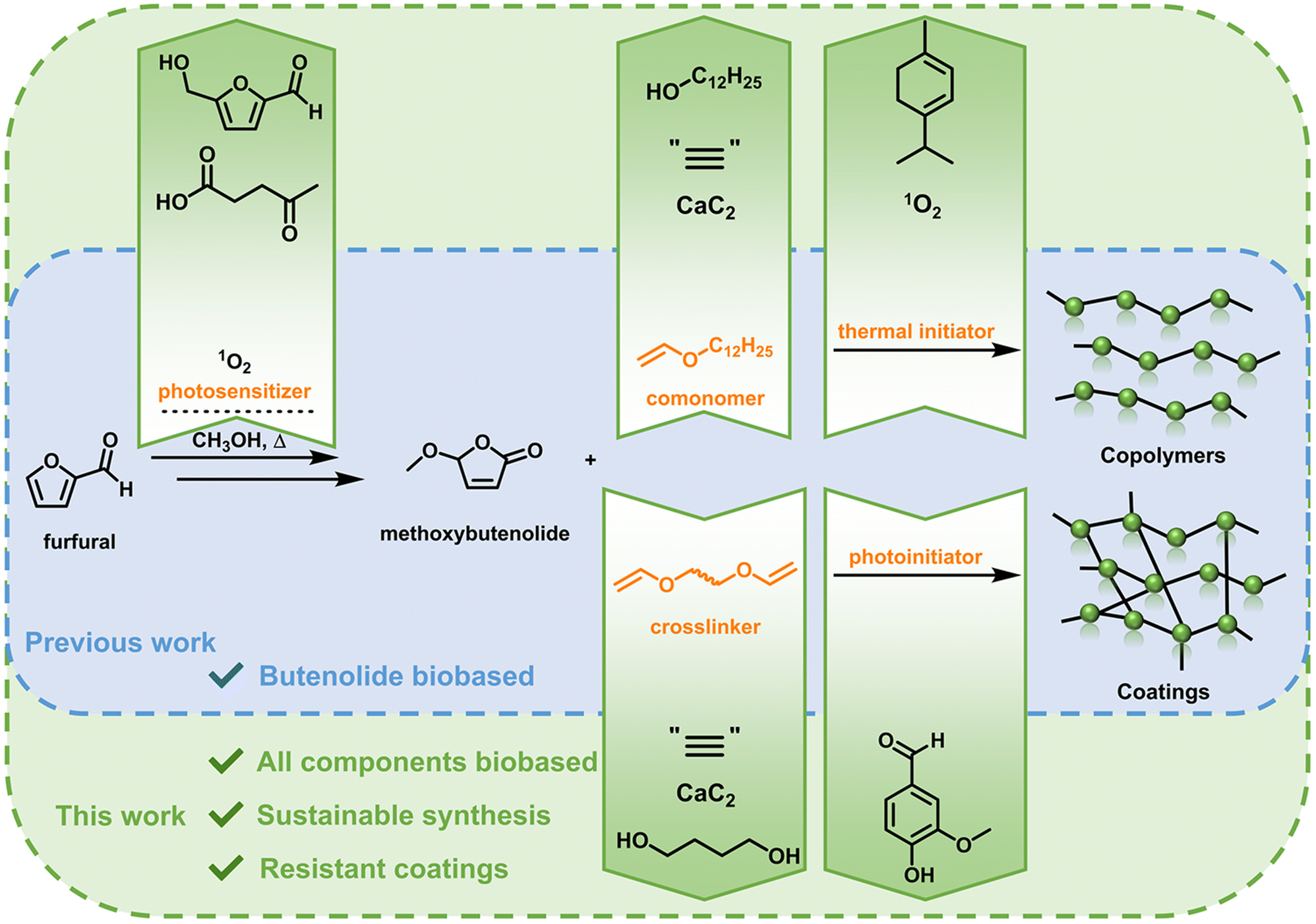 | ||
| Scheme 1 Previously described synthesis of partially biobased polymers and coatings (blue). Integrated biobased polymer and coating system with all components derived from biorenewable resources (green). | ||
Results and discussion
At the outset, to develop entirely biobased polymers and coatings, we sought to design a sustainable photosensitizer to transform furfural into hydroxybutenolide using catalytic photooxygenation with molecular oxygen, providing the precursor for the butenolide monomer. Recently, Mascal et al. described the synthesis of a biobased organic dye, which we will now refer to as FLY 450 (Furan Lactone Yellow with λmax = 450 nm), with a high molecular absorptivity (ε) derived from the platform chemicals HMF and levulinic acid (Fig. 1A).34 We envisioned that such biobased lactone-substituted furans might well generate singlet oxygen under aerobic irradiation, as similar organic dyes containing strong chromophores and high ε values often exhibit photosensitization properties.35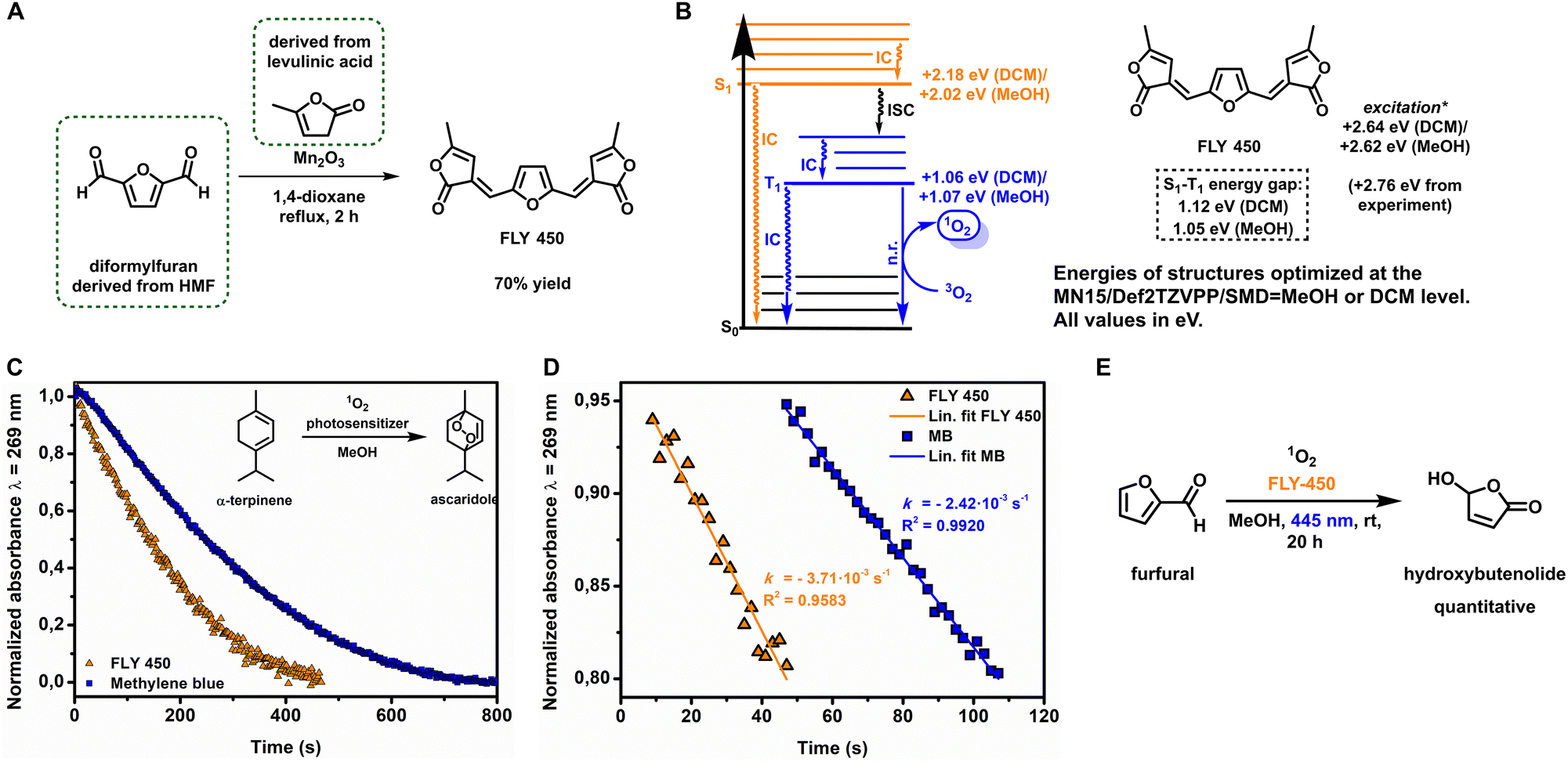 | ||
| Fig. 1 (A) Synthesis of FLY 450 from the biobased chemicals diformylfuran and α-angelicalactone. (B) DFT/TD-DFT calculation results, conducted at the MN15/Def2TZVPP/SMD = DCM/MeOH level, with the S1–T1 energy gap ΔEST being derived. (C) Decay of the singlet oxygen scavenger α-terpinene, followed by UV-Vis spectroscopy in methanol at λ = 269 nm using FLY 450 (orange triangle) or methylene blue (blue square). (D) Rate of 1O2 production (s−1) of FLY 450 and methylene blue in methanol as a function of the decay of the scavenger, followed by UV-Vis spectroscopy at λ = 269 nm, determined from 95% to 80% conversion of the scavenger using FLY 450 (orange triangle) or methylene blue (blue square). Photooxygenation conditions: α-terpinene (40 μM), photosensitizer (1 μM), pre-oxygenated methanol, 293 K, and λirr = 445 nm. (E) Photooxidation of furfural employing FLY 450 (5 mol%) as the photosensitizer yielding hydroxybutenolide quantitatively. | ||
To interrogate the potential of such dyes, we turned to computational chemistry to predict the properties of FLY 450. We hypothesized that, in order for FLY 450 to generate singlet oxygen upon irradiation, it must sufficiently populate its triplet T1 excited state after intersystem crossing (ISC) from the singlet S1 excited state initially accessed after absorption of a photon. Since the rate of ISC is inversely proportional to the obtained S1–T1 energy gap ΔEST, this value was examined using density functional theory (DFT) or time-dependent density functional theory (TD-DFT) methods.36 The FLY 450 S0, S1 and T1 energies, as well as the vertical TD-DFT excitation energies were obtained at the MN15/Def2TZVPP/SMD = DCM or MN15/Def2TZVPP/SMD = MeOH level (Fig. 1B and ESI Table S1†).37–39 From these calculations we found promising ΔEST values of 1.12 eV (DCM) or 1.05 eV (MeOH), suggesting that ISC to T1 should be facile for both solvent systems. Notably, the lower ΔEST computed for more polar MeOH (due to a more stabilized S1 state in simulated MeOH versus simulated DCM) suggests that one might predict fast intersystem crossing, and therefore strong photosensitization activity for FLY 450 in polar, protic media. Known photosensitizer dyes (e.g. methylene blue) typically perform slightly better in less polar or aprotic media,40 and these computational predictions encouraged us that FLY 450 would indeed exhibit high photosensitization activity, even in green solvents such as MeOH, or even with water as the co-solvent.
FLY 450 was synthesized in 70% yield via condensation of α-angelicalactone with diformylfuran (Fig. 1A).34 First, we were encouraged to find that the experimental values for the FLY 450 absorption wavelength λmax = 450 nm (+2.76 eV) were in very strong agreement with the computed vertical excitation energies of +2.78 or +2.79 eV, simulated in DCM or MeOH, respectively (Fig. 1). Next, in order to evaluate the oxygen photosensitization properties, FLY 450 was used in catalytic amounts (1 mol%) in the photooxidation of furfural to hydroxybutenolide in methanol using our previously described batch rotary photoreactor (see ESI Fig. S1†).26,41,42 To our delight, conversion towards the desired hydroxybutenolide was observed, experimentally confirming the computational predictions regarding the ISC rate, and thus also the photosensitization properties of FLY 450. The quantum yield (ΦΔ), the efficiency of 1O2 generation of this new photosensitizer, was determined by monitoring the decay of a 1O2 scavenger over time by UV-Vis spectroscopy upon blue light irradiation (λirr = 445 nm). The rate of 1O2 production is equal to the consumption rate of a scavenger, in our case the biobased oil α-terpinene, which is present in a large excess (40 eq.), as it is reasonable to assume that every molecule of 1O2 that is formed is captured. α-Terpinene is a diene that can undergo a [4 + 2] cycloaddition with 1O2 to yield the stable endoperoxide ascaridole. It was important for this study that the absorption maxima of the scavenger α-terpinene (λmax = 269 nm) and the resulting product ascaridole (λmax = 205 nm) had no overlap and did not absorb in the visible light region where the photosensitizer FLY 450 (λmax = 450 nm) is excited (λirr = 445 nm) (ESI Fig. S3†). The quantum yield of FLY 450 in methanol and dichloromethane was derived from the ratio of scavenger decay rates, the photosensitizer absorptions at irradiation wavelengths and the reported quantum yields43,44 using methylene blue as a reference compound, according to a modified method from a published procedure (see ESI Fig. S6–S10†).45 Using the rate calculated from the portion of the curve between 5% and 20% conversion, i.e. the linear decay of α-terpinene (Fig. 1C and D), the quantum yield of FLY 450 in methanol was found to be ΦΔ = 0.22 ± 0.02. This is a high value considering a small organic compound with no additional effects to enhance ISC (Table 1), such as the inclusion of ISC-accelerating heavy atoms. Similarly, in dichloromethane (DCM), a high quantum yield is observed for FLY 450 (ΦΔ = 0.22 ± 0.02) (Table 1). It should be noted that due to the difference in the molar extinction coefficient ε, a higher absolute rate of scavenger decay (i.e. a higher absolute 1O2 production rate) is observed for methylene blue (Table 1). However, our newly designed biobased photosensitizer FLY 450 exhibits excellent quantum yields (Table 1), particularly also in a greener alcoholic solvent (in this case, MeOH), signifying efficient singlet oxygen formation for sustainable applications.
Employing FLY 450 as a photosensitizer (5 mol%) in a batch photooxidation of furfural to hydroxybutenolide, full conversion to the desired product was achieved after 20 h (Fig. 1E). After the reaction in methanol, we noticed discoloration of the solution, which suggested that some photobleaching of our photosensitizer FLY 450 was taking place. We hypothesized that as FLY 450 contains an electron rich furan moiety, it should itself be inherently susceptible to [4 + 2] cycloadditions with 1O2. Using a DFT thermochemistry approach we propose a decomposition pathway similar to the reaction mechanism of furfural towards hydroxybutenolide by the formation of the endoperoxide and subsequent decomposition by the methanolic solvent (see ESI Scheme S1†).
With a sustainable synthesis towards hydroxybutenolide using our new biobased photosensitizer FLY 450 for 1O2 formation, the next step involved the derivatization, i.e. introduction of an alkoxy substituent. Previously we have shown the condensation of hydroxybutenolide with a variety of different alcohols allowing tuning of the properties of the ultimate polymers.26 Condensation reactions occur readily on heating in the absence of acid due to the unique tautomeric open form structure of hydroxybutenolide (Fig. 2A). In the case of the low boiling point and environmentally attractive methanol,46 hydroxybutenolide can be dissolved and heated at reflux in methanol to provide methoxybutenolide (76% yield), which was purified with distillation under reduced pressure giving us the first monomer (Fig. 2).
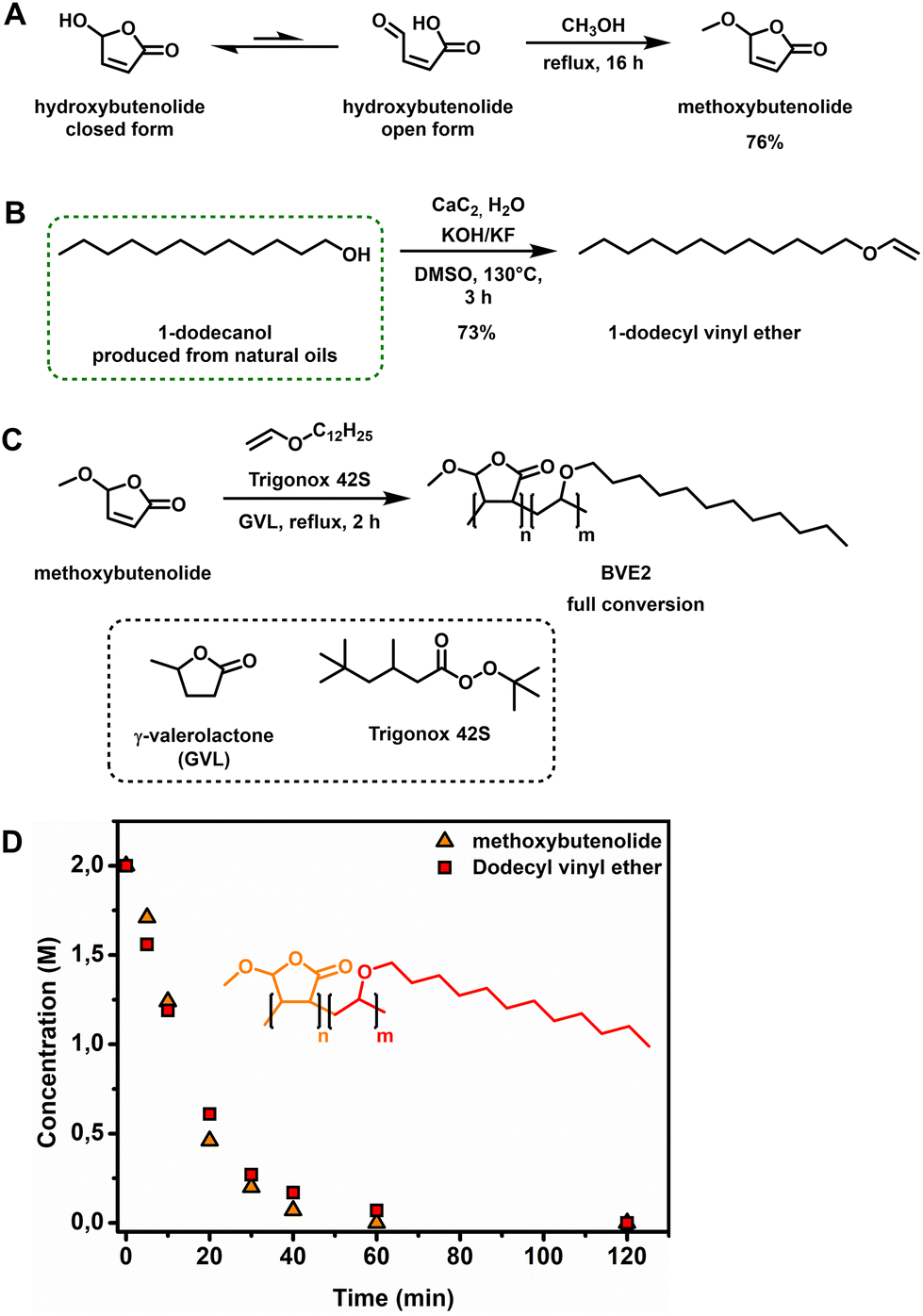 | ||
Fig. 2 (A) Hydroxybutenolide equilibrium with the closed form as the dominant species and methanol condensation of the open form towards methoxybutenolide. (B) Vinylation of 1-dodecanol using calcium carbide.47,52–54 (C) Copolymerization of methoxybutenolide with dodecyl vinyl ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) in GVL using Trigonox 42S as the radical initiator. (D) Concentration of monomers over time during the copolymerization of methoxybutenolide with dodecyl vinyl ether followed by 1H NMR spectroscopy by taking samples and flash freezing (−18 °C) them at certain timestamps using a 1,3,5-trimethoxybenzene internal standard. Reaction conditions: monomers 2 M, Trigonox 42S (6 mol%), 1,3,5-trimethoxybenzene 0.5 eq., 120 °C, and 2 h. :1 ratio) in GVL using Trigonox 42S as the radical initiator. (D) Concentration of monomers over time during the copolymerization of methoxybutenolide with dodecyl vinyl ether followed by 1H NMR spectroscopy by taking samples and flash freezing (−18 °C) them at certain timestamps using a 1,3,5-trimethoxybenzene internal standard. Reaction conditions: monomers 2 M, Trigonox 42S (6 mol%), 1,3,5-trimethoxybenzene 0.5 eq., 120 °C, and 2 h. | ||
Vinyl ethers are well known monomers used in a vast number of polymer materials47 and they copolymerize particularly well with alkoxybutenolides, easily reaching full conversion under free radical polymerization in 2 h at 120 °C.26 For almost a century, the production of these useful comonomers, by the vinylation of alcohols using acetylene, has been a well-established procedure.48 Recently, calcium carbide as the source for acetylene has been increasingly explored and is nowadays considered a green and sustainable methodology.49,50 Calcium carbide is traditionally synthesized from coal in electric furnaces at elevated temperatures. Employing biochar, obtained from the pyrolysis of biorenewable resources (and thus of biobased descent), allows for shorter reaction times and lower, but still high (1700 °C), working temperatures.51 It is important to note that with biobased calcium carbide, the original industrial Reppe process of producing acrylic acid from acetylene, carbon monoxide and water could in theory become a sustainable technology. However, this reaction remains unattractive because of safety and pollution control problems arising from the formation of toxic side products.7
Aside from being derived from renewable feedstock, an additional advantage related to using calcium carbide is the ease of handling solids rather than gases. Alcohols and even polyols, like carbohydrates, are readily vinylated in closed vessels.52–54 Fortunately, the vinylation of dodecanol (derived from natural oils like coconut and palm kernel oils)28 gives us access to our previously reported comonomer, now made in a sustainable way.
Using the previously reported optimal ratio of 1 to 4 (calcium carbide:water) dodecanol was converted to dodecyl vinyl ether and isolated in 73% yield (Fig. 2B). With both biobased monomers in hand, we commenced with the free radical copolymerization of methoxybutenolide and dodecyl vinyl ether. Copolymerization using Trigonox 42S as a thermal radical initiator (t1/2 = 30 min at 120 °C) in either 1-methoxy-2-propanol or butyl acetate as solvent (boiling points: 120 °C and 126 °C, respectively) resulted in a 1:1 copolymerization with 95% conversion (see ESI Fig. S11–S14†). As the solvent is the major component (in weight) in the reaction, it is highly important to include it in the transition to fully biobased polymers and coatings. With careful consideration, GVL was chosen as a suitable solvent replacing butyl acetate or 1-methoxy-2-propanol. GVL is derived from levulinic acid and is used as a sustainable solvent (boiling point: 205 °C) and as a platform chemical for specialty chemicals and fuels.32
Although the boiling point is considerably higher than those of its counterparts, GVL was employed here as green solvent for the copolymerization of methoxybutenolide and dodecyl vinyl ether, which resulted in a fast and full conversion of both of the monomers and a clean 1:1 copolymerization (Fig. 2C, D and ESI Fig. S15–S18†).
Although a minor component in terms of carbon content in the resulting polymer chain, we sought to replace the radical initiator with a biobased alternative in order to obtain all components in a biobased fashion. Intrigued by the structure of the singlet oxygen scavenger product from our quantum yield determination studies (Fig. 1), ascaridole, we envisioned that the thermal cleavage of the endoperoxide could initiate the copolymerization of a butenolide–vinyl ether mixture. To the best of our knowledge, only one example of free radical polymerization of methacrylonitrile initiated by thermal decomposition of ascaridole is known.55 We commenced with the larger scale synthesis and isolation of the endoperoxide using the previously described rotary photoreactor (Fig. 3A and ESI Fig. S1†).26,41,42 In order to elucidate the stability of ascaridole, the thermal decomposition was studied at different temperatures. Based on an initial DFT thermochemistry prediction of the O–O bond homolytic cleavage of Trigonox 42S and ascaridole (see ESI Scheme S2†), we calculated the thermal stability of ascaridole as a radical initiator and found that it was higher than that of Trigonox 42S (Fig. 3B), suggesting that elevated temperatures would be required to obtain similar half-life times for radical initiation. Taking advantage of the higher boiling point green solvent GVL, ascaridole was dissolved in GVL (0.5 M) and heated at 140 °C and 150 °C, while the conversion was followed over time using 1H-NMR spectroscopy by taking samples at regular intervals. As the thermal decomposition follows pseudo-first-order reaction kinetics, due to radical formation followed by presumably radical transfer reactions to the solvent, a linear relationship between conversion and time at a given temperature is found (Fig. 3B). At 140 °C, a rate constant was revealed that resulted in a half-life time of 45 min. The measured half-life for the decomposition of ascaridole was compared to that of Trigonox 42S in GVL (Fig. 3B, red diamonds). Our comparison revealed a half-life of 29 min that corresponds to the literature value,56 validating our measurements and our DFT thermochemistry predictions. At 150 °C, the half-life of ascaridole was significantly decreased to 17 min, which suggests that unfavorable termination reactions in the free radical polymerization process might have occurred.
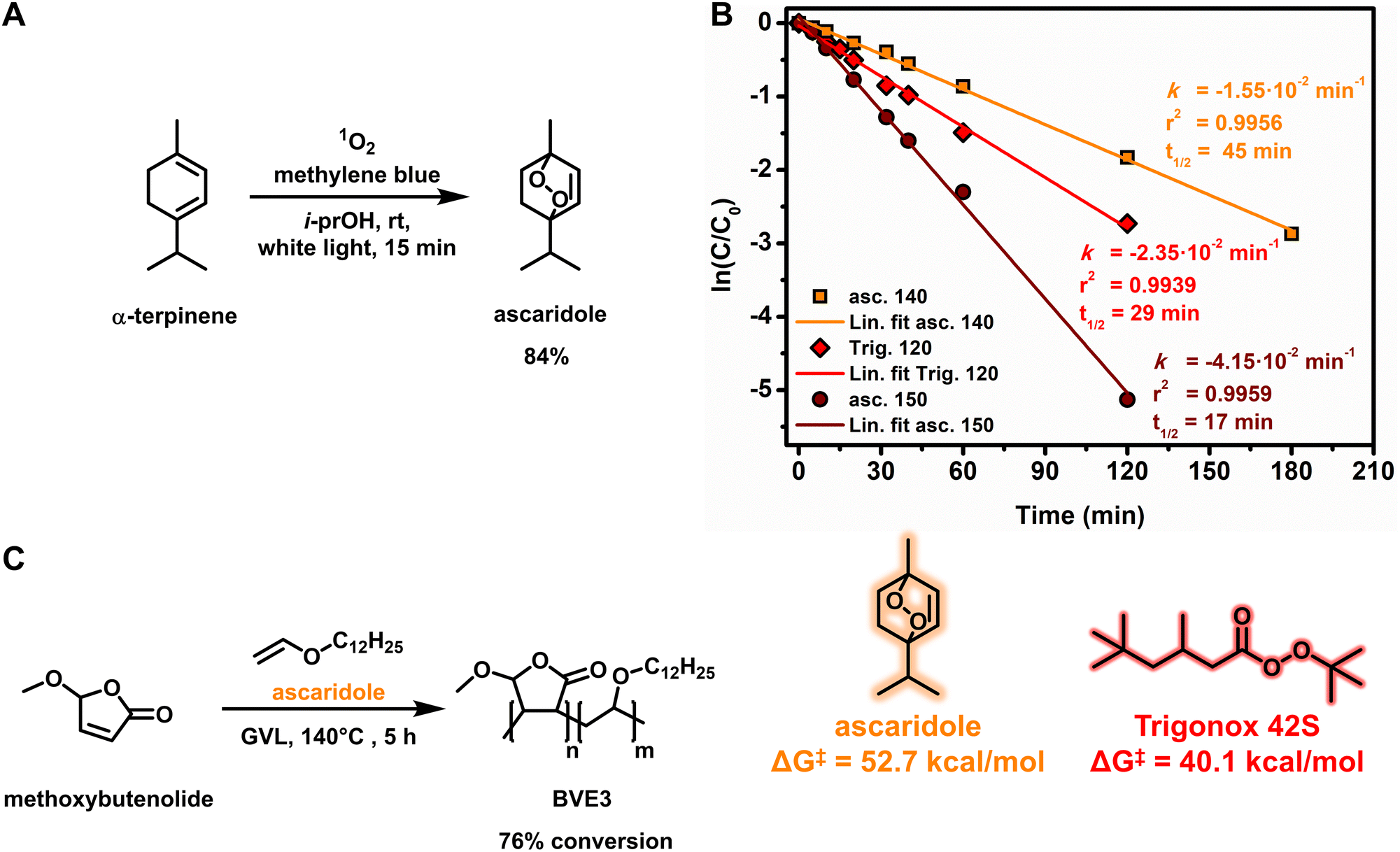 | ||
| Fig. 3 (A) Photooxidation of α-terpinene to ascaridole. (B) Rate of thermal decomposition and determination of the half-life time of ascaridole at 140 °C (orange squares) and 150 °C (brown circles) and Trigonox 42S at 120 °C (red diamonds) monitored by 1H-NMR spectroscopy by taking samples at regular intervals (top) and computationally calculated energies of radical initiators (bottom). Reaction conditions: radical initiator 0.5 M, GVL, and 3 h. (C) Copolymerization of methoxybutenolide and dodecyl vinyl ether using ascaridole (6 mol%) as the radical initiator. | ||
Performing the copolymerization of methoxybutenolide and dodecyl vinyl ether in GVL at 140 °C using ascaridole (6 mol%) resulted in 76% conversion after 5 h, clearly demonstrating the viability of ascaridole as a fully biobased thermal radical initiator for the free radical polymerization of fully biobased components (Fig. 3C). Following the reaction kinetics of the copolymerization, incorporation of both monomers was confirmed, albeit with a slightly diminished amount of methoxybutenolide. Furthermore, the copolymerization shows an initial fast conversion with a rate constant (kobs) in the same order of magnitude as determined for the copolymerization in butyl acetate, followed by a slower conversion (see ESI Fig. S19–S22†). Performing copolymerization at 150 °C did not meaningfully impact the polymerization behavior.
Focusing on the polymer properties, the molecular weights of the copolymers synthesized with ascaridole in GVL and with Trigonox 42S in butyl acetate and GVL were analyzed and are summarized in Table 2. A higher rate constant is observed upon changing the solvent from butyl acetate to GVL, attributed to a higher observed molecular weight. We propose that there is an increased compatibility of the reacting monomers with the more polar solvent GVL. Substituting Trigonox 42S with fully biobased ascaridole as the radical initiator results in a lower molecular weight being observed, in line with the incomplete conversion and the lower rate of polymerization, which indicates that termination reactions are involved to a greater extent (Table 2).
| Copolymersa | K obs (s−1) | Conversion (%) | M n (kDa) | M W (kDa) | D |
|---|---|---|---|---|---|
|
a Reaction conditions: 1:1 ratio of monomers, thermal initiator (6 mol%), 2 M in solvent at given T, and 2–5 h.
b Polydispersity (D) is calculated by dividing MW by Mn.
|
|||||
| BVE1 | 6.10 × 10−4 | 95 | 1.3 | 3.0 | 2.2 |
| BVE2 | 1.15 × 10−3 | >99 | 2.0 | 3.6 | 1.7 |
| BVE3 | 3.23 × 10−4 | 76 | 1.0 | 2.0 | 2.0 |
Having established that full biobased polymers could be formed using ascaridole as the thermal initiator in combination with two biobased monomers, we proceeded towards the formation of surface coatings. Previously we have shown that excellent hard transparent coatings could be formed through solvent-free UV-curing using alkoxybutenolides in combination with a divinyl ether based crosslinker and a phosphine oxide photoinitiator.26 Utilizing the described robust sustainable vinylation method, a biobased divinyl ether crosslinker (1,4-butane divinyl ether) was prepared effortlessly from the glucose-derived 1,4-butanediol.33 The final remaining component of the coating mixture to be made from biorenewable resources is the photoinitiator. Recently, Versace et al. described the synthesis of a vanillin-based type I photoinitiator (Fig. 4A).57 We envisioned that the biobased photoinitiator, VAPO, could be a direct replacement of the previously used BAPO photoinitiator, as they both contain a similar acyl-phosphine oxide structure and exert similar reactivity. VAPO was thus synthesized according to a literature procedure in a three-step process from vanillin via allylation, tertiary phosphine oxide formation and alcohol oxidation with high yields (Fig. 4A). The UV-Vis absorption spectrum of VAPO revealed, although showing a slightly lower intensity of absorption at λ = 395 nm compared to BAPO, that the photoinitiator is suitable for the comonomers and our current UV-curing setup (Fig. 4B). A homogeneous neat mixture of methoxybutenolide (1 eq.), 1,4-butane divinyl ether (0.5 eq.) and VAPO (6 mol%) was applied on a 20 × 10 cm glass panel and irradiated for 20 min using a UV-Flood lamp 36 (35 W) UVA lamp (Fig. 4C). A clear, hard, transparent and above all biobased coating of 50 μm was obtained on glass. Lower loading of VAPO (3 mol%) resulted in a tacky coating, justifying the higher loading of VAPO (6 mol%) (ESI Fig. S23 and S24†). Interestingly, applying 3 mol% of the original photoinitiator BAPO together with the methoxybutenolide (1 eq.) and 1,4-butane divinyl ether (0.5 eq.) resulted in a hard but semi-clear coating, presumably caused by unreacted monomers (ESI Fig. S23 and S24†).
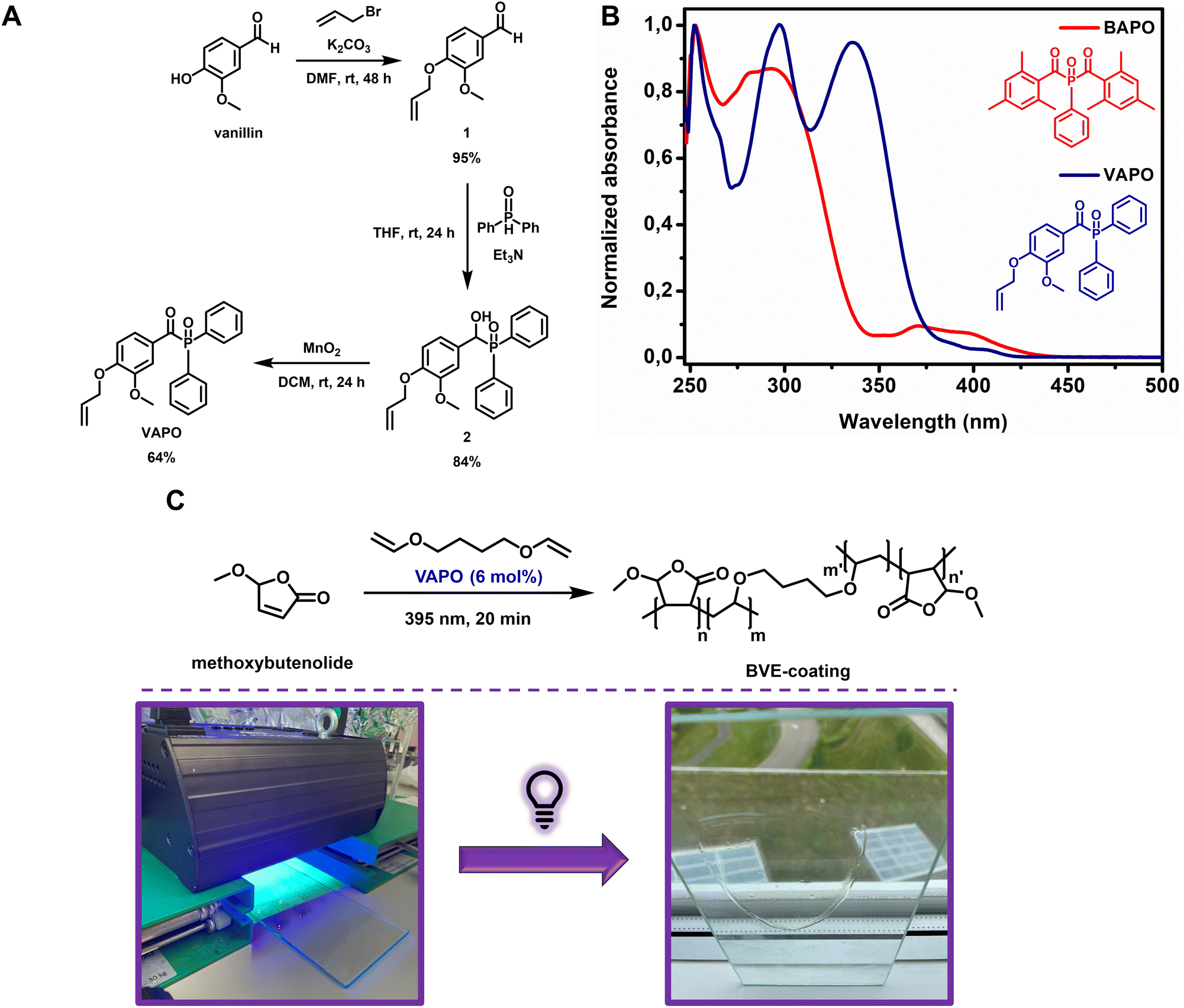 | ||
| Fig. 4 (A) Synthesis of the vanillin-based photoinitiator VAPO. (B) Normalized UV-Vis absorption spectra of the previously used BAPO (red) and the biobased VAPO (navy). (C) Crosslinking of alkoxybutenolides (1 eq.) with 1,4-butane divinyl ether (0.5 eq.), VAPO (6 mol%) as the radical initiator and UV light (λirr = 395 nm, 20 min) as the trigger (top), and UV-curing of the coating mixture on a glass panel (bottom). | ||
To determine the properties of the VAPO- and BAPO-initiated biobased coatings, we subjected the materials to a Knoop hardness test and Differential Scanning Calorimetry (DSC) to establish the hardness and the glass transition temperature (Tg), respectively. Both coatings display similar properties, showing effective replacement of BAPO with the vanillin-based VAPO (Table 3). The values found for the hardness and Tg are typically in the range of those of hard acrylate based coatings.58,59 Furthermore the coatings were subjected to a 2-butanone (MEK) double rub test where excellent solvent resistance (>200 double rubs) was observed with no deterioration of the surface except for slight whitening (ESI Fig. S25†).
| Coatings | Initiator | Dry film thickness (μm) | Knoop hardness (kg mm−2) | T g (°C) | MEK resistance (double rubs) |
|---|---|---|---|---|---|
| a 6 mol% initiator for the formation of a uniform tacky-free coating. | |||||
| BVEC1 | VAPO | 50 | 9.9 (±0.1) | 24 | >200 |
| BVEC2 | BAPO | 55 | 13.9 (±4.3) | 43 | >200 |
Conclusion
In conclusion, we have developed a polymer and coating system using starting materials derived from renewable resources and implementing sustainable synthetic steps. The main monomers used in the polymerization were synthesized from the platform chemical furfural and common biobased alcohols, using molecular oxygen (in a photosensitization process) and calcium carbide as sustainable reagents. The biobased photosensitizer FLY 450, used in the photooxidation of furfural, showed excellent photosensitization properties with high molar extinction coefficients and quantum yields. The biobased methoxybutenolide–dodecyl vinyl ether copolymer was synthesized with high conversion in the green solvent GVL using the natural oil-derived radical initiator ascaridole, resulting in a fully biobased and sustainable polymer. Finally a fully biobased coating was formed using a vanillin derived photoinitiator on a glass surface which exhibits excellent hardness and outstanding solvent resistance in line with properties of hard acrylate based coatings. This integrated sustainable polymer and coating system illustrates viable and promising sustainable alternatives for key components used in the production of materials widely abundant in modern society.Author contributions
B. L. F. conceptualized the research project and coordinated it with the help of K. J. v. d. B. J. G. H. H., T. F., M. L. L. and N. E. conducted the research and its validation. G. A. performed the DFT studies. J. G. H. H. and B. L. F. prepared the manuscript with inputs from T. F., G. A. and M. L. L. All authors reviewed the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
We thank O. K. B. Staal for technical support regarding the photooxidation setups and J. L. Sneep and J. Hekelaar for assistance with HRMS. Funding: this work is in collaboration with AkzoNobel and part of the Advanced Research Center for Chemical Building Blocks, ARC CBBC, which is cofounded and co-financed by the Netherlands Organization for Scientific Research (NWO, contract 736.000.000) and the Netherlands Ministry of Economic Affairs and Climate. We are grateful for the generous financial support to B. L. F. (the Ministry of Education, Culture and Science of the Netherlands Gravitation Programme No. 024.001.035) and to G. A. (EMBO LTF232-2020 Postdoctoral Fellowship).References
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- M. Beller, G. Centi and L. Sun, ChemSusChem, 2017, 10, 6–13 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- K. Kohli, R. Prajapati and B. K. Sharma, Energies, 2019, 12, 233 CrossRef CAS.
- A. K. Mohanty, F. Wu, R. Mincheva, M. Hakkarainen, J.-M. Raquez, D. F. Mielewski, R. Narayan, A. N. Netravali and M. Misra, Nat. Rev. Methods Primers, 2022, 2, 46 CrossRef CAS.
- T. Ohara, T. Sato, N. Shimizu, G. Prescher, H. Schwind, O. Weiberg, K. Marten, H. Greim, T. D. Shaffer and P. Nandi, Acrylic Acid and Derivatives, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2020, pp. 1–21 Search PubMed.
- M. M. Lin, Appl. Catal., A, 2001, 207, 1–16 CrossRef CAS.
- R. Höfer and M. Selig, Green Chemistry and Green Polymer Chemistry, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 5–14 Search PubMed.
- H. Lutz, H. P. Weitzel and W. Huster, Aqueous Emulsion Polymers, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 479–518 Search PubMed.
- D. Urban and D. Distler, Introduction, in Polymer Dispersions and Their Industrial Applications, Wiley-VCH, Weinheim, 2002, pp. 1–14 Search PubMed.
- R. Schwalm, Radiation-Curing Polymer Systems, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 567–579 Search PubMed.
- G. Crapper, Powder Coatings, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 541–566 Search PubMed.
- R. Höfer, History of the Sustainability Concept – Renaissance of Renewable Resources, in Sustainable Solutions for Modern Economies, The Royal Society of Chemistry, 2009, pp. 1–11 Search PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- A. Z. Yu, J. M. Sahouani and D. C. Webster, Prog. Org. Coat., 2018, 122, 219–228 CrossRef CAS.
- F. Diot-Néant, E. Rastoder, S. A. Miller and F. Allais, ACS Sustainable Chem. Eng., 2018, 6, 17284–17293 CrossRef.
- M. Moreno, M. Goikoetxea, J. C. de la Cal and M. J. Barandiaran, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3543–3549 CAS.
- J. T. Trotta, M. Jin, K. J. Stawiasz, Q. Michaudel, W.-L. Chen and B. P. Fors, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2730–2737 CrossRef CAS.
- S. Pérocheau Arnaud, E. Andreou, L. V. G. Pereira Köster and T. Robert, ACS Sustainable Chem. Eng., 2020, 8, 1583–1590 CrossRef.
- S. Okada and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 822–827 CrossRef CAS.
- T. Okuda, K. Ishimoto, H. Ohara and S. Kobayashi, Macromolecules, 2012, 45, 4166–4174 CrossRef CAS.
- G. Quintens, J. H. Vrijsen, P. Adriaensens, D. Vanderzande and T. Junkers, Polym. Chem., 2019, 10, 5555–5563 RSC.
- I. Khalil, G. Quintens, T. Junkers and M. Dusselier, Green Chem., 2020, 22, 1517–1541 RSC.
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- J. G. H. Hermens, T. Freese, K. J. van den Berg, R. van Gemert and B. L. Feringa, Sci. Adv., 2020, 6, eabe0026 CrossRef CAS PubMed.
- A. Jaswal, P. P. Singh and T. Mondal, Green Chem., 2022, 24, 510–551 RSC.
- K. Noweck and W. Grafahrend, Fatty Alcohols, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2006, pp. 117–141 Search PubMed.
- J. S. Wau, M. J. Robertson and M. Oelgemöller, Molecules, 2021, 26, 1685 CrossRef CAS PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- F. Ronzani, N. Costarramone, S. Blanc, A. K. Benabbou, M. L. Bechec, T. Pigot, M. Oelgemöller and S. Lacombe, J. Catal., 2013, 303, 164–174 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- A. Burgard, M. J. Burk, R. Osterhout, S. Van Dien and H. Yim, Curr. Opin. Biotechnol., 2016, 42, 118–125 CrossRef CAS PubMed.
- J. Saska, Z. Li, A. L. Otsuki, J. Wei, J. C. Fettinger and M. Mascal, Angew. Chem., Int. Ed., 2019, 58, 17293–17296 CrossRef CAS PubMed.
- A. Gualandi, M. Anselmi, F. Calogero, S. Potenti, E. Bassan, P. Ceroni and P. G. Cozzi, Org. Biomol. Chem., 2021, 19, 3527–3550 RSC.
- S. Xu, Y. Yuan, X. Cai, C.-J. Zhang, F. Hu, J. Liang, G. Zhang, D. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824–5830 RSC.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS.
- C. A. Clark, D. S. Lee, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2016, 20, 1792–1798 CrossRef CAS.
- J. G. H. Hermens, A. Jensma and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202112618 CrossRef CAS PubMed.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS PubMed.
- U. Yoshiharu, Chem. Lett., 1973, 2, 743–744 CrossRef.
- L. V. Lutkus, S. S. Rickenbach and T. M. McCormick, J. Photochem. Photobiol., A, 2019, 378, 131–135 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- E. Kirillov, K. Rodygin and V. Ananikov, Eur. Polym. J., 2020, 136, 109872 CrossRef CAS.
- W. Reppe, Production of vinyl ethers, US1959927A, 1931.
- S. P. Teong and Y. Zhang, J. Bioresour. Bioprod., 2020, 5, 96–100 CrossRef CAS.
- K. S. Rodygin, M. S. Ledovskaya, V. V. Voronin, K. A. Lotsman and V. P. Ananikov, Eur. J. Org. Chem., 2021, 43–52 CrossRef CAS.
- Z. Li, Z. Liu, R. Wang, X. Guo and Q. Liu, Chem. Eng. Sci., 2018, 192, 516–525 CrossRef CAS.
- S. P. Teong, A. Y. H. Chua, S. Deng, X. Li and Y. Zhang, Green Chem., 2017, 19, 1659–1662 RSC.
- K. S. Rodygin, I. Werner and V. P. Ananikov, ChemSusChem, 2018, 11, 292–298 CrossRef CAS PubMed.
- G. Werner, K. S. Rodygin, A. A. Kostin, E. G. Gordeev, A. S. Kashin and V. P. Ananikov, Green Chem., 2017, 19, 3032–3041 RSC.
- R. Zand and R. B. Mesrobian, J. Am. Chem. Soc., 1955, 77, 6523–6524 CrossRef CAS.
- Initiators for thermoplastics, Nouryon, https://www.nouryon.com/globalassets/inriver/resources/brochure-initiators-for-thermoplastics-feb2021-en_us.pdf, 2021 (accessed on 10/07/2022).
- L. Breloy, C. Negrell, A. S. Mora, W. S. J. Li, V. Brezová, S. Caillol and D. L. Versace, Eur. Polym. J., 2020, 132, 109727 CrossRef CAS.
- K. J. van den Berg, L. G. J. van der Ven and H. J. W. van den Haak, Prog. Org. Coat., 2008, 61, 110–118 CrossRef CAS.
- D. E. Fiori, D. A. Ley and R. J. Quinn, J. Coat. Technol., 2000, 72, 63–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03657f |
| This journal is © The Royal Society of Chemistry 2022 |