
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogenation of CO2 to formic acid in biphasic systems using aqueous solutions of amino acids as the product phase†
Nils
Guntermann
a,
Giancarlo
Franciò
 a and
Walter
Leitner
*ab
a and
Walter
Leitner
*ab
aInstitut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
bMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim a. d. Ruhr, Germany. E-mail: walter.leitner@cec.mpg.de
First published on 13th September 2022
Abstract
Carbon capture and utilization is considered a promising approach for introducing CO2 into the chemical value chain, especially in combination with bioenergy applications (BECCU). We report here on the catalytic hydrogenation of CO2 to formic acid in a biphasic reaction system using aqueous solutions of amino acids as the product phase and possible capture solutions for biogenic CO2. The molecular structure of the ruthenium catalyst and the catalyst phase were matched through a combined design process identifying n-dodecanol (lauryl alcohol) as the preferred “green” solvent. A total turnover number (TON) of over 100 000 mole HCOOH per mole of catalyst (46 582 g HCOOH per g of Ru) with minimal contamination of the aqueous phase with metal or organic solvent was obtained. The resulting aqueous solutions attained almost quantitative conversions with up to 0.94 mol formic acid per mol amino acid (ca. 108 g HCOOH per kg). Such solutions may find use directly, or after upgrading, in agricultural applications without the need for energy intensive and costly isolation of pure formic acid.
Introduction
Formic acid (FA) is one of the most widely discussed target products for hydrogenation of CO2 with a significant potential to reduce the carbon footprint as compared to its fossil-based production.1–8 However, major challenges are the energy demand for CO2 capture and release9,10 as well as isolation of the formic acid product from the reaction mixture.11 In the present paper, we demonstrate a concept to integrate carbon capture technologies based on proteinogenic amino acids with catalytic CO2 conversion to yield HCOOH solutions with a potential for agricultural applications, minimizing both upstream and downstream unit operations. This opens the possibility for the valorization of waste CO2 streams from biogas production directly with local and decentralized applications in the same industrial sector.The most advanced technologies for CO2 capture from flue gases rely on basic organic amines, such as monoethanolamine (MEA) or methyldiethanolamine (MDEA).12,13 In the search for bio-based alternatives, amino acids and their salts have been investigated.14–17 Their applicability in reversible carbon capture has been demonstrated with higher reaction rates and CO2-loading for the respective potassium salts as compared to the neutral amino acids.17–19 Scrubbing solutions based on amino acid salts have been applied already at the pilot plant scale using PostCap™ technology.20
Formic acid has a number of applications in relation to biological processes, e.g. as a biomass preservative in silage, or as an additive in the food or pharma industry.21 In academic research, formic acid has been shown to enhance microbial growth and/or product formation rates during fermentation processes.22–25
Most notably in the present context, mixtures of formic acid with proteinogenic amino acids have potential applications as livestock feed. Methionine26,27 and arginine28 are limiting amino acids in poultry feed, and supplements of these amino acids are commercially available.29 Similarly, arginine30–32 as well as methionine and histidine are essential amino acids for pig breeding.33,34 At the same time, formic acid and formic acid salts are used as preservatives in animal feeds.35–37 Aqueous solutions of mixtures of amino acids and formic acid may therefore constitute attractive target products for use in animal farming.
We therefore envisage the use of catalytic CO2 hydrogenation as the molecular relay integrating upstream CO2 capture in aqueous amino acid solutions directly with the potential for downstream use of the resulting product phases (Fig. 1).
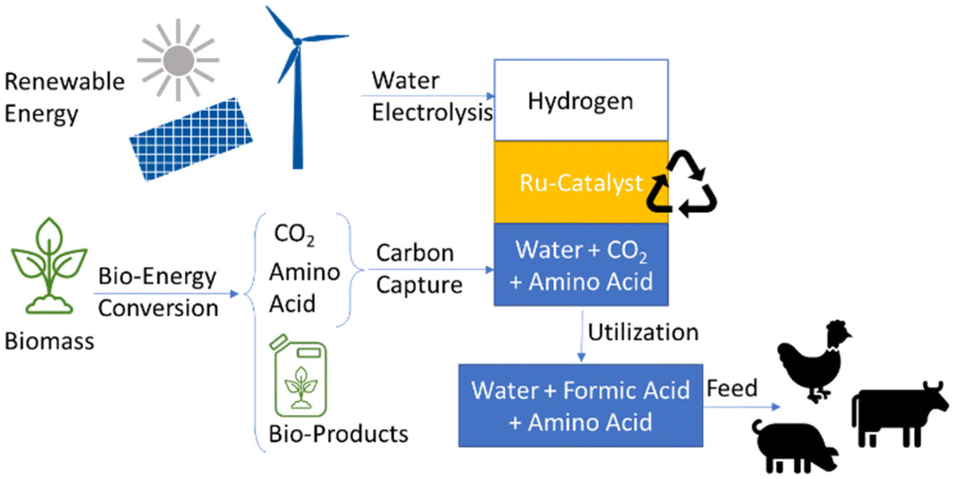 | ||
| Fig. 1 Envisaged BECCU concept from biomass and renewable energy to amino acid–formic acid solutions as livestock feed. | ||
The essential prerequisite to establishing potential value chains from biogenic CO2 sources to agricultural applications is the effective separation of the organometallic catalyst from the aqueous product solution. The catalyst phase must be chosen to avoid the leaching of metals or ligands as well as cross-contamination with organic solvents into the aqueous phase. A tailored biphasic reaction system enabling such a fully integrated process scheme is presented in this paper.
Results and discussion
The catalytic hydrogenation of CO2 in the presence of aqueous amino acid solutions has been demonstrated recently using homogeneous catalysts in THF/water mixtures in the context of reversible hydrogen storage.38,39 For the use of aqueous product solutions most directly in the applications envisaged here, essential parameters are the choices of amino acid and solvent used as the catalyst phase (Scheme 1). However, several concepts for liquid–liquid biphasic systems to separate organometallic catalysts from very polar formic acid product phases have been reported.40–49 Based on the experience in our laboratories,50–52 we initiated a systematic study to design a catalytic system combining high catalyst productivity with effective retention and separation from the amino-acid-based product phase. | ||
| Scheme 1 Reaction system for the catalytic conversion of CO2 and H2 to formic acid, featuring pre-catalysts cis-[Ru(dppm)2Cl2] and cis-[Ru(C12-dppm)2Cl2] in methyl isobutyl carbinol (MIBC) and the amino acids used in the present study. | ||
The organometallic complex cis-[Ru(dppm)2Cl2]50 (dppm = bis-diphenylphosphinomethane) and its analogous congener tagged with hydrophobic side chains cis-[Ru(C12-dppm)2Cl2]53 were selected as catalyst precursors to cover a broad range of potential solvents for catalyst separation and recycling. Previous studies showed that they form the corresponding monohydride complexes under hydrogen pressure, which can be assumed as catalytically active species in line with what is generally known about Ru–phosphine catalysts in this field.54
The proteinogenic amino acids arginine (Arg), histidine (His), lysine (Lys), and methionine (Met) were used in the aqueous phase (Scheme 1). In a first screening, the amino acids Arg, Lys, and His carrying basic side groups were studied because amines are known to capture CO2 as well as stabilize HCOOH, effectively shifting the reaction equilibrium to the formic acid side. As the catalyst phase, the solvent methyl isobutyl carbinol (MIBC) in combination with cis-[Ru(dppm)2Cl2] as catalyst precursor was evaluated due to the high activity shown in previous studies on biphasic systems using aqueous MEA solutions as the capture and product phase.50 The volume ratio between MIBC and water was adjusted to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 with amino acid concentrations of 0.86 M for Arg and Lys and 0.25 M for His, based on their respective solubilities. The catalyst concentration in the MIBC phase was 0.5 mM corresponding to loadings of 1:2580 and 1:750, respectively.
:3 with amino acid concentrations of 0.86 M for Arg and Lys and 0.25 M for His, based on their respective solubilities. The catalyst concentration in the MIBC phase was 0.5 mM corresponding to loadings of 1:2580 and 1:750, respectively.
The reactions were carried out under initial partial pressures (at room temperature) of 30 bar CO2 and 60 bar H2 and a reaction temperature of 70 °C. The reaction times were determined by monitoring the pressure drop until no further decrease of pressure was observed (ESI Fig. S3†). The final concentration of formic acid and the corresponding ratio of HCOOH to amino acid were determined by 1H NMR spectroscopy. The initial rates, as measured from the pressure drop, are given as mol HCOOH per mol ruthenium and hour (i.e., turnover frequency, TOF [h−1]). The results are shown together with the results for MEA as the benchmark in Fig. 2.
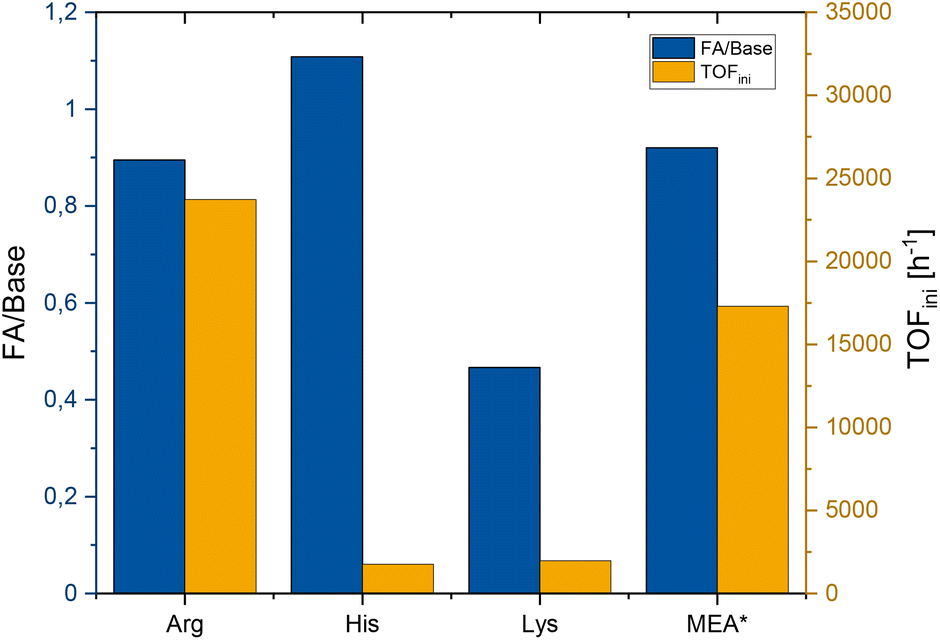 | ||
| Fig. 2 Formic acid/amino acid ratio and initial TOF for Arg, His and Lys. Conditions: 30 bar CO2, 60 bar H2, 2.58 mmol Arg/Lys or 0.75 mmol His in water (3 mL), 1 μmol cis-[Ru(dppm)2Cl2] in MIBC (2 mL), 70 °C, reaction time until the pressure was constant. *Values for comparison from Scott et al. with MEA (7.9 mmol MEA in 2 mL H2O, 4.1 μmol cis-[Ru(dppm)2Cl2] in 1.5 mL MIBC, 30 bar CO2, 60 bar H2, 70 °C).50 | ||
A formic acid/amino acid ratio of approximately 1:1 was reached with Arg and His, corresponding to the thermodynamic limit for amine bases in an aqueous solution.1,55–57 For Lys, the reaction did not proceed beyond a ratio of 0.5:1. Given the long reaction time in this case, this may reflect, at least in part, catalyst deactivation. Most notably, the productivity with Arg matched that of MEA very well and the remarkable initial rate of 23000 h−1 even surpassed the benchmark case significantly.
While the organic solvent MIBC allowed for a very fast reaction, it also resulted in significant cross-solubility into the aqueous phase of 0.32 mmol or 2.2%. In order to minimize any mutual cross-solubility, the nonpolar solvent n-tetradecane was investigated, which has also been used for CO2 hydrogenation in biphasic systems previously.58 The lipophilic-tagged ligand bis(bis(4-dodecylphenyl)phosphanyl)methane (C12-dppm) was used to ensure solubility and exclusive partitioning of the ruthenium catalyst in the organic phase (Fig. 3).11 Quantitative conversion for Arg and His was reached again with the more basic amino acid. For Lys, only small amounts of formic acid were obtained even with prolonged reaction times.
 | ||
| Fig. 3 Formic acid/base ratio and initial TOF for Arg, His and Lys. Conditions: 30 bar CO2, 60 bar H2, 2.58 mmol Arg/Lys or 0.75 mmol His, respectively, in water (3 mL), 1 μmol cis-[Ru(C12-dppm)2Cl2] in tetradecane (2 mL), 70 °C, reaction time until the pressure was constant. | ||
In general, however, the rates were nearly two orders of magnitude lower with tetradecane/cis-[Ru(C12-dppm)2Cl2] than with MIBC/cis-[Ru(dppm)2Cl2]. In order to separate the electronic effect of the ligand modification from the solvent effects, the C12-tagged complex was also used with MIBC as the catalyst phase, resulting in a TOFini of 2827 h−1. While this confirmed the expected reduced activity of the more electron rich C12-tagged complex,59,60 it also revealed a major contribution of the chosen solvent. Therefore, a detailed screening of solvents possessing a miscibility gap with water using the same pre-catalyst cis-[Ru(C12-dppm)2Cl2] under identical conditions with Arg as the amino acid was undertaken.
The resulting initial activities are shown in Fig. 4. The results demonstrate the drastic influence of the chosen organic phase spanning two orders of magnitude in the rate between the lowest initial TOF observed with hexanol and the highest activity with ethyl acetate. No obvious correlations between specific functional groups or structural units in the solvents are apparent. Also, solvatochromic and other solvent parameters (ESI Fig. S6–S10†), gas solubilities (ESI Fig. S11 and S12†) or cross-solubility into the aqueous phase (ESI, Fig S13†) did not show any clear trend. Mass transfer limitations61 were excluded by variation of the catalyst concentration. As the activity did not increase significantly at a lower catalyst loading, the capacity of all catalytically active centers was sufficiently utilized for selected solvents from either side of the reactivity range (ESI, Fig. S14†).
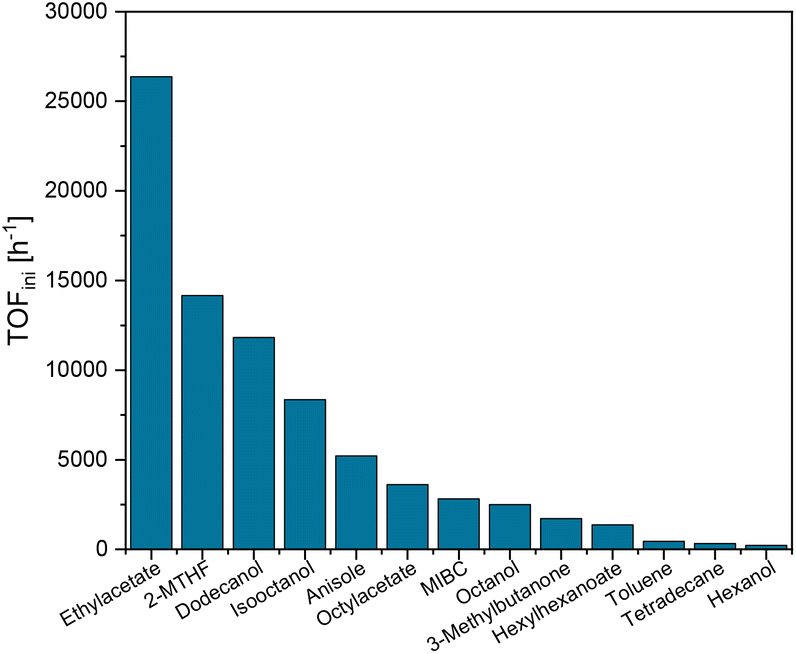 | ||
| Fig. 4 Initial TOFs for the biphasic CO2 hydrogenation using aqueous Arg and cis-[Ru(C12-dppm)2Cl2] as catalyst in various solvents. Conditions: 30 bar CO2, 60 bar H2, 2.58 mmol Arg in water (3 mL), 1 μmol cis-[Ru(C12-dppm)2Cl2] in the indicated solvent (2 mL), 70 °C. TOFini calculated from initial linear pressure drop; see the ESI, Fig. S5.† | ||
An interesting volcano-type trend results from the correlation of TOFini values with dielectric constants of the solvents, as shown in Fig. 5. Of the three top-performing solvents, n-dodecanol – also known as lauryl alcohol – best meets the design criteria for the target application: it shows practically no cross-solubility in water, it is a fatty alcohol derived from biomass, and it is compatible with biological applications, for example in consumer care products. Therefore, this “green” solvent was used for further optimization.
 | ||
| Fig. 5 Correlation between the dielectric constant and TOFini from the experiments described in Fig. 4. | ||
The solubility limit of arginine in the neutral, aqueous solution is about 1 M, due to the betaine form of the amino acid. We anticipated that the solubility would be significantly higher at low pH resulting from the presence of CO2 at the beginning and the formation of HCOOH at the end of the reaction. We therefore explored the possibility of using over-saturated CO2 solutions to achieve higher product concentrations.
In Scheme 2, pictures of the reaction mixture of 12 mmol Arg with 2 mL water (corresponding to 6 M upon dissolution) and 2 mL catalyst solution are shown before the reaction, after pressurization with CO2, and after the reaction. Before pressurization, large amounts of undissolved Arg were dispersed in water below the yellow catalyst phase. After saturation with CO2, mixture became turbid but most of the solid particles were dissolved. At the end of the hydrogenation when the pressure reached a constant value after 1.5 h, both phases had become clear and arginine was completely dissolved. Also, a phase expansion of the aqueous solution was visible. Analysis of the aqueous phase revealed a FA/base ratio of 0.77. The TOFini was estimated to be 15345 h−1 under these conditions. Since the volume and density do not remain constant, the wt% was more reliable in terms of concentration than the molar amounts. The obtained product solution consisted of 9.4 wt% FA and 46.4 wt% Arg.
 | ||
| Scheme 2 Pictures of the Arg, water and catalyst solution in the autoclave before the reaction (left), after saturation with CO2 (middle) and after the reaction with H2 (right). | ||
In order to mimic the CO2 capture step, a CO2 saturated solution with Arg, corresponding to a 6 M concentration, was prepared on a liter scale. This concentration was found to provide an optimum balance between the final concentration and the formation rate TOFini (Table S5 and Fig. S15†). The capture capacity of Arg was determined as 0.94 mol CO2 per mol Arg under 2 bar.38 The catalytic hydrogenation was performed in a largely automated semi-batch process with a 100 mL autoclave51 using 20 mL of catalyst phase containing only 10 μmol of the Ru-complex and 50 g of the separately prepared CO2 saturated aqueous solution. The autoclave was pressurized with H2 at 90 bar and heated to 70 °C while stirring using a magnetic stir bar. The set-up allowed continuous feeding of H2 gas at isobaric conditions while monitoring the H2 flow (see Fig. 6). After about 5 h, consumption of H2 stopped, the stirring was discontinued to allow phase separation, and the aqueous phase was removed for analysis under pressure at 70 °C. The autoclave was charged with a new batch of CO2-saturated arginine solution and the reaction repeated without any catalyst make-up in between. Over ten runs, the final formic acid-to-amino acid ratio reached the 1:1 limit (0.91 ± 0.03:1 on average, 9.0–10.8 wt% FA) indicating practically quantitative conversion of the captured CO2 to FA by catalytic hydrogenation. The data correspond to a total TON of 102282 corresponding to 46.6 kg HCOOH per g of ruthenium.
 | ||
| Fig. 6 Delivered H2 during the recycling experiments in the semi-continuous setup. Conditions: 90 bar H2, 70 °C, 10 μmol cis-[Ru(C12-dppm)2Cl2] in n-dodecanol (20 mL); ca. 50 g of CO2-saturated 6 M Arg-solution added and removed in each run. | ||
The rate of formation corresponded to a TOFini of 3357 h−1 in the first run, which was significantly lower than on the 10 mL scale reflecting most likely mass transfer limitations due to less effective mixing. The system proved very robust, however, and the rates only declined after eight repetitive batches. In run ten, almost 50% of the initially observed rate was still maintained and the thermodynamic concentration limit was reached upon prolonged reaction time. The contamination of the aqueous product phase with ruthenium was determined by ICP-MS measurements (Table S7†). While a notable leaching of Ru into the product phase was noted after the first reaction (0.50 ppm), only trace amounts were detected in all further runs (0.01–0.08 ppm). Over the entire process, 3.3% of the originally charged Ru was leached into the product phase with a total loss of only 1% in runs 2–10 demonstrating proficient catalyst retention. The low leaching values cannot explain the reduced catalytic activity indicating that the chemical deactivation of the catalyst also plays a role under small-scale laboratory conditions. After the last run, 15.5 g of catalyst phase was recovered corresponding to 95% of the initially charged n-dodecanol proving also the excellent phase separation and negligible cross-solubility.
While basic amino acids such as Arg have the potential to act as scrubbing agents for CO2 capture, other amino acids are also very important as nutrients in animal feed. In particular, methionine is an important amino acid for livestock feed for poultry in which formic acid is a prominent preservative.26,62 Therefore, we also examined the application of the biphasic system for CO2 hydrogenation with Met in the aqueous phase. The water phase was saturated with Met at its maximum solubility (0.33 M) and n-dodecanol containing 0.5 mM of cis-[Ru(C12-dppm)2Cl2] was used as the catalyst phase. The reaction was conducted under standard conditions (30 bar CO2, 60 bar H2, 70 °C) for 16 h led to a formic acid-to-amino acid ratio of 0.6:1, corresponding to a TON of 595. This result demonstrates that the catalytic system is also compatible with neutral amino acids, opening a broad spectrum of possible compositions for applications of these solutions.
Amino acids are often applied in combinations for feed supplements. The formation of formic acid was therefore investigated using an aqueous solution containing a mixture of the three essential amino acids, namely arginine, histidine and methionine at their saturation limits (Arg 0.86 M, His 0.25 M, Met 0.33 M).63 Under standard conditions (3 mL H2O, 30 bar CO2, 60 bar H2, 70 °C, 0.5 mM cis-[Ru(C12-dppm)2Cl2] in 2 mL dodecanol), a concentration of 0.86 M FA was achieved after 2.5 h.
Conclusion
In summary, we have demonstrated that the hydrogenation of CO2 to formic acid can be performed effectively in biphasic systems comprising aqueous solutions of amino acids as the product phase together with an organic phase containing an organometallic catalyst. Besides the choice of catalyst, a major tuning factor of these biphasic systems lies in the respective organic phase. The bio-based solvent n-dodecanol (lauryl alcohol) was identified as a preferred solvent due to a combination of high catalyst activity, negligible water miscibility, and its benign character. The use of CO2-saturated Arg solutions as would be obtained from CO2 capture was successfully demonstrated to integrate the upstream carbon dioxide source with the downstream product phase directly. Excellent retention of the catalyst was achieved leading to metal contamination in the lower ppm range in the aqueous phase. In repetitive batch experiments, a total turnover number of more than 100000 was achieved with initial turnover frequencies in the 103 h−1 range. Solutions of up to 0.94 mol FA per mol amino acid were obtained using the basic amino acid Arg corresponding to 108 g of newly formed HCOOH per kg solution.
Based on these results, aqueous solutions of amino acids and formic acid resulting from the hydrogenation of CO2 can be envisaged for use directly or by further upgrading for high value applications without the need for energy intensive isolation of the HCOOH product from the reaction mixture. This would allow the replacement of the present fossil-based formic acid with a product based on biogenic CO2 for certain applications. The possible connection of the energetic use of biomass in biogas units and the upgrading of the inevitable CO2 byproduct to produce solutions that are used in farming directly may open novel opportunities for the concept of bio-energy conversion and carbon capture and utilization (BECCU) for local value chains contributing to a reduced CO2 footprint in the agricultural sector.
Experimental
Repetitive hydrogenation in the semi-continuous reaction setup
The flow-scheme of the semi-continuous reaction setup51 controlled by LabView™ software (National Instruments) is shown in Fig. 7 and described in more detail in the ESI.† The catalyst cis-[Ru(C12-dppm)2Cl2] was weighed into a Schlenk-tube (23.0 mg) and dissolved in n-dodecanol (10 mL). This solution was then added into the 100 mL autoclave, the Schlenk-tube was rinsed twice with n-dodecanol (5 mL) and the rinsing solutions were also added to the autoclave. The autoclave was sealed and connected to the plant. The substrate solution was prepared by weighing arginine (157.0 g) for multiple runs into the 900 mL high pressure reservoir, which was then evacuated three times and flushed with argon. Afterwards, water (150 mL) was added in argon counterflow. The high-pressure reservoir was positioned on a precision scale balance, connected to the plant, pressurized with CO2 to 30 bar and stirred at rt until the pressure remained constant. | ||
| Fig. 7 Flow-scheme of the semi-continuous plant. | ||
Capillaries and a HPLC-pump were flushed with the aqueous arginine solution up to the substrate inlet of the autoclave. Then, the autoclave was filled with 50 g of the substrate solution and pressurized with hydrogen (90 bar) over a bypass. The autoclave was then heated to 70 °C and the attained pressure (105 bar) was set as the fixed pressure in LabView™. The reaction was started by turning on the stirrer while the pressure was kept constant by dosing H2via a mass-flow controller (MFC). Data recording was started simultaneously. The reaction was stopped as soon as no further flow of H2 was detected. The stirrer was stopped and the phases were allowed to settle and separate for 2 min. The product solution was removed through the bottom fine dosing valve at the reaction temperature. Through the integrated window, the phase boundary was observed to avoid losses of the catalyst phase. The removed product phase was weighed and about 0.1 g of this solution was mixed with maleic acid (10 mg) as the internal standard and analyzed by 1H NMR spectroscopy. For the subsequent run, a fresh substrate solution was added into the autoclave with the HPLC pump so that the previous reaction pressure was obtained again. Afterwards the stirrer, the H2 delivery via the MFC, and the data recording were started again. This procedure was repeated nine times. After the last run, the catalyst phase was also removed and analyzed. The ruthenium contents of the aqueous phases were quantified by ICP-MS.
Author contributions
G. Franciò and W. Leitner conceived the idea and designed the experiments; N. Guntermann conducted the experiments under the supervision of G. Franciò and W. Leitner; N. Guntermann prepared the first draft; W. Leitner and G. Franciò revised the manuscript; all the authors endorsed the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832 and by the Bundesministerium für Bildung und Forschung (BMFB, Federal Ministry of Education and Research, 031B0850A). The authors are responsible for the content of this publication. Open Access funding was provided by the Max Planck Society.References
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207 CrossRef CAS
.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef CAS
- C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254 CrossRef CAS PubMed
- P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936 CrossRef CAS
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434 CrossRef CAS PubMed
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296 CrossRef CAS PubMed
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982 RSC
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187 CrossRef PubMed
- S. Deutz and A. Bardow, Nat. Energy, 2021, 6, 203 CrossRef CAS
- C. M. Jens, M. Scott, B. Liebergesell, C. G. Westhues, P. Schäfer, G. Franciò, K. Leonhard, W. Leitner and A. Bardow, Adv. Synth. Catal., 2019, 361, 307 CrossRef CAS
- K. A. Mumford, Y. Wu, K. H. Smith and G. W. Stevens, Front. Chem. Sci. Eng., 2015, 9, 125 CrossRef CAS
- R. S. Haszeldine, Science, 2009, 325, 1647 CrossRef CAS PubMed
- J. van Holst, S. R. A. Kersten and K. J. A. Hogendoorn, J. Chem. Eng. Data, 2008, 53, 1286 CrossRef CAS
- Y. Bian, S. Shen, Y. Zhao and Y. Yang, J. Chem. Eng. Data, 2016, 61, 2391 CrossRef CAS
- X. Wang, N. G. Akhmedov, D. Hopkinson, J. Hoffman, Y. Duan, A. Egbebi, K. Resnik and B. Li, Appl. Energy, 2016, 161, 41 CrossRef CAS
- A. Syalsabila, A. S. Maulud, H. Suleman and N. A. H. M. Nordin, Int. J. Chem. Eng., 2019, 2019, 1 Search PubMed
- Z. Zhang, Y. Li, W. Zhang, J. Wang, M. R. Soltanian and A. G. Olabi, Renewable Sustainable Energy Rev., 2018, 98, 179 CrossRef CAS
- V. S. Sefidi and P. Luis, Ind. Eng. Chem. Res., 2019, 58, 20181 CrossRef
- A. E. Reichl, R. Schneider, A. Ohligschläger, T. Rogalinski and S. Hauke, Energy Procedia, 2014, 63, 6379 CrossRef CAS
-
J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, Formic Acid, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2016 Search PubMed
- N. J. Claassens, I. Sánchez-Andrea, D. Z. Sousa and A. Bar-Even, Curr. Opin. Biotechnol., 2018, 50, 195 CrossRef CAS PubMed
- J. La, G. de Cruz, F. Machens, K. Messerschmidt and A. Bar-Even, ACS Synth. Biol., 2019, 8, 911 CrossRef PubMed
- S. Zobel, J. Kuepper, B. Ebert, N. Wierckx and L. M. Blank, Eng. Life Sci., 2017, 17, 47 CrossRef CAS PubMed
- L. M. Blank, T. Narancic, J. Mampel, T. Tiso and K. O'Connor, Curr. Opin. Biotechnol., 2020, 62, 212 CrossRef CAS PubMed
- S. R. Fernandez, S. Aoyagi, Y. Han, C. M. Parsons and D. H. Baker, Poult. Sci., 1994, 73, 1887 CrossRef CAS PubMed
- R. E. Warnick and J. O. Anderson, Poult. Sci., 1968, 47, 281 CrossRef CAS
- Y. Han, H. Suzuki, C. M. Parsons and D. H. Baker, Poult. Sci., 1992, 71, 1168 CrossRef CAS PubMed
- Supplementary feed for chickens, ducks, geese, quails and other poultry, “Röhnfried Hennengold”, zu finden unter https://stengel-fussring.com/produkte-nach-marken/roehnfried/roehnfried-hennengold-mineralfutter-mit-den-aminosaeuren-methionin-l-arginin-fluessig-1l/, 2022.
- G. Wu, F. W. Bazer, T. A. Davis, L. A. Jaeger, G. A. Johnson, S. W. Kim, D. A. Knabe, C. J. Meininger, T. E. Spencer and Y.-L. Yin, Livest. Sci., 2007, 112, 8 CrossRef
- D. Che, S. Adams, B. Zhao, G. Qin and H. Jiang, Curr. Protein Pept. Sci., 2019, 20, 736 CrossRef CAS PubMed
- B. Tan, Y. Yin, Z. Liu, X. Li, H. Xu, X. Kong, R. Huang, W. Tang, I. Shinzato and S. B. Smith,
et al.
, Amino Acids, 2009, 37, 169 CrossRef CAS PubMed
-
National Research Council, Division on Earth and Life Studies, Board on Agriculture and Natural Resources, Committee on Nutrient Requirements of Swine, Nutrient requirements of swine, National Academies Press, Washington, D.C., 11th revised edn, 2012 Search PubMed
-
PIC North America, Nutrient Specifications Manual 2016, Hendersonville, TN 37075, 2016 Search PubMed
- R. E. Muck, E. M. G. Nadeau, T. A. McAllister, F. E. Contreras-Govea, M. C. Santos and L. Kung, J. Dairy Sci., 2018, 101, 3980 CrossRef CAS PubMed
- S. C. Ricke, D. K. Dittoe and K. E. Richardson, Front. Vet. Sci., 2020, 7, 563 CrossRef PubMed
- V. Bampidis, G. Azimonti, M. d. L. Bastos, H. Christensen, B. Dusemund, M. Kouba, M. Kos Durjava, M. López-Alonso, S. López Puente and F. Marcon,
et al.
, EFSA J., 2019, 17, e05645 Search PubMed
- D. Wei, H. Junge and M. Beller, Chem. Sci., 2021, 12, 6020 RSC
- D. Wei, R. Sang, P. Sponholz, H. Junge and M. Beller, Nat. Energy, 2022, 7, 438 CrossRef CAS
-
J. J. Anderson, D. J. Drury, J. E. Hamlin and A. G. Kent, WO8602066, BP Chemicals Limited, 1986
-
R. G. Beevor, D. J. Gulliver, M. Kitson and R. M. Sorrel, EP0357243, BP Chemicals Limited, 1989
-
M. J. Green, A. R. Lucy, M. Kitson and S. J. Smith, EP0329337, BP Chemicals Limited, 1989
-
T. Schaub, O. Bey, A. Meier, D. M. Fries and R. Hugo, WO2013050367, BASF, 2011
- T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278 CrossRef CAS
- A. Behr, P. Ebbinghaus and F. Naendrup, Chem. Eng. Technol., 2004, 27, 495 CrossRef CAS
- C. A. Ohlin and G. Laurenczy, High Pressure Res., 2003, 23, 239 CrossRef CAS
- Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Jiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127 CrossRef CAS PubMed
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. Surya Prakash, Green Chem., 2016, 18, 5831 RSC
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580 CrossRef CAS PubMed
- M. Scott, B. Blas-Molinos, C. Westhues, G. Franciò and W. Leitner, ChemSusChem, 2017, 10, 1085 CrossRef CAS PubMed
- W. Leitner, G. Franciò, M. Scott, C. Westhues, J. Langanke, M. Lansing, C. Hussong and E. Erdkamp, Chem. Ing. Tech., 2018, 90, 1504 CrossRef CAS
- S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. Vom Stein, U. Englert, M. Hölscher and J. Klankermayer,
et al.
, Chem. Sci., 2015, 6, 693 RSC
- M. Scott, C. G. Westhues, T. Kaiser, J. C. Baums, A. Jupke, G. Franciò and W. Leitner, Green Chem., 2019, 21, 6307 RSC
-
M. Scott, Dissertation, RWTH Aachen, 2019
- W. L. F. Gassner, J. Chem. Soc., Chem. Commun., 1993, 1465 RSC
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168 CrossRef CAS PubMed
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, J. Am. Chem. Soc., 2005, 127, 13118 CrossRef CAS
- N. Guntermann, H. G. Mengers, G. Franciò, L. M. Blank and W. Leitner, Green Chem., 2021, 23, 9860 RSC
- P. Zhang, S.-F. Ni and L. Dang, Chem. – Asian J., 2016, 11, 2528 CrossRef CAS PubMed
- S.-F. Ni and L. Dang, Phys. Chem. Chem. Phys., 2016, 18, 4860 RSC
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302 RSC
- P. T. H. van To, K. Masagounder and M. E. Loewen, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2021, 255, 110908 CrossRef
- We thank one of the reviewers for suggesting this pertinent experiment during revision.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02598a |
| This journal is © The Royal Society of Chemistry 2022 |