
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShort electrochemical asymmetric synthesis of (+)-N-acetylcolchinol†
Yi
Du
a,
Adelaide
Lunga
a,
Aleksandr E.
Rubtsov
 ab and
Andrei V.
Malkov
*a
ab and
Andrei V.
Malkov
*a
aDepartment of Chemistry, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: A.Malkov@lboro.ac.uk
bDepartment of Chemistry, Perm State University, Bukireva 15, Perm 614990, Russia
First published on 1st September 2022
Abstract
A short synthesis of N-acetylcolchinol using a greener and step-economical pathway is reported where all the redox reactions, except for the asymmetric reduction, were carried out electrochemically, replacing protocols that employ transition metals or stoichiometric hazardous reagents. In a 4-step racemic sequence, chemoselective reduction of chalcone and intramolecular oxidative arene–arene coupling were performed in an electrochemical cell giving the target N-acetylcolchinol with an overall 41% yield. In a 7-step asymmetric variant, electrochemistry was also employed for the deprotection of p-methoxyphenyl amine. The target compound was obtained with a 33% overall yield and 99.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 er.
:0.5 er.
Introduction
Colchicine 1 (Fig. 1) is a well-known pseudo-alkaloid that has been widely used to treat gout, immune-mediated diseases, and psoriatic arthritis.1 It was shown to inhibit leukocyte-endothelial cells and T-cells by binding to intracellular tubulin monomers, which prevents their polymerization.2 Thus, colchicine has the potential to impair the process of antigen recognition and may inhibit cancer cell growth, but it proved to be toxic to normal cells. Instead, a less toxic to mammalian cells demecolcine 2, where the acetyl on the amino group is replaced with methyl, is used in chemotherapy. More recently, based on its anti-inflammatory properties, colchicine was investigated as a potential treatment for COVID-19 with some positive effects reported.3 Investigation of colchicine analogues revealed that allocolchicinoids derivatives where the 7-membered tropolone ring was replaced with a benzene ring (Fig. 1, 3–6) showed good biological activity and less toxicity compared to the parent colchicine.4N-Acetylcolchinol 3 is a known tubulin polymerisation inhibitor. Its water-soluble phosphate ZD6126 (4) was developed as the prodrug.5 The phosphate is converted in vivo into the active N-acetylcolchinol 3, which binds to the tubulin cytoskeleton of the endothelial cells in tumour blood vessels. ZD6126 selectively induced tumour vascular damage and tumour necrosis at well-tolerated doses in animal models, but it was found to be toxic, therefore, was discontinued. However, the pronounced biological activity of colchicine alkaloids continues to attract the attention of researchers to this class of compounds, thus maintaining the need for robust methodology to access their synthetic analogues. Herein, we present a simple, short and efficient sequence for the synthesis of N-acetylcolchinol 3 taking advantage of electrochemical methods.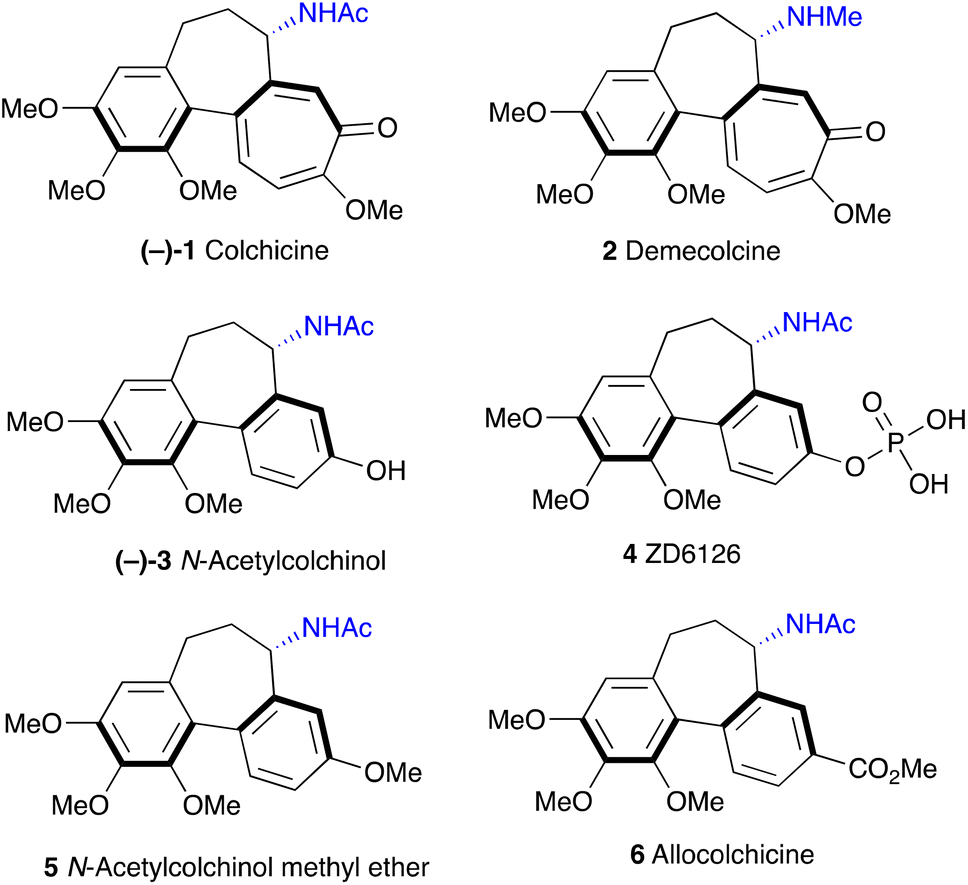 | ||
| Fig. 1 Colchicine and its analogues. | ||
In the middle of the 20th century, Rapoport6,7 and Cook8 reported the first syntheses of N-acetylcolchinol methyl ether 5 where the 7-membered ring of the colchinol framework was constructed by the sequence of transformations involving oxidative scission of the respective phenanthrene derivatives. Wulff and co-workers9 in their synthesis of allocolchicine 6 designed a strategy that centred on a Diels–Alder cycloaddition to assemble the methyl benzoate ring of 6, followed by aromatisation. Since then, a selection of diverse strategies toward colchicine alkaloids were disclosed.10–15 Among the plethora of synthetic approaches toward the structural core of the colchicine alkaloids, the routes involving coupling of the two aromatic fragments look most advantageous (Scheme 1),16–20 especially considering that the immediate precursors 7–9 can be prepared by trivial synthetic methods from the readily available materials.
 | ||
| Scheme 1 Cyclisation step in the synthesis of allocolchinoids. | ||
Macdonald16 reported a non-phenolic intramolecular oxidative coupling of the protected acetamide 8 using stoichiometric quantities of toxic Tl(O2CCF3)3. The same protocol was later employed by Chong.17 Fagnou18 proposed synthesis of allocolchicine 6 from the intermediate 9 by a Pd(0)-catalysed biaryl coupling, though high catalyst loading (10 mol%) was required. The toxicity of Tl(III) salts prompted a search for safer alternatives. Kocienski19 synthesised the N-acetylcolchinol 3 by intramolecular oxidative coupling of precursor 8 (R = OTBS) using hypervalent iodine(III) reagent PhI(O2CCF3)2 (PIFA). More recently, Yang20 employed PhI(OAc)2 (PIDA) in the oxidative cyclisation step in their gram scale asymmetric synthesis of colchicine 1. While our manuscript was the preparation, Waldvodgel21 reported on the electrochemical dehydrogenative coupling of 8 (R = OMe), although in modest yield (33%), which was improved to 62% through a reagent-mediated coupling employing MoCl3(HFIP)2.
However, the coupling of a simple precursor 7 to 3 remains challenging with the yields generally staying at a modest level. In this report, we present a green and atom-economical synthesis of N-acetylcolchinol 3 both in racemic and enantioselective variants, where the redox processes are accomplished electrochemically.
Results and discussion
Racemic synthesis
The reinvigorated interest in electrochemical methodology for organic synthesis,22–26 and our own recent experience in this field,27–29 prompted us to design a retrosynthetic sequence for N-acetylcolchinol 3, where two out of four steps are carried out electrochemically (Scheme 2). The first disconnection relies on the C–H/C–H coupling of electron-reach aromatic compound 7. The acetamide 7 can be obtained either from ketone 10 or from alcohol 11 through the Ritter reaction following the method reported by Doyle.30 Ketone 10 is included in this retrosynthetic scheme because it enables an enantioselective variant of the synthesis. Based on the previous reports31,32 and the work from our laboratories,27–29 we envisaged that both ketone 10 and alcohol 11 can be obtained chemoselectivity by electrochemical reduction of chalcone 12. The retrosynthetic sequence is completed by the retro aldol disconnection of 12 to the commercially available 3,4,5-trimethoxybenzaldehyde 13 and 3-hydroxyacetophone 14 (Scheme 2).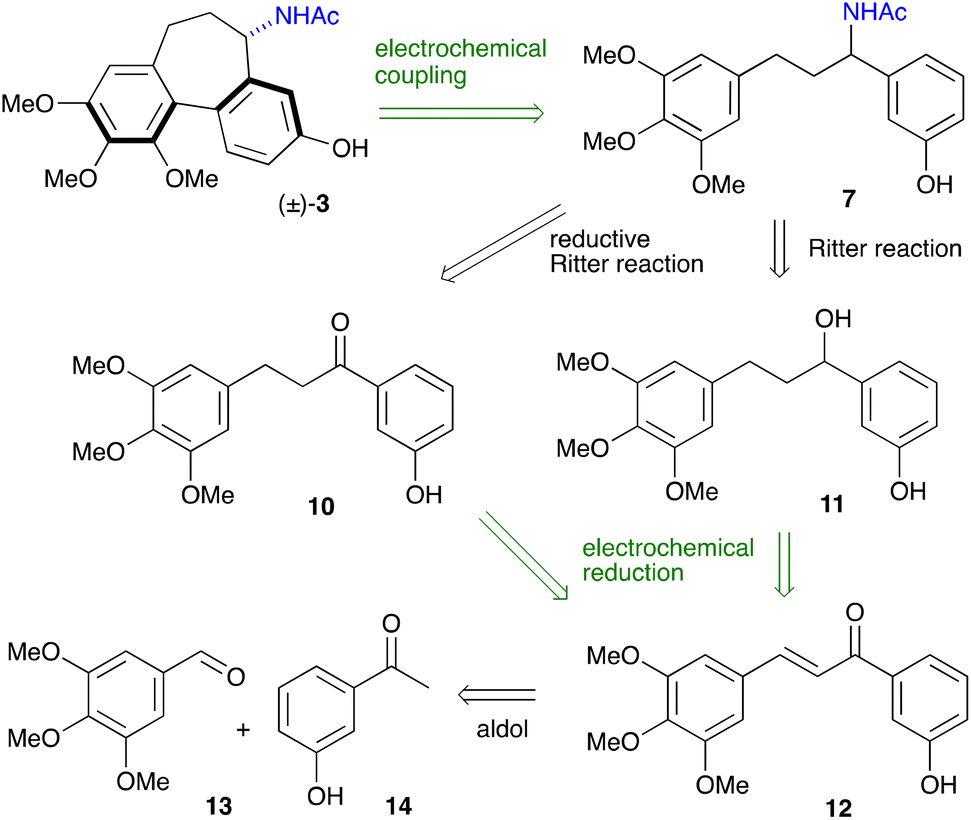 | ||
| Scheme 2 Proposed retrosynthetic scheme for N-acetylcolchinol. | ||
The synthesis commenced with the aldol condensation of the inexpensive, commercially available ketone 14 and aldehyde 13 using ethanolic alkoxide. The resulting chalcone 12 was isolated in 92% yield on a 10 mmol scale; it contains all the carbons of the target molecule skeleton.
Next, attention turned to the reduction of α,β-unsaturated ketone 12 to either ketone 10 or alcohol 11. Electrochemical reduction of the C–C double bond in unsaturated ketones has been reported recently by us29 and others,31,32 though there were no examples of the complete reduction to the saturated alcohol. Mechanistic probes by Xia31 into the electrochemical hydrogenation of the C–C double bond in chalcones carried out in a 4:1 mixture of DMSO/MeOH using Pt plate electrodes and NH4Cl as an electrolyte revealed that both ammonium cation and methanol can act as proton sources at the cathode whereas DMSO and MeOH27 are likely to serve as sacrificial reductants at the anode. Therefore, it is reasonable to assume that the double bond is reduced first followed by the reduction of the carbonyl. Taking this into account, we aimed to delineate conditions influencing the relative rates of the two processes.
Some representative optimisation experiments are collected in Table 1. For the full set of the data, see ESI.† First, we focused on the complete reduction of chalcone 12 to alcohol 11. After some experimentation, it was found that electrolysis of chalcone 12 for 5 hours at RT in a 4:1 mixture of DMSO and MeOH containing 5 equiv. of ammonium thiocyanate under the constant current of 10 mA with two carbon plate electrodes led selectively to the desired alcohol 11, which was isolated in a 92% yield; only minor quantities of 10 were detected by LCMS (Table 1, entry 1).
a
|
|
|||
|---|---|---|---|
| Entry | Variation from the standard condition | Yield of 10b (%) |
Yield of 11b (%) |
|
a Reaction conditions: Carbon plate anode, carbon plate cathode, undivided cell; substrate 0.2 mmol, electrolyte 5 equiv. in 10 mL of solvent DMSO/MeOH (4:1), constant current (10 mA), reaction time 5 hours.
b Conversions by LCMS (isolated yields given in parentheses).
c Detected by LCMS but not isolated.
d NR = no reaction.
|
|||
| 1 | None | Tracesc | (92) |
| 2 | nBu4NI instead of NH4SCN | 52 | 42 |
| 3 | nBu4NF instead of NH4SCN | 54 | 40 |
| 4 | Et4NBF4 instead of NH4SCN | 80 | 12 |
| 5 | NH4OAc instead of NH4SCN | 56 | 41 |
| 6 | NH4Cl instead of NH4SCN | 60 | 38 |
| 7 | DMSO as solvent | 60 | 30 |
| 8 | MeOH as solvent | NRd | NRd |
| 9 | CH3CN as solvent | 95 (92) | Tracesc |
| 10 | DMF as solvent | 98 | Tracesc |
| 11 | Ni(+)/Ni(−) instead of C(+)/C(−) | 89 (82) | Tracesc |
| 12 | C(+)/Ni(−) instead of C(+)/C(−) | 50 | Tracesc |
| 13 | C(+)/Fe(−) instead of C(+)/C(−) | 68 | Tracesc |
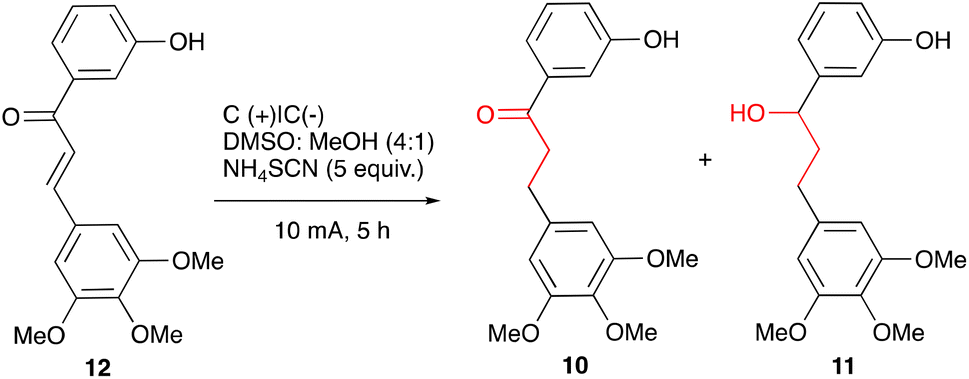
A brief screening of commercially available ammonium salts identified ammonium thiocyanate as optimal for the complete reduction of 12 to 11. Reduction of ketone 10 to alcohol 11 was distinctly slower when other common electrolytes were employed (Table 1, entries 2–6). Solvent composition proved to be an important factor in achieving high chemoselectivity in the reaction. In pure DMSO, a 2:1 mixture of ketone 10 and alcohol 11 was formed, whereas in methanol the reduction was completely suppressed (entries 7 and 8, respectively). The reduction also failed to proceed in DCM. Interestingly, in acetonitrile and DMF, only ketone 10 was obtained, with only traces of the overreduction observed (entries 9 and 10). The same level of selectivity can be attained by employing Ni electrodes instead of carbon (entry 11). A similar effect was achieved by swapping only cathode for Ni or Fe, though at the expense of the overall conversion (entries 11 and 12). In the absence of DMSO and MeOH, only thiocyanate was left to perform the role of the sacrificial reductant,33 which likely affected the rate of the reduction of 10 to 11.
Next, the conversion of alcohol 11 to acetamide 7 was investigated. Doyle and co-workers30 reported a reductive Ritter reaction where a ketone was treated with triethylsilane in aqueous acetonitrile in the presence of concentrated sulfuric acid to give the respective N-acetamide. In this process, the ketone is reduced to the alcohol first, which then undergoes Ritter substitution with acetonitrile. Following this protocol, both ketone 10, and its mixtures with alcohol 11 (see Table 1), can be converted to acetamide 7 with yields ranging from 56 to 64%. Naturally, pure alcohol 11 does not require the use of triethylsilane. Under the otherwise identical conditions, it was converted to acetamide 7 in a 73% yield.
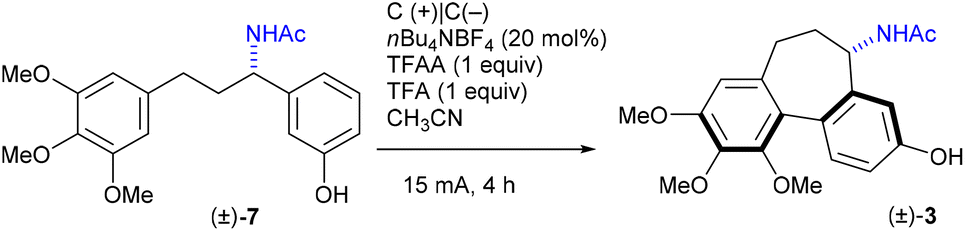
With acetamide 7 in hand, we embarked on optimising electrochemical conditions for the intramolecular oxidative coupling to furnish the desired colchinol 3 (Table 2). The electrochemical aryl–aryl coupling has been the subject of several investigations in the last two decades,21,34–38 though the conditions were found to be highly dependent on the nature of the substrates. In the set of electrochemical experiments using racemic 7 to trigger the formation of the 7-membered ring of the target (±)-3, the highest yield of 68% has been achieved in a non-divided cell equipped with two carbon electrodes using 10 mol% of nBu4NBF4, in MeCN as the solvent, with 1 equiv. each of TFFA and TFA (Table 2, entry 1). A CV curve for 7 showed one irreversible anodic oxidation peak at 0.45 V vs. Ag/AgNO3, whereas product 3 did not contain any redox peaks in the range −0.4 to 0.8 V (see ESI† for details). Other electrolytes showed inferior results (entries 2–5).
a
|
|
||
|---|---|---|
| Entry | Variation from the standard condition | Yieldb of 3 (%) |
| a Reaction conditions: Carbon plate anode, carbon plate cathode, undivided cell; substrate 0.1 mmol, 20 mol% electrolyte in 7 mL of solvent, constant current (15 mA), additives 1 equiv. each, reaction time 4 hours. b Isolated yield. c In other solvents, such as MeOH, iPrOH, THF and HFIP, only product traces were detected by LCMS. HFIP = (CF3)2CHOH. | ||
| 1 | None | 68 |
| 2 | nBu4NI instead of nBu4NBF4 | Trace |
| 3 | nBu4NBr instead of nBu4NBF4 | Trace |
| 4 | nBu4NHSO4 instead of nBu4NBF4 | NR |
| 5 | nBu4NF instead of nBu4NBF4 | 28 |
| 6 | DCM as solventc | 32 |
| 7 | Without TFAA/TFA | 46 |
| 8 | Without TFAA | 22 |
| 9 | Without TFA | 59 |
| 10 | H2O instead of TFAA/TFA | NR |
| 11 | Glassy carbon plate anode and cathode | 54 |
According to the previous reports,34–39 the solvent plays a significant role in the process by stabilising reactive intermediates and tuning nucleophilicity to favour the cross-coupling as opposed to the homo-coupling of two phenols. In our screening experiments, apart from MeCN, only dichloromethane showed some cyclisation, whereas THF and alcohols, including HFIP, appeared to shut down the reaction (entry 6).
At the same time, the addition of a 1:1 mixture of TFAA/TFA played an important role as in their absence the yield decreased (entries 7–9). No cyclisation took place in the presence of water (entry 10). Glassy carbon electrodes exhibited a slightly lower yield compared to the standard carbon plate electrodes (entry 11).
With the optimised conditions for each step, the overall 4-step synthesis of racemic colchinol 3 is presented in Scheme 3. The target compound (±)-3 was obtained in the overall yield of 41% as a single diastereoisomer confirming that the stereoselectivity of the aromatic coupling is controlled by the benzylic stereogenic centre, similar to the oxidative coupling instigated by stoichiometric oxidants.19,20,40 Next, the attention turned to the enantioselective variant of the synthesis.
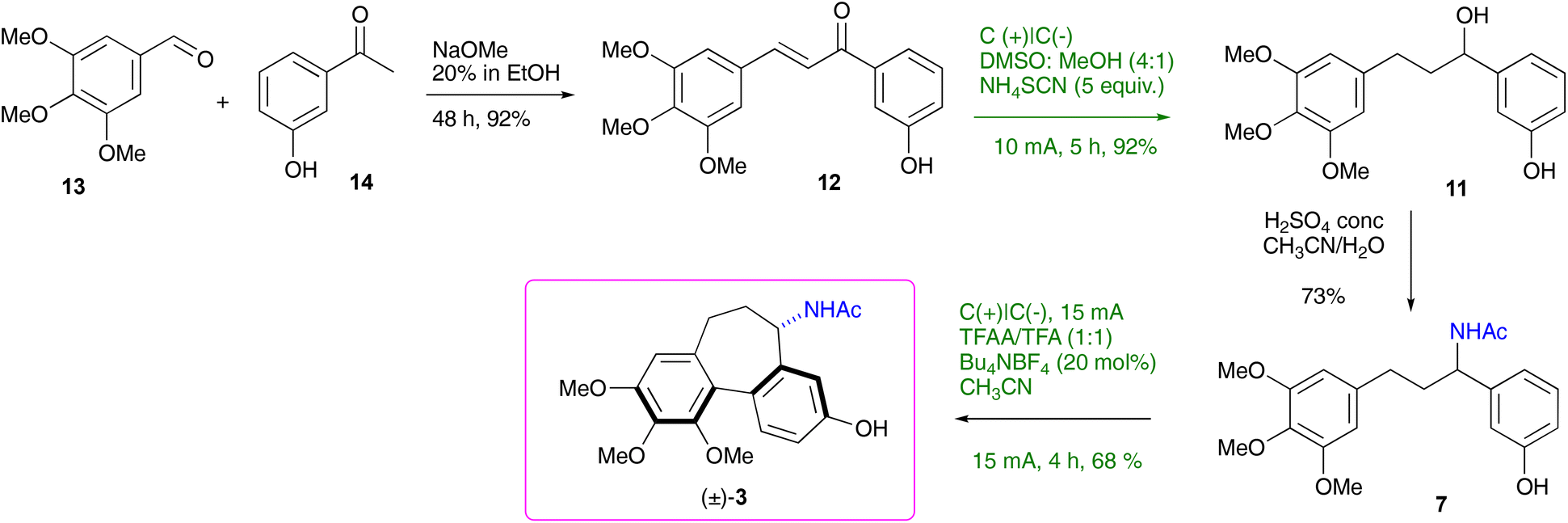 | ||
| Scheme 3 Synthesis of (±)-N-acetylcolchinol. | ||
Asymmetric variant
We envisaged that ketone 10 can be converted to imine 15 by condensation with 4-methoxyaniline followed by organocatalytic asymmetric reduction to amine 16 using the method developed in our laboratories.41–43 Reduction of a TBS-protected imine 15b with trichlorosilane in the presence of catalyst 17b was reported by us previously;40,44 catalyst 17a synthesised from unnatural D-valine performed equally well (Scheme 4). To avoid the extra silylation step of converting 15a to 15b, we investigated the reduction of the unprotected 15a. At room temperature, the enantioselectivity was modest, but it improved dramatically by lowering the reaction temperature to −30 °C. Amine 16a was obtained in a 91% yield and 99.5:0.5 er. We also attempted this reaction as a one-pot reductive amination,45,46 but combining prolonged heating required to convert ketone 10 to amine 15a with a low-temperature reduction proved challenging, therefore we settled for a two-step protocol.
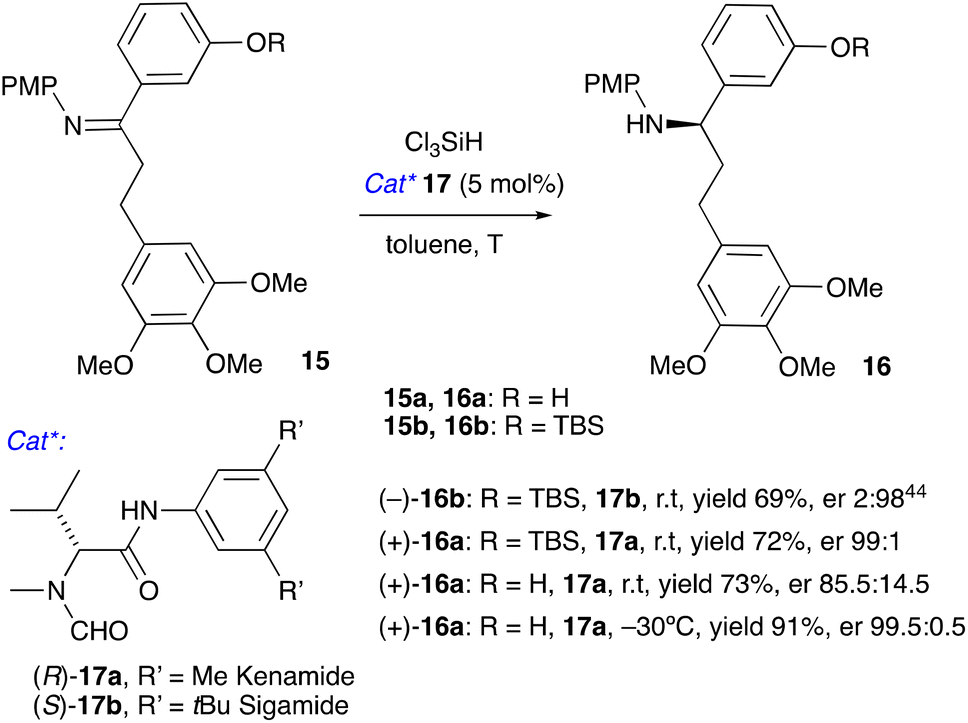 | ||
| Scheme 4 Asymmetric synthesis of chiral amines 16 and 17. (PMP = 4-MeOC6H4, TBS = tBuMe2Si). | ||
With the enantiopure 16a in hand, we next turned to electrochemical oxidative removal of the 4-methoxyphenyl group (PMP) in 16a. Mioskowski and Royer47 reported electrochemical deprotection of the PMP employing Pt electrodes in a divided cell. However, under their conditions, a low conversion in the formation of 18 was observed (Table 3, entry 5). Additional experiments were carried out to identify the optimal conditions as follows: amine 16a (0.1 mmol) was electrolysed under the constant potential of 0.85 V for 20 h at RT in a divided cell equipped with carbon plate electrodes, in a 9:1 mixture of MeCN and H2O (20 mL) containing 10 equiv. of NaClO4 and 3 equiv. of triflic acid. This led to a free amine 18, which after a standard workup (for details, see ESI†) was acylated to furnish pure (+)-7 in a 62% yield (Table 3, entry 1).
a
|
|
||
|---|---|---|
| Entry | Variation from the standard condition | Yieldb of 18 (%) |
| a Reaction conditions: Carbon plate anode, carbon plate cathode, divided cell; substrate 0.1 mmol, 10 equiv. of electrolyte in 20 mL of solvent, constant voltage (0.85 V), additive 0.5 equiv., reaction time 20 hours. b Conversion by LC-MS. c Isolated yield of (+)-7 after the treatment of 18 with acetic anhydride in DCM. | ||
| 1 | None | 76 (62c) |
| 2 | TFA instead of triflic acid | Trace |
| 3 | H2SO4 instead of triflic acid | Trace |
| 4 | Without triflic acid or H2O | Trace |
| 5 | Pt(+)/Pt(−) instead of C(+)/C(−) | 30 |
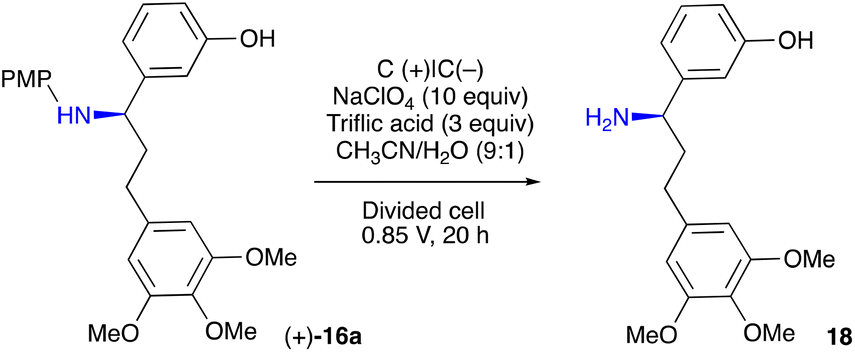
Triflic acid proved to be an important component of the mixture, as attempts to remove it or replace it with other acids failed to give any product (entries 2–4).
It was noted that among the competing side reactions of the removal of the PMP group in 16a was its oxidative cyclisation into 19, which represents an attractive alternative route to the target colchinol (+)-3. Therefore, this reaction was investigated next (Table 4).
a
|
|
||
|---|---|---|
| Entry | Variation from the standard condition | Yieldb of 19 (%) |
| a Reaction conditions: Carbon plate anode, carbon plate cathode, undivided cell; substrate 0.1 mmol, 20 mol% electrolyte in 7 mL of solvent, constant current (7 mA), additives 1 equiv. each, reaction time 4 hours. b Conversion by LC-MS. c Isolated yield. | ||
| 1 | None | 82 (74c) |
| 2 | CuSO4 instead of Cu2O | 50 |
| 3 | CuCl instead of Cu2O | 56 |
| 4 | CuBr instead of Cu2O | 60 |
| 5 | Cu(MeCN)4BF4 instead of Cu2O | 53 |
| 6 | With triflic acid | 70 (56c) |
| 7 | Without H2O | 20 |
| 8 | K2SO4 instead of Na2SO4 | 76 (63c) |
| 9 | Pt(+)/Pt(−) instead of C(+)/C(−) | 48 |
| 10 | Cu(+)/Cu(−) instead of C(+)/C(−) | Trace |

The conditions employed for the cyclisation of acetamide 7 (Table 2) have undergone some modifications. First, it was found that the addition of Cu2O played a crucial role in promoting oxidative aromatic coupling (Table 4, entry 1). Other Cu(I) and Cu(II) salts also gave the desired product but were slightly less efficient (entries 2–5). The beneficial effect of Cu is not surprising as for a long time Cu salts have been used, both stoichiometrically and catalytically, to effect biaryl cross-coupling.48,49 Switching to an aqueous solvent was another important change to the original protocol as a poor conversion was observed in anhydrous acetonitrile (entry 7). At the same time, the addition of acid was found no longer necessary (entry 6). In the aqueous reaction medium, simple inorganic salt can be used as supporting electrolytes (entries 1 and 8). The reaction is best carried out in an undivided cell equipped with carbon plate electrodes. Using other electrode materials gave inferior results (entries 9 and 10). It is worth noting that when the optimal conditions for the oxidative coupling of 16a were applied to acetamide 7, low conversion was observed (see ESI, Table S2†), suggesting that the substituents on the nitrogen play a significant role.
For the overall enantioselective synthesis of (+)-N-acetylcolchinol 3 we opted for the route involving cyclisation of (+)-16a into (+)-19. The synthetic sequence that commences with the unsaturated ketone 12 is presented in Scheme 5. Chemoselective electrochemical hydrogenation of 12 (Table 1, entries 9 and 10) furnished ketone 10 in 92% yield, which was converted to imine 15a (82%) by heating in toluene with 4-methoxyaniline in the presence of molecular sieves 5Å. Catalytic asymmetric reduction of 15a with trichlorosilane in the presence of 17a gave rise to a highly enantioenriched amine (+)-16a (91%, er 99.5:0.5), which was subjected to electrochemical cyclisation (Table 4, entry 1) to afford tricyclic (+)-19 (76%). Finally, electrochemical deprotection of the N-PMP group (a slightly lower voltage and a shorter time were used compared to those shown in Table 3) followed by acylation furnished the target (+)-N-acetylcolchinol 3 in a 70% yield over the two steps. The complete synthetic route from the commercial starting reagents 13 and 14 was accomplished in 7 steps with a 33% overall yield.
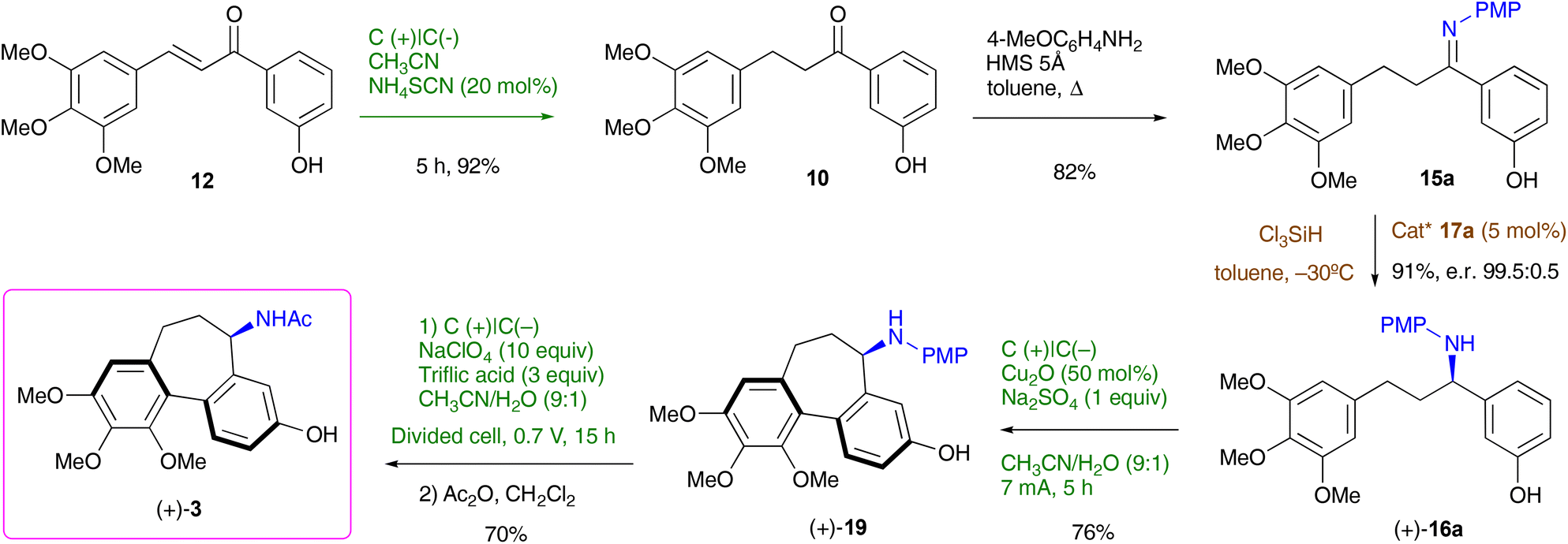 | ||
| Scheme 5 Synthesis of (+)-N-acetylcolchinol 3. | ||
Conclusions
In summary, we have developed two variants of stereoselective synthesis of N-acetylcolchinol where the reagent-based redox steps were replaced with mild electrochemical protocols. In a 4-step racemic variant, we first developed an efficient electrochemical procedure for the reduction of α,β-unsaturated ketone chemoselectively either to saturated alcohol or ketone. Then, the electrochemical conditions were optimised to achieve phenolic oxidative cyclisation of N-acetamide into N-acetylcolchinol. In the entire sequence, all the reactions were completed at room temperature with an overall yield of 41%.In a 7-step enantioselective version, the saturated ketone was converted to the imine by the reaction with 4-methoxyaniline. The imine was reduced to the respective enantioenriched amine by organocatalytic reduction with trichlorosilane with er 99.5:0.5. The next two redox steps, oxidative phenolic coupling and removal of the N-PMP group, were carried out electrochemically followed by acylation of the free amine to furnish the dextrorotatory enantiomer of N-acetylcolchinol in a 33% overall yield. Overall, this work showcases the enabling power of electrochemical redox methods in application to the stereoselective synthesis of complex molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
YD and AVM thank H2020 BBI JU for the PHENOLEXA Grant 101023225. RAE thanks the Russian Science Foundation for Grant 18-73-10156. YD and AL acknowledge additional support from Loughborough University.References
- P. G. Shekelle, S. J. Newberry, J. D. FitzGerald, A. Motala, C. E. O'Hanlon, A. Tariq, A. Okunogbe, D. Han and R. Shanman, Ann. Intern. Med., 2017, 166, 37–51 CrossRef PubMed.
- G. Taraboletti, G. Micheletti, R. Dossi, P. Borsotti, M. Martinelli, F. Fiordaliso, A. J. Ryan and R. Giavazzi, Clin. Cancer Res., 2005, 11, 2720–2726 CrossRef CAS PubMed.
- A. Z. Reyes, K. A. Hu, J. Teperman, T. L. Wampler Muskardin, J.-C. Tardif, B. Shah and M. H. Pillinger, Ann. Rheum. Dis., 2021, 80, 550–557 CrossRef CAS PubMed.
- A. Brossi, J. Med. Chem., 1990, 33, 2311–2319 CrossRef CAS.
- P. M. LoRusso, S. M. Gadgeel, A. Wozniak, A. J. Barge, H. K. Jones, Z. S. DelProposto, P. A. DeLuca, J. L. Evelhoch, S. A. Boerner and C. Wheeler, Invest. New Drugs, 2008, 26, 159–167 CrossRef CAS.
- H. Rapoport, A. R. Williams and M. E. Cisney, J. Am. Chem. Soc., 1950, 72, 3324–3325 CrossRef.
- H. Rapoport, A. R. Williams and M. E. Cisney, J. Am. Chem. Soc., 1951, 73, 1414–1421 CrossRef CAS.
- J. Cook, J. Jack, J. Loudon, G. Buchanan and J. MacMillan, J. Chem. Soc., 1951, 1397–1403 RSC.
- A. V. Vorogushin, A. V. Predeus, W. D. Wulff and H.-J. Hansen, J. Org. Chem., 2003, 68, 5826–5831 CrossRef CAS.
- T. Graening and H. G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 3230–3256 CrossRef CAS PubMed.
- M. G. Banwell, J. N. Lambert, M. F. Mackay and R. J. Greenwood, J. Chem. Soc., Chem. Commun., 1992, 974–975 RSC.
- M. G. Banwell, M.-A. Fam, R. W. Gable and E. Hamel, J. Chem. Soc., Chem. Commun., 1994, 2647–2649 RSC.
- S. Djurdjevic and J. R. Green, Org. Lett., 2007, 9, 5505–5508 CrossRef CAS.
- S. D. Broady, M. D. Golden, J. Leonard, J. C. Muir and M. Maudet, Tetrahedron Lett., 2007, 48, 4627–4630 CrossRef CAS.
- G. Besong, D. Billen, I. Dager, P. Kocienski, E. Sliwinski, L. R. Tai and F. T. Boyle, Tetrahedron, 2008, 64, 4700–4710 CrossRef CAS.
- J. S. Sawyer and T. L. Macdonald, Tetrahedron Lett., 1988, 29, 4839–4842 CrossRef CAS.
- T. R. Wu and J. M. Chong, Org. Lett., 2006, 8, 15–18 CrossRef CAS PubMed.
- M. Leblanc and K. Fagnou, Org. Lett., 2005, 7, 2849–2852 CrossRef CAS.
- G. Besong, K. Jarowicki, P. J. Kocienski, E. Sliwinski and F. T. Boyle, Org. Biomol. Chem., 2006, 4, 2193–2207 RSC.
- X. Liang, L. Li, K. Wei and Y.-R. Yang, Org. Lett., 2021, 23, 2731–2735 CrossRef PubMed.
- D. Pollok, F. U. Rausch, S. B. Beil, P. Franzmann and S. R. Waldvogel, Org. Lett., 2022, 24, 3760–3765 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- A. Alkayal, V. Tabas, S. Montanaro, I. A. Wright, A. V. Malkov and B. R. Buckley, J. Am. Chem. Soc., 2020, 142, 1780–1785 CrossRef CAS PubMed.
- A. M. Sheta, M. A. Mashaly, S. B. Said, S. S. Elmorsy, A. V. Malkov and B. R. Buckley, Chem. Sci., 2020, 11, 9109–9114 RSC.
- A. M. Sheta, A. Alkayal, M. A. Mashaly, S. B. Said, S. S. Elmorsy, A. V. Malkov and B. R. Buckley, Angew. Chem., Int. Ed., 2021, 60, 21832–21837 CrossRef CAS.
- M. P. Doyle, D. J. DeBruyn, S. J. Donnelly, D. A. Kooistra, A. A. Odubela, C. T. West and S. M. Zonnebelt, J. Org. Chem., 1974, 39, 2740–2747 CrossRef CAS.
- B. Huang, Y. Li, C. Yang and W. Xia, Chem. Commun., 2019, 55, 6731–6734 RSC.
- Y. Qin, J. Lu, Z. Zou, H. Hong, Y. Li, Y. Li, L. Chen, J. Hu and Y. Huang, Org. Chem. Front., 2020, 7, 1817–1822 RSC.
- V. A. Kokorekin, V. L. Sigacheva and V. A. Petrosyan, Tetrahedron Lett., 2014, 55, 4306–4309 CrossRef CAS.
- J. L. Röckl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef.
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed.
- K. Okamoto and K. Chiba, Org. Lett., 2020, 22, 3613–3617 CrossRef CAS.
- A. Lipp, M. Selt, D. Ferenc, D. Schollmeyer, S. R. Waldvogel and T. Opatz, Org. Lett., 2019, 21, 1828–1831 CrossRef CAS PubMed.
- A. Lipp, D. Ferenc, C. Gütz, M. Geffe, N. Vierengel, D. Schollmeyer, H. J. Schäfer, S. R. Waldvogel and T. Opatz, Angew. Chem., Int. Ed., 2018, 57, 11055–11059 CrossRef CAS PubMed.
- A. Kirste, M. Nieger, I. M. Malkowsky, F. Stecker, A. Fischer and S. R. Waldvogel, Chem. – Eur. J., 2009, 15, 2273–2277 CrossRef CAS PubMed.
- A. V. Malkov and P. Kočovský, in Lewis Base Catalysis in Organic Synthesis, ed. E. Vedejs and S. E. Denmark, Wiley-VCH Verlag GmbH & Co, Weinheim, Germany, 2016, vol. 3, ch. 20, pp. 1011–1038 Search PubMed.
- A. V. Malkov, A. Mariani, K. N. MacDougall and P. Kočovský, Org. Lett., 2004, 6, 2253–2256 CrossRef CAS.
- A. V. Malkov, S. Stončius, K. N. MacDougall, A. Mariani, G. D. McGeoch and P. Kočovský, Tetrahedron, 2006, 62, 264–284 CrossRef CAS.
- A. V. Malkov, K. Vrankova, R. C. Sigerson, S. Stončius and P. Kočovský, Tetrahedron, 2009, 65, 9481–9486 CrossRef CAS.
- A. V. Malkov, K. Vrankova, S. Stoncius and P. Kocovsky, J. Org. Chem., 2009, 74, 5839–5849 CrossRef CAS PubMed.
- A. V. Malkov, S. Stončius and P. Kočovský, Angew. Chem., Int. Ed., 2007, 46, 3722–3724 CrossRef CAS PubMed.
- K. K. Popov, J. L. P. Campbell, O. Kysilka, J. Hošek, C. D. Davies, M. Pour and P. Kočovský, J. Org. Chem., 2022, 87, 920–943 CrossRef CAS PubMed.
- S. De Lamo Marin, T. Martens, C. Mioskowski and J. Royer, J. Org. Chem., 2005, 70, 10592–10595 CrossRef CAS PubMed.
- P. Kočovský, Š. Vyskočil and M. Smrčina, Chem. Rev., 2003, 103, 3213–3246 CrossRef.
- Y. E. Lee, T. Cao, C. Torruellas and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 6782–6785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02321k |
| This journal is © The Royal Society of Chemistry 2022 |