
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst-free site-selective cross-aldol bioconjugations†
Nicholas D. J.
Yates‡
 a,
Saeed
Akkad‡
a,
Amanda
Noble
a,
Tessa
Keenan
a,
Natasha E.
Hatton
a,
Nathalie
Signoret
*b and
Martin A.
Fascione
*a
a,
Saeed
Akkad‡
a,
Amanda
Noble
a,
Tessa
Keenan
a,
Natasha E.
Hatton
a,
Nathalie
Signoret
*b and
Martin A.
Fascione
*a
aDepartment of Chemistry, University of York, York, UK YO10 5DD. E-mail: martin.fascione@york.ac.uk
bHull York Medical School, University of York, York, UK YO10 5DD. E-mail: nathalie.signoret@york.ac.uk
First published on 26th September 2022
Abstract
The bioconjugation of proteins to small molecules serves as an invaluable tool for probing biological mechanisms and creating biomaterials. The most powerful bioconjugation stratagems are those which have rapid kinetics, are site-selective, can be conducted in aqueous solvent under mild conditions and do not require the use of additional potentially toxic catalysts. Herein we present spontaneous coupling via aldol ligation of proteins (SCALP) a catalyst-free “green” site-selective protein ligation between α-oxo aldehyde functionalised proteins and enolisable aldehyde probes at neutral pH, and demonstrate the utility of this system in the targeting of prostate cancer cells with functionalised nanobodies.
Introduction
The field of protein bioconjugation has developed rapidly over the past three decades and a plethora of techniques now exist for modifying the side-chains of canonical amino acid residues such as lysine,1,2 tyrosine,3,4 tryptophan,5 methionine,6–8 histidine,9 and cysteine.1,10–12 However, the targeting of canonical amino acids in proteins is seldom site-specific, as multiple copies of the same amino acid may be solvent-exposed.13 To overcome these limitations, methods have been developed that enable site-specific installation of bioorthogonal functionalities, such as alkynes,14–17 azides,14,15 tetrazines18,19 and aldehydes,20–24 into proteins. These bioorthogonal motifs can subsequently be conjugated to, rapidly and selectively.25C–C bond formation represents a gold-standard in the formation of stable bioconjugates. Aldol condensations are one of the most fundamental tools for C–C bond formation in organic synthesis,26 and accordingly conjugation to protein aldehyde functionalities via aldol/enolate-type chemistries represents a popular method via which C–C bond formation between small molecule probes and proteins can be achieved.20,27–30 Existing examples of such bioconjugation chemistries include Knoevenagel condensation,27,28 aldol condensation using 2,4-thiazolidinediones as nucleophilic donors29 and organocatalyst-mediated protein aldol ligation (OPAL).20 Herein we introduce spontaneous coupling via aldol ligation of proteins (SCALP), a facile and gentle site-selective C–C bond forming bioconjugation method between reactive α-oxo aldehyde functionalities on proteins and enolisable aldehyde probes (Fig. 1). Whereas many aldol-ligations require the usage of organocatalysts based on proline, catalysing aldol condensation via enamine formation,31–34 SCALP adds to the repertoire of catalyst-free aldol C–C bond ligations. SCALP is simple, clean, cheap, site-selective, and can be performed in aqueous solution at neutral pH, making it ideal for use in the preparation of small molecule–protein bioconjugate therapeutics35,36 such as antibody or nanobody drug conjugates.37,38 Due to its catalyst-free nature, SCALP reactions also have a far greater reaction mass efficiency compared to many existing aldehyde bioconjugation reactions.20,39 SCALP could therefore represent a greener alternative to other popular established aldehyde bioconjugation methodologies, such as iso-Pictet Spengler ligation,40–42 OPAL20 or oxime/hydrazone ligation,39 which can either be slow at neutral pH, necessitating the usage of more equivalents of probe,40–42 or require the usage of super-stoichiometric quantities of catalyst in order to perform effectively.20,39
 | ||
| Fig. 1 Bioconjugation via SCALP to form stable conjugates linked by C–C bonds. | ||
Nanobodies are promising scaffolds for antibody-based therapies and can potentially reduce the costs and bottlenecks arising from monoclonal antibody production by obviating the need for complex mammalian expression systems.43 JVZ007 is a nanobody capable of high affinity binding to the extracellular domain of surface prostate-specific membrane antigen (PSMA),44 which is overexpressed on prostate cancer cells.45–48 We showcase the SCALP platform by modifying the JVZ007 nanobody site-selectively to yield a functional agent capable of labelling prostate cancer cells. We also demonstrate that bioconjugates labelled using SCALP can be subjected to further downstream bioconjugation reactions, such as azide–alkyne Huisgen cycloadditions.
Results and discussion
As a test-bed for the reaction a series of potentially enolisable aldehydes 1 were trialled for SCALP reactivity using non-enolisable α-oxo aldehyde LYRAG 2 (Fig. 2, ESI Fig. S35†).20,39,49,50 We hypothesised that phenylacetaldehydes would represent good nucleophilic SCALP-donors as the conjugated nature of the enol/enolates formed from these aldehydes should serve to make the enol/enolate more energetically accessible, thereby facilitating the synthesis of β-hydroxyaldehyde bioconjugates 3 at neutral pH (Fig. 2). Our study in aqueous conditions supports this hypothesis, demonstrating aliphatic aldehydes, like butyraldehyde 12, have no appreciable SCALP reactivity, whilst with the exception of diphenylacetaldehyde 8, all phenylacetaldehydes afforded SCALP products in high conversion (Table 1). Predictably benzaldehyde 11, which does not have an α-proton, showed no SCALP reactivity. Competition experiments were also conducted in which various SCALP-donors 5–15 and phenylacetaldehyde 4 were pre-mixed and delivered simultaneously to α-oxo aldehyde LYRAG 2. The relative conversions to SCALP–LYRAG products 3 were determined in order to establish the rates of reaction of the various SCALP-donors relative to phenylacetaldehyde 4. These experiments reinforce the hypothesis that the accessibility of the enolate at neutral pH determines the rate of the SCALP reaction, as the phenylacetaldehydes with strongly electron-withdrawing substituents, (4-nitrophenyl)acetaldehyde 10 and (4-fluorophenyl)acetaldehyde 14, demonstrated the highest relative reactivities (see Table 1 and Fig. 3). | ||
| Fig. 2 SCALP reactions between α-oxo aldehyde–LYRAG 2 and SCALP-donors 1 to yield (SCALP)–LYRAG products 3. | ||
 | ||
| Fig. 3 The percentage conversions of α-oxo aldehyde LYRAG 2 to SCALP–LYRAG 3 for various SCALP donors 1 (black), and the relative rate of reactions of various SCALP donors relative to phenylacetaldehyde 4 (grey). | ||
| Entry | Structure | Mean conversion | Relative rate of reaction compared to 4 |
|---|---|---|---|
| 1 |

|
74% | 1.0 |
| 2 |

|
0% | 0.0 |
| 3 |
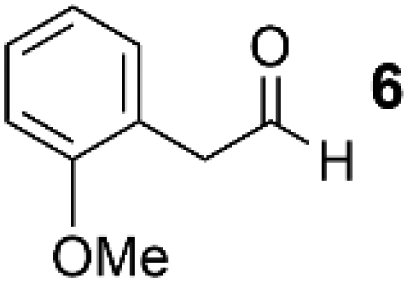
|
67% | 0.65 |
| 4 |

|
0% | 0.0 |
| 5 |

|
0% | 0.0 |
| 6 |

|
83% | 1.0 |
| 7 |

|
100% | 21 |
| 8 |

|
0% | 0.0 |
| 9 |

|
0% | 0.0 |
| 10 |

|
50% | 0.30 |
| 11 |
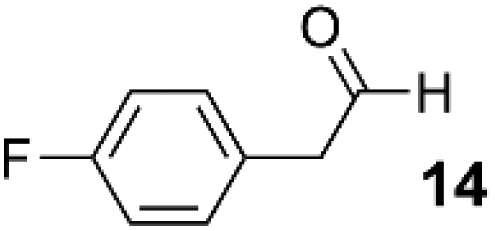
|
77% | 2.0 |
| 12 |
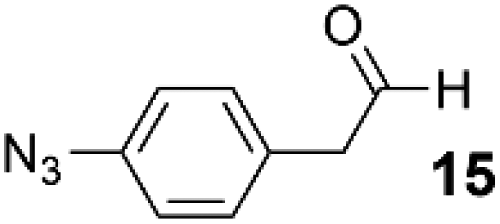
|
87% | 0.80 |
We chose to showcase the utility of SCALP bioconjugation using the JVZ007 nanobody, which can be used for the selective targeting of the prostate-specific membrane antigen (PSMA) on prostate cancer cells.44,53 As JVZ007 has yet to be structurally characterised, the structure of JVZ007 was predicted using Phyre2.51 Comparison of the predicted structure of JVZ007 with the crystal structures of the nanobodies NB7 and NB37 (which also bind PSMA)52 revealed that many conserved residues (which are thought to mediate the binding of NB7 and NB37 to PSMA)52 are distal from the N-terminus of JVZ007 (Fig. 4). We therefore selected the N-terminus for bioconjugation and selectively introduced an α-oxo aldehyde functionality at this site via oxidative cleavage of the N-terminal serine with NaIO4 (ESI, Fig. S36†).20,21,24,44
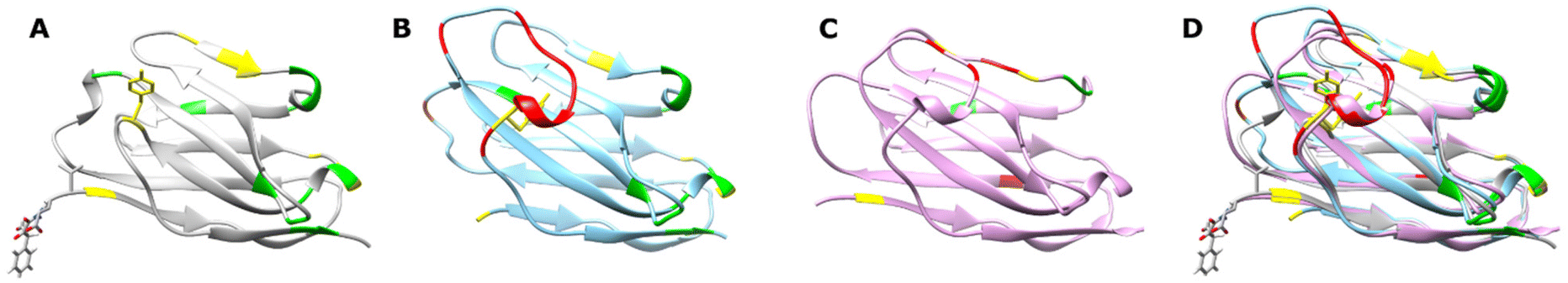 | ||
| Fig. 4 Comparison of suspected fold of JVZ007 with that of other PSMA binding nanobodies. (A) Predicted fold of JVZ007 generated using Phyre2,51 showing an N-terminal modification. (B) Nanobody NB7, PDB: 6XXN.52 (C) Nanobody NB37, PDB: 6XXP.52 (D) Overlay of the structures of JVZ007, NB7 and NB37, showing the highly conserved nature of the fold of the different nanobodies known to bind PSMA. Amino acid residues that are postulated to be involved in the binding of NB7 and NB37 to PSMA that are conversed in JVZ007 are coloured green, whereas those that are similar (i.e. a phenylaniline in place of a tyrosine or a threonine in place of a serine) are depicted in yellow. Residues postulated to in involved in binding PSMA in NB7 and NB37 that are absent in JVZ007 are coloured red. | ||
In order to demonstrate that SCALP bioconjugation could be used in conjunction with existing popular bioconjugation strategies, an azide motif was installed onto α-oxo aldehyde JVZ007 via SCALP with (4-azidophenyl)acetaldehyde 15, affording azide–JVZ007 (Fig. 5, ESI Fig. S36†). Azide–JVZ007 was characterised via protein mass spectrometry and was observed in several forms; the expected β-hydroxy aldehyde cross-aldol product, the corresponding hydrate, and hemiacetal (Fig. 5). No residual α-oxo aldehyde functionalised JVZ007 was detected by mass spectrometry after the SCALP ligation, verifying that SCALP donors with a rate of reaction comparable to phenylacetaldehyde are still reactive enough to label α-oxo aldehyde proteins. The site-selective nature of the technique was verified via trypsin digestion (ESI, Fig. S42–S45†). In addition, use of SCALP rather than classic OPAL conditions20 to prepare azide–JVZ007 was calculated to result in a 46% improvement in the E-factor and a 64% increase in reaction mass efficiency (see ESI†).
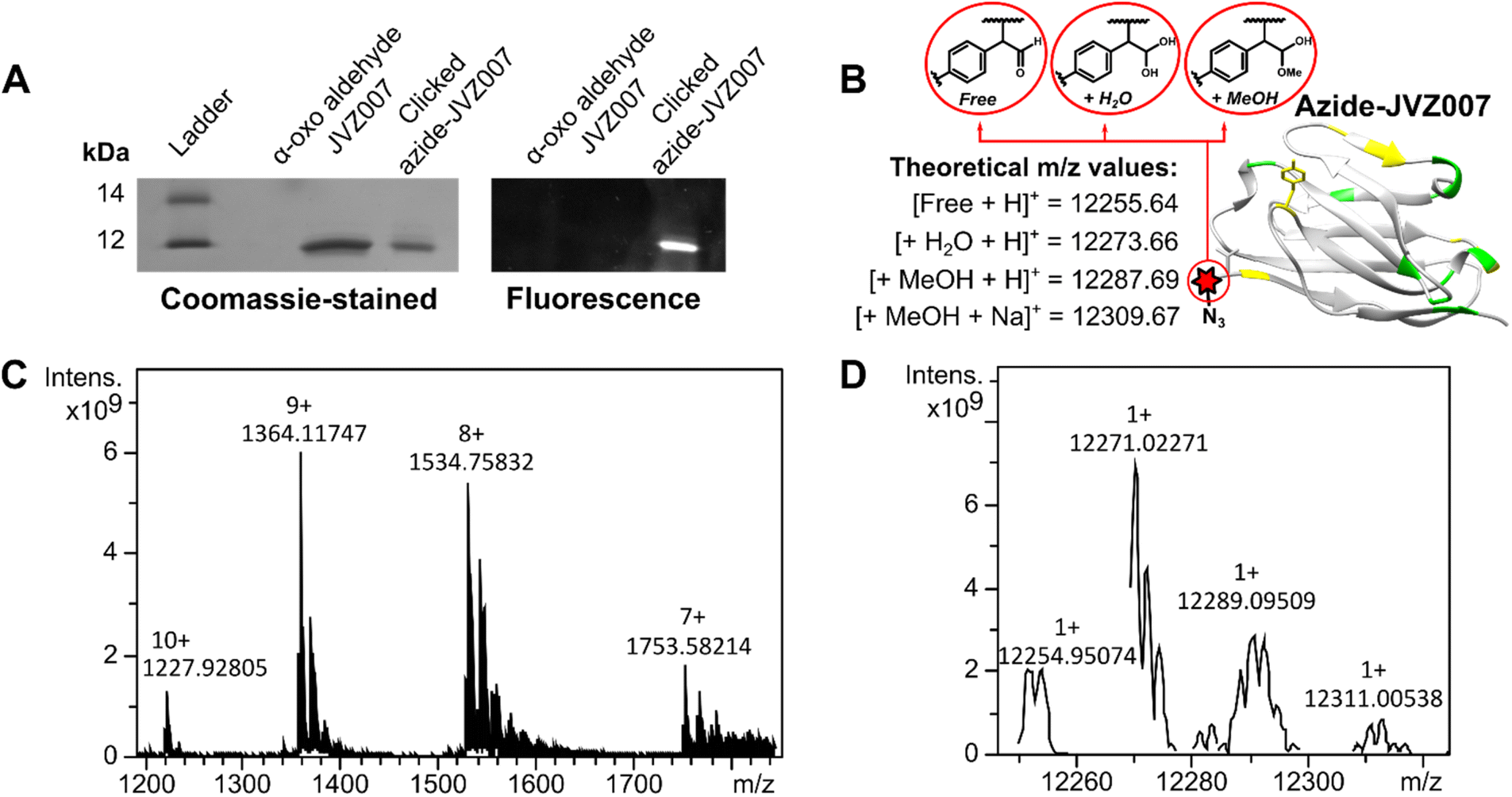 | ||
| Fig. 5 Azide–JVZ007 characterisation. SDS-PAGE gel analysis (A) shows successful fluorescent labelling of azide–JVZ007 via a strategy that utilises azide–alkyne Huisgen cycloaddition with dansylated azide probe S9 (see ESI, Fig. S34†). Protein mass spectrometry analysis shows protein charge-state ladders (C) that, upon deconvolution, yield peaks (D) consistent with azide–JVZ007 bearing either a free aldehyde (Free), a hydrated aldehyde (+H2O), and hemiacetal (+MeOH) (B). The discrepancies between the experimentally determined and theoretical m/z values are less than 3 m/z, and similar degrees of discrepancy are commonly observed during protein mass spectrometry analysis.20,23,24,27,54 | ||
We then fluorescently-labelled azide–JVZ007 via a methodology that utilised both a copper-catalysed Huisgen cycloaddition reaction between an azide and a terminal alkyne and a copper-free strain-promoted azide–alkyne Huisgen cycloaddition between an azide and a dibenzocyclooctyne (DBCO) motif (ESI, Fig. S37†).55 As expected, SDS-PAGE gel analysis demonstrated only azide–JVZ007 was fluorescently labelled, whereas α-oxo aldehyde JVZ007 remained unlabelled (Fig. 5).
Subsequently, 2-(4-methyoxyphenyl)acetaldehyde 9 was selected as a scaffold upon which to construct a more complicated functionalised SCALP probe, as the phenol moiety could easily be derivatised. Accordingly, biotinylated SCALP donor probe S5 (ESI, Fig. S24†) was synthesised using solid phase peptide synthesis.
SCALP bioconjugation between α-oxo aldehyde JVZ007 and biotinylated-SCALP donor probe S5 yielded biotin–JVZ007 (Fig. S36†), and the successful biotinylation was verified by both western-blot and protein mass spectrometry analyses (Fig. 6). Protein mass spectrometry again showed the biotin–JVZ007 SCALP product to be present in several forms derived from the expected β-hydroxy aldehyde cross-aldol product; as a hydrate and a hemiacetal (Fig. 6). Finally, we used the biotin–JVZ007 bioconjugate in flow cytometry experiments to demonstrate that JVZ007 still bound to PSMA selectively when modified at its N-terminus using PSMA-positive LNCaP cells and PSMA-negative PC3 cells56 (Fig. 7A), verifying that this labelling was indeed PSMA-specific via a competition experiment with unlabelled JVZ007 (Fig. 7B).
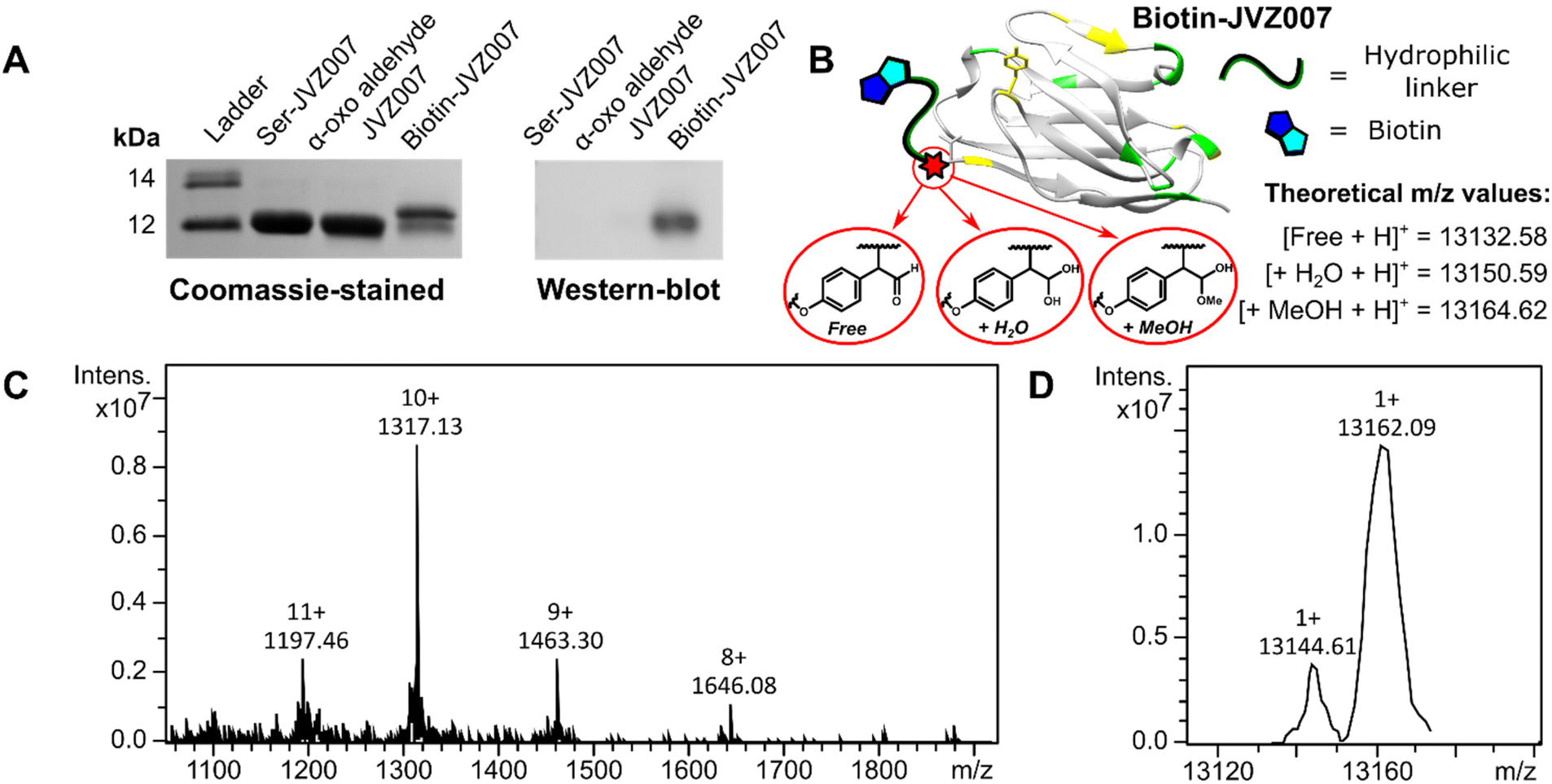 | ||
| Fig. 6 Biotin–JVZ007 characterisation. SDS-PAGE gel and western blot analyses (A) show successful biotinylation of JVZ007 via SCALP. Protein mass spectrometry analysis also shows protein charge-state ladders (C) that, upon deconvolution (D), yield peaks consistent with both biotin–JVZ007 hydrate (+H2O) and biotin–JVZ007 hemiacetal (+MeOH) (B). The discrepancies between the experimentally determined and theoretical m/z values are less than 6 and 3 m/z respectively, and similar degrees of discrepancy are commonly observed during protein mass spectrometry analysis.20,23,24,27,54 | ||
 | ||
Fig. 7 Biotin–JVZ007 binding. Representative histogram overlays from cells subsequently incubated with biotin–JVZ007 and streptavidin R-phycoerythrin conjugate (SAPE). (A) Experiments with LNCaP PSMA +ive (red) and PC3 PSMA −ive (black) cells show an increase in fluorescence for LNCaP cells due to the binding of the biotinylated JVZ007 nanobody to PSMA. (B) LNCaP cells incubated with the biotin–JVZ007 (pink) show increased fluorescence relative to LNCaP cells incubated with a mixture of non-labelled JVZ007 and biotin–JVZ (black) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), as unlabelled JVZ007 can compete with biotin–JVZ007 for accessible PSMA. :1), as unlabelled JVZ007 can compete with biotin–JVZ007 for accessible PSMA. | ||
Conclusions
To conclude, we have demonstrated that SCALP can be used as a mild site-selective C–C bond forming bioconjugation for α-oxo aldehydes biomolecules and simple aldehyde probes that bear acidic α-protons, without the need for catalysts. SCALP donors that bear electron-withdrawing substituents undergo conjugation more rapidly than SCALP donors bearing electron-donating substituents, which we attribute to the lowering of the pKa of the α-proton by electron-withdrawing groups making the enolate form of the SCALP donor more accessible. We also demonstrate the potential utility of SCALP for the preparation of protein bioconjugate therapeutics by selectively biotinylating the PSMA binding nanobody JVZ007. Furthermore, we demonstrate the suitability of SCALP in conjunction with established downstream bioconjugation methodologies by installing azide functionality onto JVZ007 and subsequently subjecting the resultant azide–JVZ007 bioconjugate to successive copper-catalysed Huisgen cycloaddition and copper-free strain-promoted azide–alkyne Huisgen cycloadditions. We anticipate that SCALP may find wide applications for the construction of the protein-small molecule bioconjugates where “greener” aldehyde-targeting bioconjugation reactions with greater reaction mass efficiency/reduced E-factors are desired, or where the stability of the targeted protein scaffold is compromised in the presence of excess small reagents such as catalysts, or by acidic/basic pH.Experimental
General methods and materials
All commercially available reagents were used as received. 4-Fluorophenylacetaldehyde dimethyl acetal, 2,4-dimethoxybenzaldehyde, sodium hydride, 4-nitrobenzaldehyde, ethylene glycol, p-toluenesulfonic acid monohydrate, 10% palladium on activated carbon, nitrosonium tetrafluoroborate, phenylacetaldehyde, 3-(4-methoxyphenyl)propionaldehyde, benzaldehyde and lithium bis(trimethylsilyl)amide were purchased from Sigma Aldrich. (Methoxymethyl)triphenylphosphonium chloride was purchased from Acros Organics. Potassium tert-butoxide, butyraldehyde and sodium azide were purchased from Alfa Aesar. (4-Methoxyphenyl)acetaldehyde, diphenylacetaldehyde and (2-methoxyphenyl)acetaldehyde were purchased from Fluorochem. Fmoc-protected amino acids were purchased from Fluorochem.Anhydrous solvents were dried prior to use according to standard methods. GPR-grade solvents were used for flash chromatography purposes. Solution-phase synthetic reactions were carried out using oven-dried glassware. All concentrations were performed in vacuo unless otherwise stated. Thin layer chromatography was carried out on Merck silica gel 60 F254 precoated aluminium foil sheets and these were visualized using UV light (254 nm).
Mass spectrometry
Small-molecule high resolution mass spectrometry (HRMS) data were obtained at RT on a Bruker Daltonics microTOF mass spectrometer coupled to an Agilent 1200 series LC system at The University York Centre of Excellence in Mass Spectrometry (CoEMS).High Performance Liquid Chromatography-Electrospray Ionization Mass Spectrometry (LC-MS) of peptides and biotin–JVZ007 was performed using a Dionex UltiMate® 3000 Ci Rapid Separation LC system equipped with an UltiMate® 3000 photodiode array detector probing at 250–400 nm, coupled to a HCT ultra ETD II (Bruker Daltonics) ion trap spectrometer, using Chromeleon® 6.80 SR12 software (ThermoScientific), esquireControl version 6.2, Build 62.24 software (Bruker Daltonics), and Bruker compass HyStar 3.2-SR2, HyStar version 3.2, Build 44 software (Bruker Daltonics) at CoEMS. Protein electrospray ionization (ESI) mass spectra of azide–JVZ007 and DBCO-JVZ007 were obtained on a Bruker Solarix XR 9.4 T FTICR mass spectrometer at CoEMS. Mass spectrometry data analysis was performed using ESI Compass 1.3 DataAnalysis, version 4.4 software (Bruker Daltonics). All mass spectrometry was conducted in positive ion mode unless stated otherwise.
Prior to analysis by LC-MS, peptides were diluted 1:3 in water and then further diluted 1:1 in acetonitrile with 1% (v/v) formic acid. Peptide samples were chromatographically analysed using an Ascentis Express C18, 2.7 μm (5 cm × 4.6 mm, Supelco). Water with 0.1% (v/v) formic acid (solvent A) and acetonitrile with 0.1% (v/v) formic acid (solvent B) were used as the mobile phase at a flow rate of 0.3 mL min−1 at RT (RT). A multistep gradient of 9.2 min was programmed as follows: 5% B for 0 min, a linear gradient to 10% B over 1 min, 10% B for 1 min, a linear gradient to 50% B over 2 min, a linear gradient to 90% B over 1 min, 90% B for 2 min, linear gradient to 5% B over 1 min, 5% B for 1 min. Note that the multistep gradient finishes in 5% B in order to re-equilibrate the column. Under these conditions, all peptides typically eluted between 2 and 7 min.
Prior to analysis, protein samples were desalted and analysed adjusted to a final concentration of 0.3–10 μM in 1:1 water:acetonitrile + 1% (v/v) formic acid. Protein samples were analysed without the use of a column at RT.
Flow cytometry
LNCaP and PC3 cells were detached using 10 mM EDTA in PBS, washed, resuspended in ice cold FACS buffer and plated in a 96 well plate at 75000 cells per well. Cells were incubated for 1 hour at 4 °C with JVZ–biotin at different concentrations (25–0.5 μg mL−1). For the competition binding assay the concentration of biotin–JVZ007 was kept constant and JVZ007 was added to a molar ratio of 1:1 relative to biotin–JVZ007. After nanobody incubation the cells were spun, washed with FACS buffer and incubated with secondary streptavidin R-phycoerythrin conjugate (SAPE) (BD554061 0.5 mg mL−1, 500 equivalents relative to biotin–JVZ007) for 1 hour at 4 °C. Cells were then washed, fixed overnight in FACS buffer containing 1% formaldehyde and resuspended in FACS buffer for analysis on a Cytoflex S.
Protein/peptide bioconjugation
α-oxo aldehyde LYRAG was prepared and characterised exactly as previously reported.20α-oxo aldehyde JVZ007 was prepared and characterised exactly as previously reported.24
To DCBO-terminal alkyne linker S7 (1.0 mg, 1.1 μmol) was added (MeCN)4Cu(I)BF4 (1.5 mg, 4.8 μmol). The mixture was then dissolved in 40 μL of anhydrous DMF under nitrogen. The resultant solution was incubated in darkness under nitrogen at room temperature for 30 minutes, to ensure coordination of Cu(I) to the DBCO unit of S7, thereby prohibiting its ability to partake in strain-promoted azide–alkyne cycloaddition reactions.55
1.8 μL of the S7/(MeCN)4Cu(I)BF4 mixture was then delivered to the azide–JVZ007 solution, delivering an ∼10-fold excess of copper-pretreated S7 relative to azide–JVZ007. 200 equivalents of (+)-sodium L-ascorbate was then delivered (using 10 μL of 100 mM stock solution). The reaction vessel was sealed shut to prevent excess oxygen diffusing into the sample, and the sample was incubated for 3 hours at 37 °C while the copper catalysed click reaction between the terminal alkyne of S7 and the aryl azide of azide–JVZ007 ensued. After this time, the sample was diluted to a volume of 1 mL using pH 7100 mM sodium EDTA solution and then dialysed overnight into pH 7100 mM sodium EDTA solution using 3.5 kDa dialysis tubing. During this step any copper coordinated to DBCO is removed, restoring the ability of the DBCO motif to partake in strain-promoted azide alkyne cycloaddition reaction. The sample was then dialysed into HPLC-grade water. This yielded DBCO-JVZ007 (ESI, Fig. S37†), which was characterised by UV-vis and mass spectrometry analyses (ESI, Fig. S38†). The DBCO–JVZ007 sample was then flash-frozen using liquid nitrogen and lyophilised.
To a 19 μM sample of DBCO–JVZ007 was delivered 10 equivalents of dansylated azide probe S8 and an appropriate volume of 5× concentrated reducing buffer (10% SDS, 10 mM 2-mercaptoethanol, 20% glycerol, 200 mM Tris-HCl pH 6.8, 0.05% bromophenol blue). The resulting sample was boiled for 5 minutes prior to analysis via SDS-PAGE, during which time strain-promoted azide–alkyne cycloaddition between the DBCO motif of DBCO–JVZ007 and the azide motif of S8 can be assumed to have reached completion.
Author contributions
Nicholas D. J. Yates: Conceptualization, methodology, investigation, writing – original draft preparation. Writing – reviewing and editing Saeed Akkad: Conceptualization, methodology, investigation, writing – original draft preparation. Amanda Noble: Methodology, investigation. Tessa Keenan: Methodology, investigation. Natasha E. Hatton: Methodology, investigation. Nathalie Signoret: Conceptualization, supervision, writing – reviewing and editing. Martin A. Fascione: Conceptualization, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Adam A. Dowle, Tony Larson, and Chris J. Taylor from the Bioscience Technology Facility, Metabolomics and Proteomics lab, Department of Biology, Dr Ed Bergstrom and The York Centre of Excellence in Mass Spectrometry. The York Centre of Excellence in Mass Spectrometry was created thanks to a major capital investment through Science City York, supported by Yorkshire Forward with funds from the Northern Way Initiative, and subsequent support from the Engineering and Physical Sciences Research Council (EP/K039660/1; EP/M028127/1). This work was supported by The University of York (S. A. and A. N.), The Engineering and Physical Sciences Research Council (EP/S013741/1, T. K., EP/V044303/1, N. D. Y., and EPSRC-NPIF PhD studentship (1944852) to N. E. H.), and York Against Cancer (A. N.).References
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS.
- J. S. Nanda and J. R. Lorsch, in Methods Enzymol, ed. J. Lorsch, Academic Press, 2014, vol. 536, pp. 87–94 Search PubMed.
- B. X. Li, D. K. Kim, S. Bloom, R. Y. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes and D. W. C. MacMillan, Nat. Chem., 2021, 13, 902–908 CrossRef CAS PubMed.
- S. Leier, S. Richter, R. Bergmann, M. Wuest and F. Wuest, ACS Omega, 2019, 4, 22101–22107 CrossRef CAS PubMed.
- K. Maruyama, K. J. Malawska, N. Konoue, K. Oisaki and M. Kanai, Synlett, 2020, 784–787 CAS.
- J. Kim, B. X. Li, R. Y. Huang, J. X. Qiao, W. R. Ewing and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 21260–21266 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, M. Weeks Amy, M. Hornsby, S. Lee Peter, V. Nichiporuk Rita, T. Iavarone Anthony, A. Wells James, F. D. Toste and J. Chang Christopher, Science, 2017, 355, 597–602 CrossRef CAS PubMed.
- A. H. Christian, S. Jia, W. Cao, P. Zhang, A. T. Meza, M. S. Sigman, C. J. Chang and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 12657–12662 CrossRef CAS PubMed.
- S. Jia, D. He and C. J. Chang, J. Am. Chem. Soc., 2019, 141, 7294–7301 CrossRef CAS PubMed.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed.
- F. A. Al-Lolage, M. Meneghello, S. Ma, R. Ludwig and P. N. Bartlett, ChemElectroChem, 2017, 4, 1528–1534 CrossRef CAS.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Eur. J. Chem., 2019, 25, 43–59 CrossRef CAS PubMed.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS.
- S. I. Presolski, V. P. Hong and M. G. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 CrossRef PubMed.
- L. Kiick Kristi, E. Saxon, A. Tirrell David and R. Bertozzi Carolyn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- S. Singh, I. S. Dubinsky-Davidchik and R. Kluger, Org. Biomol. Chem., 2016, 14, 10011–10017 RSC.
- J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898–2901 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- R. J. Spears, R. L. Brabham, D. Budhadev, T. Keenan, S. McKenna, J. Walton, J. A. Brannigan, A. M. Brzozowski, A. J. Wilkinson, M. Plevin and M. A. Fascione, Chem. Sci., 2018, 9, 5585–5593 RSC.
- R. J. Spears and M. A. Fascione, Org. Biomol. Chem., 2016, 14, 7622–7638 RSC.
- M. J. Appel and C. R. Bertozzi, ACS Chem. Biol., 2015, 10, 72–84 CrossRef CAS PubMed.
- R. L. Brabham, T. Keenan, A. Husken, J. Bilsborrow, R. McBerney, V. Kumar, W. B. Turnbull and M. A. Fascione, Org. Biomol. Chem., 2020, 18, 4000–4003 RSC.
- T. Keenan, R. J. Spears, S. Akkad, C. S. Mahon, N. E. Hatton, J. Walton, A. Noble, N. D. Yates, C. G. Baumann, A. Parkin, N. Signoret and M. A. Fascione, ACS Chem. Biol., 2021, 16, 2387–2400 CrossRef CAS PubMed.
- R. Brabham and M. A. Fascione, ChemBioChem, 2017, 18, 1973–1983 CrossRef CAS PubMed.
- S. Paladhi, A. Chauhan, K. Dhara, A. K. Tiwari and J. Dash, Green Chem., 2012, 14, 2990–2995 RSC.
- R. A. Kudirka, R. M. Barfield, J. M. McFarland, P. M. Drake, A. Carlson, S. Banas, W. Zmolek, A. W. Garofalo and D. Rabuka, ACS Med. Chem. Lett., 2016, 7, 994–998 CrossRef CAS.
- J. Yu, D. Shen, H. Zhang and Z. Yin, Bioconjugate Chem., 2018, 29, 1016–1020 CrossRef CAS PubMed.
- P. Wang, S. Zhang, Q. Meng, Y. Liu, L. Shang and Z. Yin, Org. Lett., 2015, 17, 1361–1364 CrossRef CAS PubMed.
- T. S. Howard, R. D. Cohen, O. Nwajiobi, Z. P. Muneeswaran, Y. E. Sim, N. N. Lahankar, J. T. H. Yeh and M. Raj, Org. Lett., 2018, 20, 5344–5347 CrossRef CAS PubMed.
- D. W. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- R. Ian Storer and D. W. C. MacMillan, Tetrahedron, 2004, 60, 7705–7714 CrossRef.
- A. B. Northrup, I. K. Mangion, F. Hettche and D. W. MacMillan, Angew. Chem., Int. Ed., 2004, 43, 2152–2154 CrossRef CAS PubMed.
- S. P. Brown, M. P. Brochu, C. J. Sinz and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 10808–10809 CrossRef CAS PubMed.
- F. Li and R. I. Mahato, Mol. Pharmaceutics, 2017, 14, 1321–1324 CrossRef.
- Y. Feng, Z. Zhu, W. Chen, P. Prabakaran, K. Lin and D. S. Dimitrov, Biomedicines, 2014, 2, 1–13 CrossRef PubMed.
- E. Y. Yang and K. Shah, Front. Oncol., 2020, 10, 1182 CrossRef PubMed.
- J. Z. Drago, S. Modi and S. Chandarlapaty, Nat. Rev. Clin. Oncol., 2021, 18, 327–344 CrossRef PubMed.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- S. Pomplun, M. Y. H. Mohamed, T. Oelschlaegel, C. Wellner and F. Bergmann, Angew. Chem., Int. Ed., 2019, 58, 3542–3547 CrossRef CAS PubMed.
- P. Agarwal, R. Kudirka, A. E. Albers, R. M. Barfield, G. W. de Hart, P. M. Drake, L. C. Jones and D. Rabuka, Bioconjugate Chem., 2013, 24, 846–851 CrossRef CAS PubMed.
- P. Agarwal, J. van der Weijden, M. Sletten Ellen, D. Rabuka and R. Bertozzi Carolyn, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed.
- S. Muyldermans, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS.
- K. L. S. Chatalic, J. Veldhoven-Zweistra, M. Bolkestein, S. Hoeben, G. A. Koning, O. C. Boerman, M. de Jong and W. M. van Weerden, J. Nucl. Med., 2015, 56, 1094 CrossRef CAS PubMed.
- S. S. Chang, Rev. Urol., 2004, 6(Suppl 10), S13–S18 Search PubMed.
- A. Cimadamore, M. Cheng, M. Santoni, A. Lopez-Beltran, N. Battelli, F. Massari, A. B. Galosi, M. Scarpelli and R. Montironi, Front. Oncol., 2018, 8, 653 CrossRef.
- S. Perner, M. D. Hofer, R. Kim, R. B. Shah, H. Li, P. Möller, R. E. Hautmann, J. E. Gschwend, R. Kuefer and M. A. Rubin, Hum. Pathol., 2007, 38, 696–701 CrossRef CAS PubMed.
- B. T. Ristau, D. S. O'Keefe and D. J. Bacich, Urol. Oncol., 2014, 32, 272–279 CrossRef PubMed.
- R. J. Spears, C. McMahon, M. Shamsabadi, C. Bahou, I. A. Thanasi, L. N. C. Rochet, N. Forte, F. Thoreau, J. R. Baker and V. Chudasama, Chem. Commun., 2022, 58, 645–648 RSC.
- N. Nepomniaschiy, V. Grimminger, A. Cohen, S. DiGiovanni, H. A. Lashuel and A. Brik, Org. Lett., 2008, 10, 5243–5246 CrossRef CAS PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS.
- L. Rosenfeld, A. Sananes, Y. Zur, S. Cohen, K. Dhara, S. Gelkop, E. Ben Zeev, A. Shahar, L. Lobel, B. Akabayov, E. Arbely and N. Papo, J. Med. Chem., 2020, 63, 7601–7615 CrossRef CAS PubMed.
- E. A. M. Ruigrok, N. van Vliet, S. U. Dalm, E. de Blois, D. C. van Gent, J. Haeck, C. de Ridder, D. Stuurman, M. W. Konijnenberg, W. M. van Weerden, M. de Jong and J. Nonnekens, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 1339–1350 CrossRef CAS PubMed.
- R. L. Brabham, R. J. Spears, J. Walton, S. Tyagi, E. A. Lemke and M. A. Fascione, Chem. Commun., 2018, 54, 1501–1504 RSC.
- S. Yoshida, T. Kuribara, H. Ito, T. Meguro, Y. Nishiyama, F. Karaki, Y. Hatakeyama, Y. Koike, I. Kii and T. Hosoya, Acta Chem. Scand., Ser. A, 2019, 55, 3556–3559 RSC.
- A. Ghosh, X. Wang, E. Klein and W. D. W. Heston, Cancer Res., 2005, 65, 727–731 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details the preparation of JVZ007, the synthesis and characterisation of organic compounds/probes and the method used for SDS-PAGE and western blot analyses. See DOI: https://doi.org/10.1039/d2gc02292c |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |