
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-free photochemical decomposition of sulfur hexafluoride by phosphines: formation of difluorophosphoranes as versatile fluorination reagents†
Philipp
Rotering
ab,
Christian
Mück-Lichtenfeld
 c and
Fabian
Dielmann
*a
c and
Fabian
Dielmann
*a
aInstitute of General, Inorganic and Theoretical Chemistry, Leopold-Franzens-Universität Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: fabian.dielmann@uibk.ac.at
bInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster Corrensstrasse 30, 48149 Münster, Germany
cOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster Corrensstrasse 40, 48149 Münster, Germany
First published on 27th September 2022
Abstract
The chemical activation of SF6 has garnered considerable attention because of its possible utilization as a cheap and safe reagent in chemical synthesis. Such a process becomes particularly attractive when combined with the disposal of the potent greenhouse gas after its technical application. Herein, we report on the photochemical reaction of SF6 with phosphines, which selectively produces difluorophosphoranes and phosphine sulfides. Computational and experimental studies show that the π(Ar) → σ*(SF6) charge-transfer excitation of a preformed R3P⋯SF6 complex is the initial activation step. Using triphenylphosphine, the decomposition of SF6 was carried out in a solvent-free, scalable process, giving a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of difluorotriphenylphosphorane and triphenylphosphine sulfide (TPP-Fluor), which was utilized for the deoxyfluorination of alcohols and carboxylic acids and for the preparation of common hexafluorophosphate salts.
:1 mixture of difluorotriphenylphosphorane and triphenylphosphine sulfide (TPP-Fluor), which was utilized for the deoxyfluorination of alcohols and carboxylic acids and for the preparation of common hexafluorophosphate salts.
Introduction
Sulfur hexafluoride (SF6) has been recognized as the most potent greenhouse gas among the industrial gases with a global warming potential of 23500 relative to CO2 over a 100-year time horizon.1 It is therefore listed as one of the six greenhouse gases in the 1997 Kyoto Protocol, which implemented regulative measures for SF6 usage in various industries.2 Today, technical uses of SF6 are largely limited to a few industries, including the electrical industry, which accounts for 80% of SF6 consumption, and other sectors such as metal casting, semiconductor manufacturing, and medical applications.3 The unique physical and chemical properties of the gas make its use in these areas particularly attractive, sometimes even indispensable.4 Despite all efforts to avoid emissions by managing SF6 in closed cycles or by replacing the greenhouse gas, the concentration of SF6 in the atmosphere is still on the rise.5 The reasons for this trend are diverse, but related to the fact that the disposal of used SF6 is challenging due to the extreme chemical inertness of the gas.6
The common decomposition process at the end of life of SF6 in electrical equipment is pyrolysis at temperatures above 1100 °C, whereby the sulfur and fluorine elements are converted to CaSO4 and CaF2 by chemical reaction with CaCO3.7 Because this process has high energy requirements and is not suitable for all SF6 applications, the development of alternative abatement methods is a current field of research.8 Particularly worthwhile in this context would be to obtain value-added compounds from the disposal process. Several different approaches exist which we herein divide into two main categories according to the initial activation strategy.
The first class of methods involves the direct fragmentation of SF6 under forcing conditions, e.g., via pyrolysis, photolysis or by various plasma methods.8 Since the inertness of SF6 arises from kinetic barriers to dissociation rather than its high thermodynamic stability, a clear advantage of the direct fragmentation strategy is that cheap reactants such as CaCO3, H2S, H2O or O2 can be applied to capture the SF6 fragments and no additional reagents or solvents are required. Notably, the use of catalysts can significantly alleviate the harsh conditions, but is associated with additional costs for catalyst replacement.9 The decomposition products are usually toxic and corrosive and require appropriate secondary treatment before they can be released. Moreover, the utilization of the decomposition products in fluorinations is hampered by the extreme reaction conditions and the low selectivity of the methods.
In the second class of methods, SF6 is chemically activated in solution under mild conditions, which involves a single-electron transfer or the nucleophilic attack on SF6 and therefore requires strongly reducing or highly nucleophilic substances, respectively. Compounds that react with SF6 at ambient conditions include alkali metals in liquid ammonia,10 strong organic reductants,11–13 electron-rich phosphines,14 N-heterocyclic carbenes,15,16 reactive anions,17,18 aluminium(I) compounds,19 transition metal complexes20–22 and organic radicals generated photolytically.23 These transformations are often highly selective and afford well-defined products which can be considered as potential fluorination agents.11,13,15,18,22 A disadvantage, however, is the need to conduct the reaction under inert gas due to the high reactivity of the reductants. A milestone towards the use of less sensitive and easier to handle stoichiometric reducing agents was the rhodium-catalyzed degradation of SF6 using silanes as reducing agents and phosphines as sulfur scavenger.20 Using photosensitizers for the reductive activation of SF6, the reaction was carried out in the presence of suitable substrates, which enabled deoxyfluorination of alcohols and pentafluorosulfanation of styrene derivatives.24–27 Despite the promising achievements in using SF6 as a fluorination reagent, when it comes to the disposal of surplus SF6 on a large scale in a cost-effective manner, these solution-phase strategies have the drawback of requiring expensive reagents, catalysts, and hazardous organic solvents. Although only carried out on a small scale, the electrochemical reduction of SF6 has the potential to overcome some of these drawbacks.28 Given these considerations, our intention was to combine the advantages of both categories and develop a solvent-free process that would yield well-defined products which could be used as fluorination reagents in chemical synthesis.
We have recently shown that the electron-releasing character of phosphines is significantly enhanced by attaching strong π-donor substituents to the phosphorus atom.29 Phosphines equipped with three substituents are ranked among the strongest nonionic superbases and are characterized by extreme reactivity towards electrophiles.30 Accordingly, unlike commercially available alkyl or aryl phosphines, phosphines modified in this way can activate SF6 at ambient conditions and convert it into potential fluorinating reagents.14 The recent report by Braun and coworkers on the photochemical activation of SF6 by N-heterocyclic carbenes inspired us to consider the reaction of more simple phosphines with SF6 triggered by irradiation with light, which we report herein.15
Results and discussion
Among commercially available phosphines, triphenylphosphine has the most attractive properties for a scalable degradation of SF6 because it is an inexpensive, nontoxic, air-stable solid that is produced on a large scale. However, as demonstrated previously, Ph3P is not nucleophilic enough to activate SF6 even at elevated temperature.14 To investigate whether light can induce the reaction, we first recorded the UV-vis spectra of Ph3P in THF under an atmosphere of argon or SF6. Both spectra were identical and only showed an absorption band at 240–310 nm (Fig. 1). Previous studies on the photolysis of Ph3P showed that this absorption involves the n → π* excitation leading to the homolytic cleavage of a P–Ph bond.31,32 We therefore irradiated 0.2 M solutions of Ph3P in THF under an atmosphere of 1 bar SF6 using narrow-band LED light sources (see the SI for details). Exposure of the solution to UV light at 310 nm gave several P–F species, including Ph4P+ and Ph2PF3, in agreement with the photolytic cleavage of P–Ph bonds. However, the formation of P–F species likewise indicates the successful activation of SF6 under these conditions. Irradiation of the solution with light at 365 nm or 405 nm led to the clean conversion of Ph3P into a 3:1 mixture of difluorotriphenylphosphorane and triphenylphosphine sulfide within 6 hours or 16 hours, respectively. Note that the reaction is significantly faster using light at 365 nm, despite the lower relative irradiance (365 nm LED: 9 mW cm−2, 405 nm LED: 28 mW cm−2). No reaction between Ph3P and SF6 was observed with blue (450 nm) or orange (585 nm) light.
 | ||
| Fig. 1 Experimental UV-vis spectra of selected triarylphosphines (c = 3.8 × 10–5 M in THF). | ||
We next irradiated solid Ph3P under an SF6 atmosphere for 24 hours using the 365 nm LEDs. Although the solid material turned light brown at the solid–gas interface facing the light source, the 31P and 19F NMR spectra of the dissolved solid product revealed that less than 1% of the Ph3P had been converted to fluorinated species. We then carried out the photoreaction using a Ph3P melt to increase the direct contact between the reactants. Irradiation of a Ph3P melt at 80 °C under 1 bar SF6 atmosphere resulted in the quantitative formation of difluorotriphenylphosphorane and triphenylphosphine sulfide in a ratio of 3:1 within 8 hours (Scheme 1). The difluorophosphorane can be separated from the phosphine sulfide by recrystallization from α,α,α-trifluorotoluene in 81% yield. However, separation of the products proved unnecessary for applications of Ph3PF2 in fluorination reactions owing to the chemical inertness of Ph3PS (vide infra). The obtained 3:1 mixture of Ph3PF2 and Ph3PS is therefore referred to as TPP-Fluor in the following. To demonstrate the scalability of the process, a flat-bottomed glass vessel containing 100 g of Ph3P under 1 bar SF6 pressure was placed above an LED array (see the ESI for details†). Upon irradiation with light at 365 nm, Ph3P started to melt in the vessel due to heat uptake from the LED array, producing TPP-Fluor within 9 hours in quantitative yield. It is noteworthy that the reaction time increases only slightly despite the tenfold scale of the reaction, suggesting that the reaction rate is limited by the solubility and diffusion of SF6 in Ph3P.
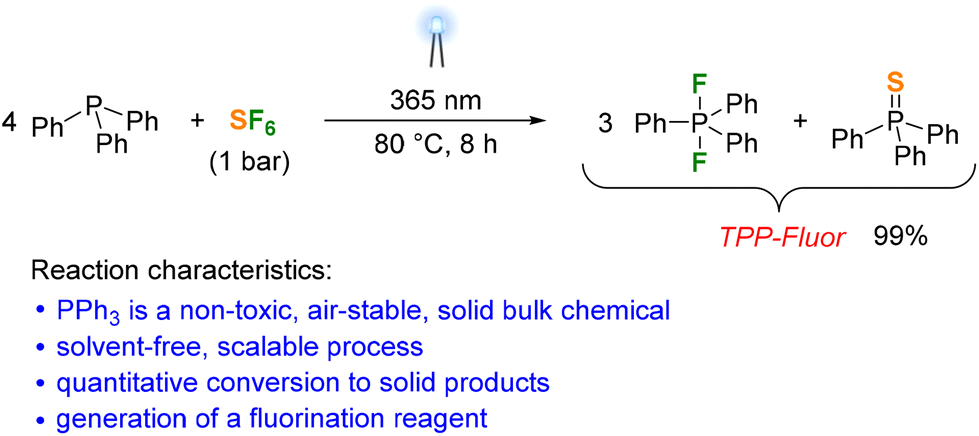 | ||
| Scheme 1 Photoreduction of SF6 with triphenylphosphine affording a 3:1 mixture of difluorotriphenylphosphorane and triphenylphosphine sulfide. | ||
The scope of the photochemical reaction between phosphines and SF6 was investigated by irradiating THF solutions of various phosphines under 1 bar SF6 atmosphere with light at 365 nm (Scheme 2). Several phosphines were smoothly converted into the difluorophosphorane derivatives in excellent yield, while other phosphines did not react with SF6 under these conditions. While this behaviour can be rationalized by the electron-poor character of perfluorinated phosphines (12) and phosphites (8, 9), it is unexpected for the alkylphosphines and aminophosphines (5–7), because they are stronger reducing agents than triphenylphosphine. In addition, electron-rich arylphosphines were readily converted into the difluorophosphoranes (13, 15, 16). An explanation for this behaviour can be derived from the series of aminophosphines (2–5), which show a trend of decreasing reactivity with the number of phenyl groups and suggests that the photochemical reaction with SF6 requires at least one phenyl group at the phosphorus atom. Finally, ortho substituents appear to hamper the reaction (14) or lead to decomposition reactions (10, 11). Note that the decomposition of the sterically more encumbered phosphines (10, 11) was also observed upon irradiation with light at 365 nm in the absence of SF6 and is attributed to the red-shifted n(P) → π*(Mes) absorption band (Fig. 1).
 | ||
| Scheme 2 Scope of the photochemical activation of SF6 by phosphines affording a mixture of phosphine sulfides and difluorophosphoranes, of which only the latter are depicted. Yields determined by 31P NMR spectroscopy. aPhosphine present in reaction mixture. bDecomposition of the phosphine upon irradiation. | ||
Previous studies showed that the photoexcitation of Ph3P with light at λ = 266 nm leads to the homolytic cleavage of a phosphorus-phenyl bond to form the diphenylphosphinyl radical Ph2P˙ and phenyl radical Ph˙.31–34 This P−C bond cleavage has been reported to occur either from the singlet (1Ph3P*) or the triplet excited state (3Ph3P*).33,34 However, when Ph3P is irradiated in the presence of molecular oxygen, an electron transfer to O2 occurs from the photoexcited state of Ph3P to give the radical ion pair Ph3P˙+/O2˙−.35 The reaction produces triphenylphosphine oxide in excellent yield upon irradiation with light of wavelengths over 310 nm. However, very sluggish oxidation of Ph3P was observed using light with longer wavelength than 350 nm. By contrast, the reaction between Ph3P and SF6 is triggered by light of λ > 350 nm and produces only products with intact P−Ph bonds. We therefore consider an initial n → π* excitation of Ph3P followed by subsequent electron transfer to SF6 unlikely, especially since no decomposition of Ph3P with light at 365 nm or 405 nm was observed in the absence of SF6 (Fig. S58†).
To gain an insight into the reaction pathway of arylphosphines with SF6 focussing on the initial activation step, we performed DFT calculations at the PW6B95-D3//TPSS-D3/def2-TZVP level of theory using the COSMO-RS solvation model with THF for 298 K (see the ESI†). The proposed mechanism for the formation of Ph3PF2 from Ph3P and SF6 is illustrated in Fig. 2. Starting from an endergonic encounter complex [Ph3P⋯SF6], electronic excitation occurs with a calculated wavelength of 360 nm (step A in Fig. 2) and corresponds to a charge-transfer state in which one electron is transferred from a π orbital of the arene (HOMO−1, Fig. 4a) to the delocalized σ* orbital of SF6 (LUMO, Fig. 4b). According to the TD-DFT result, the n(P) → σ*(SF6) transition occurs at a higher wavelength (525 nm) with low oscillator strength, and thus it is not expected to be involved in the reaction (Fig. 3). This conclusion agrees with the experimental observation that light at 585 nm does not induce the photochemical reaction and that phosphines lacking aryl substituents do not react with SF6.
 | ||
| Fig. 2 Energy diagram of the proposed mechanism for the photochemical reaction of Ph3P with SF6. ΔG: PW6B95-D3//TPSS-D3/def2-TZVP + COSMO-RS(THF). TDDFT: B3LYP/def2-TZVP + CPCM(THF). Free energies are reported with respect to isolated Ph3P and SF6 and include implicit solvation energies in THF. | ||
 | ||
| Fig. 3 Calculated absorption spectrum of [Ph3P⋯SF6] using TD-DFT at the B3LYP/def2-TZVP level of theory and the implicit solvation model CPCM(THF). | ||
 | ||
| Fig. 4 (a and b) Frontier orbitals of the encounter complex [Ph3P⋯SF6]. Orbital energies calculated with B3LYP/def2-TZVP + CPCM(THF). Isosurface value 0.05 a.u. (c) Spin density ρα–ρβ (isosurface value = 0.005 a.u.) and (d) electrostatic potential (isodensity value 0.02 a.u.) of the optimized triplet state 3[Ph3PF⋯SF5] (PW6B95//TPSS-D3/def2-TZVP). | ||
To get a qualitative insight into the photochemical processes that may occur after the charge transfer excitation, we have located the conical intersection that connects the S0 and S* state of the complex using the range-separated hybrid functional CAM-B3LYP36 and the def2-SVP basis set.37 In the ionic complex, the distance between the two ions (d(P⋯S) = 3.74 Å) is significantly smaller than in the ground state complex (d(P⋯S) = 4.81 Å), while the SF bonds are elongated by about 0.07 Å. This indicates that the charge-transfer excitation promotes the reaction by driving the reactants to a distance short enough for electron and/or fluorine transfer.
In agreement with this observation, we were not able to locate a transition structure for F transfer with our (ground state) DFT calculations. Only when we optimized the triplet state of the complex (3[Ph3P⋯SF6]), which could be formed by intersystem crossing from the charge transfer state (S*), we observed immediate fluoride ion transfer to give the radical ion pair 3[Ph3PF⋯SF5] (step B). Therein, the spin density is evenly distributed over both fragments of the triplet complex (Fig. 4c), and the unpaired electron in (Ph3PF)˙ occupies the σ* orbital of the P–F bond, partially delocalized in one phenyl ring. The radical ion pair 3[Ph3PF⋯SF5] can either dissociate or instantaneously transfer an electron after ISC to form the ion pair [Ph3PF]+/[SF5]− (step C). Note that the radical ion pair [Ph3P]˙+/[SF6]˙− representing the “dissociated” charge-transfer state, has a relative free energy of 49.5 kcal mol−1, which is well below the photoexcited state. We therefore cannot distinguish whether a consecutive fluoride (B/B′) and electron transfer (C) or a direct fluorine atom transfer (D) occurs after photoexcitation. These processes are presumably fast and involve closely associated ion pairs. Overall, the first reaction step is exergonic (−32.7 kcal mol−1) and involves a formal “F+ transfer” resulting in the ion pair [Ph3PF]+/[SF5]−, which is strongly stabilized by solvation in THF. The subsequent fluoride transfer (step E) is exergonic by more than −20 kcal mol−1 and expected to have a low energy barrier.14
The UV-vis spectra of the free phosphines Ph3P, Mes3P, (2-OMe-C6H4)3P, (4-OMe-C6H4)3P and of the corresponding noncovalent SF6 complexes were calculated using TD-DFT at the B3LYP/def2-TZVP level of theory and the implicit solvation model CPCM for THF. The free phosphines exhibit a strong absorption band of the n(P) → π*(Ar) excitation (Fig. S63†). In agreement with the experimental spectra (Fig. 1), the absorption band appears at 290 nm for Ph3P and is blue-shifted for (4-OMe-C6H4)3P, but red-shifted for the arylphosphines with substituted ortho positions. The n → π* excitation of Mes3P covers the region of irradiation (365 nm), which explains the observed decomposition reaction. Although we tend to be cautious with the interpretation of the excitation spectra obtained with the hybrid functional B3LYP, a comparison of the UV region of the noncovalent SF6 complexes is qualitatively in good agreement with the experimental observations (Fig. S64†): only [Ph3P⋯SF6] and [(4-OMe-C6H4)3P⋯SF6] absorb with significant intensity at 365 nm due to the π(Ar) → σ*(SF6) charge transfer excitation. The ortho-substituted aryl phosphines either have a different absorption maximum ((2-OMe-C6H4)3P: 385 nm), or the n(P) → π*(Ar) band is extended into the region of the charge-transfer excitation (Mes3P). Irradiation of THF solutions of Mes3P, (o-tol)3P or (2-OMe-C6H4)3P for 3 hours under an SF6 atmosphere with light at 405 nm did not cause the phosphines to decompose or react with SF6. For Mes3P, this is consistent with the computational results. However, the lack of reactivity in the case of (2-OMe-C6H4)3P suggests that the steric bulk of the phosphine preventing the formation of the encounter complex must also be considered.
Since our experimental and computational results suggest that the initial SF6 activation step corresponds to a π(Ar) → σ*(SF6) charge transfer excitation, we further investigated the versatility of our approach by using an external π system as photosensitizer combined with an alkyl phosphine as reductant. Tri-n-butylphosphine does not react with SF6 upon irradiation with light at 365 nm (cf.Scheme 2). However, when the reaction is performed in the presence of stochiometric amounts of benzophenone or acetophenone as photosensitizer, (nBu)3P reacts with SF6 within 24 h to give a mixture of tri-n-butylphosphine sulfide and tri-n-butyldifluorophosphorane (Scheme 3). The reaction is more selective with acetophenone than with benzophenone. With catalytic amounts of the photosensitizer (20 mol%), the reaction rate is significantly lower (Table S1†). No reaction with SF6 was observed when THF solutions of triethyl phosphite or triphenyl phosphite were irradiated under the same conditions using acetophenone as photosensitizer.
 | ||
| Scheme 3 Photochemical reaction of SF6 with an alkylphosphine using photosensitizers. | ||
Collectively, the photochemical reaction of SF6 with phosphines proves to be general enough to afford alkyl-, aryl-, and heteroaryl-substituted phosphine sulphides and difluorophosphoranes. The latter are important starting materials in the context of Lewis acid-catalyzed transformations38 or CO2 sequestration.39 They are usually synthesized by oxidation of the phosphines using harsh reagents such as XeF2, SF4, HgF2 or N2F4,40 but more convenient protocols have been recently developed.41
Owing to the straight-forward synthetic access, the utilization of TPP-Fluor as reagent in chemical synthesis is particularly interesting in terms of a chemical valorization of SF6 after its technical application. In fact, difluorotriphenylphosphorane has been successfully applied as deoxyfluorination reagent to convert primary and secondary alcohols into fluoroalkanes at reaction temperatures above 140 °C.42 As a proof of principle, the deoxyfluorination of 1-hexanol was performed with the TPP-Fluor reagent and gave 1-fluorohexane in 22% yield. We also used TPP-Fluor for the preparation of acyl fluorides directly from carboxylic acids. Acyl fluorides are versatile reagents in chemical synthesis that can be prepared from carboxylic acids using cyanuric fluoride,43 BrF3,44 SeF4,45 (Me4N)SCF346 or sulfur-based fluorination reagents.47 Recently, Prakash and co-workers disclosed a protocol for the stepwise conversion of carboxylic acids into acyl fluorides using Ph3P/NBS for the activation of the carboxylic acid and Et3N-3HF as fluoride ion source.48
Lauric acid was selected as model substrate to optimize the reaction conditions (Table 1). The progress of the deoxyfluorination was monitored by 31P and 19F NMR spectroscopy, confirming the formation of Ph3PO, lauroyl fluoride and HF/FHF−. We suspect that the liberation of HF during the reaction is the reason why three equivalents of difluorophosphorane (1 eq. TPP-Fluor) are required to achieve good conversion (Table 1, entries 1–4), albeit the addition of 2,6-lutidinium triflate as proton source had little influence on the yield (Table 1, entry 7). The reaction was inhibited under Brønsted basic conditions by using sodium carboxylate as substrate or by adding CsF as an additional nucleophile (Table 1, entries 6 and 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | R | Additive | TPP-Fluor (equiv.) | Yielda (%) |
| a As determined by quantitative 19F NMR spectroscopy using α,α,α-trifluorotoluene as internal standard. | |||||
| 1 | 80 °C, 16 h | H | — | 0.17 | 21 |
| 2 | 80 °C, 16 h | H | — | 0.33 | 36 |
| 3 | 80 °C, 16 h | H | — | 0.66 | 69 |
| 4 | 80 °C, 16 h | H | — | 1 | 88 |
| 5 | 60 °C, 16 h | H | — | 1 | 19 |
| 6 | 80 °C, 16 h | H | 2 CsF | 0.33 | <5 |
| 7 | 80 °C, 16 h | H | 2 Lut·HOTf | 0.33 | 44 |
| 8 | 80 °C, 16 h | Na | — | 1 | <5 |

Application of the optimized conditions to the deoxyfluorination of various carboxylic acids illustrates its synthetic capabilities (Scheme 4). Aliphatic carboxylic (17, 19, 20) acids were readily converted to the corresponding products independent of the steric bulk of the alkyl group. Among the aromatic carboxylic acids only the electron-rich benzoic acids (22, 23) underwent the desired transformation, while the reaction is sluggish for benzoic acids bearing electron-neutral (18, 21) or electron-withdrawing functionalities (25, 26). Ester functions (24) were not tolerated because the liberated HF/FHF cleaves the ester bond, concomitant with further deoxyfluorinations.
 | ||
| Scheme 4 Direct deoxyfluorination of carboxylic acids using TPP-Fluor. Yields determined by quantitative 19F NMR spectroscopy using α,α,α-trifluorotoluene as internal standard. | ||
We also considered using TPP-Fluor for the fluorination of inorganic substrates (Scheme 5). Treatment of phosphorus pentachloride with TPP-Fluor resulted in complete exchange of fluoride and chloride atoms to give a mixture of chlorotriphenylphosphonium chloride and hexafluorophosphate salts. As already observed in the deoxyfluorination reactions, Ph3PS did not participate in the reaction. The hexafluorophosphate ion can be readily precipitated from the mixture in good yield as Bu4NPF6 after aqueous workup and addition of Bu4NBr. Furthermore, [PPN]PF6 was assembled from the chlorotriphenylphosphonium ions following the [PPN]+ cation synthesis of Ruff and Schlientz by treating the reaction mixture with hydroxylamine hydrochloride and additional Ph3P.49 Both reactions demonstrate novel, straightforward routes to salts consisting of weakly coordinating ions, of which Bu4NPF6 is a common electrolyte in electrochemistry. It is noteworthy that chlorotriphenyl-phosphonium salts are the key intermediates in the large-scale industrial recycling process of triphenylphosphine oxide, which is based on the chlorination of Ph3PO with phosgene and subsequent reduction with aluminium powder.50 Moreover, there are several elegant methods for the recycling of Ph3PO and Ph3PS to Ph3P,51 including electrochemical methods50,52 and methods based on the use of dihydrogen gas as reducing agent.53
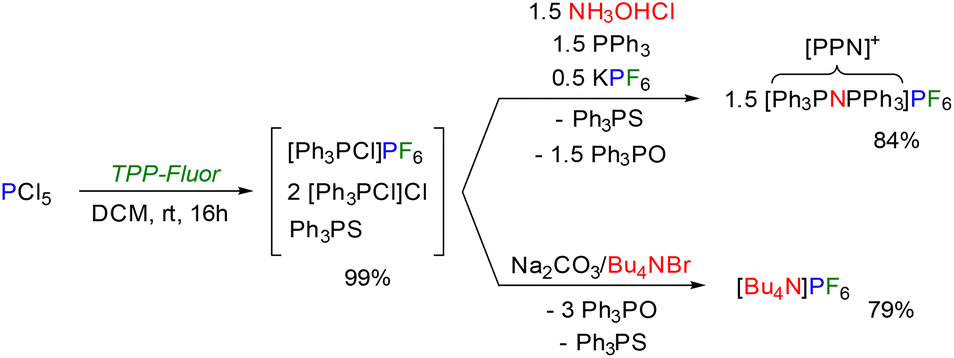 | ||
| Scheme 5 Direct conversion of PCl5 into PF6 salts using TPP-Fluor. Isolated yields are reported. | ||
Conclusions
In conclusion, this report discloses a user-friendly, scalable protocol for the photochemical degradation of SF6. The solvent-free process produces a well-defined solid fluorinating reagent consisting of a 3:1 mixture of difluorotriphenylphosphorane and triphenylphosphine sulfide, referred to as TPP-Fluor. Utilization of TPP-Fluor for the deoxyfluorination of alcohols and carboxylic acids and for the preparation of common hexafluorophosphate salts from PCl5 was established. Both reactions highlight possibilities to generate value-added products from the decomposition of the greenhouse gas SF6 after its technical application by using inexpensive commodity chemicals.
The photochemical reaction between phosphines and SF6 can also be performed in solution providing a convenient approach to various difluorophosphoranes without the need for hazardous fluorination reagents. Reaction screening with phosphines bearing different substitution patterns revealed that at least one aryl substituent is required for the photochemical SF6 activation. Alternatively, acetophenone can be used as photosensitizer to drive the fragmentation of SF6 with alkyl phosphines. Computational studies indicate that the reaction proceeds through a π(Ar) → σ*(SF6) charge-transfer excitation as the initial activation step, followed by a fluorine/electron or a direct fluoride transfer. While it is generally assumed that the photochemical activation of SF6 occurs via excitation of the reductant, which then facilitates the electron transfer to SF6,15,24–27 our study suggests that direct charge-transfer excitation of a preformed SF6 complex must also be considered. This result implies that substances susceptible to decomposition upon irradiation with short wavelength light can still react with SF6 upon charge-transfer excitation by light with a longer wavelength and thus opens new avenues for the photochemical derivatization of SF6, which is under current investigation in our laboratory.
Author contributions
P. R. carried out the synthetic experiments and analysed the experimental data. C. M.-L. performed the computational investigations. F. D., P. R. and C. M.-L. wrote the manuscript. F. D. directed the investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the DFG (Emmy Noether program: DI 2054/1-1, SFB 858). We thank DILO GmbH for the generous donation of SF6 gas and Jonas Franzen for helping with experiments for the revision.References
- Ø. Hodnebrog, M. Etminan, J. S. Fuglestvedt, G. Marston, G. Myhre, C. J. Nielsen, K. P. Shine and T. J. Wallington, Rev. Geophys., 2013, 51, 300–378 CrossRef.
- C. Breidenich, D. Magraw, A. Rowley and J. W. Rubin, Am. J. Int. Law, 1998, 92, 315–331 CrossRef.
- (a) B. K. Sovacool, S. Griffiths, J. Kim and M. Bazilian, Renewable Sustainable Energy Rev., 2021, 141, 110759 CrossRef CAS; (b) C. T. Dervos and P. Vassiliou, J. Air Waste Manage. Assoc., 2000, 50, 137–141 CrossRef CAS.
- (a) M. Rabie and C. M. Franck, Environ. Sci. Technol., 2018, 52, 369–380 CrossRef CAS PubMed; (b) P. Billen, B. Maes, M. Larrain and J. Braet, Energies, 2020, 13, 1807 CrossRef CAS.
- (a) ESRL Global Monitoring Laboratory, Halocarbons and other Atmospheric Trace Species, Sulfur hexafluoride (SF6) – Combined Data Set, 2021, Available: https://www.esrl.noaa.gov/gmd/hats/combined/SF6.html Search PubMed; (b) P. G. Simmonds, M. Rigby, A. J. Manning, S. Park, K. M. Stanley, A. McCulloch, S. Henne, F. Graziosi, M. Maione, J. Arduini, S. Reimann, M. K. Vollmer, J. Mühle, S. O'Doherty, D. Young, P. B. Krummel, P. J. Fraser, R. F. Weiss, P. K. Salameh, C. M. Harth, M.-K. Park, H. Park, T. Arnold, C. Rennick, L. P. Steele, B. Mitrevski, R. H. J. Wang and R. G. Prinn, Atmos. Chem. Phys., 2020, 20, 7271–7290 CrossRef CAS.
- (a) J. R. Case and F. Nyman, Nature, 1962, 193, 473 CrossRef CAS; (b) K. Seppelt, Chem. Rev., 2015, 115, 1296–1306 CrossRef CAS.
- G. Mauthe, L. Niemeyer, B. M. Pryor, R. Probst, H. Bräutigam, P. A. O’Connell, K. Pettersson, H. D. Morrison, J. Poblotzki and D. Koenig, Electra, 1996, 164, 121–131 Search PubMed.
- X. Zhang, H. Xiao, J. Tang, Z. Cui and Y. Zhang, Crit. Rev. Environ. Sci. Technol., 2017, 47, 1763–1782 CrossRef CAS.
- (a) J. Zhang, J. Z. Zhou, Z. P. Xu, Y. Li, T. Cao, J. Zhao, X. Ruan, Q. Liu and G. Qian, Environ. Sci. Technol., 2014, 48, 599–606 CrossRef CAS; (b) J. Zhang, J. Z. Zhou, Q. Liu, G. Qian and Z. P. Xu, Environ. Sci. Technol., 2013, 47, 6493–6499 CrossRef CAS PubMed; (c) N.-K. Park, H.-G. Park, T. J. Lee, W.-C. Chang and W.-T. Kwon, Catal. Today, 2012, 185, 247–252 CrossRef CAS; (d) D. Kashiwagi, A. Takai, T. Takubo, H. Yamada, T. Inoue, K. Nagaoka and Y. Takita, J. Colloid Interface Sci., 2009, 332, 136–144 CrossRef CAS; (e) D. Kashiwagi, A. Takai, T. Takubo, K. Nagaoka, T. Inoue and Y. Takita, Ind. Eng. Chem. Res., 2009, 48, 632–640 CrossRef CAS; (f) Y. Zhang, Y. Li, Z. Cui, D. Chen and X. Zhang, AIP Adv., 2018, 8, 55215 CrossRef.
- (a) H. Deubner and F. Kraus, Inorganics, 2017, 5, 68 CrossRef; (b) H. C. Cowen, F. Riding and W. Warhurst, J. Chem. Soc., 1953, 4168–4169 RSC; (c) G. C. Demitras and A. G. MacDiarmid, Inorg. Chem., 1964, 3, 1198–1199 CrossRef CAS.
- M. Rueping, P. Nikolaienko, Y. Lebedev and A. Adams, Green Chem., 2017, 19, 2571–2575 RSC.
- D. Sevenard, P. Kirsch, A. A. Kolomeitsev and G.-V. Roschenthaler, Patent DE10220901, 2004.
- A. Taponard, T. Jarrosson, L. Khrouz, M. Médebielle, J. Broggi and A. Tlili, Angew. Chem., Int. Ed., 2022 DOI:10.1002/anie.202204623.
- F. Buß, C. Mück-Lichtenfeld, P. Mehlmann and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 4951–4955 CrossRef PubMed.
- P. Tomar, T. Braun and E. Kemnitz, Chem. Commun., 2018, 54, 9753–9756 RSC.
- S. Huang, Y. Wang, C. Hu and X. Yan, Chem.: Asian J., 2021, 16, 2687–2693 CrossRef CAS.
- (a) B. S. N. Huchenski and A. W. H. Speed, Chem. Commun., 2021, 57, 7128–7131 RSC; (b) R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Chem. – Eur. J., 2021, 27, 6465–6478 CrossRef CAS PubMed.
- G. Iakobson, M. Pošta and P. Beier, J. Fluorine Chem., 2018, 213, 51–55 CrossRef CAS.
- D. J. Sheldon and M. R. Crimmin, Chem. Commun., 2021, 57, 7096–7099 RSC.
- L. Zámostná and T. Braun, Angew. Chem., Int. Ed., 2015, 54, 10652–10656 CrossRef PubMed.
- (a) L. Zámostná, T. Braun and B. Braun, Angew. Chem., Int. Ed., 2014, 53, 2745–2749 CrossRef PubMed; (b) P. Holze, B. Horn, C. Limberg, C. Matlachowski and S. Mebs, Angew. Chem., Int. Ed., 2014, 53, 2750–2753 CrossRef CAS PubMed; (c) B. G. Harvey, A. M. Arif, A. Glöckner and R. D. Ernst, Organometallics, 2007, 26, 2872–2879 CrossRef CAS; (d) R. Basta, B. G. Harvey, A. M. Arif and R. D. Ernst, J. Am. Chem. Soc., 2005, 127, 11924–11925 CrossRef CAS PubMed.
- C. Berg, T. Braun, M. Ahrens, P. Wittwer and R. Herrmann, Angew. Chem., Int. Ed., 2017, 56, 4300–4304 CrossRef CAS PubMed.
- (a) X. Song, X. Liu, Z. Ye, J. He, R. Zhang and H. Hou, J. Hazard. Mater., 2009, 168, 493–500 CrossRef CAS PubMed; (b) L. Huang, Y. Shen, W. Dong, R. Zhang, J. Zhang and H. Hou, J. Hazard. Mater., 2008, 151, 323–330 CrossRef CAS PubMed; (c) L. Huang, D. Gu, L. Yang, L. Xia, R. Zhang and H. Hou, J. Environ. Sci., 2008, 20, 183–188 CrossRef CAS; (d) L. Huang, W. Dong, R. Zhang and H. Hou, Chemosphere, 2007, 66, 833–840 CrossRef CAS PubMed.
- D. Rombach and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2020, 59, 300–303 CrossRef CAS PubMed.
- D. Rombach and H.-A. Wagenknecht, ChemCatChem, 2018, 10, 2955–2961 CrossRef CAS.
- T. A. McTeague and T. F. Jamison, Angew. Chem., Int. Ed., 2016, 55, 15072–15075 CrossRef CAS PubMed.
- S. Kim, Y. Khomutnyk, A. Bannykh and P. Nagorny, Org. Lett., 2021, 23, 190–194 CrossRef CAS PubMed.
- (a) S. Bouvet, B. Pégot, S. Sengmany, E. Le Gall, E. Léonel, A.-M. Goncalves and E. Magnier, Beilstein J. Org. Chem., 2020, 16, 2948–2953 CrossRef CAS PubMed; (b) M. Govindan, R. Adam Gopal and I. S. Moon, Chem. Eng. J., 2020, 382, 122881 CrossRef CAS; (c) Y. Li, A. Khurram and B. M. Gallant, J. Phys. Chem. C, 2018, 122, 7128–7138 CrossRef CAS.
- (a) M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857–11860 CrossRef PubMed; (b) P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem. – Eur. J., 2017, 23, 5929–5933 CrossRef CAS PubMed.
- (a) S. Ullrich, B. Kovačević, X. Xie and J. Sundermeyer, Angew. Chem., Int. Ed., 2019, 58, 10335–10339 CrossRef CAS; (b) M. D. Böhme, T. Eder, M. B. Röthel, P. D. Dutschke, L. F. B. Wilm, E. Hahn and F. Dielmann, Angew. Chem., Int. Ed., 2022 DOI:10.1002/anie.202202190; (c) F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840–1843 CrossRef PubMed; (d) F. Buß, P. Rotering, C. Mück-Lichtenfeld and F. Dielmann, Dalton Trans., 2018, 47, 10420–10424 RSC; (e) F. Buß, M. B. Röthel, J. A. Werra, P. Rotering, L. F. B. Wilm, C. G. Daniliuc, P. Löwe and F. Dielmann, Chem. – Eur. J., 2022, 28, e202104021 CrossRef PubMed; (f) P. Rotering, L. F. B. Wilm, J. A. Werra and F. Dielmann, Chem. – Eur. J., 2020, 26, 406–411 CrossRef CAS.
- M. L. Kaufman and C. E. Griffin, Tetrahedron Lett., 1965, 12, 767–772 Search PubMed.
- J. D. L. Horner, Tetrahedron Lett., 1965, 12, 763–767 CrossRef.
- Y. Sakaguchi and H. Hayashi, J. Phys. Chem. A, 2004, 108, 3421–3429 CrossRef CAS.
- Y. Sakaguchi and H. Hayashi, Chem. Phys. Lett., 1995, 245, 591–597 CrossRef CAS.
- (a) S. Yasui, Y. Ogawa, K. Shioji, M. Mishima and S. Yamazaki, Bull. Chem. Soc. Jpn., 2014, 87, 988–996 CrossRef CAS; (b) S. Yasui, M. M. R. Badal, S. Kobayashi and M. Mishima, Chem. Lett., 2013, 42, 866–868 CrossRef CAS; (c) S. Yasui, Y. Ogawa, K. Shioji and S. Yamazaki, Chem. Lett., 2013, 42, 1478–1480 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) C. B. Caputo, D. Winkelhaus, R. Dobrovetsky, L. J. Hounjet and D. W. Stephan, Dalton Trans., 2015, 44, 12256–12264 RSC; (b) J. Möbus, T. vom Stein and D. W. Stephan, Chem. Commun., 2016, 52, 6387–6390 RSC; (c) S. Postle, V. Podgorny and D. W. Stephan, Dalton Trans., 2016, 45, 14651–14657 RSC; (d) J. M. Bayne, M. H. Holthausen and D. W. Stephan, Dalton Trans., 2016, 45, 5949–5957 RSC; (e) M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016–2021 RSC; (f) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef CAS; (g) M. Mehta, M. H. Holthausen, I. Mallov, M. Pérez, Z.-W. Qu, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8250–8254 CrossRef CAS PubMed; (h) M. Tress, E. U. Mapesa, W. Kossack, W. K. Kipnusu, M. Reiche and F. Kremer, Science, 2013, 341, 1371–1374 CrossRef CAS.
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4714–4717 CrossRef CAS PubMed.
- (a) W. C. Firth, S. Frank, M. Garber and V. P. Wystrach, Inorg. Chem., 1965, 4, 765 CrossRef CAS; (b) W. C. Smith, J. Am. Chem. Soc., 1960, 82, 6176 CrossRef CAS; (c) K. M. Doxsee, E. M. Hanawalt and T. J. R. Weakley, Inorg. Chem., 1992, 31, 4420 CrossRef CAS.
- (a) D. Bornemann, C. R. Pitts, L. Wettstein, F. Brüning, S. Küng, L. Guan, N. Trapp, H. Grützmacher and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 22790–22795 CrossRef CAS PubMed; (b) D. Bornemann, F. Brüning, N. Bartalucci, L. Wettstein and C. R. Pitts, Helv. Chim. Acta, 2021, 104 DOI:10.1002/hlca.202000218.
- (a) J. Eljo, M. Carle and G. Murphy, Synlett, 2017, 2871–2875 Search PubMed; (b) Z. Yang and X. Shen, in Fluorination, ed. J. Hu and T. Umemoto, Springer Singapore, Singapore, 2018, pp. 1–7 Search PubMed; (c) Y. Kobaqashi and C. Akashi, Chem. Pharm. Bull., 1968, 16, 1009 CrossRef.
- G. A. Olah, M. Nojima and I. Kerekes, Synthesis, 1973, 487 CAS.
- O. Cohen, R. Sasson and S. Rozen, J. Fluorine Chem., 2006, 127, 433–436 CrossRef CAS.
- G. A. Olah, M. Nojima and I. Kerekes, J. Am. Chem. Soc., 1974, 96, 925 CrossRef CAS.
- T. Scattolin, K. Deckers and F. Schoenebeck, Org. Lett., 2017, 19, 5740–5743 CrossRef CAS PubMed.
- (a) G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048–7054 CrossRef CAS; (b) F. Beaulieu, L.-P. Beauregard, G. Courchesne, M. Couturier, F. LaFlamme and A. L'Heureux, Org. Lett., 2009, 11, 5050–5053 CrossRef CAS; (c) C. Kaduk, H. Wenschuh, M. Beyermann, K. Forner, L. A. Carpino and M. Bienert, Lett. Pept. Sci., 1996, 2, 285–288 CrossRef CAS; (d) A. L'Heureux, F. Beaulieu, C. Bennett, D. R. Bill, S. Clayton, F. LaFlamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, J. Org. Chem., 2010, 75, 3401–3411 CrossRef PubMed.
- S. B. Munoz, H. Dang, X. Ispizua-Rodriguez, T. Mathew and G. K. S. Prakash, Org. Lett., 2019, 21, 1659–1663 CrossRef CAS PubMed.
- J. K. Ruff, W. J. Schlientz, R. E. Dessy, J. M. Malm, G. R. Dobson and M. N. Memering, Inorg. Synth., 1974, 84–90 CAS.
- A. Rajput, M. Soni and B. Chakraborty, ChemElectroChem, 2022 DOI:10.1002/celc.202101658.
- E. Podyacheva, E. Kuchuk and D. Chusov, Tetrahedron Lett., 2019, 60, 575–582 CrossRef CAS.
- B. Chakraborty, P. W. Menezes and M. Driess, J. Am. Chem. Soc., 2020, 142, 14772–14788 CrossRef CAS PubMed.
- (a) O. M. Demchuk, W. Świerczyńska, K. Dziuba, S. Frynas, A. Flis and K. M. Pietrusiewicz, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 64–68 CrossRef CAS; (b) A. J. Stepen, M. Bursch, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2018, 57, 15253–15256 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02172b |
| This journal is © The Royal Society of Chemistry 2022 |