
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCholine chloride–ethylene glycol based deep-eutectic solvents as lixiviants for cobalt recovery from lithium-ion battery cathode materials: are these solvents really green in high-temperature processes?†
Nand
Peeters
 a,
Kwinten
Janssens
b,
Dirk
de Vos
b,
Koen
Binnemans
a and
Sofía
Riaño
*a
a,
Kwinten
Janssens
b,
Dirk
de Vos
b,
Koen
Binnemans
a and
Sofía
Riaño
*a
aKU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. box 2404, B-3001 Leuven, Belgium. E-mail: sofia.riano@kuleuven.be
bCentre for Membrane separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), Department of Microbial and Molecular Systems (M2S), KU Leuven, 3001 Leuven, Belgium
First published on 18th August 2022
Abstract
Deep-eutectic solvents (DESs) are often considered to be safe, eco-friendly and non-toxic solvents. Due to these green credentials, they are increasingly being studied for application in metal recycling processes. One example is their use as lixiviants for the recovery of cobalt from lithium cobalt oxide (LiCoO2, LCO), which is a common cathode material in lithium-ion batteries. Here, leaching of cobalt is facilitated by reduction of cobalt(III) to cobalt(II) in the presence of a reducing agent. However, several recent publications report on the use of DESs as lixiviants at high temperatures (180 °C) without addition of a reducing agent. Typical DESs for these applications are based on mixtures of choline chloride and ethylene glycol (ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EG). Unfortunately, these studies ignore the limited thermal stability of ChCl:EG at high temperatures, which limits the recyclability of this DES. In this work, the drawbacks of using ChCl:EG as the lixiviant in high-temperature ionometallurgical processes are demonstrated. Structural analysis confirmed that ChCl:EG is not stable at 180 °C, forming hazardous and toxic decomposition products such as trimethylamine and 2-chloroethanol. It was hypothesized that choline chloride reduces cobalt(III) while simultaneously undergoing a radical β-hydrogen abstraction reaction, thereby decomposing to trimethylamine and other degradation products. The main conclusion is that this type of DES should not be used for high-temperature leaching processes due to their limited stability under such conditions.
:EG). Unfortunately, these studies ignore the limited thermal stability of ChCl:EG at high temperatures, which limits the recyclability of this DES. In this work, the drawbacks of using ChCl:EG as the lixiviant in high-temperature ionometallurgical processes are demonstrated. Structural analysis confirmed that ChCl:EG is not stable at 180 °C, forming hazardous and toxic decomposition products such as trimethylamine and 2-chloroethanol. It was hypothesized that choline chloride reduces cobalt(III) while simultaneously undergoing a radical β-hydrogen abstraction reaction, thereby decomposing to trimethylamine and other degradation products. The main conclusion is that this type of DES should not be used for high-temperature leaching processes due to their limited stability under such conditions.
Introduction
Deep-eutectic solvents (DESs) were first reported about 20 years ago by Abbott et al.1 and are enjoying increasing interest as potential greener alternatives to molecular organic solvents, with the additional advantage of being much cheaper than ionic liquids (ILs).2 Although the definition of a DES is still under debate, we define DESs as mixtures of pure compounds that have a lower melting temperature than their constituents.3,4 The depression of the melting temperature, which peaks at the so-called eutectic point, is partially explained by a decrease in lattice energy due to the formation of hydrogen bonds between the components of the mixture.4,5 Hence, a DES is composed of compounds that are hydrogen-bond donors (HBDs) and hydrogen-bond acceptors (HBAs). The composition of a DES is usually represented by abbreviating the names of the HBAs and HBDs, together with the molar ratio in which they are present. DESs are quite similar to ILs in the sense that their physicochemical properties can also be fine-tuned by combining different starting materials and that they also have ionic components.2 The advantages of DESs over ILs include their easier and more environmentally friendly synthesis, and their cheaper and less toxic components.6 For example, a commonly used DES is synthesized by simply mixing choline chloride (ChCl) as HBA with ethylene glycol (EG) as HBD in a molar ratio of 1:2. This DES was first reported by Abbott et al.7 and is nowadays called “Ethaline”, usually abbreviated as ChCl:EG (1:2).8,9 Both choline chloride and ethylene glycol are relatively cheap, relatively non-toxic and biodegradable, and are produced at industrial scales.10,11 DESs have been used in a broad range of applications, including extractive metallurgy.6,12
Recently, many researchers have been interested in replacing the aqueous phase in hydrometallurgy by non-aqueous media.12 As shown in Fig. 1, DESs can act as non-aqueous lixiviants during solvoleaching (i.e. leaching with non-aqueous solvents), as more polar phases in non-aqueous solvent extraction and as non-aqueous electrolytes during electrowinning.9,13–15 Solvometallurgy with DESs or ILs is also often called “ionometallurgy”.16
 | ||
| Fig. 1 Schematic comparison of a hydrometallurgical process (left) versus a DES-based solvometallurgical process (right). With solvent extraction being abbreviated as SX. | ||
The recovery of cobalt from secondary waste streams is a trending topic since cobalt is a critical raw material and is abundantly present in the cathode material of lithium-ion batteries (LIBs).17,18 Two cobalt-rich LIB cathode materials that are frequently used in fundamental studies for cobalt recovery are lithium cobalt oxide (LiCoO2, LCO) and lithium nickel manganese cobalt oxide (LiNiMnCoO2, NMC). The leaching of cobalt is achieved by the reduction of cobalt(III) in LCO or NMC to the more soluble cobalt(II).19 Recently, we investigated the use of a choline chloride–citric acid based DES as lixiviant for the leaching of LCO. Quantitative cobalt leaching was achieved after one hour at 40 °C using metallic copper (already present as LIB-current collector) as reducing agent for cobalt(III).20
However, a number of publications use DESs as lixiviants to leach cobalt from LCO at high temperatures without additional reducing agents for cobalt(III). Cobalt was quantitatively leached from LCO with ChCl:EG (1:2) by Tran et al., and with ChCl:EG (1:2) from NMC by Schiavi et al., both at a temperature of 180 °C and a leaching time of 24 hours.21,22 Wang et al. reported quantitative leaching of cobalt from LCO at 180 °C after 12 hours in a choline chloride–urea based DES (ChCl:urea 1:2).23 Chen et al. investigated a polyethylene glycol–thiourea based DES (PEG:thiourea 2:1) with the same objective, but could recover only ca. 60% of cobalt after 24 hours at 160 °C.24 In all these studies, it is explicitly stated that no reducing agents were added for the cobalt(III) reduction and that the HBAs or HBDs of the used DESs act as reducing agents themselves. If this hypothesis was correct, the DES components would be oxidized themselves upon reduction of cobalt(III). This conversion into oxidation products, which are no longer able to reduce cobalt(III), implies that the cobalt leaching efficiency will progressive decrease upon repeated recycling and reuse of the DES. Furthermore, the high leaching temperatures also result in the thermal decomposition of the DESs, further impeding their recyclability. Chen et al. and Delgado-Mellado et al. confirmed that the thermal decomposition of ChCl:EG (1:2) and ChCl:urea (1:2) starts at ca. 100 °C and 172 °C, respectively.25,26 However, ChCl:EG (1:2) and ChCl:urea (1:2) have been used as lixiviants at 180 °C by the abovementioned authors.21–23 In a truly environmentally friendly and cost-effective leaching process, the DES must be recycled efficiently. Thus, the exact mechanism of cobalt(III) reduction must be known. Deeper mechanistic insight into what is exactly happening at the molecular level is required before one claims about the green characters of these solvent systems can be made or before these processes can be further upscaled or used in industry.
In the present work, we demonstrate the drawbacks of using DESs to leach cobalt from LCO at high temperatures. In particular, the function of ChCl:EG (1:2) as lixiviant is emphasized, due to its recent ubiquity in solvometallurgy/ionometallurgy.9,21,22 The thermal stability of ChCl:EG (1:2) was studied by thermogravimetric analysis (TGA). Gas chromatography-mass spectrometry (GC-MS) was used to identify the decomposition products after heating at 180 °C for 24 hours, both in the presence and absence of LCO. Possible functional group transformations of ChCl:EG (1:2) during leaching of LCO were tracked using infrared spectroscopy. More insights on the leaching mechanism were gained, whereby the effective reducing agent of cobalt(III) in ChCl:EG (1:2) was revealed. The leaching efficiencies by non-DES lixiviant systems were also evaluated and compared with those of the DESs. This comparison is used to critically reflect on the further use of DESs as lixiviants for LCO or similar cathode materials at high temperatures.
Experimental
Synthesis of DESs and lixiviants
DESs were prepared by adding HBAs and HBDs at certain molar ratios into a 250 mL glass beaker. A magnetic stirring bar was added and the DES was stirred at 40 °C on a heating plate while covered by a watch glass until a homogenous solution was obtained. Other lixiviants were prepared in a similar manner by mixing one component with a diluent using the same procedure. If the solution was still turbid after 48 hours, the temperature was increased to 75 °C until a homogeneous solution was obtained (maximally for 24 hours). Hereafter, the solution was transferred into a vial while still hot, and immediately used to leach in a pre-heated sand bath.Leaching experiments
Unless stated otherwise, 100 mg of LCO and 5 mL of lixiviant were added into a 10 mL glass vial. This solid-to-liquid ratio (i.e. S:L) of 20 g L−1 has also been used by other authors, and was kept constant during this work.21–24 A magnetic stirring bar was added, the vial was sealed with a septum cap and placed into a sand bath on a temperature-controlled heating plate. Leaching was subsequently carried out at 180 °C or 140 °C for 24 hours while stirring at 900 rpm. After leaching, the vial was removed from the sand bath with a wooden clamp and cooled for 2 min. The vial was hereafter opened while wearing heat-resistant gloves, the leachate was filtered through a syringe filter (Chromafil® pore size 0.45 μm, diameter 25 mm) and 50 μL of the filtrate was immediately diluted 2000 times with 2 vol% nitric acid for metal analysis with ICP-OES. The leaching efficiency of cobalt was calculated as described previously.20,27 When leaching with methyl-trioctylammonium chloride (TOMAC), ChCl:urea and chlorochlorine chloride–ethylene glycol (ChCl2:EG); the same procedure was followed, except that the leaching was carried out but in round bottomed flasks (50 mL) attached to a condenser instead of 10 mL vials. This was done to avoid pressure build-up and possible explosions. It is highly recommended to take extra safety precautions when leaching LCO with TOMAC, ChCl:urea and ChCl2:EG at high temperatures. The used chemicals, instrumentation and procedures for both metal and structural analysis, and a list with all the abbreviations and structures of all the mentioned chemicals are given in the ESI.†
Results and discussion
Thermal decomposition of ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :EG (1:2)
:EG (1:2)
The stability of a lixiviant is often a drawback of solvoleaching, especially at high temperatures.4 It is therefore highly recommended to first evaluate the thermal stability of the chosen lixiviant. The TGA curve of ChCl:EG (1:2) is given in Fig. 2, showing a mass loss of ca. 42% when heated at 180 °C.
 | ||
| Fig. 2 TGA curve of ChCl:EG (1:2) recorded under a flow of air at a heating rate of 5 K min−1. | ||
This significant mass loss is already an indication that the DES has thermal stability issues when leaching LCO at 180 °C for 24 hours. Although the mass loss is the lowest at temperatures below 50 °C, Tran et al. confirmed that the cobalt leaching efficiency was negligible from temperatures of 25 °C until ca. 150 °C during leaching of LCO by ChCl:EG (1:2) for 24 hours.21Fig. 2 gives thus the first indication that ChCl:EG (1:2) is probably not suited to leach LCO at conditions that ensure no decomposition. Furthermore, the flash point of EG is about 115 °C,28 which is much lower than the leaching temperature of 180 °C. Exceeding the flashpoint represents an explosion risk and extra safety measures must be taken when upscaling a process based on this DES. Hence, issues connected with leaching using ChCl:EG (1:2) at high temperatures contradict the claims that DESs are stable, safe and eco-friendly.29,30
Decomposition products of ChCl:EG (1:2) at 180 °C after 24 hours
More insights in the cobalt leaching mechanism by ChCl:EG (1:2) could be obtained by identifying the decomposition products. Fig. 3 shows the decomposition products obtained after heating the DES in the presence and absence of LCO, at 180 °C for 24 hours, as identified in the GC-MS chromatograms. This comparison clearly shows that the DES exhibits increased decomposition into dimethylaminoethanol (Fig. 3F) and 2-methoxyethanol (Fig. 3C) when it is heated in the absence of LCO.
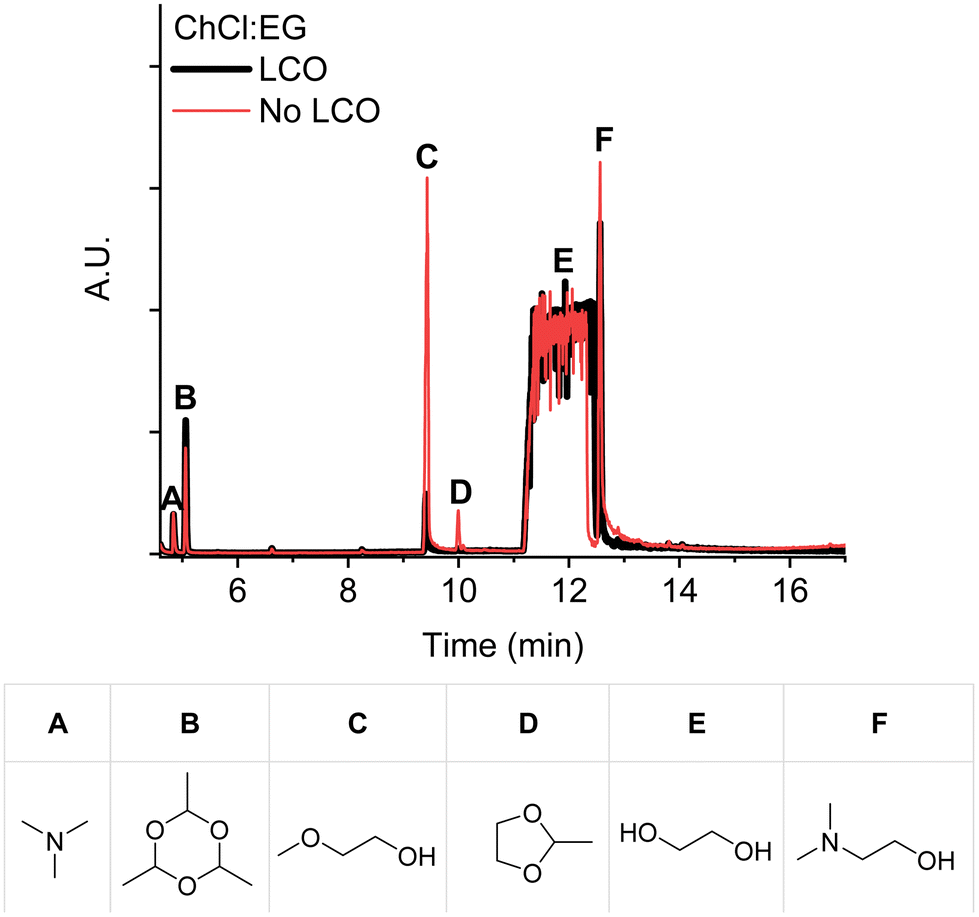 | ||
| Fig. 3 GC-MS chromatograms of ChCl:EG (1:2) after heating (no LCO) and after leaching LCO, both at 180 °C for 24 hours. The chemical structures of the identified decomposition products are shown below the GC chromatogram. | ||
Dimethylaminoethanol is presumably formed via the reversed Menshutkin reaction of choline, as described by Haerens et al. and shown in Scheme 1.31 Additionally, the formed methyl chloride could undergo an etherification reaction with ethylene glycol (Fig. 3E) resulting in 2-methoxyethanol, which is in agreement with the observations of Schiavi et al.22
 | ||
| Scheme 1 Possible decomposition reactions of choline chloride as HBA and reactions with ethylene glycol as HBD in ChCl:EG (1:2) during heating (in absence of LCO) and leaching of LCO both at 180 °C for 24 hours. | ||
Scheme 1 also shows the Hoffmann elimination of choline chloride.22,31 This reaction results in the formation of acetaldehyde and trimethylamine (Fig. 3A), which is a toxic gas.32 Acetaldehyde reacts with ethylene glycol to form the cyclic acetal 2-methyl-1,3-dioxolane (Fig. 3D), which was also detected in larger quantities when the DES is heated in absence of LCO. Although several authors report free acetaldehyde as a major decomposition product of choline chloride, only its cyclic trimer paraldehyde (Fig. 3B) was detected.22,31,33–35 The authors who detected free acetaldehyde used a polar GC-column, while our column was less polar. In our case, acetaldehyde might exit the column too fast to enable detection.
Another explanation for the un-detected free acetaldehyde is its immediate conversion into 2-methyl-1,3-dioxolane. This product was also detected after the direct injection of the headspace gasses into the GC (Fig. 4). Although these chromatograms had a lower signal-to-noise ratio, the highly toxic 2-chloroethanol gas (Fig. 4A) was detected as well, which was also reported by Schiavi et al.22,36,37
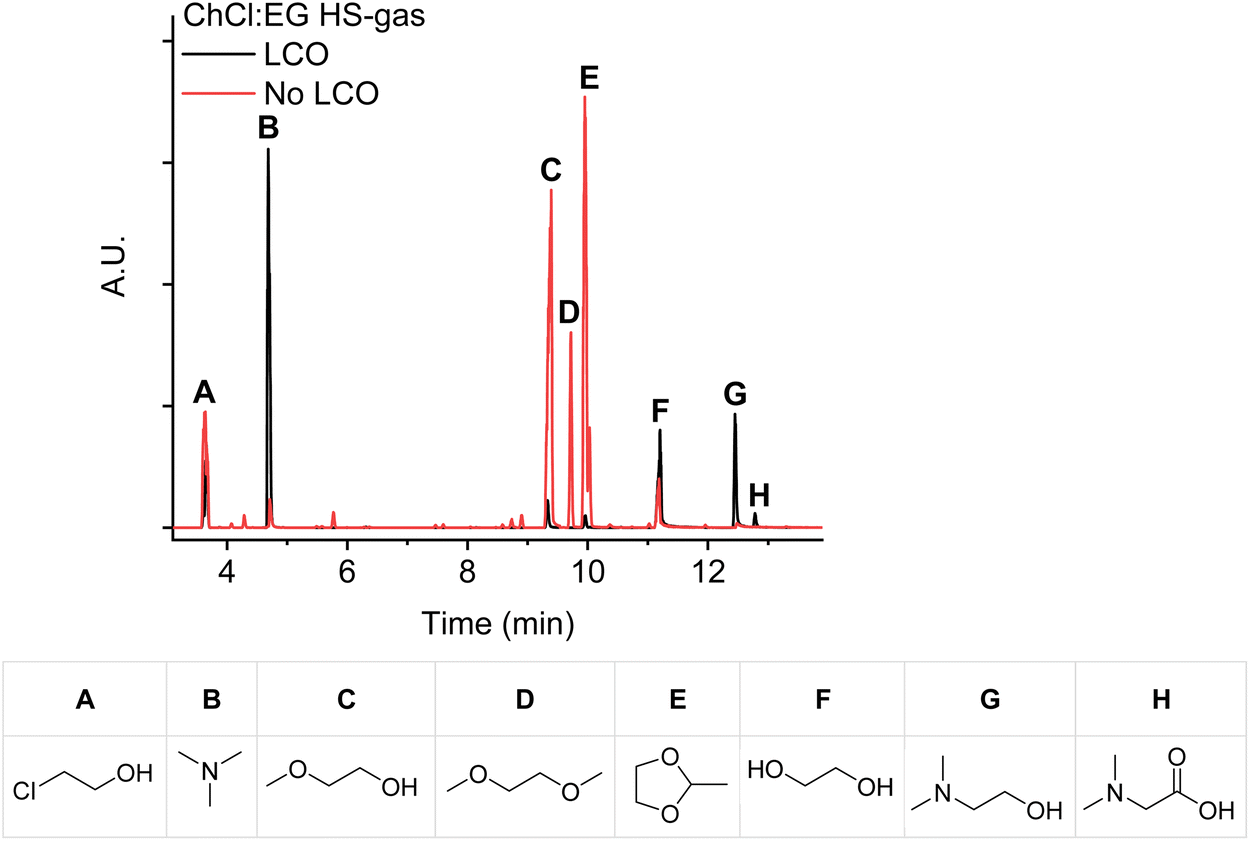 | ||
| Fig. 4 GC-MS chromatograms after direct gas injection of the headspace gasses of ChCl:EG (1:2) after heating and after leaching LCO, both at 180 °C for 24 hours. The chemical structures of the identified decomposition products are shown below the GC chromatogram. | ||
Fig. 3 and 4 also show a significant degree of decomposition of ChCl:EG (1:2) after leaching LCO at 180 °C for 24 hours. It clearly confirms that the DES progressively degrades after each leaching cycle. Schiavi et al. removed these decomposition products by bubbling nitrogen gas through the cobalt-depleted DES, before successfully leaching LCO in the second cycle.22 However, this approach only removes the decomposition products, but cannot prevent the DES from decomposing further. Hence, the decomposition reactions, the formed toxic products, and the bubbling of nitrogen gas should make the common conception that DESs are stable, non-toxic and cheap questionable.38,39
Identification of the reducing agent during leaching of LCO by ChCl:EG (1:2)
Initial insights about the reducing agent in the LCO leaching reactions were obtained by investigating which of the two DES components, choline chloride or ethylene glycol, is (directly or indirectly) responsible for the effective cobalt leaching. The cobalt leaching efficiency by each DES component was investigated separately. First, the chloride concentration in ChCl:EG (1:2) was determined by argentometry, being approximately 3.9 mol L−1. Subsequently, a solution was prepared by dissolving 3.9 mol L−1 of lithium chloride in ethylene glycol, in order to evaluate a solvent system with a similar chloride concentration as ChCl:EG (1:2), but without the choline cation. Lithium chloride was selected because of its very high solubility in ethylene glycol.40 This approach impeded an accurate determination of the lithium leaching efficiency, due to the high amount of lithium present in the solvent matrix. For this reason, and because the main focus was on cobalt, the leaching efficiency of lithium was not considered in the rest of this work. Since choline chloride has a melting point of 302 °C,41 it could not be directly evaluated as lixiviant in reactions at 180 °C. Several ChCl:EG DESs with different molar ratios between choline chloride and ethylene glycol were used to leach LCO. Table 1 shows that the cobalt leaching efficiency is significantly lower when the lixiviant is richer in ethylene glycol. This is in contradiction with both the works of Tran et al. and Schiavi et al., both stating that ethylene glycol is the cobalt(III) reducing agent during leaching.21,22 Nevertheless, the results shown in Table 1 convincingly demonstrate that choline and/or its decomposition products contribute to the efficient leaching of cobalt.
:EG DESs and ethylene glycola
Decomposition products of choline chloride as potential cobalt(III) reducing agents
To the best of our knowledge, there are no reports on the leaching of cobalt-containing materials by choline chloride, although two if its decomposition products (trimethylamine and acetaldehyde) are potentially reactive towards cobalt(III). Fig. 3 shows the formation of both trimethylamine and acetaldehyde (not directly detected but probably formed) during leaching with the DES at high temperatures. Cobalt(III) has an affinity for N-donor atom ligands.42 Hence, trimethylamine might solubilize cobalt by forming soluble cobalt(III) complexes during leaching, without the need for cobalt(III) reduction.43 On the other hand, aldehydes are known to be reducing agents towards cobalt(III) species, which makes acetaldehyde a possible cobalt(III) reducing agent during leaching of LCO.44 The potential cobalt leaching ability of trimethylamine or acetaldehyde was evaluated by adding these components in two different concentrations to ethylene glycol and comparing the cobalt leaching efficiencies. These concentrations corresponded to the stoichiometric amount with respect to the amount of cobalt in LCO, or a tenfold stoichiometric excess. Furthermore, for each tested system, 3.9 mol L−1 lithium chloride dissolved in ethylene glycol was also present to maintain a similar chloride concentration as in ChCl:EG (1:2), as described previously. However, both trimethylamine and acetaldehyde are gasses at room temperature and could evaporate or decompose when the leaching reaction is performed at 180 °C.45 Since these compounds could get lost during the leaching reaction, they may not be contributing to the leaching of LCO. To verify this, tributylamine and decanal were also evaluated as possible lixiviants, since these homologues of trimethylamine and acetaldehyde, both have a boiling point higher than 180 °C.45,46 From Table 2, it is evident that amines have no significant effect on the leaching cobalt efficiency from LCO. This conclusion is also supported by the predominance of cobalt(II) chloride complexes in the ChCl:EG (1:2) leachate, as observed by Schiavi et al. using UV-Vis absorption spectroscopy.22 While acetaldehyde had no significant effect on the leaching efficiency, decanal increased the cobalt leaching efficiency remarkably. This was evident from the detection of decanoic acid, i.e. the oxidation product of decanal, after leaching (see ESI, Fig. S1†). However, the high cobalt leaching efficiency of ca. 92% by ChCl:EG (1:2) (see Table 1) was not achieved by decanal, not even after extending the leaching time to 48 hours.
| Lixiviant | Equimolar amount added | %L Co |
|---|---|---|
|
a Leaching conditions: 180 °C, 24 h, 900 rpm, S:L = 20 g L−1.
b 3.9 mol L−1 LiCl was dissolved.
|
||
| Trimethylamine in EGb | 1 | 6.8 |
| Trimethylamine in EGb | 10 | 7.3 |
| Tributylamine in EGb | 1 | 8.1 |
| Tributylamine in EGb | 10 | 7.9 |
| Tributylamine | Pure | 8.7 |
| Acetaldehyde in EGb | 1 | 6.7 |
| Acetaldehyde in EGb | 10 | 6.5 |
| Decanal in EGb | 1 | 17.5 |
| Decanal in EGb | 10 | 18.9 |
| Decanal | Pure | 20.2 |
| ChCl:EG (1:2) |
— | 91.6 |
Although decanal can reduce cobalt(III) to cobalt(II), the only possible similar aldehyde that could be formed during leaching of LCO with ChCl:EG (1:2) is acetaldehyde. There is thus still a possibility that acetaldehyde reduces cobalt(III) after it is formed in situ by the thermal decomposition of choline chloride. In this case, acetic acid should be formed. However, no acetic acid could be detected by GC-MS after leaching LCO with ChCl:EG (1:2). Additionally, infrared spectra did not show the typical carbonyl stretching band around ca. 1760–1665 cm−1 after leaching LCO with ChCl:EG (1:2) (see Fig. S2, ESI†). In fact, no significant differences are evident at all between the three ChCl:EG (1:2) spectra.
In order to completely exclude acetaldehyde as a cobalt(III) reducing agent, gas phase FT-IR spectra were also recorded from the collected headspaces. These were collected at different time intervals during leaching of LCO by ChCl:EG (1:2) at 180 °C (see ESI, Fig. S3†). The typical sharp carbonyl stretching vibration around ca. 1760–1665 cm−1 did not appear or disappear during the entire leaching period. This excludes the possible decomposition of acetaldehyde by oxidation to acetic acid. So it is unlikely that acetaldehyde is the reducing agent of cobalt(III). Furthermore, although the leaching of cobalt increases over time (see ESI, Fig. S4†), no clear functional group could be unambiguously identified in Fig. S2 and S3.† The band increasing in intensity at the infrared spectra shown in Fig. S3† located around ca. 3000–2800 cm−1 are probably related to alkyl C–H stretching vibrations.45 These peaks could be assigned to many of the formed decomposition products, but not to one in particular.
The remaining decomposition products 2-methoxyethanol and dimethylaminoethanol were also evaluated as lixiviants to leach cobalt from LCO at 180 °C for 24 hours. Both products were mixed with LiCl-containing ethylene glycol in 1:2 molar ratio, as in ChCl:EG (1:2). These mixtures are referred as 2ME:EG (1:2) and DMAE:EG (1:2), respectively. Table 3 shows that dimethylaminoethanol slightly increased the cobalt leaching efficiency, while 2-methoxyethanol had no significant effect. GC-MS analysis detected dimethylglycine as oxidation product of dimethylaminoethanol in case of DMAE:EG (1:2). However, the maximum leaching yield in this solvent system was much lower than the high cobalt leaching efficiency by ChCl:EG (1:2).
Choline chloride as potential cobalt(III) reducing agent
Since dimethylglycine was detected as the oxidation product of dimethylaminoethanol, a similar reaction can be predicted for choline chloride as well. Reports of this reaction can be found in the literature. For example, NMR studies by Bednarz et al. on the oxidation of choline chloride by a persulfate solution confirmed the presence of betaine aldehyde and betaine as oxidation products.35 Sinclair et al. investigated the electrochemical decomposition of ChCl:EG (1:2) and reported that choline chloride is electrochemically decomposed, but they could not detect oxidation products of choline chloride by GC-MS due to their low volatility.47 In our present work, no NMR studies were performed since the paramagnetic nature of cobalt(II) in the ChCl:EG (1:2) leachate caused too severe line broadening. Therefore, evidence for the presence of betaine (and its derivatives) were sought in the GC-MS chromatograms and infrared spectra.
Only small amounts of dimethylglycine could be detected in the GC-MS chromatograms after leaching cobalt from LCO with ChCl:EG (1:2) at 180 °C for 24 hours (Fig. 4H). Hence, the decomposition of choline chloride to dimethylaminoethanol predominates, and a small quantity is oxidized to dimethylglycine (Scheme 2).
 | ||
| Scheme 2 Formation of dimethylaminoethanol and dimethylglycine as thermal decomposition products of choline chloride during leaching of cobalt from LCO by ChCl:EG (1:2) at 180 °C for 24 hours. | ||
However, the low concentration of dimethylglycine indicates that dimethylaminoethanol is probably not the decisive cobalt(III) reducing agent, as mentioned previously. As Sinclair et al. correctly indicated, other analysing techniques are required to detect betaine or betaine aldehyde.47 The FT-IR comparison in Fig. S5 (ESI†) already shows that betaine is probably not formed in the ChCl:EG (1:2) leachate. Suuronen et al. studied the thermal decomposition of betaine and confirmed that first significant degradation reactions start around 245 °C.48 Since both betaine and betaine aldehyde have boiling points above 180 °C, no gas-phase FT-IR spectra were investigated.45
Rhee et al. used a derivatization method to detect betaine in the presence of choline chloride by LC-MS using a UV-detector. Herein, betaine was transformed by 2-bromoacetophenone into a strongly UV-absorbing derivative, whereas choline chloride does not react at all with this derivatizing reagent.49 We applied this method to a reference sample that contained both choline chloride and betaine, and on the ChCl:EG (1:2) leachate. However, comparison of the LC-MS data in Fig. S6 (ESI†) confirmed that the ChCl:EG (1:2) leachate did not contain betaine. Therefore, the investigations on the potential cobalt(III) reducing ability of the hydroxyl function of choline chloride were aborted.
Another possibility is that cobalt(III) is reduced by the chloride anion of choline chloride in ChCl:EG (1:2). The mechanism would be similar to the leaching of LCO by hydrochloric acid, in which the chloride anion is oxidized to chlorine gas while reducing cobalt(III) to cobalt(II).20,50 Therefore, a tetramethylammonium chloride–ethylene glycol based DES was prepared at a 1:2 molar ratio (i.e. Me4NCl:EG 1:2) and used to leach LCO at 180 °C for 24 hours. The comparison with ChCl:EG (1:2) as lixiviant is given in Table 4. Although Me4NCl:EG (1:2) has approximately the same chloride concentration as ChCl:EG (1:2), no significant effect on the cobalt leaching efficiency was observed. Moreover, for both Me4NCl:EG and ChCl:EG, the gasses evolved during leaching were collected in balloons and subsequently transferred via a tube over a chlorine detector and further guided through a 4 mol L−1 potassium iodide solution. In neither case, no chlorine gas was detected and no brown iodine was formed. Hence, the high cobalt leaching efficiency cannot be attributed to the chloride anion of choline chloride when ChCl:EG (1:2) was used to leach LCO at 180 °C for 24 hours.
| Lixiviant |
n
HBA:nEG |
%L Co |
|---|---|---|
|
a Leaching conditions: 180 °C, 24 h, 900 rpm, S:L = 20 g L−1.
|
||
| Me4NCl:EG |
1:2 |
8.8 |
| ChCl:EG |
1:2 |
91.6 |
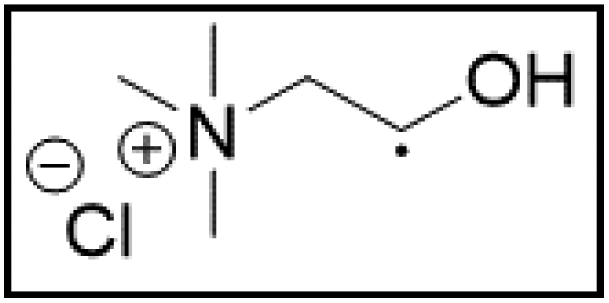
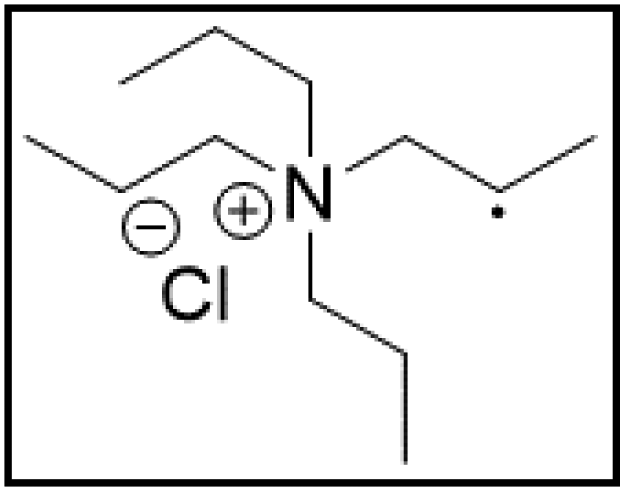
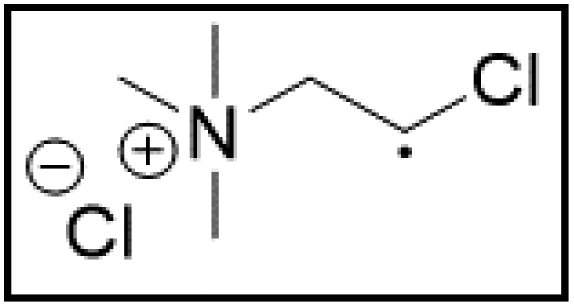
However, choline chloride can also undergo a radical oxidation reaction via a hydrogen abstraction at its β-position. Foster et al. confirmed the dominant abundance of the (CH3)3NCH2CHOH radical when choline chloride was treated with a hydrogen peroxide solution.51 This β-hydrogen abstraction of choline chloride was also reported by Bednarz et al., Ackerman et al. and Shkrob et al.34,35,52 Radical reactions are frequently initiated at higher temperatures and/or by redox reactions.53 Both these conditions are met in our studied case. In order to investigate whether the radical oxidation reaction takes place, various quaternary ammonium salts were compared, including both salts that are prone to the radical oxidation and those that are less prone to undergo this reaction. In these experiments, several quaternary ammonium salts as HBAs were mixed with ethylene glycol as HBD in a 1:2 molar ratio and then used to leach LCO. The cobalt leaching efficiencies are compared in Table 5. In this table, the HBAs are ranked according to the stability of their formed radicals (shown in the right column), with those forming the most stable radicals at the bottom. As Table 5 shows, the cobalt leaching efficiencies also increase from top to bottom. This indicates that cobalt is more efficiently leached if the radical formed after abstraction of the β-hydrogen is more stable. For example, Me4NCl has no β-carbon atoms, and therefore the cobalt leaching efficiency is poor. Although Et4NCl has a carbon atom in the β-position, a primary and hence less stable radical is formed after hydrogen abstraction. This results in a poor cobalt leaching efficiency.
| HBA | %L Co (140 °C) | %L Co (180 °C) | Radical | |
|---|---|---|---|---|
|
a Leaching conditions: 140 °C and 180 °C, 24 h, 900 rpm, S:L = 20 g L−1. All HBAs were mixed with ethylene glycol as HBD at nHBA:nHBD = 1:2.
|
||||
| ChCl:EG |
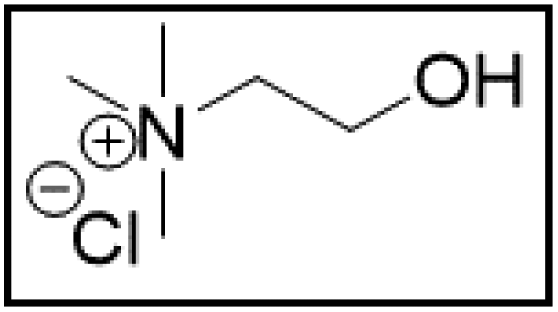
|
19.7 | 91.6 |
|
| Me4NCl:EG |
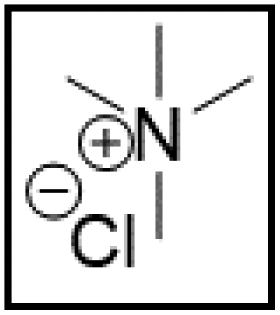
|
0.5 | 8.8 |

|
| Et4NCl:EG |

|
2.6 | 7.7 |

|
| Me3NBuCl:EG |
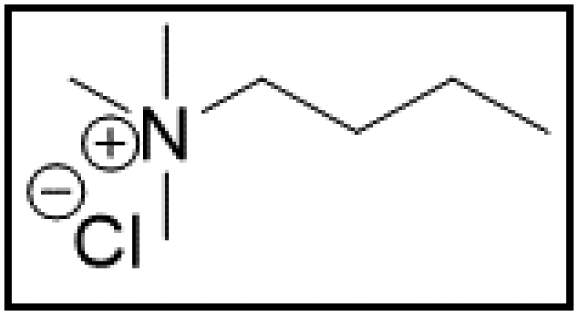
|
2.9 | 78.9 |

|
| Pr4NCl:EG |

|
13.5 | 97.9 |
|
| Bu4NCl:EG |

|
16.8 | 95.5 |

|
| M-ChCl:EG |
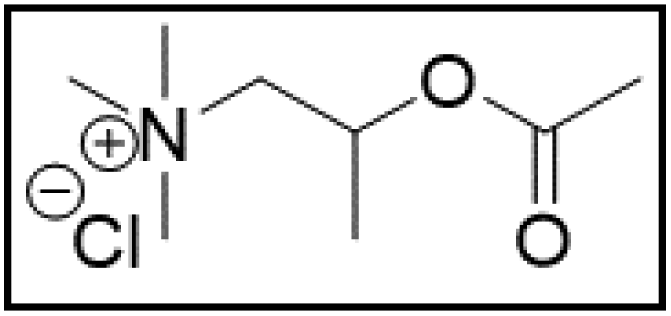
|
32.5 | 102.2 |

|
| ChCl2:EG |

|
101.2 | 103.8 |
|
Me3NBuCl, Pr4NCl and Bu4NCl have β-carbon atoms and form a more stable secondary radical.54 Therefore, these quaternary ammonium salts can leach cobalt more efficiently than Me4NCl and Et4NCl. However, Pr4NCl and Bu4NCl have four alkyl chains that can form a secondary β-radical, while Me3NBuCl only has one. This is most likely the reason why Pr4NCl and Bu4NCl leach cobalt more efficiently than Me3NBuCl. In addition, M-ChCl forms a tertiary β-radical, and has an ester group next to this radical. A tertiary radical is more stable than a secondary radical, and the adjacent ester group also stabilizes the radical by the resonance effect. As a result, M-ChCl leaches cobalt quantitatively at 180 °C and ca. 33% is already leached at 140 °C. The radical of ChCl2 has an adjacent atom with lone electron pairs which is highly stabilizing.54 Consequently, quantitative cobalt leaching is already achieved at 140 °C.
However, radical reactions are well-known to be dangerous due to severe explosion risks, especially if they occur without researchers being aware of them.55,56 These safety risks were evident throughout the experimental work, as certain vials exploded and condensers had to be used afterwards (see experimental).
The results shown in Table 5 suggest that leaching of cobalt by ChCl:EG (1:2) is facilitated by the formation of a secondary radical on the β-carbon of choline. This takes place at 180 °C and the radical is further stabilized by the oxygen lone electron pair of the adjacent hydroxyl function. Hence, the decisive factors for quaternary ammonium salts to act as efficient lixiviants are: (1) the possibility to form a stable radical at the β-carbon atom and (2) being solubilized by a component that does not undergo undesired side reactions at 180 °C. For example, TOMAC could potentially form these stable radicals and is a liquid at 180 °C. Sulfolane is a polar solvent, just like ethylene glycol, and has an even higher boiling temperature (285 °C vs. 197 °C for EG).57,58 Hence, TOMAC could directly be used as liquid quaternary ammonium salt to leach LCO while sulfolane could function to solubilize choline chloride like ethylene glycol in ChCl:EG (1:2). Sulfolane was therefore mixed with choline chloride at 1:2 and used to leach LCO as well. The performance of TOMAC and choline chloride–sulfolane with ChCl:EG (1:2) as LCO lixiviants are compared in Table 6. TOMAC is an ionic liquid and sulfolane cannot donate hydrogen bonds to choline chloride; therefore, neither system can be considered as a DES. However, Table 6 shows that both TOMAC and ChCl–sulfolane are able to leach cobalt quantitatively as well. These results support our hypothesis and also confirm that the formation of a DES is not a requirement at all to obtain the high cobalt leaching efficiency.
:EG (1:2), TOMAC and choline chloride diluted in sulfolanea




Mechanism of choline chloride as cobalt(III) reducing agent
Confirming an unambiguous leaching mechanism of cobalt from LCO by ChCl:EG (1:2) remains challenging due to the formation of numerous decomposition products. Tentative reaction and decomposition mechanisms are presented in Scheme 3, showing that the cobalt(III) reduction is mainly caused by radical β-hydrogen abstraction from choline chloride. Hence, cobalt(III) in LCO acts as a radical initiator. The generated secondary radical further decays to trimethylamine and acetaldehyde or its enol product (i.e. ethenol or vinyl alcohol). Much more trimethylamine was detected by GC-MS when ChCl:EG (1:2) was used to leach LCO than when the neat DES was heated (in the absence of LCO) (see Fig. 4B). As mentioned before, it was impossible to detect acetaldehyde as the other degradation product. However, since additions on the anti-Markovnikov position can occur at high temperatures, its enol product could be chlorinated and/or hydrated, as shown in Scheme 3.59,60 The chloride anions of choline chloride could be responsible for the chlorination of ethenol, while its hydration could be related to the presence of water. This is because water is produced during leaching of LCO, and extracted from the air due to the hygroscopic nature of choline chloride.27 The chlorinated product of ethenol (i.e. 2-chloroethanol) was detected by GC-MS (see Fig. 4A). However, it was detected in higher concentrations after ChCl:EG (1:2) was heated (in the absence of LCO) than after it was used as lixiviant. This is probably because ethylene glycol can also be transformed into 2-chloroethanol at high temperatures in the presence of chloride-rich media, such as in ChCl:EG (1:2).61 Furthermore, the hydrated product of ethenol at the anti-Markovnikov position would be ethylene glycol, which generally gives broad peaks in GC-MS chromatograms because it has the tendency to stick to the GC-columns, as shown in Fig. 3.62 This drawback, and the fact that ethylene glycol is a component of the DES itself, makes the comparison between ChCl:EG (1:2) after leaching and heating difficult. Only the GC-MS analysis of the headspace gas (Fig. 4F) confirmed that ethylene glycol was detected in higher concentrations after ChCl:EG (1:2) was used as lixiviant than after it was heated. Choline chloride also decomposes to dimethylaminoethanol, which in turn is oxidized to dimethylglycine by cobalt(III). The results reported in Table 3 confirmed that cobalt is not quantitatively leached by this reaction and thereby indicated that this is not the decisive reaction. However, more detailed investigations are necessary to complete the reaction scheme and determine the conversion rates of each reaction.
 | ||
| Scheme 3 Proposed leaching mechanism of cobalt from LCO by ChCl:EG (1:2) at 180 °C for 24 hours.52 | ||
Degradation pathways as those shown in Scheme 3 were also observed for other lixiviant systems reported in Table 5. For example, GC-MS chromatograms of Bu4NCl:EG (see ESI Fig. S7†) confirmed the presence of the degradation products butanal, 1-chlorobutane and butanol, which were detected in higher concentrations after leaching than after just heating the solvent in the absence of LCO. These observations are in agreement with the proposed reaction mechanism in Scheme 3.
ChCl:urea (1:2) and PEG:thiourea (2:1) as lixiviants for LCO at elevated temperatures
ChCl:urea (1:2) was also used to leach LCO at 180 °C for 18 hours, as described by Wang et al.23 Unfortunately, an extremely viscous DES leachate was obtained after leaching. Wang et al. stated in their Experimental section that the leachates were filtered at 100 °C, without mentioning any difficulties with sample handling resulting from the high viscosity.23 Furthermore, as mentioned above, Delgado-Mellado et al. confirmed that ChCl:urea (1:2) shows signs of thermal degradation at a temperature of about 172 °C.26 The high viscosity of the ChCl:urea (1:2) leachate makes it difficult to upscale the process of Wang et al.23 Furthermore, a leaching temperature of 180 °C would probably cause the degradation of ChCl:urea (1:2) as well, similarly to what was observed for ChCl:EG (1:2).
Chen et al., observed that using PEG:thiourea (2:1) to leach LCO, only 60% of cobalt was leached at 160 °C after 24 hours.24 These energy-intensive leaching conditions and the poor cobalt leaching efficiency renders this process cost-ineffective when upscaled. Since our focus is on ChCl:EG (1:2), a more detailed discussion of ChCl:urea (1:2) and PEG:thiourea (1:2) as lixiviants is omitted.
Conclusion
The experimental results presented in this paper demonstrate that one should be cautious when using DESs based on choline chloride and ethylene glycol, such as ChCl:EG (1:2). In recent studies, this DES was used as a lixiviant at 180 °C, for up to 24 hours to leach cobalt from LCO or NMC cathodes of LIBs, while stressing its green nature for these applications. However, we show that ChCl:EG (1:2) is not thermally stable at these elevated temperatures and that toxic decomposition products are even formed, such as trimethylamine or 2-chloroethanol. Additionally, we have shown that choline chloride, rather than ethylene glycol, is the reducing agent for cobalt(III) in the reductive leaching reaction of LCO. Therefore, we recommend monitoring the thermal stability of DESs based on choline chloride in detail before using them in high-temperature ionometallurgical processes. If DESs decompose during leaching reactions at elevated temperatures, one cannot state that DESs are green nor safe solvents in these applications. One should refrain from claims about the greenness of DESs if no experimental evidence is provided for their thermal and chemical stability when heated to higher temperatures for extended periods of time.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program: Grant Agreement 694078—Solvometallurgy for critical metals (SOLCRIMET). The authors thank Christine Wouters for the support in the GC/MS analysis and Galahad O'Rourke for the help with the TGA.References
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 1, 70–71 RSC.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- T. El Achkar, H. Greige-Gerges and S. Fourmentin, Environ. Chem. Lett., 2021, 19, 3397–3408 CrossRef CAS.
- N. Rodriguez Rodriguez, A. Van Den Bruinhorst, L. J. B. M. Kollau, M. C. Kroon and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 11521–11528 CrossRef CAS.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- A. P. Abbott, S. Nandhra, S. Postlethwaite, E. L. Smith and K. S. Ryder, Phys. Chem. Chem. Phys., 2007, 9, 3735–3743 RSC.
- S. Ghosh and S. Roy, Surf. Coat. Technol., 2014, 238, 165–173 CrossRef CAS.
- S. Spathariotis, N. Peeters, K. S. Ryder, A. P. Abbott, K. Binnemans and S. Riaño, RSC Adv., 2020, 10, 33161–33170 RSC.
- L. Li, J. Lu, Y. Ren, X. X. Zhang, R. J. Chen, F. Wu and K. Amine, J. Power Sources, 2012, 218, 21–27 CrossRef CAS.
- X. W. Wei, C. Y. Gong, M. L. Gou, S. Z. Fu, Q. F. Guo, S. Shi, F. Luo, G. Guo, L. Y. Qiu and Z. Y. Qian, Int. J. Pharm., 2009, 381, 1–18 CrossRef CAS PubMed.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2017, 3, 570–600 CrossRef.
- A. H. Whitehead, M. Pölzler and B. Gollas, J. Electrochem. Soc., 2010, 157, 328–334 CrossRef.
- R. Bernasconi, G. Panzeri, G. Firtin, B. Kahyaoglu, L. Nobili and L. Magagnin, J. Phys. Chem. B, 2020, 124, 10739–10751 CrossRef CAS PubMed.
- Y. Wang, Y. Xue and C. Zhang, Green Chem., 2021, 23, 6301–6321 RSC.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC.
- D. L. Thompson, J. M. Hartley, S. M. Lambert, M. Shiref, G. D. J. Harper, E. Kendrick, P. Anderson, K. S. Ryder, L. Gaines and A. P. Abbott, Green Chem., 2020, 22, 7585–7603 RSC.
- I. Vural Gürsel, T. Noël, Q. Wang and V. Hessel, Green Chem., 2015, 17, 2012–2026 RSC.
- Z. Yuan, H. Liu, W. F. Yong, Q. She and J. Esteban, Green Chem., 2022, 24, 1895–1929 RSC.
- N. Peeters, K. Binnemans and S. Riaño, Green Chem., 2020, 22, 4210–4221 RSC.
- M. K. Tran, M. T. F. Rodrigues, K. Kato, G. Babu and P. M. Ajayan, Nat. Energy, 2019, 4, 339–345 CrossRef CAS.
- P. G. Schiavi, P. Altimari, M. Branchi, R. Zanoni, G. Simonetti, M. A. Navarra and F. Pagnanelli, Chem. Eng. J., 2021, 417, 129249 CrossRef CAS.
- S. Wang, Z. Zhang, Z. Lu and Z. Xu, Green Chem., 2020, 22, 4473–4482 RSC.
- Y. Chen, Y. Lu, Z. Liu, L. Zhou, Z. Li, J. Jiang, L. Wei, P. Ren and T. Mu, ACS Sustainable Chem. Eng., 2020, 8, 11713–11720 CrossRef CAS.
- W. Chen, Z. Xue, J. Wang, J. Jiang, X. Zhao and T. Mu, Acta Phys.-Chim. Sin., 2018, 34, 904–911 CAS.
- N. Delgado-Mellado, M. Larriba, P. Navarro, V. Rigual, M. Ayuso, J. García and F. Rodríguez, J. Mol. Liq., 2018, 260, 37–43 CrossRef CAS.
- N. Peeters, K. Binnemans and S. Riaño, Green Chem., 2022, 24, 2839–2852 RSC.
- R. F. Dye, Korean J. Chem. Eng., 2001, 18, 571–579 CrossRef CAS.
- B. S. Đorđević, D. Z. Troter, Z. B. Todorović, I. G. Đalović, L. P. Stanojević, P. M. Mitrović and V. B. Veljković, J. Food Meas. Charact., 2020, 14, 2570–2577 CrossRef.
- J. Wu, Q. Liang, X. Yu, L. Qiu-Feng, L. Ma, X. Qin, G. Chen and B. Li, Adv. Funct. Mater., 2021, 31, 1–25 Search PubMed.
- K. Haerens, E. Matthijs, K. Binnemans and B. Van der Bruggen, Green Chem., 2009, 11, 1357–1365 RSC.
- E. X. Chen, H. R. Fu, R. Lin, Y. X. Tan and J. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 22871–22875 CrossRef CAS PubMed.
- J. González-Rivera, E. Husanu, A. Mero, C. Ferrari, C. Duce, M. R. Tinè, F. D. Andrea, C. S. Pomelli and L. Guazzelli, J. Mol. Liq., 2020, 300, 1–10 CrossRef.
- M. Ackerman and R. M. Lemmon, J. Phys. Chem., 1967, 71, 3350–3352 CrossRef CAS PubMed.
- S. Bednarz, A. Błaszczyk and D. Bła, Catal. Today, 2015, 257, 297–304 CrossRef CAS.
- D. Winfield, J. Ring, J. Horn, E. M. White and J. Locklin, Green Chem., 2021, 23, 9658–9668 RSC.
- L. Jia, M. Makha, C. X. Du, Z. J. Quan, X. C. Wang and Y. Li, Green Chem., 2019, 21, 3127–3132 RSC.
- Y. Murakami, S. K. Das, Y. Himuro and S. Maeda, Phys. Chem. Chem. Phys., 2017, 19, 30603–30615 RSC.
- C. Mukesh, S. G. Khokarale, P. Virtanen and J. P. Mikkola, Sustainable Energy Fuels, 2019, 3, 2125–2134 RSC.
- H. S. Isbin and K. A. Kobe, J. Am. Chem. Soc., 1945, 67, 464–465 CrossRef CAS.
- A. P. Abbott, A. D. Ballantyne, J. P. Conde, K. S. Ryder and W. R. Wise, Green Chem., 2012, 14, 1302–1307 RSC.
- A. K. Ghosh, C. S. Purohit and R. Ghosh, Polyhedron, 2018, 155, 194–201 CrossRef CAS.
- V. M. Norwood and K. W. Morse, Inorg. Chem., 1987, 26, 284–288 CrossRef CAS.
- C. F. Hendriks, H. C. A. van Beek and P. M. Heertjes, Ind. Eng. Chem. Prod. Res. Dev., 1978, 17, 260–264 CrossRef CAS.
- J. A. Dean, Lange's handbook of chemistry, McGraw-Hill, New York, 5th edn, 1990 Search PubMed.
- H. Tang, Y. Shen, B. G. Li and M. Radosz, Macromol. Rapid Commun., 2008, 29, 1834–1838 CrossRef CAS.
- N. S. Sinclair, X. Shen, E. Guarr, R. F. Savinell and J. S. Wainright, J. Electrochem. Soc., 2021, 168, 1–9 Search PubMed.
- J. Suuronen, I. Pitkänen, H. Halttunen and R. Moilanen, J. Therm. Anal. Calorim., 2002, 69, 359–369 CrossRef CAS.
- I. Rhee and K.-J. Paeng, Anal. Sci. Technol., 2015, 28, 112–116 CrossRef.
- X. Chen, J. Li, D. Kang, T. Zhou and H. Ma, Green Chem., 2019, 21, 6342–6352 RSC.
- T. Foster and P. R. West, Can. J. Chem., 1973, 51, 4009–4017 CrossRef CAS.
- I. A. Shkrob, T. W. Marin and J. F. Wishart, in Applications of EPR in Radiation Research, Springer International Publishing, Cham, 2014, pp. 453–485 Search PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- H. Zipse, Top. Curr. Chem., 2006, 263, 163–189 CrossRef CAS.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- Y. Wu, J. Y. Chen, J. Ning, X. Jiang, J. Deng, Y. Deng, R. Xu and W. M. He, Green Chem., 2021, 23, 3950–3954 RSC.
- P. S. Metkar, E. J. Till, D. R. Corbin, C. J. Pereira, K. W. Hutchenson and S. K. Sengupta, Green Chem., 2015, 17, 1453–1466 RSC.
- Y. Gu and F. Jérôme, Green Chem., 2010, 12, 1127–1138 RSC.
- Y. Chen, J. Zhang, Z. Tang and Y. Sun, J. Photochem. Photobiol., A, 2020, 392, 112340–112350 CrossRef CAS.
- S. Sitha and L. L. Jewell, in Chemistry for Sustainable Development, Springer Netherlands, Dordrecht, 2012, pp. 375–387 Search PubMed.
- M. Orefice, V. T. Nguyen, S. Raiguel, P. T. Jones and K. Binnemans, Ind. Eng. Chem. Res., 2022, 61, 754–764 CrossRef CAS.
- Y. H. Zhang, Y. A. Feng and W. K. Lu, J. Chromatogr. A, 2000, 904, 87–97 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section with list of chemical and analytical instrumentation, list of abbreviations and structures of all the mentioned chemicals, GC-MS chromatograms of decanal as LCO lixiviant, comparison of FT-IR spectra between the ChCl:EG (1:2) and acetic acid, gas phase FT-IR spectra at different time intervals during leaching of LCO by ChCl:EG (1:2), the cobalt leaching efficiency from LCO versus time by ChCl:EG (1:2), comparison of FT-IR spectra between ChCl:EG (1:2) and betaine, LC-MS chromatograms of the betaine derivatization reaction and GC-MS chromatograms of Bu4NCl:EG (1:2) as LCO lixiviant. See DOI: https://doi.org/10.1039/d2gc02075k |
| This journal is © The Royal Society of Chemistry 2022 |