
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-promoted oxidation of aldehydes to carboxylic acids under aerobic and photocatalyst-free conditions†‡
Charikleia S.
Batsika
ab,
Charalampos
Koutsilieris§
ab,
Giorgos S.
Koutoulogenis§
ab,
Maroula G.
Kokotou§
 ab,
Christoforos G.
Kokotos
*ab and
George
Kokotos
*ab
ab,
Christoforos G.
Kokotos
*ab and
George
Kokotos
*ab
aLaboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, Athens 15771, Greece. E-mail: ckokotos@chem.uoa.gr; gkokotos@chem.uoa.gr
bCenter of Excellence for Drug Design and Discovery, National and Kapodistrian University of Athens, Panepistimiopolis, Athens 15771, Greece
First published on 26th July 2022
Abstract
The development of green and sustainable methods for the conversion of aldehydes to carboxylic acids is of high academic and industrial interest. We demonstrate herein a practical and efficient light-promoted oxidation of aldehydes, utilising atmospheric oxygen as the sole oxidant. The transformation is promoted by either sunlight or LED 370 nm irradiation, without the use of a catalyst. A solvent mixture of acetone and water proved to be the most effective solvent medium. A wide range of aliphatic or aromatic aldehydes were successfully employed, providing the corresponding carboxylic acids in high to excellent yields. A mechanism was proposed based on direct infusion-high resolution mass spectrometry (DI-HRMS) studies, suggesting a solvent-assisted oxidative transformation. The method is green, sustainable and operationally simple under very mild conditions.
Introduction
The oxidation of aldehydes to the corresponding carboxylic acids is a fundamental, textbook and popular oxidative organic transformation.1 It has attracted high industrial interest, since several industrially important aldehydes are obtained via the hydroformylation reaction of alkenes,2 and then serve as starting materials for high-added value chemicals. Most of the general synthetic processes for the oxidation of aldehydes rely upon stoichiometric amounts of highly hazardous oxidants.1,3 A mild oxidant, widely employed in oxidation protocols, is oxygen, since it is an ambient, abundant and harmless gas, which can be considered the “greenest” approach towards oxidation reactions.3c,4 The “green” character of molecular oxygen as the oxidant, either deriving from ambient air or being supplied as a gas to the reaction mixture, was very early realized.4a Thus, in the past decade, aerobic oxidations have been recognized as highly attractive oxidative processes for various applications in organic chemistry, due to their low cost and environmental friendliness,3c,4 especially for the conversion of aldehydes to carboxylic acids.3c,4 Furthermore, the aerobic oxidation of aldehydes into the corresponding carboxylic acid is an industrial process and up to millions of tons per year of aliphatic carboxylic acids, among them, 2-ethylhexanoic acid, are produced by this method.3b These acids have various industrial uses, such as paint driers, vinyl stabilizers and cosmetic products.Although it is known that aldehydes are prone to oxidation, synthetic methods for their oxidation to carboxylic acids under green and mild conditions are still scarce. Except from stoichiometric methods employing a variety of oxidants, there is a number of catalytic approaches that demonstrate the aerobic oxidation of aldehydes (Scheme 1A and B).5–13 In 2016 and 2017, two methods using atmospheric oxygen as the sole oxidant were demonstrated, employing either a copper or an iron(III) catalyst, respectively,5,6 while an interesting contribution, employing Mn was reported in 1990.7 In 2018, a method based on nickel catalysis was reported, however a limited number of examples was demonstrated,8 while a metal-free-catalysed oxidation of aldehydes has been also reported using an N-heterocyclic carbene (NHC) (Scheme 1A).9 In 2019, N-hydroxyphthalimide was used as the catalyst (5 mol%) for the aerobic oxidation of aldehydes either in acetonitrile or water,10a and the mechanism of this reaction was thoroughly investigated (Scheme 1A).10b Photochemistry has also provided some elegant catalytic procedures (Scheme 1B).11–13 In 2010, Safari and coworkers employed visible light with a porphyrin catalyst for the oxidation of aldehydes,11 in 2013, Cho et al. reported the oxidation of aldehydes using oxygen bubbling and oxygen balloon in the presence of Ru or Ir photocatalyst under blue LED irradiation,12 while Favre-Reguillon and coworkers presented a continuous flow photochemical oxidation with camphorquinone (Scheme 1B).13 However, it has been demonstrated that the conversion of aldehydes to carboxylic acids can be also performed without a catalyst (Scheme 1C).14,15,17–21 In 2000, Lehtinen and coworkers described the aerobic (experimental and computational study) oxidation of 9 aliphatic aldehydes in chlorinated solvents.14 In 2008, Vigalok and Shapiro reported the oxidation of aldehydes “on water” and in “water”, however in a limited number of substrates.15 In that work, benzoic acid was obtained in 83% yield from benzaldehyde after a reaction time of 12 h, while the aerobic oxidation results served as the platform for the authors to introduce other aerobic oxidations “on water”.16 Caddick et al. studied the aerobic autooxidation of aldehydes under neat reaction conditions, as a part of a study directed to the metal-free hydroacylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and NN bonds.17 The reaction time was only 2 h, but low conversions were observed (0–60%).17 Favre-Reguillon and Bellefon et al. studied the autooxidation of a limited number of aldehydes in comparison to a Mn(II)-catalysed reaction18 and proposed a continuous flow process for the aerobic oxidation of aldehydes, however with a limited substrate scope, while oxygen under pressure was required.19 In 2018, the aerobic oxidation of aldehydes in water was reported, but the study was limited only in aromatic aldehydes, while a reaction time of 24 h was usually employed.20 The authors also demonstrated that they could reduce the reaction time down to 8 h (from 24 h, yield for benzoic acid 99%), when 0.5 mol% of a Fe catalyst was added.20 Also, in 2018, an aerobic oxidation of aldehydes in the eco-friendly solvent dipropylene glycol dimethyl ether (DPDME) was reported, but an elevated temperature of 80 °C and a reaction time of 10 h were necessary for the completion of the reaction.21
C and NN bonds.17 The reaction time was only 2 h, but low conversions were observed (0–60%).17 Favre-Reguillon and Bellefon et al. studied the autooxidation of a limited number of aldehydes in comparison to a Mn(II)-catalysed reaction18 and proposed a continuous flow process for the aerobic oxidation of aldehydes, however with a limited substrate scope, while oxygen under pressure was required.19 In 2018, the aerobic oxidation of aldehydes in water was reported, but the study was limited only in aromatic aldehydes, while a reaction time of 24 h was usually employed.20 The authors also demonstrated that they could reduce the reaction time down to 8 h (from 24 h, yield for benzoic acid 99%), when 0.5 mol% of a Fe catalyst was added.20 Also, in 2018, an aerobic oxidation of aldehydes in the eco-friendly solvent dipropylene glycol dimethyl ether (DPDME) was reported, but an elevated temperature of 80 °C and a reaction time of 10 h were necessary for the completion of the reaction.21
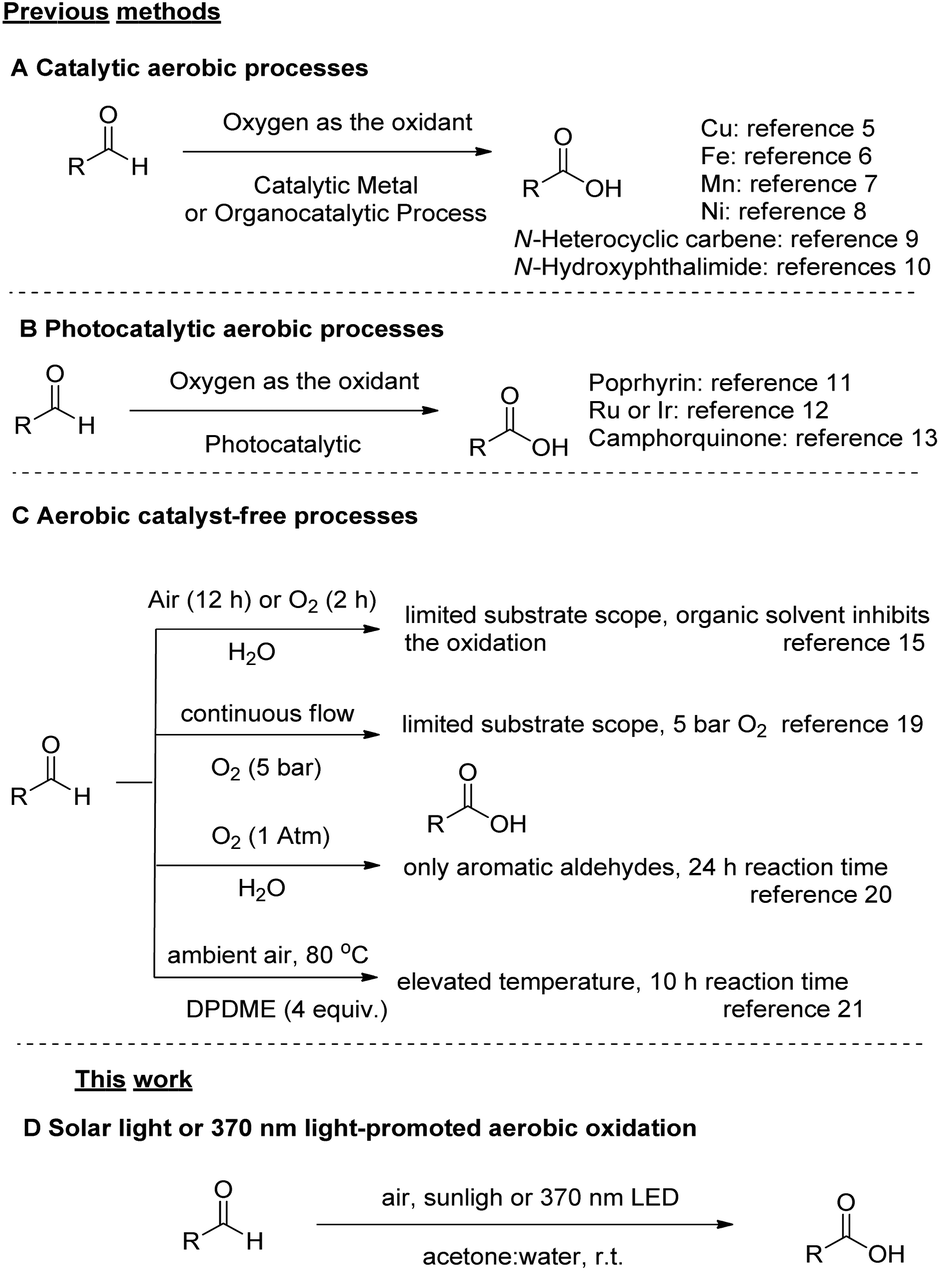 | ||
| Scheme 1 Aerobic oxidation of aldehydes to acids: previous methods and this work. | ||
The direct use of atmospheric oxygen as the oxidant is the ideal methodology for the development of green and sustainable oxidative organic transformations, since molecular oxygen offers high atom economy and production of water as the only by-product. From another point of view, visible-light photocatalysis is an emerging energy-saving platform providing attractive green chemical transformation under mild reaction conditions.22,23 In this work, we merge aerobic oxidation with light irradiation and we demonstrate an efficient sunlight or UVA-light promoted oxidation of aldehydes to carboxylic acids under aerobic conditions, without the use of any photocatalyst (Scheme 1D). A mixture of acetone and water was proven to be the most effective solvent system. This environmentally benign oxidation protocol is characterised by operational simplicity and it has been successfully applied in a variety of aliphatic and aromatic aldehydes. The mechanism for this oxidative transformation was studied by Direct Infusion-High Resolution Mass Spectrometry (DI-HRMS), revealing a solvent-assisted phenomenon.
Results and discussion
Among the methods reported for the aerobic oxidation of aldehydes, only three employ light for the activation of the catalyst.11–13 However, no catalyst-free method combined with light irradiation has been investigated so far for the practical synthesis of carboxylic acids from aldehydes, although the photochemical oxidation of heptanal has been known since 1954.24 The aim of our study was the development of a mild, green oxidative transformation of aldehydes, taking advantage of the use of atmospheric oxygen and sunlight or UVA-light as the source of energy. Dodecanal (1a) was employed as a model substrate and the reaction conditions for its oxidation were first investigated by evaluating the light source, the solvent, and the time. The results of this optimization study are summarized in Table 1.25 Ethyl acetate was employed as the solvent, in order to study different irradiation sources (Table 1, entries 1–6). Dodecanal was converted into acid 2a in a 22% yield under daylight after 5 hours (Table 1, entry 2), while the background reaction in dark produced acid only in 7% yield (Table 1, entry 1). However, the effect of irradiation was profound and the acid was produced in traces and 82% (77% yield of isolated product) yield, after 5 hours of irradiation with Kessil LED 456 nm and 370 nm, respectively (Table 1, entries 3 and 4 vs. entries 1 and 2). No product was observed in the presence of 4-AcNH-TEMPO, verifying the radical character of the reaction (Table 1, entry 5). When the reaction was left under an argon atmosphere, the reaction practically did not take place (traces, Table 1, entry 6), highlighting the importance of atmospheric oxygen as the oxidant. Switching solvent from ethyl acetate to acetone, under irradiation at 370 nm, led to a faster reaction, since following the reaction by 1H-NMR revealed the formation of acid 2a in 94% yield after only 3 hours (Table 1, entry 7). When the reaction was performed on water, dodecanoic acid was obtained in 82% conversion after 3 hours (Table 1, entry 8). Furthermore, the presence of water (10% v/v) afforded slightly better yield, leading to product 2a in 96% conversion (94% yield of isolated product) after 3 hours (Table 1, entries 9 vs. 7, 8 and 10). Keeping constant the presence of water (10%), we evaluated tetrahydrofuran, the green solvents 2-methyl-tetrahydrofuran and γ-valerolactone, as well as EtOAc as the potential solvent, however, none of them led to better results than acetone in 3 hours (Table 1, entries 11–14). Thus, an obvious acceleration of the reaction takes place in the presence of acetone, water or their mixture versus the other solvents tested (Table 1, entries 7–10 vs. 4, 11–14). Having defined a mixture of acetone–water as the optimum medium, we next evaluated LEDs of different wavelengths: 390 nm, 427 nm, 440 nm, 456 nm, 467 nm and 525 nm. Only in the case of 390 nm, a similar yield was obtained (Table 1, entries 15–20), suggesting that 370 nm was the optimum wavelength. Interestingly, when the reaction was left for 3 h under sunlight, the product was obtained in 87% (82% yield of isolated product, Table 1, entry 21). Collectively, acetone–water (10%) was proven to be the optimum reaction medium, while either sunlight or UVA-light at 370 nm were found to be the optimum light sources.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Light | Time (h) | Yielda (%) |
| a Conversion determined by NMR. Yield of isolated product after base–acid wash in parenthesis. b Caution: ethereal solvents, such as THF and 2-Me-THF, can undergo autoxidation under normal storage conditions to form unstable and potentially dangerous peroxide by-products. | ||||
| 1 | EtOAc | Dark | 5 | 7 |
| 2 | EtOAc | Daylight | 5 | 22 |
| 3 | EtOAc | 456 nm | 5 | Traces |
| 4 | EtOAc | 370 nm | 5 | 82 (77) |
| 5 | EtOAc | 370 nm 4-AcNH-TEMPO | 5 | 0 |
| 6 | EtOAc | 370 nm under Argon | 5 | Traces |
| 7 | Acetone | 370 nm | 3 | 94 |
| 8 | H2O | 370 nm | 3 | 82 |
| 9 | Acetone + 10% v/v H 2 O | 370 nm | 3 | 96 (94) |
| 10 | Acetone + 3% v/v H2O | 370 nm | 3 | 94 |
| 11 | THFb + 10% v/v H2O | 370 nm | 3 | 49 |
| 12 | 2-Me-THFb + 10% v/v H2O | 370 nm | 3 | Traces |
| 13 | γ-Valerolactone + 10% v/v H2O | 370 nm | 3 | 31 |
| 14 | EtOAc + 10% v/v H2O | 370 nm | 3 | 64 |
| 15 | Acetone + 10% v/v H2O | 390 nm | 3 | 80 |
| 16 | Acetone + 10% v/v H2O | 427 nm | 3 | Traces |
| 17 | Acetone + 10% v/v H2O | 440 nm | 3 | 14 |
| 18 | Acetone + 10% v/v H2O | 456 nm | 3 | Traces |
| 19 | Acetone + 10% v/v H2O | 467 nm | 3 | 0 |
| 20 | Acetone + 10% v/v H2O | 525 nm | 3 | 0 |
| 21 | Acetone + 10% v/v H 2 O | Sunlight | 3 | 87 (82) |
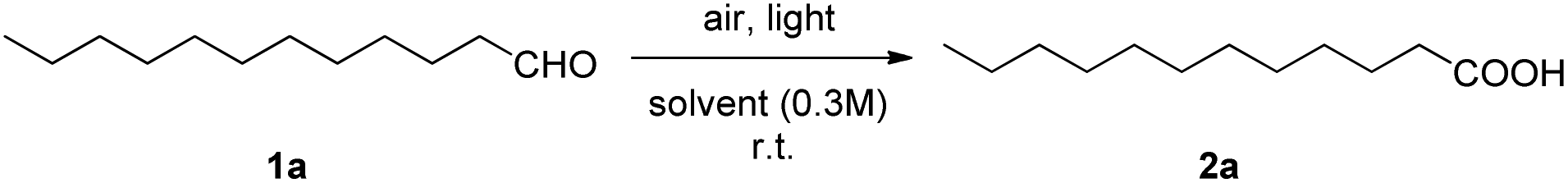
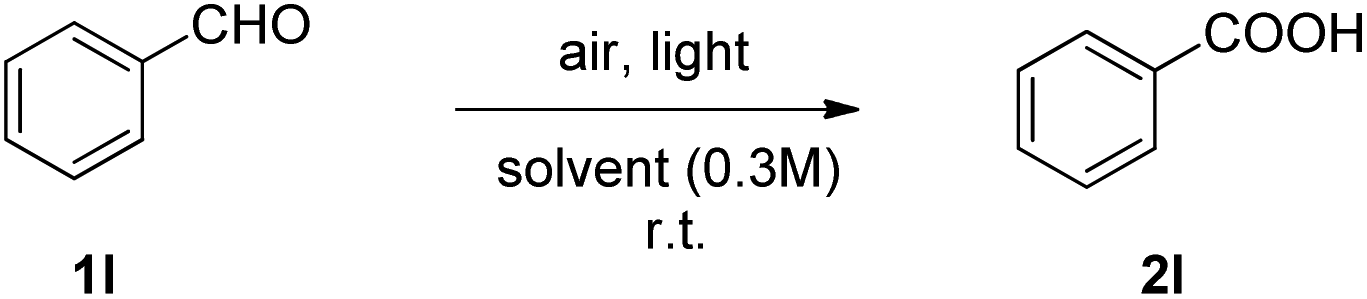
Then, we moved our interest in the oxidation of aromatic aldehydes, such as benzaldehyde (1l). The results of this optimization study are summarized in Table 2, where the conversion of the light-promoted aerobic oxidation was determined by HPLC.25 When the reaction was performed at 370 nm and ethyl acetate was employed as the solvent, a longer reaction time was necessary (Table 2, entry 1). When acetone was employed as the solvent, this time, 85% yield was obtained after 9 hours (Table 2, entry 2). Switching solvent to water, under irradiation at 370 nm, led to a lower yield of isolated product (Table 2, entry 3). However, when the mixed solvent mixture of acetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (10% v/v) was employed, an excellent conversion was observed, leading to a 87% yield of isolated 2l, after base–acid extractions in a slightly decreased reaction time (7 h) at 370 nm (Table 2, entry 4). Extending the reaction time to 9 h did not lead to higher yields.25 We also tested the influence of the ratio of acetone:water to the reaction outcome, but none of them led to better results.25 Keeping constant the mixture of acetone–water as the optimum medium, we next evaluated LEDs of different wavelengths: 390 nm, 427 nm, 456 nm or 525 nm (Table 2, entries 5–8). In all cases, keeping the reaction time at 9 hours, except for the case of 390 nm, lower yields were observed (Table 2, entries 5–8), suggesting that 370 nm was the optimum wavelength. Interestingly, when the reaction was left for 8 h under sunlight, the product was obtained in 82% yield (Table 2, entry 9). However, we observed the formation of a mixture of acid:peracid (1:1). In order to fully convert the peracid to acid, we employed a simple post-reaction work-up that led to the isolation of pure acid 2l.25 We also observed a difference between performing the reaction in summer (approx. temp. 35 °C) vs. winter (approx. temp. 5 °C). In sharp contrast, when the reaction was performed at 370 nm, mainly acid 2l was observed. Monitoring the reaction by 13C-NMR, a very low amount of peracid was detected.25
:water (10% v/v) was employed, an excellent conversion was observed, leading to a 87% yield of isolated 2l, after base–acid extractions in a slightly decreased reaction time (7 h) at 370 nm (Table 2, entry 4). Extending the reaction time to 9 h did not lead to higher yields.25 We also tested the influence of the ratio of acetone:water to the reaction outcome, but none of them led to better results.25 Keeping constant the mixture of acetone–water as the optimum medium, we next evaluated LEDs of different wavelengths: 390 nm, 427 nm, 456 nm or 525 nm (Table 2, entries 5–8). In all cases, keeping the reaction time at 9 hours, except for the case of 390 nm, lower yields were observed (Table 2, entries 5–8), suggesting that 370 nm was the optimum wavelength. Interestingly, when the reaction was left for 8 h under sunlight, the product was obtained in 82% yield (Table 2, entry 9). However, we observed the formation of a mixture of acid:peracid (1:1). In order to fully convert the peracid to acid, we employed a simple post-reaction work-up that led to the isolation of pure acid 2l.25 We also observed a difference between performing the reaction in summer (approx. temp. 35 °C) vs. winter (approx. temp. 5 °C). In sharp contrast, when the reaction was performed at 370 nm, mainly acid 2l was observed. Monitoring the reaction by 13C-NMR, a very low amount of peracid was detected.25
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Light | Time (h) | Yielda (%) |
|
a Conversion determined by NMR or HPLC, while the reaction was monitored by HPLC. Yield of isolated product after base–acid wash in parenthesis.
b A mixture of acid:peracid in a ratio of 1:1 was obtained.
|
||||
| 1 | EtOAc | 370 nm | 9 | 83 |
| 2 | Acetone | 370 nm | 9 | 85 |
| 3 | H2O | 370 nm | 9 | (38) |
| 4 | Acetone + 10% v/v H 2 O | 370 nm | 7 | 87 (87) |
| 5 | Acetone + 10% v/v H2O | 390 nm | 9 | 86 |
| 6 | Acetone + 10% v/v H2O | 427 nm | 9 | 25 |
| 7 | Acetone + 10% v/v H2O | 456 nm | 9 | 28 |
| 8 | Acetone + 10% v/v H2O | 525 nm | 9 | 14 |
| 9 | Acetone + 10% v/v H2O | Sunlight | 8 | 82 (1:1)b |
| 10 | Acetone + 10% v/v H2O | Dark | 9 | <5% |
Light acceleration is evident (as in the case of dodecanal), since when the reaction was performed under identical conditions under dark, traces of acid were detected (<5% yield) (Table 2, entry 10). From the UV-Vis spectra of dodecanal or benzaldehyde at concentrations similar to those used for the synthetic experiments25 one can identify the absorbance of the reactants at UVA wavelengths (350–400 nm).
The substrate scope of the methodology is presented in Scheme 2. Saturated aliphatic aldehydes 1a and 1b, as well as those containing either an alicyclic (1c) or an aryl moiety on the aliphatic chain (1d), were converted into carboxylic acids in high yields ranging from 85% to 94%, when the reactions were irradiated with LED 370 nm. Similar yields were observed using sunlight as the source of irradiation. The oxidation proved to be selective, since aldehydes bearing a terminal triple bond (1e) or a terminal double bond (like 1f) afforded the desired carboxylic acids 2e and 2f, with no oxidation occurring on the triple or double bond. Lower yields were obtained for 2e and 2f, when the reaction was performed under sunlight. However, in the case of oleyl aldehyde (1g), both under 370 nm-mediated reaction and under sunlight, the desired oleic acid (2g) was obtained in yield around 30%. Also, an amount of the corresponding epoxide of the oleic acid was isolated, proving that more reactive towards oxidation internal double bonds are susceptible to oxidation. Moreover, 10-hydroxydecanal (1h) was converted into acid 2h in high yields (81% and 74%). Thus, aldehydes are oxidised selectively in the presence of primary aliphatic alcohols. In all these cases, no peracid formation was detected. When α,α-disubstituted aldehydes are employed under photochemical conditions or radical chemistry conditions, in general, the produced acyl radical can be fragmented to the corresponding secondary aliphatic radical with CO extrusion, which usually leads to lower yields.7,26 Herein, when the aldehyde group was at a secondary carbon atom, products 2i–k were produced in slightly lower yields (53–75%) under either LED 370 nm or sunlight. It is interesting to note that 2-ethyl-hexanoic acid (2j), which is industrially synthesised employing an aerobic oxidation process,3b can be produced in good yield (66%) at 370 nm. When the same reaction was performed under sunlight, the desired product was obtained in 53% yield. In both cases (370 nm or sunlight), the corresponding percarboxylic acid, [as well as 3-hydroxyheptane (from 3-heptyl radical and oxygen)], were produced. Again, using a simple work-up procedure, the peracid could be transformed into the desired acid.25 Thus, we can generalise, that in the cases of 2i–k, the slightly lower yields can be due to the above-mentioned fragmentation,26 while a mixture of acid:peracid was observed and it can be transformed to the desired acid, upon the appropriate treatment. Aromatic aldehydes were found to require longer reaction times, usually 5–8 hours. Benzaldehyde was converted to benzoic acid (2l) under LED 370 nm and sunlight in 87% and 74% yield, respectively. The presence of an electron donor or an electron acceptor group at the para position led to the production of acids in high yields. Compounds 2m–r were obtained in 62–95% yields, under LED 370 nm or sunlight. However, para-nitrobenzaldehyde (1s) proved to be problematic, under these conditions. Fortunately, extending the reaction time up to 96 h at 370 nm, the desired acid 2s could be obtained in 87% yield. Unfortunately, para-hydroxybenzaldehyde resulted in 2t in low yields (44% and 26%, respectively). 2-Naphthaldehyde required a long reaction time (26 h) to afford the acid in 77% yield under LED 370 nm. Under sunlight, acid 2u was obtained in 38% yield after 8 hours. Finally, heterocyclic aldehydes 1v and 1w were tested and afforded the desired acids in good to high yields, proving that no nitrogen or sulfur oxidation is occurring. Unfortunately, the easily oxidized furylaldehyde could not be employed. As in the cases of α,α-disubstituted aldehydes, some aromatic aldehydes in the sunlight-promoted reactions led to a mixture of peracid:acid, which could be transformed to the desired acid by work-up. Monitoring the 370 nm- or the sunlight-mediated processes of different aldehydes (primary aliphatic, α,α-disubstituted aliphatic or aromatic) by NMR,25 in some cases a mixture of acid:peracid was observed, while in other cases the amount of peracid was minimal.25 Thus, the amount of peracid generated is substrate dependent. It seems that in the cases of α,α-disubstituted aliphatic aldehydes, a mixture of acid:peracid is observed in both reaction conditions (370 nm or sunlight), while for primary aliphatic aldehydes, the amount of generated peracid is low in both cases. In aromatic aldehydes, the amount of peracid depends on the aldehyde structure and the conditions. It is of importance to notice that column chromatography is generally not necessary for the purification of the synthesized carboxylic acids. Simple extractions combined with base/acid wash may lead to the desired products in high purity.
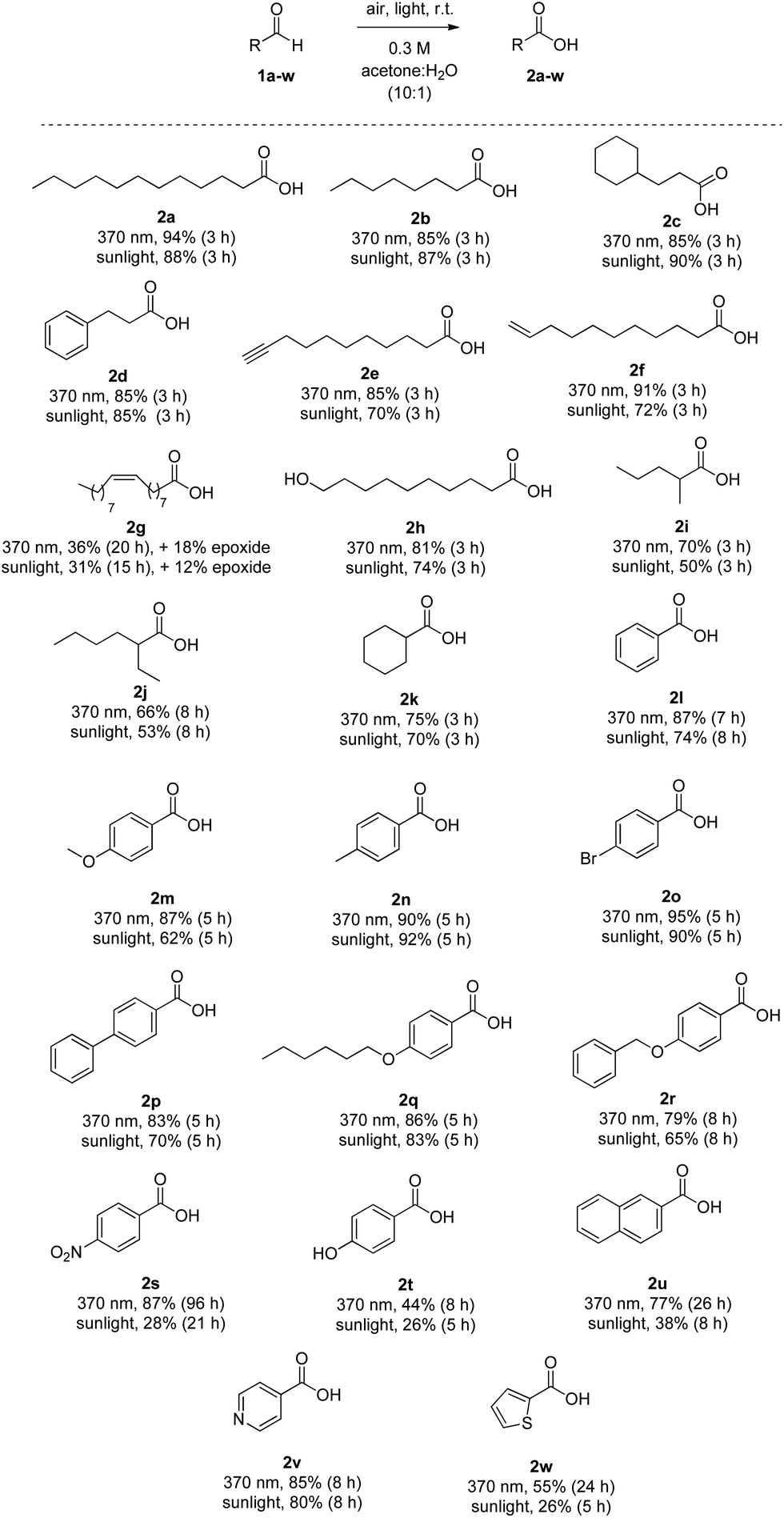 | ||
| Scheme 2 Substrate scope for aliphatic and aromatic aldehydes. | ||
A proposed reaction mechanism for this aerobic and light-promoted transformation is shown in Scheme 3. Based on previous reports on the catalyst-free aerobic oxidation of aldehydes to carboxylic acids,15–21 as well as our previous work on oxidative processes,27 the starting aldehyde is transformed to the corresponding acyl radical I in the presence of air. This process is spontaneous in the presence of air,16,17,27 while literature suggests an acceleration of the process in the presence of water.15–17,20,27 We have a long interest in photochemistry and we have been involved in the use of aldehydes as potential photoinitiators.23f,26b,28 Under irradiation and after aldehyde excitation, aldehydes can lead to radicals I and II, which are known to take part in various processes, usually involving Hydrogen Atom Transfer (HAT) events, either with different reagents or with oxygen.23f,26b,28 Thus, irradiating aldehydes with either UVA or solar irradiation, promotes the initial generation of radicals I and II, which now exist in higher concentration in the reaction mixture, compared to non-irradiated conditions. In order to strengthen and support these hypotheses by experimental data, the course of the oxidation of benzaldehyde was monitored by DI-HRMS.27,29 Selected HRMS spectra are shown in Scheme 3 (see ESI‡ for full HRMS data).25 Furthermore, we studied the trapping of acyl radical I with Tempo, using LC-HRMS25 and we quantified the significant differences in concentration of the adduct under light vs. dark reaction conditions.25 Once radical I is formed, it reacts with oxygen from air and forms peroxy radical III (Scheme 3). Hydrogen atom transfer (HAT) either from another molecule of aldehyde, generating I, or from the solvent leads to peracid IV, which was verified by HRMS and observed under sunlight conditions (Scheme 3). Then, IV reacts with another molecule of aldehyde, leading to Criegee intermediate V,17–19,30 which may collapse and afford two molecules of acid (Scheme 3). The presence of Criegee intermediate V was verified by HRMS (Scheme 3). Herein, we also observed that the use of a solvent mixture of acetone:water further accelerates this process (vs. other organic solvents or non-irradiated conditions). These claims are supported by the results of Table 1, entries 7–10 vs. 11–15 and entries 3 and 4 vs. entries 1 and 2, respectively. Furthermore, monitoring the full scan spectrum of the reaction mixture, we observed after 1 hour the generation of a peak at m/z 219.0625, whose intensity became much stronger after 3 or 4 hours of reaction (Fig. 1).25 This unexpected observation prompted us to explore an additional parallel mechanistic pathway. Using Smart Formula application of Data Analysis from Bruker Daltonics (version4.1), this measured exact mass was found to correspond to C10H12NaO4 (excellent mass error and isotopic fit) (Fig. 1). Thus, it seems that when acetone is employed as the solvent, then a second possible pathway is in place. Peracid IV can react with acetone, instead of a molecule of the starting aldehyde, forming the mixed Criegee intermediate VI (Scheme 3). The generation and the presence of VI were verified by HRMS (Scheme 3 and Fig. 1).25 Intermediate VI can collapse affording a molecule of the product acid, but at the same time a molecule of dimethyl dioxirane VII is also generated (Scheme 3). The presence of dioxirane was also verified by HRMS (Scheme 3).25 However, dioxiranes are known to promote the oxidation of aldehydes to carboxylic acids.31 To confirm these results, the oxidation of benzaldehyde was studied by DI-HRMS in a mixture of deuterated acetone-d6:water (10% v/v). Ions corresponding to mixed Criegee intermediate with deuterated acetone-d6 and deuterated-d6 dioxirane were again observed, in addition to peracid and Criegee intermediate with aldehyde (ESI, Fig. S4‡). This unprecedented finding, which was identified after careful monitoring and examination of the HRMS spectra, reveals the generation of an acetone-mixed Criegee intermediate and dioxirane oxidant under aerobic and visible light irradiation. These findings, that are proposed for the first time, can explain the acceleration of the reaction in the presence of acetone. In addition, in recent literature, it is suggested that the formation of the Criegee intermediate V is a slow process and alternative pathways are proposed for the generation of acid under non-irradiating conditions, however, we could not detect such intermediates in our study.10b,32,33 Thus, fast trapping of IV by acetone to VI could explain the faster formation of acid. Collectively, the study of the reaction mechanism by DI-HRMS suggests a solvent-accelerating process, which involves a dioxirane oxidant in situ generated under these reaction conditions, highlighting the importance of DI-HRMS as a tool to study organic chemistry reactions. In addition, such in-depth mechanistic studies and valuable insights on the generation of particular intermediates may provide inspiration and directions for future investigations. Furthermore, radical intermediate II can also react with oxygen, leading to radical VIII, which similarly can react with either the aldehyde, leading to IX (a HAT event is also taking place with an aldehyde, affording acyl radical I) or acetone, leading to XI (a HAT event is also taking place with an aldehyde, affording acyl radical I). In both cases, IX and XI are collapsing finally to the desired product and X or VII. Intermediates IX, X and XI were also detected by HRMS.
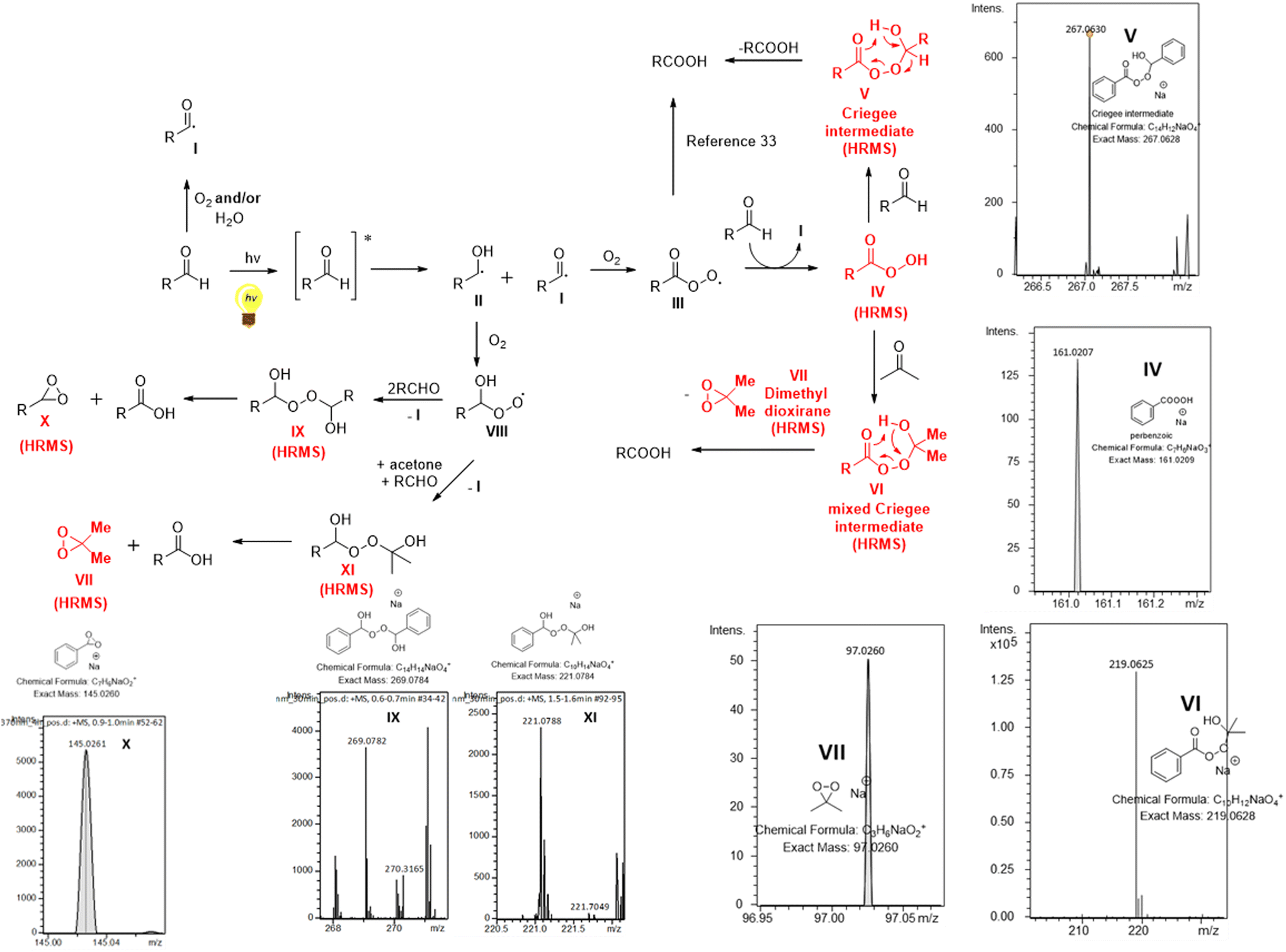 | ||
| Scheme 3 Proposed mechanism for the oxidation of aldehydes to carboxylic acids in acetone/water and selected HRMS spectra of the oxidation of benzaldehyde, verifying the generation of peracid IV, Criegee intermediate V, mixed Criegee intermediate with acetone VI, dimethyl dioxirane VII and intermediates IX, X and XI. | ||
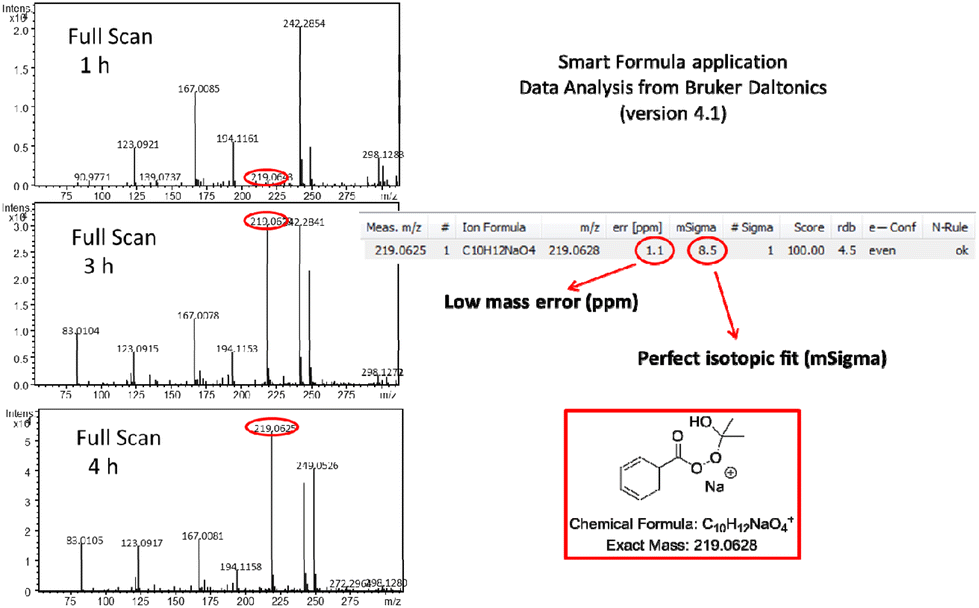 | ||
| Fig. 1 Monitoring the oxidation of benzaldehyde by DI-HRMS. Full scan HRMS spectra taken at 1 h, 3 h and 4 h, verifying the generation of mixed Criegee intermediate with acetone (m/z 219.0628). | ||
Conclusions
In conclusion, atmospheric oxygen, a clean and sustainable oxidant, was proven to be an efficient oxidative agent merged with solar or UVA-light irradiation. Both the irradiation sources LED 370 nm and sunlight, were found efficient to promote the oxidative transformation of aldehydes to carboxylic acids in short reaction time (3 to 8 hours), affording the products in high to excellent yields. Someone may use sunlight (depending on the availability of sun) or a cheap LED lamp for this very mild and operational simple transformation. Thus, a convenient, green and sustainable method for the oxidation of aldehydes to carboxylic acids has been developed.Experimental section
General
Kessil lamps PR160L were used as the irradiation source (https://kessil.com/science/PR160L.php). For all experiments, the intensity of Kessil lamps was controlled in the maximum level with power consumption: 370 nm (max 43 W), 390 nm (max 52 W), 427 nm & 440 nm (max 45 W), 456 nm (max 50 W), 467 nm (max 40 W), 525 nm (max 44 W).General procedure for the photochemical aerobic catalyst-free oxidation of aldehydes to carboxylic acids
In a test tube containing acetone (3 mL) and H2O (0.3 mL, HPLC grade), aldehyde (1.00 mmol) was added and the reaction mixture was left stirring under irradiation (370 nm or sunlight) for 3–96 h. The reaction mixture was evaporated to give a crude mixture, which was diluted in EtOAc (10 mL). Then, the crude reaction mixture was treated with an aqueous solution of NaOH 1 N (10 mL) and the aqueous layer was extracted with EtOAc (2 × 10 mL). The aqueous basic phase was then acidified with an aqueous solution of HCl 1 N (until pH = 1) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with H2O (20 mL) and dried over Na2SO4. The solvent was removed in vacuo to afford the desired carboxylic acid without the need for further purification. Carboxylic acids 2m, 2p and 2r were isolated by trituration with petroleum ether (4–6 mL) (cooled with external ice bath). Compounds 2q and 2g were purified by flash chromatography on silica gel eluting with petroleum ether (bp 40–60 °C)/ethyl acetate 7/3.General procedure for the conversion of peracid-containing mixture to the corresponding carboxylic acid
For the reactions where a mixture of peracid:acid was detected, after irradiation, the reaction mixture was diluted with acetone:H2O (3 mL:9 mL) and stirred at 40 °C (water bath) for 3 h. The solvent was evaporated to give a crude mixture, which was diluted in EtOAc (10 mL). Then, the crude reaction mixture was treated with an aqueous solution of NaOH 1 N (10 mL) and the aqueous layer was extracted with EtOAc (2 × 10 mL). The aqueous basic phase was then acidified with an aqueous solution of HCl 1 N (until pH = 1) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with H2O (20 mL) and dried over Na2SO4. The solvent was removed in vacuo to afford the desired carboxylic acid without the need for further purification.
Author contributions
G. K. and C. G. K. conceived the study, designed the experiments and analysed the data. C. S. B., C. K. and G. S. K. performed the synthesis, optimization experiments and substrate scope and M. G. K. the HRMS studies. M. G. K., G. K. and C. G. K. wrote the first draft and G. K. and C. G. K. edited the final version of the manuscript. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research presented was carried out within the framework of a Stavros Niarchos Foundation grant to the National and Kapodistrian University of Athens (G. K.). C. G. K. gratefully acknowledge the Hellenic Foundation for Research and Innovation (HFRI) for financial support through a grant, which is financed by 1st Call for H.F.R.I. Research Projects to Support Faculty Members & Researchers and the procurement of high-cost research equipment grant (grant number 655). M. G. K. would like to thank State Scholarships Foundation (IKY) for financial support through a post-doctoral research scholarship funded by the “Supporting Post-Doctoral Researchers-Call B” (MIS 5033021), action of the Operational Programme “Human Resources Development, Education and Lifelong Learning” and co-funded by the European Social Fund (ESF) and Greek National Resources.Notes and references
- For selected textbooks, see: (a) M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, Hoboken, 6th edn, 2007 Search PubMed; (b) J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, Oxford, 2nd edn, 2012 Search PubMed; (c) D. Klein, Organic Chemistry, Wiley, Hoboken, 4th edn, 2020 Search PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed.
- (a) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS PubMed; (b) J. Kubitschke, H. Lange and H. Strutz, Carboxylic Acids, Aliphatic, in Ullmann's Encyclopedia of Industrial Chemistry, 2014, pp. 1–18 Search PubMed. For a recent chapter of a book highlighting green methods, see: (c) M. Liu and C.-J. Li, Green Oxidative Synthesis of Carboxylic Acids, Wiley, 2019, pp. 159–180 Search PubMed.
- For selected reviews, see: (a) J. R. McNesby and C. A. Heller Jr., Chem. Rev., 1954, 54, 325 CrossRef CAS; (b) N. Gunasekaran, Adv. Synth. Catal., 2015, 357, 1990 CrossRef CAS; (c) X. Zhang, K. P. Rakesh, L. Ravindar and H.-L. Qin, Green Chem., 2018, 20, 4790 RSC; (d) C. A. Hone and O. C. Kappe, Top. Curr. Chem., 2019, 377, 2 CrossRef PubMed.
- M. Liu and C.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 10806 CrossRef CAS PubMed.
- H. Yu, S. Ru, G. Dai, Y. Zhai, H. Lin, S. Han and Y. Wei, Angew. Chem., Int. Ed., 2017, 56, 3867 CrossRef CAS PubMed.
- D. R. Larkin, J. Org. Chem., 1990, 55, 1563 CrossRef CAS.
- X. Yan, Y.-H. Lai and R. N. Zare, Chem. Sci., 2018, 9, 5207 RSC.
- A. K. Khatanaa, V. Singh, M. K. Gupta and B. Tiwari, Synthesis, 2018, 50, 4290 CrossRef.
- (a) P.-F. Dai, J.-P. Qu and Y.-B. Kang, Org. Lett., 2019, 21, 1393 CrossRef CAS PubMed; (b) L. Vanoye, M. Abdelaal, K. Grundhauser, B. Guicheret, P. Fongarland, C. De Bellefon and A. Favre-Réguillon, Org. Lett., 2019, 21, 10134 CrossRef CAS PubMed.
- (a) M. Hajimohammadi, N. Safari, H. Mofakham and A. Shaabani, Tetrahedron Lett., 2010, 51, 4061 CrossRef CAS; (b) M. Hajimohammadi, H. Mofakham, N. Safari and A. Mortazavi Manesh, J. Porphyrins Phthalocyanines, 2012, 16, 93 CrossRef CAS.
- N. Iqbal, S. Choi, Y. You and E. J. Cho, Tetrahedron Lett., 2013, 54, 6222 CrossRef CAS.
- Z. E. Hamami, L. Vanoye, P. Fongarland, C. de Bellefon and A. Favre-Reguillon, J. Flow Chem., 2016, 6, 206 CrossRef CAS.
- C. Lehtinen, V. Nevalainen and G. Brunow, Tetrahedron, 2000, 56, 9375 CrossRef CAS.
- N. Shapiro and A. Vigalok, Angew. Chem., Int. Ed., 2008, 47, 2849 CrossRef CAS PubMed.
- N. Shapiro, M. Kramer, I. Goldberg and A. Vigalok, Green Chem., 2010, 12, 582 RSC.
- V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301 RSC.
- L. Vanoye, A. Favre-Réguillon, A. Aloui, R. Philippe and C. Bellefon, RSC Adv., 2013, 3, 18931 RSC.
- L. Vanoye, A. Aloui, M. Pablos, R. Philippe, A. Percheron, A. Favre-Réguillon and C. Bellefon, Org. Lett., 2013, 15, 5978–5981 CrossRef CAS PubMed.
- Y. Zhang, Y. Cheng, H. Cai, S. He, Q. Shan, H. Zhao, Y. Chen and B. Wang, Green Chem., 2017, 19, 5708 RSC.
- K.-J. Liu, Y.-L. Fu, L.-Y. Xie, C. Wu, W.-B. He, S. Peng, Z. Wang, W.-H. Bao, Z. Cao, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 4916 CrossRef CAS.
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (c) K. L. Scubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef PubMed; (d) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef PubMed; (e) S. Reischauer and B. Pieber, iScience, 2021, 24, 102209 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed; (b) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC; (c) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (e) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596 RSC; (f) M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833 CrossRef CAS PubMed; (g) N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Org. Biomol. Chem., 2021, 19, 5237 RSC.
- J. R. McNesby and T. W. Davis, J. Am. Chem. Soc., 1954, 76, 2148 CrossRef CAS.
- For detailed mechanistic studies and results, see ESI.‡.
- (a) G. N. Papadopoulos, E. Voutyritsa, N. Kaplaneris and C. G. Kokotos, Chem. – Eur. J., 2018, 24, 1726 CrossRef CAS PubMed; (b) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, ChemSusChem, 2019, 12, 4194 CrossRef CAS PubMed; (c) E. Voutyritsa and C. G. Kokotos, Angew. Chem., Int. Ed., 2020, 59, 1735 CrossRef CAS PubMed; (d) N. Spiliopoulou, P. L. Gkizis, I. Triandafillidi, N. F. Nikitas, C. S. Batsika, A. Bisticha and C. G. Kokotos, Chem. – Eur. J., 2022, 28, e202200023 CrossRef CAS PubMed.
- I. Triandafillidi, M. G. Kokotou, D. Lotter, C. Sparr and C. G. Kokotos, Chem. Sci., 2021, 12, 10191 RSC.
- N. F. Nikitas, M. A. Theodoropoulou and C. G. Kokotos, Eur. J. Org. Chem., 2021, 1168–1173 CrossRef CAS.
- (a) I. Triandafillidi, M. G. Kokotou and C. G. Kokotos, Org. Lett., 2018, 20, 36 CrossRef CAS PubMed; (b) E. Voutyritsa, A. Theodorou, M. G. Kokotou and C. G. Kokotos, Green Chem., 2017, 19, 1291 RSC.
- (a) Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Bräse, Angew. Chem., Int. Ed., 2021, 60, 15138 CrossRef CAS PubMed. This Criegee intermediate is not to be confused with the Criegee zwitterion formed in alkene ozonolysis: (b) J. Criegee, Justus Liebigs Ann. Chem., 1948, 560, 127 CrossRef.
- (a) A. L. Baumstark, M. Beeson and P. C. Vasquez, Tetrahedron Lett., 1989, 30, 5567 CrossRef CAS; (b) R. Curci, A. Dinoi and M. F. Rubino, Pure Appl. Chem., 1995, 67, 811 CrossRef CAS.
- B. S. Nagy, O. C. Kappe and S. B. Ötvös, Adv. Synth. Catal., 2022, 364, 1998 CrossRef CAS.
- L. Vanoye and A. Favre-Réguillon, Org. Process Res. Dev., 2022, 26, 335 CrossRef CAS.
Footnotes |
| † First manuscript submission date: August 24th, 2021. |
| ‡ Electronic supplementary information (ESI) available: Experimental data, optimisation studies, mechanistic and HRMS studies. See DOI: https://doi.org/10.1039/d2gc02074b |
| § Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |