
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-demand continuous H2 release by methanol dehydrogenation and reforming via photocatalysis in a membrane reactor†
Haimiao
Jiao
 a,
Jianlong
Yang
b,
Xiyi
Li
a,
Chao
Wang
a and
Junwang
Tang
*a
a,
Jianlong
Yang
b,
Xiyi
Li
a,
Chao
Wang
a and
Junwang
Tang
*a
aSolar Energy and Advanced Materials Research Group, Department of Chemical Engineering, University College London, London, WC1E 7JE, UK. E-mail: junwang.tang@ucl.ac.uk
bKey Lab of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, The Energy and Catalysis Hub, College of Chemistry and Materials Science, Northwest University, Xi'an, P. R. China
First published on 30th September 2022
Abstract
Photocatalytic methanol dehydrogenation and reforming is viewed as a promising strategy to realize H2 production on demand. Herein, we report highly dispersed CuxO nanoparticles on TiO2 (PC50) for continuous H2 production from aqueous methanol solution by photocatalysis in a flow reactor at a low temperature and under atmospheric pressure. The flow membrane reactor improves the H2 production rate by a factor of 1.63 compared with the widely used batch reactor thanks to enhanced mass transfer. Furthermore, the optimized 1% Cu/PC50 exhibits a 17-times higher H2 yield (33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 702 μmol g−1 h−1) than pristine PC50. The apparent activation energy on 1% Cu/PC50 is found to be halved to as low as 4.0 kJ mol−1, which is much less than those in other methanol reforming processes. The diverse characterisation proposes that CuxO as electron acceptors could effectively promote charge separation and work as active sites for the reduction reaction, together with the improved mass transfer in the reactor leading to enhanced photocatalytic performance.
702 μmol g−1 h−1) than pristine PC50. The apparent activation energy on 1% Cu/PC50 is found to be halved to as low as 4.0 kJ mol−1, which is much less than those in other methanol reforming processes. The diverse characterisation proposes that CuxO as electron acceptors could effectively promote charge separation and work as active sites for the reduction reaction, together with the improved mass transfer in the reactor leading to enhanced photocatalytic performance.
1. Introduction
Hydrogen has widely been viewed as a vital chemical raw material in many fields such as ammonia synthesis, petroleum processing, metallurgical industry and so on.1 More importantly, hydrogen is regarded as the most promising clean energy to effectively mitigate the greenhouse gas effect.2 However, in the traditional hydrogen production processes, harsh reaction conditions are usually required. Nowadays, 96% of hydrogen is produced from the steam reforming process using fossil fuels (e.g. methane), which not only requires a high temperature (700–1000 °C) and pressure (3–25 bar), but also emits substantial CO and CO2.3 Moreover, due to the unfavorable physical (e.g., low critical temperature of 33 K and low energy density) characteristics,4 its storage and transport are difficult using the current infrastructure.5 Thus, it is of great significance to explore environmentally friendly and economical strategies for continuous hydrogen production onsite and on demand.Methanol, a liquid at room temperature with a large H2 storage capacity of 12.6 wt%, is one of the best liquid H2 storage media, which can be stored and transported using the current infrastructure and more importantly can potentially release H2 on demand.6 Thus, methanol economy has been long proposed by George A. Olah.7 Methanol dehydrogenation was reported by steam reforming in the presence of heterogeneous catalysts (e.g. CuO/ZnO/Al2O3) at a relatively high temperature (200–300 °C).8 Notably, a Pt/Al2O3-based catalyst was recently reported to achieve aqueous-phase reforming of methanol at a relatively lower temperature (200–225 °C) and pressure (25–50 bar).9 Using renewable energy to release H2 from methanol aqueous solution at low temperature is more preferable in particular without the emission of CO2.
Photocatalysis is a promising and feasible technology for sustainable H2 production from methanol and water by taking advantage of the renewable and abundant solar energy. Compared with the thermocatalytic steam reforming of methanol process, photocatalysis can effectively reduce the apparent activation energy of reactions, achieving H2 production under very moderate reaction conditions.10 To date, many efforts have been made for photocatalytic H2 production from methanol in the aqueous phase using batch reactors, but with very moderate efficiency caused by the following reasons. First, the fast recombination of photoinduced charge carriers in the semiconductors severely retards the quantum efficiency, thus leading to a poor H2 yield.11 Furthermore, the limited mass transfer in the batch reactor also restricts its photocatalytic efficiency.12 Finally, the limited light penetration depth in the batch reactor greatly restricts the light absorption and utilization of photocatalysts.13
In this study, the highly dispersed CuxO nanoparticles were prepared on TiO2 (PC50) by the molten salt method (MSM). Together with the designed flow membrane reactor, photocatalytic methanol dehydrogenation and reforming was investigated. The uniform distribution of CuxO exposes abundant active sites for H2 production and charge separation, together with the improved mass transfer in a flow membrane reactor, the optimal 1% Cu/PC50 shows a superior H2 yield of 25487 μmol g−1 h−1, which is 13 times higher than that (1934 μmol g−1 h−1) of pristine PC50. This performance also remarkably surpasses those of other reported photocatalysts used in methanol reforming or dehydrogenation. The apparent activation energy (4.0 kJ mol−1) on 1% Cu/PC50 is much smaller than that either on the PC50 photocatalyst or by thermocatalytic processes (e.g., 82.9 kJ mol−1 on Pt/α-MoC).14 The possible reaction mechanism was further studied by photoluminescence (PL), photocurrent response and in situ X-ray photoelectron spectroscopy (XPS).
2. Materials and methods
2.1. Synthesis of CuxO/TiO2
Copper chloride dihydrate (CuCl2·2H2O, 99.0%) from Sigma-Aldrich, anhydrous lithium chloride (LiCl, 99.0%) from Acros Organics and potassium chloride (KCl, analysis grade) from Merck KGaA were purchased and used without any further purification. Commercial titanium dioxide (anatase TiO2, PC50) was ordered from Millennium and used directly. Methanol (99.8%, Fisher Scientific) and deionized water (>15 MΩ) were utilized in this work.A series of CuxO decorated TiO2 was prepared by a modified molten-salt method (MSM).15 In a typical procedure, 26.6 mg of CuCl2·2H2O, 0.9 g of LiCl, 1.0 g KCl and 1.0 g of PC50 were ground in a mortar for 30 min to obtain a homogeneous mixture. Next, the mixture was transferred into an alumina crucible, which was then placed into a tube furnace and calcined at 773 K for 2 hours under an argon gas atmosphere. After cooling to room temperature, the mixture was pestled and washed with deionized water 5 times to remove the excess fluxing agent (LiCl and KCl). Then, the obtained sample was dried in an oven at 343 K overnight. The samples synthesized by MSM with different loading weight percentages of copper were named xCu/TiO2 (x = 0.05, 0.1, 0.5, 1, 2 wt%). For a comparison, PC50 without Cu loading under the same MSM method treatment was named PC50 (MSM).
2.2. Photocatalytic activity test
The photocatalytic methanol dehydrogenation and reforming in the two-phase system was investigated in a flow system (Fig. 1), which consisted of a reactant vessel, a membrane reactor and a product collector. The detailed information and the photograph of the flow membrane reactor are presented in the ESI and Fig. S1.† Generally, 10 mg of photocatalyst was dispersed onto a glass fiber membrane by a vacuum-filtering method to prepare a robust and uniform film. After vacuum-filtering, the as-prepared membrane was dried at 70 °C for 12 h. Then, the membrane was incorporated into the membrane reactor for the following reaction. The ratio of gaseous reactants (water vapor and methanol gas) introduced into the reactor was adjusted by the flow rate of argon via a mass flow controller (MFC, Bronkhorst) and the concentration of methanol solution in the reactant vessel. Before each run, the integrated system was purged with argon at a fixed flow rate under dark conditions to achieve the adsorption–desorption equilibrium. Then, the membrane reactor was irradiated with a 300 W xenon lamp (Beijing Perfect Light). The gaseous products were collected in the product collector and measured at a regular interval (1 h). One GC (Varian 430) equipped with thermal conductivity detectors (TCD) was used for the quantification of H2. Another GC (Varian 450) equipped with a methanizer and a flame ionization detector (FID) was used for the quantification of methanol, CO and CO2.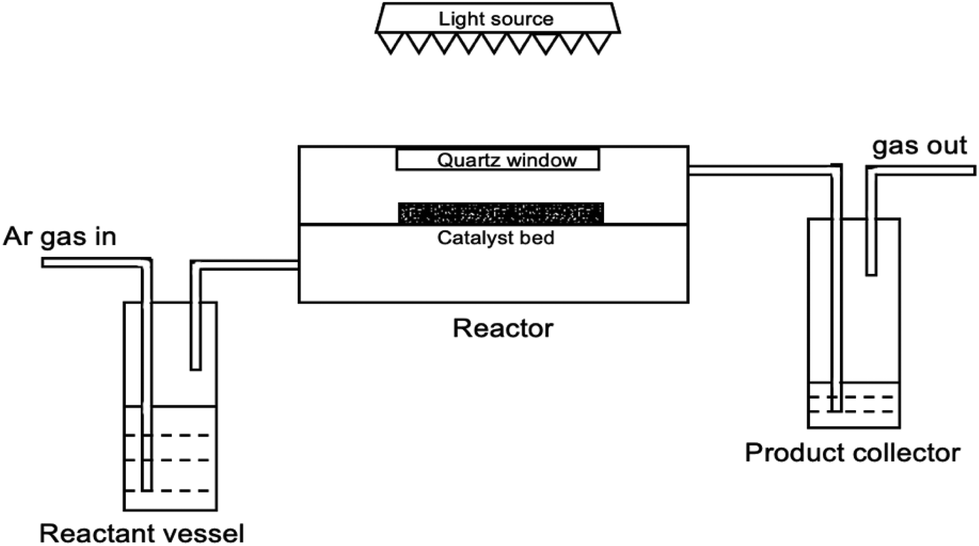 | ||
| Fig. 1 The diagram of a flow membrane reactor for photocatalytic methanol dehydrogenation and reforming. | ||
In order to detect the produced formaldehyde and formic acid, 10 mL of deionized water in the gas collector was used to capture all soluble byproducts. In detail, the concentration of HCHO was quantified by the colorimetric method with the Nash reagent.16 Typically, 100 mL of fresh Nash reagent was prepared by dissolving 30.0 g of ammonium acetate and 0.4 mL of acetylacetone in deionized water. Then, 50 μL of liquid product was mixed with 5.0 mL of water and 2 mL of Nash reagent, followed by maintaining at 60 °C in a water bath for 30 min. The absorption was measured at 412 nm with a UV-Vis spectrophotometer (Shimadzu, UV-2550). Besides, the concentration of HCOOH was analyzed by ion chromatography (Echo IC). Typically, 1 mL of solution in the product collector was taken and injected into the ion chromatography system directly. The retention time of the formate ion was measured at 6.66 min.
For a comparison, the photocatalytic methanol dehydrogenation and reforming were carried out in a batch reactor. Generally, 10 mg of the fresh photocatalyst was dispersed in 20 mL of methanol aqueous solution (50 vol%) in a quartz reactor. Then, the batch reactor was sealed and purged with argon for 30 min in the dark before irradiation under a 300 W xenon lamp. During this experiment, the batch reactor was maintained at 30 °C in a water bath. The products in the batch reactor for the following comparison were collected in the first hour run when the photocatalyst still remained active. The remaining testing procedures were identical to the measurements in the flow system. Before the quantification of HCHO and HCOOH, a specified amount of reactant solution was collected and filtered to remove the photocatalysts.
2.3 Photo-electrochemical measurements
The photoelectrode was prepared by a dispersion method.17 The detailed procedure is described as follows: 20 mg of TiO2 or Cu-modified TiO2 was dispersed in a mixture solution containing 4 mL of water, 1 mL of ethanol and 400 μL of Nafion (5% solution). Then, the mixture was sonicated for 1 h to obtain a homogeneous solution. Next, 50 μL of this uniform solution was dropped onto a FTO and dried at 80 °C on the hot plate to increase their adhesion, and the procedure was repeated three times totally. The area of samples coated on FTO was around 1 cm2.All tests were carried out with a three-electrode system in an electrochemical workstation (IVIUM) where the FTO electrode coated with catalysts was used as the working electrode, the Pt plate and Ag/AgCl were used as the counter electrode and reference electrode in a 0.1 M Na2SO4 electrolyte, respectively. A 150 W xenon lamp was used as a simulated light source and the interval switching time was set at 10 s. The chronoamperometry (CA) measurements were conducted in a 10 vol% methanol solution (pH = 7) at an applied potential of 0.10 V.
2.4 Materials characterization
The powder X-ray diffraction (PXRD) patterns were obtained on a Stoe STADI-P instrument equipped with a Mo source (Kα1 = 0.70930 Å). The UV-vis DRS spectra were recorded on a Shimadzu UV-2550 UV-vis spectrophotometer with a diffuse reflectance unit. Steady-state PL spectra were collected on an F4500 spectrofluorometer with a 325 nm excitation laser. The Raman spectra were obtained on a DXR 2SXR2 instrument (Thermo Fisher Scientific, Co., Ltd). In situ XPS spectra were collected on a PHI 5000 VersaProbe III instrument. HRTEM was conducted on a Talos F200X instrument (FEI Co., Ltd). SEM was performed on a Hitachi SU-8010 instrument. The ICP-AES was conducted on the MP-AES instrument (Agilent 4100).3. Results and discussion
3.1 Photocatalytic performance
The photocatalytic activity for methanol dehydrogenation and reforming over the as-prepared samples was evaluated in the flow membrane reactor (Fig. 1) under 300 W Xe light irradiation (T: 303 K, P: 1 atm), and the results are shown in Fig. 2a. The pristine PC50 without any cocatalyst shows a small amount of H2 (1934 μmol g−1 h−1) from methanol aqueous solution, probably due to the high recombination rate of photoinduced electrons and holes and a lack of active reaction sites. Besides, the MSM treatment does not affect its activity. It should be noted that the introduction of copper significantly increases the H2 yield rate, from 1934 μmol g−1 h−1 to 25487 μmol g−1 h−1 with the Cu content from 0 wt% to 1 wt%, reaching the highest yield rate of 25487 μmol g−1 h−1 at 1 wt%. This is around 13 times higher than that of pristine PC50. The enhanced photocatalytic methanol dehydrogenation and reforming for H2 production over 1 wt% Cu/PC50 is due to both the increased charge separation and catalytic effect by copper species, which would be discussed later. Besides, the highly dispersive fine CuxO nanoparticles could also expose a large number of active sites, thereby resulting in enhanced performance.18 However, the excessive loading of Cu (e.g. 2 wt% Cu/PC50) might have blocked light harvesting, leading to decreased photoactivity.19
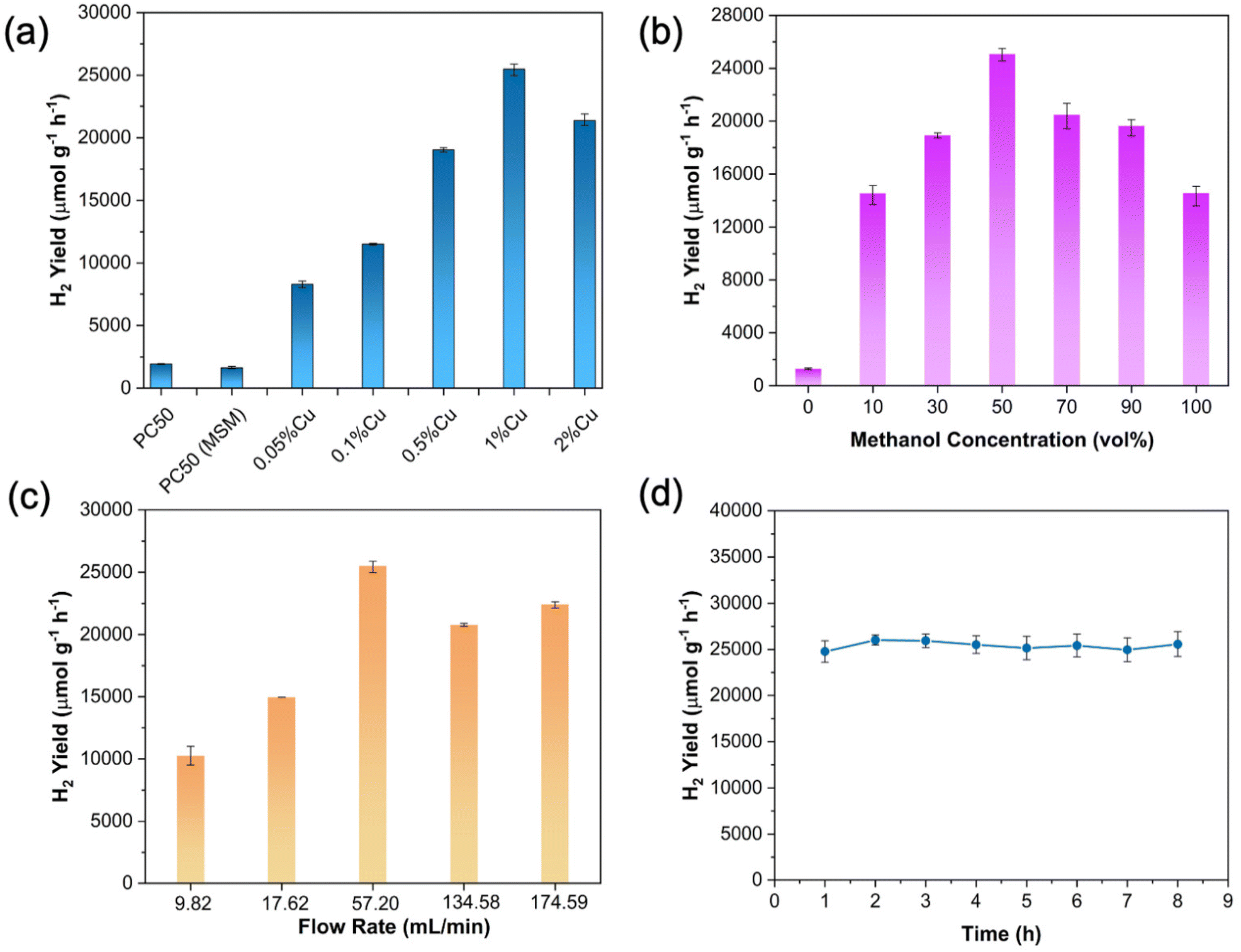 | ||
| Fig. 2 (a) H2 yield over different Cu wt% on PC50 (experimental conditions: 50 vol% of methanol aqueous solution, 57.20 ml min−1 of argon gas). H2 yield from methanol by photocatalysis over 1% Cu/PC50 with (b) different methanol concentrations at a carrier gas flow rate of 57.20 ml min−1 and (c) different flow rates of argon gas with 50 vol% of methanol aqueous solution. (d) Stability test for photocatalytic methanol dehydrogenation and reforming on 1% Cu/PC50. | ||
To further optimize the reaction conditions in the flow membrane reactor, the effects of the concentration of methanol in the feedstock and the flow rate of argon as a carrier gas were then investigated on the optimal sample 1% Cu/PC50 (Fig. 2b and c). The H2 yield rate exhibits a volcanic profile with the increasing volume ratio of methanol in the feedstock and reaches the maximum H2 yield (25487 μmol g−1 h−1) with 50 vol% of methanol in the aqueous solution (Fig. 2b). At a low concentration of methanol, the H2 production rate is primarily restricted by the mass transfer of methanol from the gas phase to the catalyst surface in the flow membrane reactor. However, with over 50 vol% of methanol in the feedstock, the H2 yield decreases significantly. The possible reason was that the presence of water in the reactant mixture could facilitate the desorption of the oxidation products of methanol.20,21 Less water would reduce the desorption of oxidation products from the surface of photocatalysts, thus resulting in a decreased photocatalytic activity. It is worth noting that the H2 yield is only 14565 μmol g−1 h−1 with 100 vol% of the methanol in the reactant vessel, which is approximately half of H2 produced with 50 vol% of the methanol in the reactant vessel. This indicates that only photocatalytic methanol dehydrogenation shows less efficiency for H2 production compared to the integration of photocatalytic methanol reforming and dehydrogenation. This also indicates the importance of the involved water vapor towards increasing the H2 yield in the flow membrane reactor. In addition, a small amount of H2 is yielded over 1% Cu/PC50 using pure water as a feedstock because water is much more difficult to be oxidized compared to methanol.22 Furthermore, the effects of flow rates (from 9.82 to 174.59 mL min−1) on H2 yield were then investigated (Fig. 2c). The flow rate of argon gas has a significant influence on the mass transfer in the flow membrane reactor as the reactants (methanol gas and water vapor) were carried into the reactor chamber by argon gas. The hydrogen yield increases along with the increase of the argon flow rate (from 9.82 to 57.20 mL min−1). The hydrogen production rate is in part dominated by the mass transfer of gaseous reactants to the catalyst surface. The methanol molar fraction in the methanol–water gas phase with different argon flow rates at the inlet of the flow membrane reactor was determined by the detailed procedure as described in the ESI and Fig. S2.† It shows that the methanol molar fraction in the methanol–water gas phase could be greatly increased from 52.3 to 66.7% with the increasing argon flow rate from 9.82 to 17.62 ml min−1 (Fig. S3†). Besides, the methanol molar fraction remains relatively stable while further increasing the argon flow rate. This indicates that the improved H2 yield with the rising argon flow rate from 9.82 ml min−1 to 17.62 ml min−1 might be caused by the increased methanol molar fraction in the methanol–water gas phase and the enhanced mass transfer between gaseous reactants and photocatalysts. When the flow rate is further increased, the H2 production rate is principally dominated by the mass transfer of gaseous reactants to the catalyst surface. Higher flow rates and faster mass transfer, leads to a higher H2 production rate. On further increasing the flow rate, the hydrogen production rate somewhat remains similar since the quantities of gaseous reactants adsorbed on the catalyst surface reached saturation.23 Combined with previous investigations, the optimum experimental conditions have been achieved, including 50 vol% of methanol in the feedstock and a 57.20 ml min−1 flow rate, which were used for the further studies. Under the optimal reaction conditions, the molar ratio of methanol to water in the gas phase at the inlet of the flow membrane reactor was determined to be 2.8:1 and the detailed procedure is described in the ESI and Fig. S2.†
To investigate the reason for such a high H2 production rate on 1% Cu/PC50, the reaction kinetics of the photocatalytic methanol dehydrogenation and reforming over PC50 and 1% Cu/PC50 under 300 W Xe light irradiation at different reaction temperatures were studied experimentally. The derived Arrhenius equation (lnr = −Ea/RT + C) was applied to calculate the apparent activation energy (Ea) by fitting the measured hydrogen production rate (r), where C is a constant, as shown in Fig. S4.† The apparent activation energy on 1% Cu/PC50 is 4.0 kJ mol−1, which decreases by 50% as compared to that on pure PC50 (8.0 kJ mol−1), demonstrating that the introduction of copper species could greatly reduce the barrier to photocatalytic activation of methanol. Notably, the apparent activation energy obtained over 1% Cu/PC50 by photocatalysis in the flow system is much less than that reported in the thermo-catalytic (82.9 kJ mol−1 on Pt/α-MoC)14 or photothermo-catalytic (53.6 kJ mol−1 on ZnCu alloy)24 methanol reforming processes, indicating that methanol could be activated more efficiently on our catalyst in the flow system. We also calculated the apparent quantum efficiency (AQE) based on the photocatalytic methanol dehydrogenation and reforming over 1% Cu/PC50, which is 7.75% at 365 nm (the detailed calculation steps are described in the ESI†).
The stability test over 1% Cu/PC50 was then conducted in the flow membrane reactor. No significant decay of H2 yield is observed except a slight fluctuation over 1% Cu/PC50 during an 8-hour continuous run, confirming the highly stable activity (Fig. 2d). To investigate the influence of mass transfer on the photocatalytic methanol dehydrogenation and reforming, the photocatalytic performance was also undertaken in a batch reactor while keeping other experimental conditions identical (Fig. 3a). The yield of H2 on 1% Cu/PC50 in the batch reactor with 50 vol% methanol aqueous solution is shown in Fig. S5,† which exhibits a linear increase of H2 with the reaction time. It also proves that the photocatalyst could still remain active in the batch reactor when the reaction is stopped for the following product analysis. The batch reactor exhibits a much lower H2 production rate (15625 μmol g−1 h−1) compared to the flow reactor (25487 μmol g−1 h−1) operated at room temperature although the latter with a closely packed catalyst bed could scatter more light. The enhanced photocatalytic performance for H2 production in the flow membrane reactor is attributed to two major reasons. Firstly, the flow reactor overcame the limitation of mass transfer in the batch reactor, enhancing the diffusion and adsorption of gaseous reactants on the surface of photocatalysts, thereby resulting in an increased photocatalytic efficiency.25 Secondly, the improved mass transfer in a flow system also facilitated the desorption of products. In detail, there was less resistance for hydrogen gas release in the flow reactor where only two-phase (gas–solid) reactions occurred, rather than three-phase reactions (gas–liquid–solid) involved in the batch reactor. In other words, hydrogen molecules must overcome the resistance of the liquid environment before being released in the batch reactor. In contrast, the resistance for hydrogen release was much smaller as a result of the absence of liquid solvent in the flow reactor, thus leading to a high H2 yield.23
 | ||
| Fig. 3 (a) The yield of H2 and oxidation products by a flow (left panel) and a batch reactor (right panel). (b) Comparison of the H2 yield over different photocatalysts for H2 production from methanol reported to date. All results were achieved at room temperature. (c) H2 yield from a methanol and water mixture by photocatalysis over 1% Cu/PC50 at different temperatures. | ||
Furthermore, the oxidized products of methanol by photocatalysis over PC50 and 1% Cu/PC50 in the batch and flow reactor were also analyzed in the study (Fig. 3a). The major oxidized products over PC50 and 1% Cu/PC50 in different reaction systems are C1 chemicals, including formaldehyde (HCHO), formic acid (HCOOH), carbon monoxide (CO) and carbon dioxide (CO2). Besides, the selectivity towards each C1 product over PC50 and 1% Cu/PC50 in various reaction systems is redrawn in Fig. S6.† The quantification of formaldehyde and formic acid was carried out by UV-vis spectroscopy and ion chromatography methods, respectively. The standard calibration curves used for the determination of these two products are shown in Fig. S7 and S8.† For the batch system, 100% selectivity to formaldehyde has been achieved, indicating a complete dehydrogenation process, while the methanol reforming process could happen over both PC50 and 1% Cu/PC50 with the byproduct of CO2 and HCOOH in the flow reaction system. The possible chemical reactions that occurred in the flow system might follow the following equations including photocatalytic gaseous methanol dehydrogenation (eqn (1)) and reforming (eqn (2) and (3)):
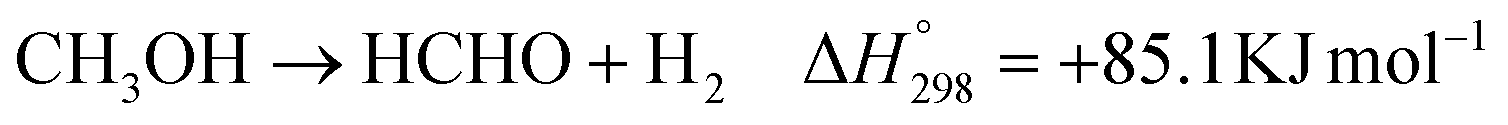 | (1) |
 | (2) |
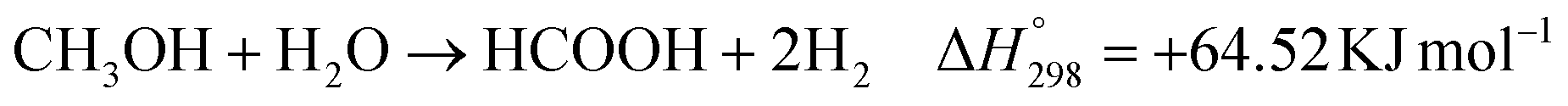 | (3) |
All the above equations are endothermic reactions in the gaseous phase, which indicates that an external energy input is required to activate these processes.
With the appearance of substantial CO2 and HCOOH as oxidation products in the flow membrane reactor, based on the chemical equation of methanol reforming (eqn (2) and (3)), water is involved in the photocatalytic H2 production from methanol, leading to a water reforming process. According to the yield of produced CO2 (382.0 μmol g−1 h−1) and HCOOH (264.5 μmol g−1 h−1) on 1% Cu/PC50 in the flow membrane reactor, around 7% of total produced H2 is from the methanol reforming process. Thus, it can be concluded that the photocatalytic methanol dehydrogenation is the major process in the flow reactor, accompanied by the methanol reforming process. However, due to the relatively fast flow rate of the reaction gas in the flow membrane reactor, part of formaldehyde could not be collected in the product vessel, leading to underestimated formaldehyde production in the flow reactor. The methanol conversion was determined to be 6.7% experimentally and theoretically and the detailed procedure is described in the ESI.† The evaluated methanol conversion in this study is much higher than other reported values in photocatalytic H2 production from methanol with a flow system (e.g. 3.4% on Pt/TiO2),26 which can be attributed to the improved mass transfer in the flow membrane reactor and the enhanced charge separation on 1% Cu/PC50.
This highlights the superiority of the flow reaction system, in which more photoinduced active carriers (h+) could be used to reform methanol, which is another reason for the highest H2 production rate in the flow reactor. Besides, thermodynamically both the methanol dehydrogenation process (eqn (1), ΔG° = +52.29 KJ mol−1) and one of the methanol reforming processes (eqn (3), ΔG° = +40.04 KJ mol−1) are uphill reactions with positive standard Gibbs free energy, whereas the other reforming process with CO2 production is a downhill reaction (eqn (2), ΔG° = −3.51 KJ mol−1), and the aforementioned relatively low apparent activation energy on 1% Cu/PC50 in the flow system indicates that incident photons can effectively provide energy enough to overcome the reaction barriers. Meanwhile, kinetically the flow process can promptly take H2 produced out of the reactor, avoiding H2 accumulation in the reactor. The above reactions including methanol dehydrogenation (eqn (1)) and reforming (eqn (2) and (3)) thus favor a shift towards the right, leading to a higher H2 yield as compared to that in the batch reactor. However, there is no obvious difference of selectivity towards C1 products over PC50 and 1% Cu/PC50 in the flow membrane reactor, suggesting that the copper species over TiO2 only serve as electron trapping sites for proton reduction as discussed below, rather than hole trapping sites to control the oxidation product selectivity. Fig. 3b summarizes the H2 production from methanol over representative photocatalysts in various photocatalytic reaction systems reported to date (also see Table S1†).27–39 This shows that 1% Cu/PC50 with a H2 yield of 25487 μmol g−1 h−1 is much higher than the best previously reported under ambient reaction conditions, indicating that 1% Cu/PC50 is an outstanding candidate for effective photocatalytic H2 production from methanol in the flow membrane reactor.
The reaction temperature was another key factor governing the activity of photocatalytic methanol dehydrogenation and reforming. The temperature-dependent photocatalytic hydrogen yield over 1% Cu/PC50 is shown in Fig. 3c. As the temperature of the reaction system increases from 30 to 60 °C, the hydrogen production rate is further improved from 25487 to 33702 μmol g−1 h−1. Besides, it should be noted that no hydrogen is produced without light irradiation at 60 °C, indicating that this catalytic process is triggered via photons rather than thermal energy. Notably, the average turnover frequency (TOF) based on the Cu active sites at 60 °C was calculated to be 227 h−1, which is higher than that (180 h−1) achieved on one benchmark noble metal catalyst (2 wt% (Pt1–Ptn)/α-MoC) at a similar temperature.40
3.2 Composition, structure and morphology characterization
The best photocatalyst was prepared by a modified MSM process, where commercial TiO2 (PC-50) was surrounded in a liquid environment generated by the molten salts (LiCl and KCl) at high temperature (500 °C), which enabled a highly even contact of TiO2 with the Cu2+ ions, thus anchoring uniform copper species on its surface. Since the sample with 1 wt% Cu loading amount exhibits the highest H2 yield rate, 1% Cu/PC-50 was fully characterized. The powder X-ray diffraction (PXRD) patterns were used to identify crystalline structures (Fig. 4a). Only anatase TiO2 (JCPDS no. 84-1286) is observed before and after the introduction of copper species, which indicates that the structure of TiO2 remained unchanged during the MSM process. Besides, no new diffraction peaks attributed to copper species could be identified over 1% Cu/PC50, suggesting the high dispersion of Cu species.41 Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was used to confirm the quantity of Cu on TiO2. It is found that the actual amount (0.95 wt%) is quite close to the nominal value (1 wt%) (Table S2†), indicating that the amount of copper could be well controlled via this robust synthesis method. The structure of the photocatalyst after the reaction was also confirmed by XRD spectra, which is the same as that of the fresh sample (Fig. 4a).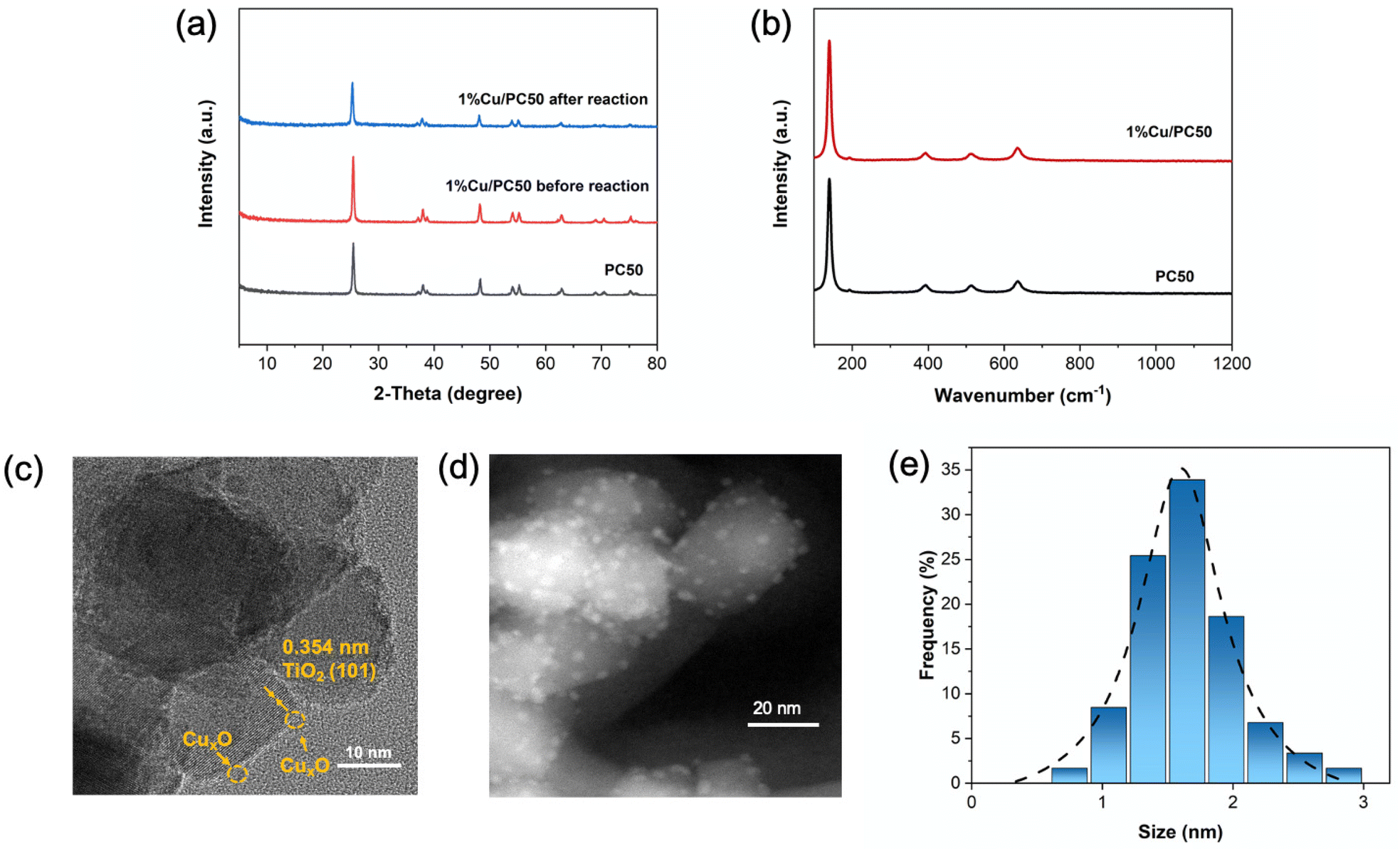 | ||
| Fig. 4 (a) PXRD and (b) Raman spectra of 1% Cu/PC50 and PC50. (c) HR-TEM and (d) HAADF images of 1% Cu/PC50 in which highly dispersed CuxO particles are very clear. (e) The size distribution of CuxO nanoparticles on a PC50 surface obtained by counting of 60 particles from the HAADF picture of 4d. | ||
The anatase phase of TiO2 was further confirmed by Raman spectra (Fig. 4b). The Raman peaks are observed at 139, 192, 393, 513 and 636 cm−1, corresponding to the Eg(1), Eg(2), B1g(1), A1g + B1g(2) and Eg(3) of modes of anatase, respectively.42 Besides, no shift of Raman peaks was detected on 1% Cu/PC50 as compared to that on PC50, which further indicates that the introduction of copper species by the MSM method could not change the TiO2 lattice.
Scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM) were employed to observe the morphology of the as-prepared samples. From the microstructure of all the samples as shown in Fig. S9,† 1% Cu/PC50 remains with an unchanged morphology after the heat treatment at 500 °C compared with pristine PC50, indicating that the molten salts in the MSM process could effectively prevent the agglomeration of TiO2 particles. Fig. 4c shows the HR-TEM image of 1% Cu/PC50, where the interplane distance of d was calculated and found to be 0.354 nm, corresponding to the (101) plane of TiO2.43 CuxO particles could be readily observed as indicated by the circled ones. A high angle annular dark field scanning transmission electron microscope (HAADF-STEM) was used to further observe the uniform dispersion of Cu species on TiO2 (Fig. 4d). The corresponding size distribution of the CuxO species over TiO2 is shown in Fig. 4e, which is 1.5 nm to 2.0 nm. From these results, it is suggested that the liquid environment formed in the MSM synthesis procedure could effectively prevent the aggregation of CuxO nanoparticles and provide a well-controlled method to disperse CuxO on the TiO2 surface.
3.3 Photocatalytic reaction mechanism
Ultraviolet-visible diffuse reflectance spectroscopy (UV-Vis DRS) was used to observe the light absorption properties of PC50 and 1% Cu/PC50. The absorption edge has only a slight red shift over 1% Cu/PC50 as compared to that over pure PC50 (Fig. 5a), indicating that the two photocatalysts had similar bandgaps.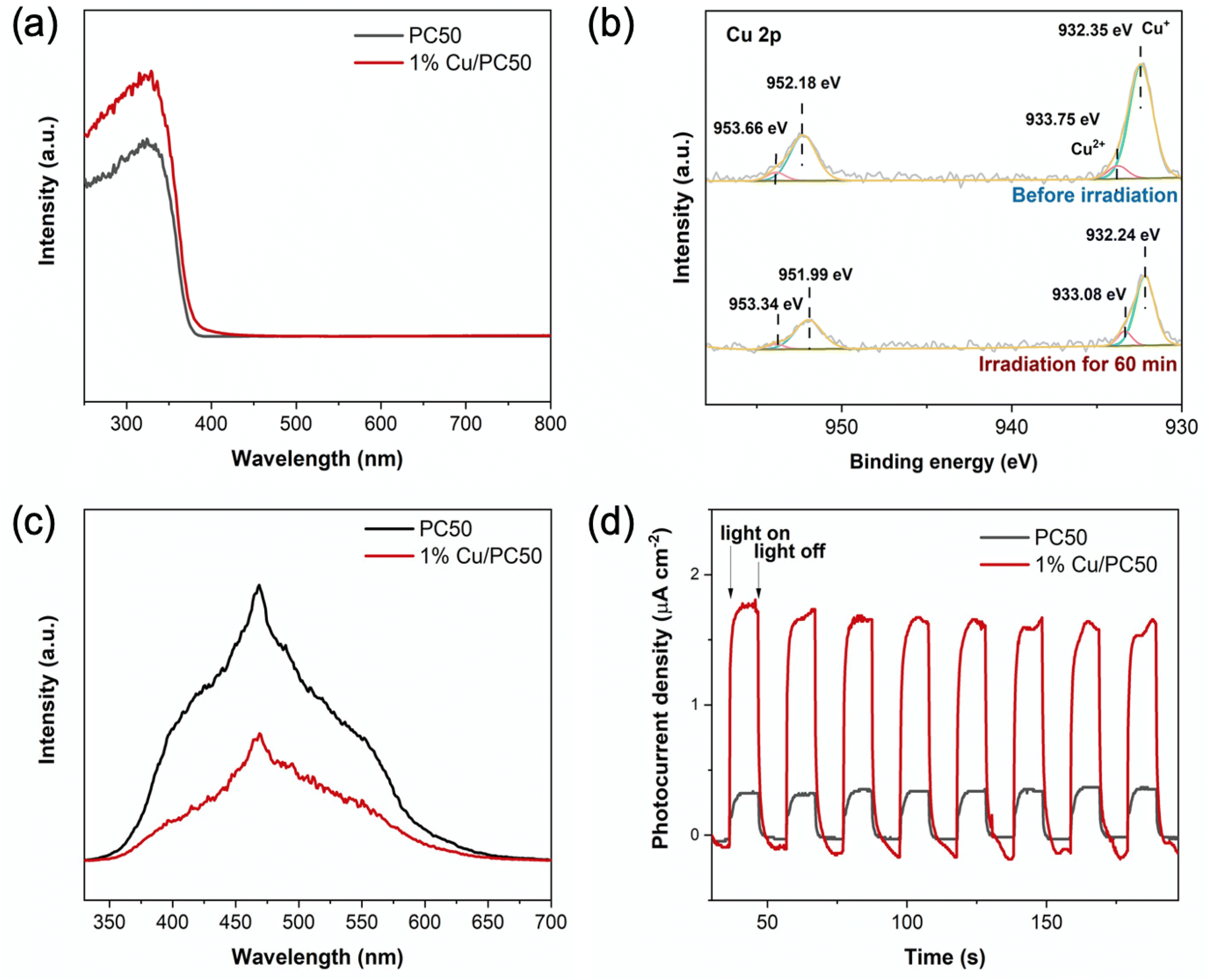 | ||
| Fig. 5 (a) UV-Vis-DRS spectra of PC50 and 1% Cu/PC50. (b) In situ XPS spectra of Cu 2p on 1% Cu/PC50 in the dark and under light illumination. (c) PL spectra of PC50 and 1% Cu/PC50. (d) Photocurrent response curve of 1% Cu/PC50 and PC50 in 0.1 M Na2SO4 with 10 vol% methanol and 0.1 V bias versus Ag/AgCl. | ||
To further unravel the function and chemical states of copper species, in situ X-ray photoelectron spectroscopy (XPS) was carried out on 1% Cu/PC50 in the dark and under light irradiation (Fig. 5b). Fig. S10† also shows the XPS survey spectrum of 1% Cu/PC50 in the dark, which demonstrates the presence of Cu, Ti and O elements in the sample. The very small C 1s peak is associated with the carbon substances in the external environment. In Fig. 5b, before light irradiation, the two Gaussian peaks at 932.35 eV and 952.18 eV are attributed to the Cu 2p1/2 and 2p3/2 of Cu+, respectively. Two other peaks at 933.75 eV and 953.66 eV are associated with the Cu2+ species.44 The formation of Cu+ on 1% Cu/PC50 might be caused by the following procedure. Firstly the precursor CuCl2·2H2O was decomposed to CuCl2 and H2O. Then CuCl2 was partially decomposed to CuCl and simultaneously released Cl2 at a high temperature under the inert atmosphere,45 forming Cu2O.46 Under light irradiation for 60 min, the ratio of Cu+ to Cu2+ increases from 5:1 to 6.25:1, which clearly indicates that CuxO serves as an electron acceptor on 1% Cu/PC50, thus resulting in an enhanced charge separation.
Photoluminescence (PL) spectroscopy was carried out to investigate the behavior of photogenerated charge carriers in the as-prepared samples. A broad band centered at around 475 nm is observed over pristine PC50 (Fig. 5c). After introducing copper species, 1% Cu/PC50 exhibits a similar emission peak profile, but with a much lower intensity compared with pristine PC50. These results indicate an efficient separation of photoinduced electrons and holes on 1% Cu/PC50. The photocurrent response with several on–off cycles of irradiation was used to demonstrate the ability of charge separation over the as-prepared materials (Fig. 5d). The photocurrent of 1% Cu/PC50 was measured and found to be 1.84 μA cm−2, which is 5.1 times higher than that (0.36 μA cm−2) of pristine PC50. These results indicate efficient charge separation on 1% Cu/PC50, which was in agreement with the PL results.47 Benefitting from these advantages, 1% Cu/PC50 exhibits an excellent performance in photocatalytic methanol dehydrogenation and reforming.
Based on the above characterization and analysis, a possible mechanism for photocatalytic methanol dehydrogenation and reforming on 1% Cu/PC50 in the flow membrane reactor is proposed (Scheme 1). As compared to the conventional batch reactor, the improved mass transfer in the gas–solid flow system enhances the reactant adsorption and product desorption, thereby leading to a superior photocatalytic H2 production from the methanol aqueous solution. Upon light irradiation, photogenerated electrons and holes appear and lie in the conduction band and the valence band of PC50, respectively. Then, the electrons at the conduction band transfer to CuxO nanoparticles, where Cu2+ is reduced to Cu+, as proved by the in situ XPS. Next, Cu+ further reduces H+ to produce H2, and simultaneously Cu+ returns to Cu2+. In parallel, the holes at the valence band oxidize methanol with water to produce HCHO, HCOOH, CO2, CO and so on.
 | ||
| Scheme 1 Proposed schematic illustration of photocatalytic methanol dehydrogenation and reforming on 1% Cu/PC50 in the flow membrane reactor. | ||
4. Conclusions
In summary, we have successfully achieved continuous H2 production by the photocatalytic methanol dehydrogenation and reforming process in the flow membrane reactor. The highly dispersed CuxO nanoparticles were loaded on PC50 by a molten salt method. The CuxO, serving as an electron sink as proved by in situ XPS, could effectively promote charge separation, together with the catalytic effect leading to improved H2 production. The optimal 1% Cu/PC50 exhibits a 13-times higher H2 yield (25487 μmol g−1 h−1) as compared to that (1934 μmol g−1 h−1) on PC50, with an AQE of 7.75% at 365 nm. The H2 production is further improved to 33702 μmol g−1 h−1 at 60 °C. The activation energy as low as 4.0 kJ mol−1 on 1% Cu/PC50 is much smaller than that of other catalysts used in the methanol reforming process, and the high photocatalytic activity remains stable. Overall, this work provides an effective and green route towards continuous H2 production on demand by photocatalytic methanol dehydrogenation and reforming.
Author contributions
H. Jiao: methodology, formal analysis, investigation, and writing – original draft, review & editing; J. Yang: materials characterisation and writing – review and editing; X. Li: writing – review and editing; C. Wang: formal analysis, and writing – review and editing; J. Tang: conceptualization, funding, resources, writing – review and editing, and supervision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by UK EPSRC (EP/S018204/2), Leverhulme Trust (RPG-2017-122), Royal Society Newton Advanced Fellowship grant (NAF\R1\191163), and Royal Society Leverhulme Trust Senior Research Fellowship (SRF\R1\21000153). H. J. and C. W. acknowledge the UCL Dean's prize and China CSC scholarship.References
- R. Ramachandran and R. K. Menon, Int. J. Hydrogen Energy, 1998, 23, 593–598 CrossRef CAS.
- W.-H. Chen and C.-Y. Chen, Appl. Energy, 2020, 258, 114078 CrossRef CAS.
- A. Pareek, R. Dom, J. Gupta, J. Chandran, V. Adepu and P. H. Borse, Mater. Sci. Energy Technol., 2020, 3, 319–327 CAS.
- I. P. Jain, P. Jain and A. Jain, J. Alloys Compd., 2010, 503, 303–339 CrossRef CAS.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS.
- D. R. Palo, R. A. Dagle and J. D. Holladay, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS.
- G. A. Olah, Catal. Lett., 2004, 93, 1–2 CrossRef CAS.
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS.
- J. W. Shabaker, R. R. Davda, G. W. Huber, R. D. Cortright and J. A. Dumesic, J. Catal., 2003, 215, 344–352 CrossRef CAS.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS.
- K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2017, 9, 1523–1544 CrossRef CAS.
- A. G. Variar, R. M. Srinivasa, V. U. Ail, S. P. Selvanathan, S. Kumarasamy and M. Tahir, J. Ind. Eng. Chem., 2021, 99, 19–47 CrossRef CAS.
- C. McCullagh, N. Skillen, M. Adams and P. K. J. Robertson, J. Chem. Technol. Biotechnol., 2011, 86, 1002–1017 CrossRef CAS.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS.
- M. Xiao, L. Zhang, B. Luo, M. Lyu, Z. Wang, H. Huang, S. Wang, A. Du and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 7230–7234 CrossRef CAS.
- R. V. Smith and P. W. Erhardt, Anal. Chem., 1975, 47, 2462–2464 CrossRef CAS.
- R. Xu, L. Kang, J. Knossalla, J. Mielby, Q. Wang, B. Wang, J. Feng, G. He, Y. Qin, J. Xie, A.-C. Swertz, Q. He, S. Kegnæs, D. J. L. Brett, F. Schüth and F. R. Wang, ACS Nano, 2019, 13, 2463–2472 CAS.
- C. Fan, X. Jiang, J. Chen, X. Wang, S. Qian, C. Zhao, L. Ding, D. Sun and Y. Tang, Small Struct., 2021, 2, 2000017 CrossRef CAS.
- J. Ran, B. Zhu and S.-Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS.
- A. J. Maira, J. M. Coronado, V. Augugliaro, K. L. Yeung, J. C. Conesa and J. Soria, J. Catal., 2001, 202, 413–420 CrossRef CAS.
- N. González-García, J. A. Ayllón, X. Doménech and J. Peral, Appl. Catal., B, 2004, 52, 69–77 CrossRef.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- S. Guo, X. Li, J. Li and B. Wei, Nat. Commun., 2021, 12, 1343 CrossRef CAS PubMed.
- S. Luo, H. Lin, Q. Wang, X. Ren, D. Hernández-Pinilla, T. Nagao, Y. Xie, G. Yang, S. Li, H. Song, M. Oshikiri and J. Ye, J. Am. Chem. Soc., 2021, 143, 12145–12153 CrossRef CAS PubMed.
- J. Oliveira de Brito Lira, H. G. Riella, N. Padoin and C. Soares, J. Environ. Chem. Eng., 2021, 9, 105068 CrossRef CAS.
- W. Cui, L. Feng, C. Xu, S. Lü and F. Qiu, Catal. Commun., 2004, 5, 533–536 CrossRef CAS.
- G. L. Chiarello, M. H. Aguirre and E. Selli, J. Catal., 2010, 273, 182–190 CrossRef CAS.
- G. L. Chiarello, L. Forni and E. Selli, Catal. Today, 2009, 144, 69–74 CrossRef CAS.
- S. Fang, Y. Liu, Z. Sun, J. Lang, C. Bao and Y. H. Hu, Appl. Catal., B, 2020, 278, 119316 CrossRef CAS.
- Z. Liu, Z. Yin, C. Cox, M. Bosman, X. Qian, N. Li, H. Zhao, Y. Du, J. Li and G. Nocera Daniel, Sci. Adv., 2016, 2, e1501425 CrossRef PubMed.
- Y. Liu, S. Yang, S.-N. Yin, L. Feng, Y. Zang and H. Xue, Chem. Eng. J., 2018, 334, 2401–2407 CrossRef CAS.
- Y. Pang, M. N. Uddin, W. Chen, S. Javaid, E. Barker, Y. Li, A. Suvorova, M. Saunders, Z. Yin and G. Jia, Adv. Mater., 2019, 31, 1905540 CrossRef CAS PubMed.
- Y. Chao, J. Lai, Y. Yang, P. Zhou, Y. Zhang, Z. Mu, S. Li, J. Zheng, Z. Zhu and Y. Tan, Catal. Sci. Technol., 2018, 8, 3372–3378 RSC.
- Z. Chai, T.-T. Zeng, Q. Li, L.-Q. Lu, W.-J. Xiao and D. Xu, J. Am. Chem. Soc., 2016, 138, 10128–10131 CrossRef CAS PubMed.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef.
- A. L. Luna, E. Novoseltceva, E. Louarn, P. Beaunier, E. Kowalska, B. Ohtani, M. A. Valenzuela, H. Remita and C. Colbeau-Justin, Appl. Catal., B, 2016, 191, 18–28 CrossRef CAS.
- F. Yu, L. Chen, X. Li, X. Shen, H. Zhao, C. Duan and Q. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 18619–18626 CrossRef CAS PubMed.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim, S.-P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS.
- X. Chen, L. Liu, Y. Yu Peter and S. Mao Samuel, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y.-W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, Nature, 2021, 589, 396–401 CrossRef CAS PubMed.
- Y. Li, W.-N. Wang, Z. Zhan, M.-H. Woo, C.-Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS.
- Z. Dong, F. Xiao, A. Zhao, L. Liu, T.-K. Sham and Y. Song, RSC Adv., 2016, 6, 76142–76150 RSC.
- S. Farsinezhad, H. Sharma and K. Shankar, Phys. Chem. Chem. Phys., 2015, 17, 29723–29733 RSC.
- L. Luo, Z. Gong, Y. Xu, J. Ma, H. Liu, J. Xing and J. Tang, J. Am. Chem. Soc., 2022, 144, 740–750 CrossRef CAS PubMed.
- Y. Yin, J. Zhu, X.-Q. Liu, P. Tan, D.-M. Xue, Z.-M. Xing and L.-B. Sun, RSC Adv., 2016, 6, 70446–70451 RSC.
- D. A. Scott, J. Am. Inst. Conserv., 1990, 29, 193–206 CrossRef.
- D. Kong, X. Han, J. Xie, Q. Ruan, C. D. Windle, S. Gadipelli, K. Shen, Z. Bai, Z. Guo and J. Tang, ACS Catal., 2019, 9, 7697–7707 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc01553f |
| This journal is © The Royal Society of Chemistry 2022 |