
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn-water synthesis of isocyanides under micellar conditions†
Francesca
Brunelli
 a,
Silvio
Aprile
a,
Camilla
Russo
b,
Mariateresa
Giustiniano
b and
Gian Cesare
Tron
*a
a,
Silvio
Aprile
a,
Camilla
Russo
b,
Mariateresa
Giustiniano
b and
Gian Cesare
Tron
*a
aDipartimento di Scienze del Farmaco, Università degli Studi del Piemonte Orientale “A. Avogadro”, Largo Donegani 2, 28100 Novara, Italy. E-mail: giancesare.tron@uniupo.it
bDipartimento di Farmacia, Università di Napoli Federico II, via D. Montesano 49, 80131, Napoli, Italy
First published on 23rd August 2022
Abstract
An in-water dehydration of N-formamides to afford isocyanides using micellar conditions at room temperature is reported. This method allows for the preparation of aliphatic isocyanides in an environmental friendly manner. The replacement of undesirable components such as phosphorous oxychloride, triethyl amine and dichloromethane (the classical combination used for the dehydration of N-formamides), by p-toluen sulphonyl chloride, sodium hydrogen carbonate and water makes this transformation really sustainable and safe.
Introduction
The search for synthetic methods with lower impact on the environment has become a priority not only for the young generations of chemists but also for previous ones.1 There is an increasing awareness of using green methods, not only in preparative chemistry, but also in pharmaceutical research.2 Medicinal chemists are increasingly aware green methods are preferable even for the preparation of a few grams of substances. Avoidance of toxic reagents and solvents should become a rule rather than a choice given the current global environmental emergency, and it should apply as well to small scale reactions.2The isocyanide is an extremely fascinating functional group above all for its chameleonic reactivity.3 Over the last century isocyanides have been widely exploited as chemical reagents, pivotal in multicomponent reactions.4 For this reason, numerous synthetic methodologies have been reported for their synthesis.5 The most versatile ones remain those that involve the dehydration of formamides. Unfortunately, the best method in terms of yield and applicability is based on the Ugi protocol which employs phosphorus oxychloride along with an excess of a tertiary nitrogen base (e.g., at least 3 equivalent of triethylamine) in dichloromethane.6 The exothermicity of this reaction often requires cooling the reaction mixture below zero degree Celsius. It is emblematic that none of the three reagents listed above can be considered green and safe. Phosphorus oxychloride is toxic and corrosive, moisture sensitive and requires to be periodically distilled before the use to remove phosphoric acid and hydrogen chloride. Triethylamine is flammable and corrosive, while dichloromethane is an ozone depleting gas in the atmosphere.7 Recently at least three important contributions have been published aimed at improving existing methodologies. The first one is based on the use of tosyl chloride as dehydrating agents; however, it holds the use of an excess of base (e.g., pyridine) and dichloromethane.8 The second one is conceptually a simple modification of the original Ugi protocol, where the reaction mixture is directly loaded and eluted with diethyl ether onto a chromatographic column, sparing solvents by skipping the traditional work up step.9 The third one devised a novel electrochemical method for the synthesis of isocyanides and it is based on the anodic oxidation of aminotetrazoles.10 Each of these methods has merits and increases the arsenal available to chemists needing to synthesize isocyanides. Although all these procedures represent a contribution toward the greenness of isocyanide synthesis, there is still room for improvement for this methodology and lowering its environmental impact. Furthermore, despite the novelty of the electrochemical method, the galvanostatic conditions used might render difficult the preparation of highly functionalized isocyanides as demonstrated by the impossibility to prepare with this method the simple p-methoxy benzyl isocyanide. After recently demonstrating that certain isocyanides are metabolically stable11 and may be innovative pharmacophoric groups,12 we have started a medicinal chemistry campaign aimed at identifying new biologically active molecules containing the isocyanide group as warhead and decided to find a new and versatile green methodology operating at room temperature for the synthesis of isocyanides starting from formamides, avoiding the use of phosphorus oxychloride, the excess of tertiary amine and dichloromethane. This idea arises from our extensive experience with the chemistry of isocyanides. Indeed, although the isocyanide is considered a very reactive functional group prone to rapidly hydrolyze in water, this is not the case. Isocyanides are stable in basic medium13 and start to hydrolyze at pH ≤ 5.14
Results and discussion
Exploiting the hydrolytic stability of isocyanides, we thought it would be possible to attempt an “in-water” synthesis using the micellar strategy developed by Professor Lipshutz. Fifteen years ago, he showed that surfactants like TPGS-750-M when dissolved in water can self-aggregates in micelles with a diameter of 40–50 nm.15,16 These micelles are not static, but there is a dynamic exchanging equilibrium between the surfactant molecules forming the micelles and those there are free in the medium, allowing for the incorporation of reagents and products into the micelles.The inner side of these micelles is hydrophobic, and it acts as the reaction “solvent”, virtually allowing for the dissolution of the water-insoluble reagents. Micelles behave like nanoreactors operating at high concentration of reagents, thus dramatically increasing the probability of reactive events.17
In Scheme 1, we reported the general mechanism for the dehydration of formamides (1) to isocyanides (4) with a phosphorous or sulfur-based dehydrating agent.
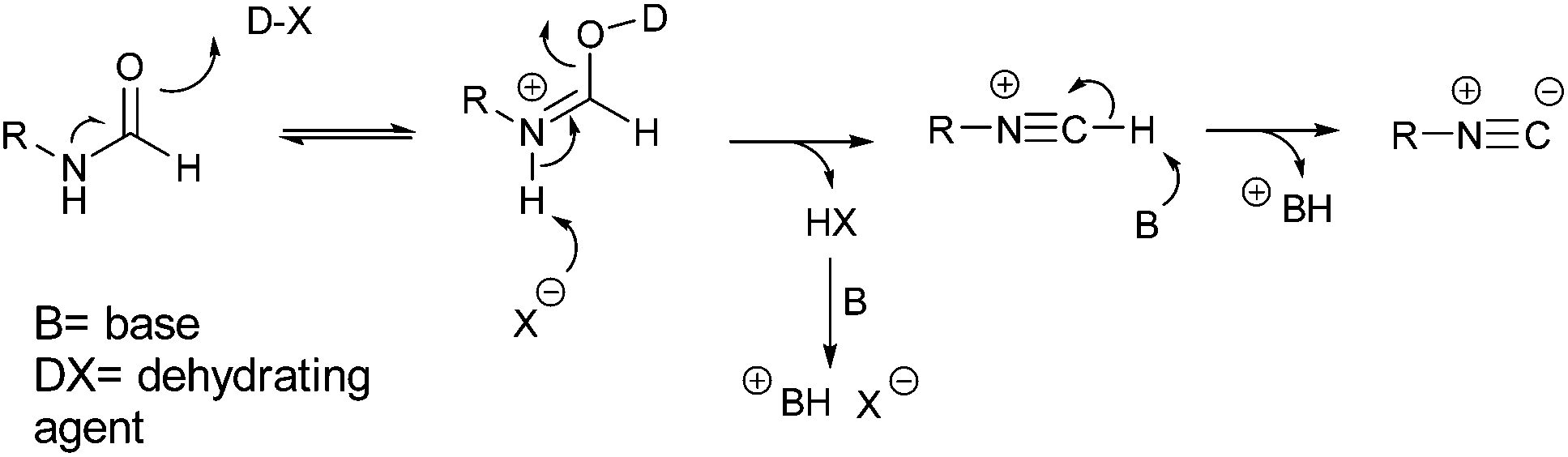 | ||
| Scheme 1 Mechanism for the dehydration of formamides to isocyanides. | ||
It is evident that the base does not take an active part in the reaction between the oxygen of the formamide and the electrophilic group of the dehydrating agent. Its role is simply to act as neutralizing agent favoring the formation of the isocyanide group and maintaining a neutral pH of the medium.18 Therefore, we reasoned that a weak, non-hazardous, and cheap water-soluble base was more than enough to perform this task. For this reason, we opted to evaluate the reaction using sodium hydrogen carbonate. Finally, as dehydrating agent, we focused our attention on tosyl chloride because it is: insoluble in water, and therefore relatively stable to hydrolysis in an aqueous environment; cheap, as it is continuously produced on large scales being a waste in the synthesis of saccharin. To test our idea, we studied the dehydration of N-phenethylformamide (5) to (2-isocyanoethyl)benzene (6) as model reaction (Scheme 2). To note that before undertaking this study, we evaluated the hydrolytic stability of tosyl chloride, (2-isocyanoethyl)benzene and N-phenethylformamide in 5 wt% TPGS-750-M/H2O and sodium bicarbonate. Formamide and isocyanide compounds were found to be fully stable up to 24 h, while tosyl chloride was hydrolyzed in full very slowly beyond 16 h (see ESI†).
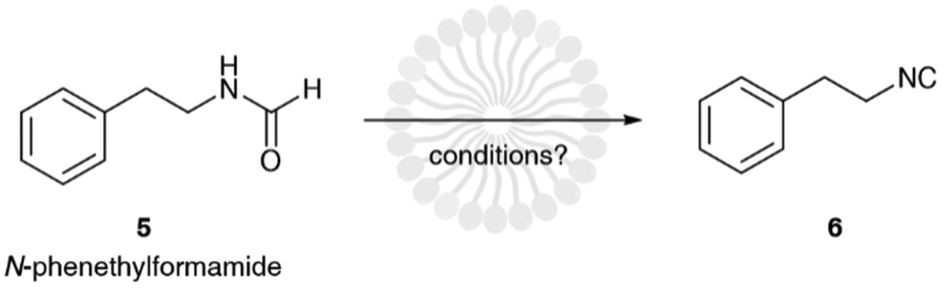 | ||
| Scheme 2 Model reaction. | ||
We started by using TPGS-750-M as surfactant and trialed different bases, while comparing the yields with respect to the formation of the isocyanide using either the Ugi (see Table 1, entry 1) or the Meier procedure (see Table 1, entry 2), carrying out the reaction on 100 mg scale of formamide (Table 1). From this first round of screening, we confirmed that sodium hydrogen carbonate is the best base and increasing the concentration of the reaction medium had a positive effect on the yield (see Table 1, entries 6 and 10). The use of nitrogen bases such as TEA (see Table 1, entries 4 and 5), morpholine (see Table 1, entries 8 and 9), collidine (see Table 1, entry 12) or pyridine (see Table 1, entry 3) did not afford the desired product. This can be ascribed to the addition reaction between the base and tosyl chloride, followed by hydrolysis to tosic acid. When the reaction with sodium hydrogen carbonate was conducted without the surfactant (see Table 1, entry 7) we did not observe the formation of the isocyanide as the tosyl chloride did not dissolve in the medium. Finally, the use of a carbonate base (see Table 1, entry 11) reduces the yield indicating that a weak base as sodium hydrogen carbonate is the best option.
| Entry | Dehydrating agent | Base | Solvent | Time (H) | Yield |
|---|---|---|---|---|---|
| a [Formamide] = 1 M; T = 0 °C. b [Formamide] = 1 M; T = r.t. c [Formamide] = 0.3 M; T = r.t. d [Formamide] = 0.45 M; T = r.t. | |||||
| 1 | POCl3 1 equiv. | TEA 3 equiv. | DCM dry | 1 | 82%a |
| 2 | p-TsCl 1.5 equiv. | Py 3 equiv. | DCM dry | 5 | 69%b |
| 3 | p-TsCl 1.5 equiv. | Py 3 equiv. | 5 wt% TPGS-750-M/H2O | 16 | Tracesc |
| 4 | p-TsCl 1.5 equiv. | TEA 3 equiv. | 5 wt% TPGS-750-M/H2O | 16 | Tracesc |
| 5 | p-TsCl 1.5 equiv. | TEA 3 equiv. | Water | 16 | No reactionc |
| 6 | p-TsCl 1.5 equiv. | NaHCO3 3 equiv. | 5 wt% TPGS-750-M/H2O | 16 | 24%c |
| 7 | p-TsCl 1.5 equiv. | NaHCO3 3 equiv. | Water | 16 | No reactionc |
| 8 | p-TsCl 1.5 equiv. | Morpholine 3 equiv. | 5 wt% TPGS-750-M/H2O | 16 | No reactionc |
| 9 | p-TsCl 1.5 equiv. | Morpholine 3 equiv. | Water | 16 | No reactionc |
| 10 | p-TsCl 1.5 equiv. | NaHCO3 3 equiv. | 5 wt% TPGS-750-M/H2O | 48 | 47%d |
| 11 | p-TsCl 1.5 equiv. | K2CO3 3 equiv. | 5 wt% TPGS-750-M/H2O | 48 | 28%d |
| 12 | p-TsCl 1.5 equiv. | Collidine 3 equiv. | 5 wt% TPGS-750-M/H2O | 48 | No reactiond |
We then carried out a new round of reaction explorations by locking sodium hydrogen carbonate as the base, while changing concentration and other parameters such as temperature (Table 2). Interestingly, when the reaction was run at 60 °C using sodium hydrogen carbonate (see Table 2, entry 1) we did not observe the formation of the product but the complete hydrolysis of the tosyl chloride. It is reasonable to think that the heating accelerates the hydrolysis rate of tosyl chloride.
| Entry | [Formamide] | Yield |
|---|---|---|
| a Reaction conditions: formamide (1 equiv.), p-TsCl (1.5 equiv.), NaHCO3 (3 equiv.), 5 wt% TPGS-750-M/H2O, 1 h, T = 60 °C. b Reaction conditions: formamide (1 equiv.), p-TsCl (1.5 equiv.), NaHCO3 (3 equiv.), 5 wt% TPGS-750-M/H2O, 16 h, T = r.t. | ||
| 1 | 0.45 M | No reactiona |
| 2 | 0.2 M | No reactionb |
| 3 | 0.67 M | 58%b |
| 4 | 1 M | 62%b |
| 5 | 1.5 M | 42%b |
The optimal conditions were mainly a function of concentration, with the best result obtained at 1 M concentration of formamide (see Table 2, entry 4). Further increasing the concentration to 1.5 M was detrimental (see Table 2, entry 5).
Finally, we proceeded to a third round of optimization by locking sodium hydrogen carbonate, molarity, and temperature (25 °C), while changing the equivalents of tosyl chloride and sodium hydrogen carbonate with a relative constant 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (Table 3) on a 300 mg scale of formamide. An optimal molar equivalence for the said reagents was identified (see Table 3, entry 2) (Fig. 1).
:2 ratio (Table 3) on a 300 mg scale of formamide. An optimal molar equivalence for the said reagents was identified (see Table 3, entry 2) (Fig. 1).
 | ||
| Fig. 1 On the left: reaction mixture immediately after the addition of the reagents; On the right: reaction mixture after 16 h. | ||
:2 ratio
As the isocyanides could have similar polarity to tosyl chloride, we used a 0.5% solution of py-tetrazine in methanol as TLC staining reagent, capitalizing on the [4 + 1] cycloaddition reaction between isocyanides and py-tetrazine to form yellow-coloured spots after gentle heating (Fig. 2).19
 | ||
| Fig. 2 (A) TLC under UV before the work up; (B) TLC using 0.5% of py-tetrazine in methanol as staining reagent, before the work up; (C) TLC under UV after the work up; (D) TLC using 0.5% of py-tetrazine in methanol as staining reagent, after the work up. (A) and (B) from left to right: p-TsCl, cospot, reaction mixture (C) and (D) from left to right: p-TsCl, cospot, organic phase. | ||
The work up procedure deserves a comment. Indeed, to remove the excess of tosyl chloride, we added NH4OH 10% in the same volume with respect to the water used for the reaction, stirring for 15 minutes, and then extracting the mixture with EtOAc. In this case the tosyl chloride is converted into the highly polar p-toluene sulfonamide facilitating the column chromatography purification.
Using the optimized reaction conditions from Table 3, entry 2, we then evaluated the use of other surfactants (Fig. 3) without observing any significant improvement (Table 4). To note that the use of Triton X raised concerns due to its environmental toxicity.20
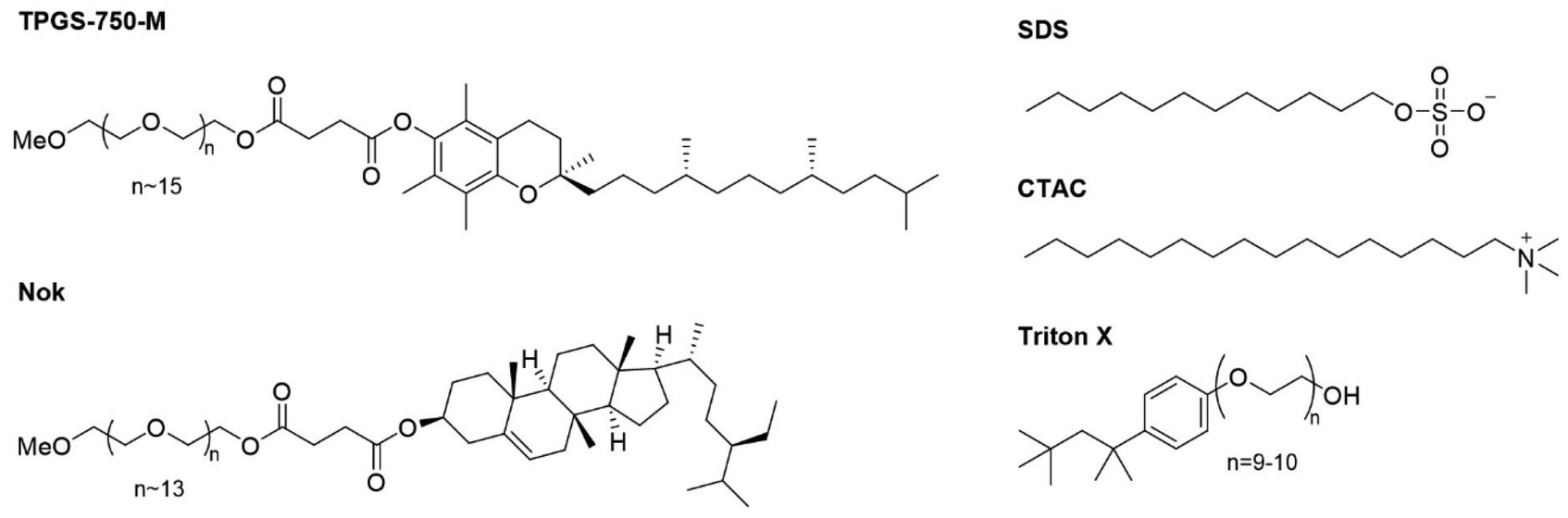 | ||
| Fig. 3 Structures of the surfactants used for reaction conditions optimization. | ||
The generality of this procedure was then evaluated by preparing different isocyanides as depicted in Fig. 4.
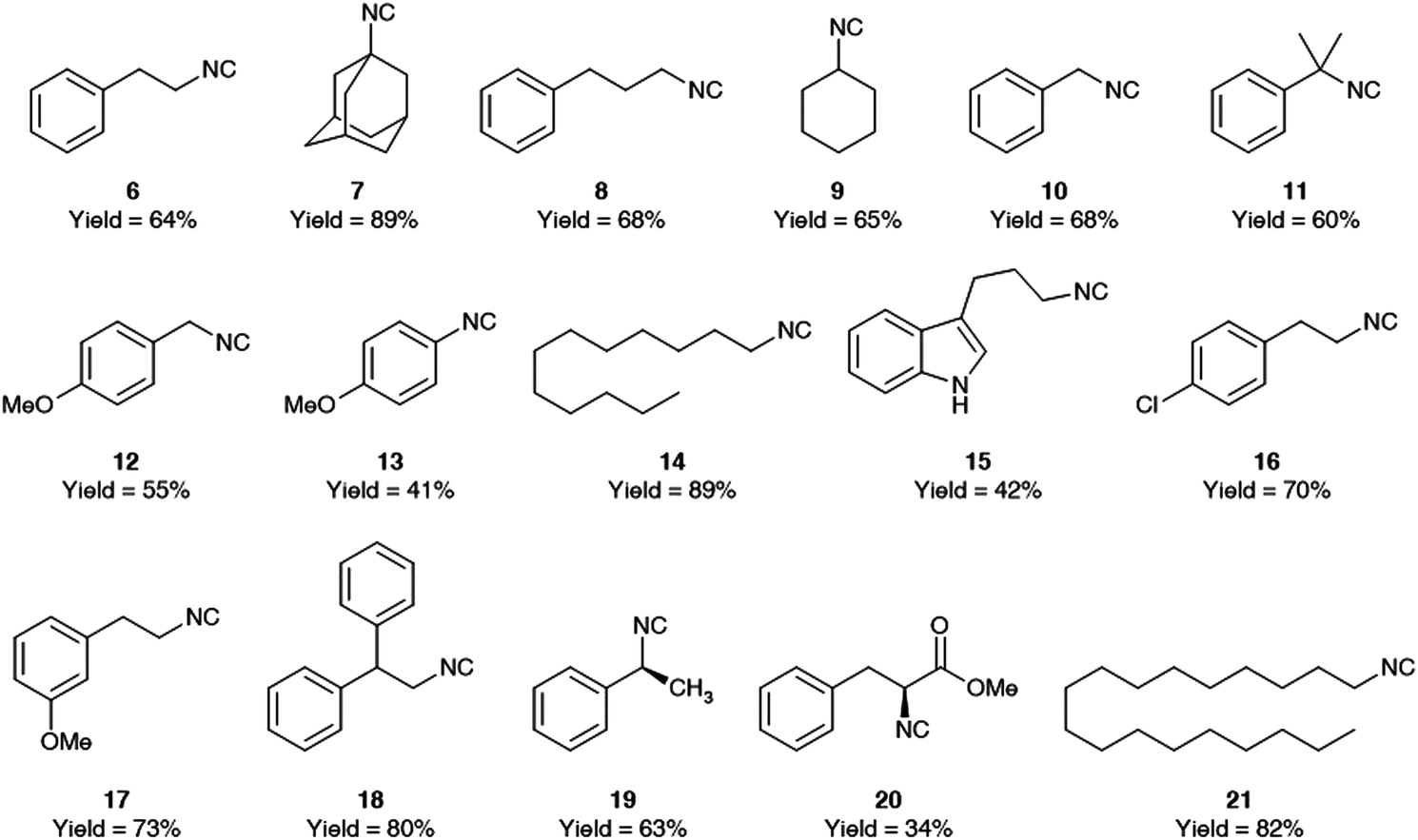 | ||
| Fig. 4 Isocyanides synthesized using aqueous micelles, with their isolated yields after column chromatography. | ||
This methodology works with primary, secondary, and tertiary aliphatic isocyanides as well as benzylic isocyanides. It is also evident that the more the isocyanide is lipophilic, the higher is the yield of the reaction. As expected, and already reported in literature,8 formamide dehydration triggered by tosyl chloride works only when electron donating groups are present on the aromatic ring and we succeed to form p-methoxyphenyl isocyanide albeit in low yield, while we could not prepare phenyl isocyanide as well as p-chloro phenyl isocyanides. For this type of isocyanides is indeed required a stronger dehydrating agent like POCl3.
One advantage of this procedure is the possibility to re-use the micellar aqueous solution by reloading fresh amounts of all reagents and reactants to the aqueous solution after extraction of the desired product with EtOAc. We have recycled the micellar solution on a scale of 500 mg of N-dodecylformamide, observing only a slight decrease in yield for the isocyanide 14 (initial reaction: 89%; 1st recycle: 82%; 2nd recycle: 79%). We have evaluated the amount of TPGS-750-M which was extracted during the recycling experiments and also its stability upon the reaction conditions (see ESI†).21
Finally, we synthesized 1-isocyanododecane (14) on a 10 mmol scale (2.13 g), using a mechanical stirrer (Fig. 5). We obtained the desired product in 82% yield.
 | ||
| Fig. 5 Synthesis of isocyanide 14 in a 10 mmol scale using a mechanical stirrer. | ||
We have also verified the possibility of running a Passerini multicomponent reaction on the crude obtained after quenching with ammonium hydroxide and extraction with EtOAc. Although the crude contains amounts of p-toluen sulphonamide and N-dodecylformamide, we succeed in forming the desired multicomponent adduct in a 70% yield (Scheme 3).
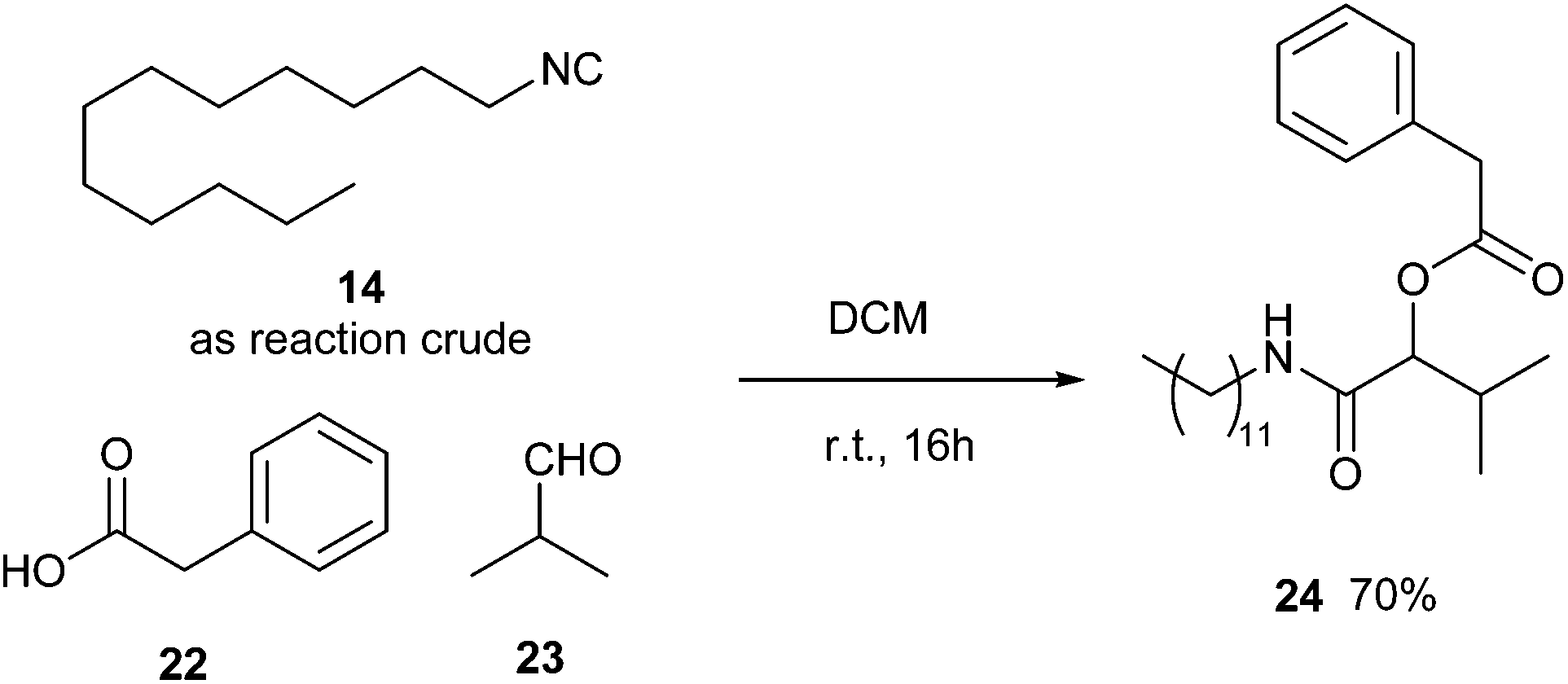 | ||
| Scheme 3 Passerini reaction without purifying isocyanide 14. | ||
Furthermore, we carried out an MCR in situ directly under micellar conditions, without isolating the isocyanide. Even in this case, we were able to form and isolate the multicomponent product in good yield (Scheme 4). This is particularly remarkable in our opinion for three reasons: (a) the possibility not to isolate the isocyanide, useful above all for those characterized by an unpleasant smell and a low boiling point; (b) a reduced consume of solvent; (c) to confirm the possibility to carry out multicomponent reactions under aqueous micellar conditions.22
 | ||
| Scheme 4 One pot tandem isocyanide synthesis and Passerini reaction. | ||
Having established the best synthetic protocol, in order to compare the different synthetic procedures, we decided to calculate various PMI values (Table 5), with or without water. PMI values were calculated using the “Process Mass Intensity Calculator Tool” developed by the ACS GCI Pharmaceutical Roundtable;23E-factor values can be obtained just doing PMI-1. In entries 1 and 2 are reported the values obtained by considering all the reactants and solvents used for the reaction and during the quenching (the mass of each reactant is reported in the ESI†). The amount of TPGS-750-M which cannot be recycled, due to degradation during the reaction or because it has been lost during extraction (46.1% in total) has been included in the calculation. Regarding our protocol, the amount of water used is the sum of those employed for the TPGS-750-M 5 wt% solution (1M with respect to the formamide) and during the quenching (500 mg on a 0.5 mmol scale). For the Ugi protocol, the amount of water used is those employed during the quenching (1000 mg); the amount of solvent is those employed during the reaction (891.1 mg of DCM). For the Meier protocol, the amount of water used is those employed during the quenching (670 mg); the amount of solvent is those employed during the reaction (891.1 mg of DCM). In entries 3 and 4 we also considered the quantity of solvent used for the extraction (1 mL of EtOAc on a 0.5 mmol scale; this quantity is the same for Ugi and Meier procedures, with the difference that the solvent used is dichloromethane). In entry 5 and 6 we considered also the solvent used for the chromatography purification. The quantity of solvent used is the same for Ugi, Meier and our procedure. Having very similar Rf, all the isocyanides required approximately 45 mL of petroleum ether/EtOAc 8:2, which was then distilled and recovered (we assumed a recovery of 90%). In entries 7, 8 and 9 we evaluated the contribution of each step of this process, calculating the values of mass intensity for the reaction (MIR), for the workup (MIW, also including the quenching procedure), and for the chromatography purification (MIP).24 For the PMI values of all the isocyanide synthesized, see ESI.† From entries 1–4, it is possible to notice that our procedure is characterized by a minor use of reactants and so it generates few organic wastes. This is particularly evident looking at the values calculated without considering water. Nevertheless, our procedure is more favourable also considering water and has the advantage that the most part of the waste generated is aqueous and so it does not require an incineration process for its discarding, thus producing less CO2. As one can see from entry 9, the greatest contribution to PMI comes from the purification procedures, which generate more wastes and leads to an increase in the PMI values (entries 5 and 6). Notwithstanding, we have demonstrated that in our protocol this step in not mandatory, but the isocyanide synthesized can be directly used in situ or isolated without chromatography purification and used as such.
| Entry | PMI values for compound 6 | Ugi procedurea | Meier procedurea | Our procedure |
|---|---|---|---|---|
| a According to our results following Meier and Ugi procedures (see Table 1, entries 1 and 2). b For the purification, we took into account the solvent for chromatography, assuming a recovery of 90%. c Water is included in this calculation. | ||||
| 1 | PMI substrate, reagents, solvents, water (reaction and quenching) | 35,1 | 35,4 | 29,1 |
| 2 | PMI substrate, reagents, solvents (reaction and quenching) | 21,3 | 24,3 | 8,4 |
| 3 | PMI substrate, reagents, solvents, water (reaction, quenching and workup) | 53,6 | 57,3 | 45,2 |
| 4 | PMI substrate, reagents, solvents (reaction, quenching and workup) | 39,7 | 46,2 | 24,4 |
| 5 | PMI substrate, reagents, solvents, water (reaction, quenching, workup and purification)b | 99,8 | 112,2 | 104,3 |
| 6 | PMI substrate, reagents, solvents (reaction, quenching, workup and purification)b | 85,9 | 101,1 | 83,6 |
| 7 | MIRc | 19,9 | 22,1 | 18,8 |
| 8 | MIWc | 33,7 | 35,2 | 26,1 |
| 9 | MIPc | 46,2 | 54,9 | 59,2 |
Experimental
General isocyanide synthesis
The corresponding N-formamide (1 equiv.) was added to 5 wt% TPGS-750-M/H2O (1 M) and the mixture was stirred for ∼5 min. Afterwards, sodium hydrogen carbonate (2.4 equiv.) and p-TsCl (1.2 equiv.) were added. The reaction was vigorously stirred for 16 hours at room temperature.Work up (0.5 mmol scale): after the addition of 0.5 mL of 10% NH4OH, the mixture was stirred for ∼15 min. Then, it was extracted with EtOAc (0.5 mL × 2), dried over sodium sulfate, and concentrated under reduced pressure. When necessary, the crude material was purified by flash column chromatography.
Procedure for the Passerini reaction with the isocyanide as a crude
The crude 1-isocyanododecane (1 equiv.) was dissolved in DCM, then isobutyraldehyde (1 equiv.) and 2-phenylacetic acid (1 equiv.) were added. The reaction was stirred for 16 hours at room temperature and then concentrated under reduced pressure. The crude material was purified by flash column chromatography (PE/EtOAc 8:2).
Procedure for the one pot tandem isocyanide synthesis and Passerini reaction
The corresponding N-formamide (1 equiv.) was added to 5 wt% TPGS-750-M/H2O (1 M) and the mixture was stirred for ∼5 min. Afterwards, sodium hydrogen carbonate (2.4 equiv.) and p-TsCl (1.2 equiv.) were added. The reaction was vigorously stirred for 16 hours at room temperature. Then isobutyraldehyde (1 equiv.) and 2-phenylacetic acid (1 equiv.) were added to the mixture and the reaction was vigorously stirred for 16 hours at room temperature. It was extracted with EtOAc, dried over sodium sulfate, and concentrated under reduced pressure. The crude material was purified by flash column chromatography (PE/EtOAc 8:2).
Conclusions
We have reported a green methodology for the synthesis of isocyanides via chemical dehydration of formamide in an aqueous environment using the cheap and nontoxic sodium hydrogen carbonate as base and tosyl chloride as dehydrating agent. Although it is very difficult to fulfill all the twelve principles of green chemistry,25 this methodology satisfies many of them. The use of water as reaction medium fulfills principle 1 as well as 5. It is indeed well-known that the use of organic solvents weights for an 80% of waste production in pharmaceutical manufactury.26 The use of 1.2 equiv. of tosyl chloride and 2.4 equiv. of sodium hydrogen carbonate can be seen as an advancement in the mass intensity of the process and as a reduction of hazardous synthesis (principles 3, 4 and 12) in comparison with the classical formamide dehydration procedures. Finally, the reaction can be carried out at room temperature avoiding the use of cooling apparatuses (principle 6), necessary when using POCl3 as dehydrating agent and during the quench of tosyl chloride.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Università del Piemonte Orientale, Novara, and Università degli Studi di Napoli “Federico II” is acknowledged. We dedicate this work to the memory of Professor Giovanni Sorba (1954-2022) who passed away shortly after his retirement.References
-
(a) N. Loste, D. Chinarro, M. Gomez, E. Roldán and B. Giner, J. Cleaner Prod., 2020, 256, 120392 CrossRef CAS
; (b) K. Grieger and A. Leontyev, J. Chem. Educ., 2020, 97, 2657–2663 CrossRef
-
(a)
Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue Jr., John Wiley & Sons, Ltd, 2018 Search PubMed
-
(a)
I. Ugi, Isonitrile Chemistry, 1st edn, Academic Press, New York, 1971, vol. 20 Search PubMed
-
(a)
Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH, 2005 Search PubMed
- M. Suginome and Y. Ito, Sci. Synth., 2004, 19, 445–530 Search PubMed
-
(a) I. Ugi and R. Meyr, Angew. Chem., 1958, 702–703 CrossRef CAS
-
(a) R. Hossaini, M. P. Chipperfield, S. A. Montzka, A. Rap, S. Dhomse and W. Feng, Nat. Geosci., 2015, 8, 186–190 CrossRef CAS
- K. A. Waibel, R. Nickisch, N. Möhl, R. Seim and M. A. R. Meier, Green Chem., 2020, 22, 933–941 RSC
- P. Patil, M. Ahmadian-Moghaddam and A. Dömling, Green Chem., 2020, 22, 6902–6911 RSC
- M. C. Leech, A. Petti, N. Tanbouza, A. Mastrodonato, I. C. A. Goodall, T. Ollevier, A. P. Dobbs and K. Lam, Org. Lett., 2021, 23(24), 9371–9375 CrossRef CAS PubMed
- U. Galli, G. C. Tron, B. Purghè, G. Grosa and S. Aprile, Chem. Res. Toxicol., 2020, 33, 955–966 Search PubMed
- A. Massarotti, F. Brunelli, S. Aprile, M. Giustiniano and G. C. Tron, Chem. Rev., 2021, 121, 10742–10788 CrossRef CAS PubMed
- I. D. Cunningham, J. Chem. Soc., Perkin Trans. 2, 1988, 1485–1490 RSC
-
(a) Y. Lim and A. Stein, Can. J. Chem., 1971, 49, 2455–2459 CrossRef
- B. H. Lipshutz, G. T. Aguinaldo, S. Ghorai and K. Voigtritter, Org. Lett., 2008, 10, 1325–1328 CrossRef CAS PubMed
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed
-
(a) B. H. Lipshutz, Synlett, 2021, 1588–1605 CrossRef CAS
-
R. B. Grossman, The Art of Writing Reasonable Organic Reaction Mechanisms, Springer, New York, 2nd edn, 2003 Search PubMed
- Y.-B. Huang, W. Cai, A. Del Rio Flores, F. F. Twigg and W. Zhang, Anal. Chem., 2020, 92, 599–602 CrossRef CAS PubMed
- V. R. Dayeha, S. L. Chowa, K. Schirmerb, D. H. Lynnc and N. C. Bolsa, Ecotoxicol. Environ. Saf., 2004, 57, 375–382 CrossRef
- C. Krell, R. Schreiber, L. Hueber, L. Sciascera, X. Zheng, A. Clarke, R. Haenggi, M. Parmentier, H. Baguia, S. Rodde and F. Gallou, Org. Process Res. Dev., 2021, 25, 900–915 CrossRef CAS
- D. Paprocki, A. Madej, D. Koszelewski, A. Brodzka and R. Ostaszewski, Front. Chem., 2018, 6, 502 CrossRef CAS PubMed
- https://www.acs.org/content/acs/en/greenchemistry/research-innovation/tools-for-green-chemistry.html .
- F. Pessel, I. Billault and M.-C. Scherrmann, Green Chem., 2016, 18, 5558–5568 RSC
-
P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed
- P. Dunn, R. Henderson, I. Mergelsberg and I. A. Wells, ACS CGI Pharmaceutical Roundtable, Moving Towards Greener Solvents for Pharmaceutical Manufacturing – An Industry Perspective (ACS Green Chemistry Institute), 2009.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc01398c |
| This journal is © The Royal Society of Chemistry 2022 |