DOI:
10.1039/D2GC01172G
(Paper)
Green Chem., 2022,
24, 6145-6155
Production of lactic acid mediated by compressed carbon dioxide on heterogeneous Ni(II) catalysts: a facile approach†
Received
28th March 2022
, Accepted 29th June 2022
First published on 30th June 2022
Abstract
Lactic acid is one of the appealing platform molecules in organic synthesis, currently in high demand after the innovation of bio-plastics. The limitations of traditional manufacturing routes (microbial process and chemical synthesis using petroleum resources) directed us to develop economically and environmentally viable catalytic strategies exploring renewable resources to meet the expected demand. In this study, we demonstrated a method of lactic acid synthesis by fixing CO2 as a waste gas into acetaldehyde used as a biobased feedstock on a highly active, recyclable heterogeneous Ni catalyst. The significance of the developed process lies in the transformation of easily available low-cost starting materials to a high-value product without harsh reaction conditions, extra sensitive reagents, and additional steps to derive the right starting material or to remove impurities, which is advantageous in terms of sustainability. Moreover, the optimization of different reaction parameters ensures achieving high yields (>80%) and an excellent production rate of 6700 μmol g−1 h−1. In addition, the characterization of the catalyst by different spectroscopic techniques suggested an interaction between the substrate and the catalyst, which provides an indication of the possible reaction path, thus a mechanism would be outlined. The successful extension of this process to various aldehydes was possible to produce their corresponding hydroxycarboxylic acid with broad functional group tolerance. Altogether, this methodology opens up a promising avenue for the production of biochemicals from renewable resources, which is a key to develop a sustainable society.
Introduction
Lactic acid (3-hydroxypropionic acid) is a bio-based “platform molecule” with a high industrial demand because it contains both carboxyl and hydroxyl moieties, which on modification can produce a vast array of compounds. Consequently, lactic acid has a wide range of applications from food processing to pharmaceuticals and in cosmetic industries1,2 as well as a feedstock for the synthesis of green solvents such as ethyl acetate.1 Lactic acid is also used as a backbone for different value-added chemicals such as lactate, pyruvic acid, pentadione, 1,2-propandiol, acetoin, and acrylic acid.2 It is likely that the demand for lactic acid as an intermediate will rise further for increasing applications in various end-use industries. Moreover, the fueled demand of bio-degradable plastics (poly-lactic acid; a polymer of lactic acid)3 to mitigate environmental issues caused by synthetic plastics significantly augmented the lactic acid market growth.
Currently, 90% of worldwide industrial-scale production of lactic acid was dominated by the microbial fermentation of suitable carbohydrates from starchy materials such as corn, rice, wheat and potato4 that compete with the food resource; however, use of high-cost raw materials (glucose and sucrose) is uneconomical.5 As an alternative, low-cost lignocellulosic biomass (which is considered as a most promising renewable resource)6 of high sugar content was used for the synthesis of lactic acid. However, the exploitation of this renewable resource requires a pretreatment step to break down the polymeric matrix into sugar using enzymes or alkali treatment or ammonia explosion, which increases the production cost. In addition, the generation of inhibitory products during the reaction decreased lactic acid productivity, whereas the formation of by-products reduced the yield, which consequently necessitate a multi-step purification process and generate enormous amounts of wastes that raise an economical and environmental concern.7
To overcome these challenges, economically appealing sustainable catalytic strategies are intriguing.8,9 In this context, heterogeneous catalysts provide a great opportunity to enhance the process efficiency via the improvement of operational conditions, catalyst design and easy product separation, thereby reducing work-up steps. Tremendous efforts have been devoted to explore several catalytic routes to lactic acid directly from renewable sugar feedstocks8,9 on heterogeneous catalysts. However, working with carbohydrate feedstocks results in poor lactic acid yield due to the generation of various side products related to the complex reaction network of isomerization, retro-aldol reaction and dehydration followed by a 1,2-hydride shift. Thus, to stimulate the lactic acid yield, effective measures are harsh reaction conditions (temperature >200 °C) and the presence of stoichiometric amounts of bases, which restricted large-scale applications.10,11 To avoid these obstacles, and to obtain an impressive yield of lactic acid or lactate, instead of carbohydrates, advances are made with glycerol (a side product of bio-diesel industry)12 as well as high-priced oxidation products of glycerol such as glycolaldehyde,13 pyruvaldehyde14 and dihydroxyacetone15 in an oxidative environment using mainly Cr and Sn salts under alkaline conditions but easy over-oxidation is a major issue.
Here, our objective is to develop an environmentally benign innovative route of lactic acid synthesis using renewable resources such as CO2. Turning CO2 as a C1 source is interesting because it is cheap, abundant and nontoxic but chemically inert and difficult to activate for bond formation. The activation of CO2 requires energy-intensive reagents and/or severe reaction conditions, which could negate the environmental benefits of the utilization of this waste gas. Moreover, use of CO2 as a reactant involves a large amount of concentrated CO2 with high purity; however, direct recovery from atmosphere (e.g., atmosphere; 0.03 vol%) or exhaust gases of transportation systems has high energy penalties and possesses economic constrain or sometimes infeasible because of very low concentrations. Indeed, opportunities exist to capture CO2 in a concentrated form from the exhaust gas of thermal power plants, oil refineries and industrial chemical plants before it enters into atmosphere.16 In this context, CO2 in a compressed form promises a steep reduction in greenhouse gas emission17 because low density allows more sequestration of CO2. Furthermore, the compressed form of CO2 is an ideal working liquid because of its critical point that is close to ambient conditions, which allows a low work compression process. The tunable hybrid properties of the gas or liquid state are advantageous to obtain products with high yields under comparatively mild conditions. Hence, catalytic deactivation and destruction of reactor systems are avoidable. Moreover, it also provides additional benefits in water to overcome the major obstruction of limited solubility of CO2 in the conventional reaction medium while being used as a reactant. Despite these beneficial properties, as a reactant, the efficiency of compressed CO2 is under-utilized.18 These exceptional beneficial properties motivated us to explore compressed CO2 in the lactic acid synthesis.
To pursue the environmentally benign innovative route, we employed most abundant acetaldehyde, which is cheap, and an important bulk chemical can be derived from biomass19 to fix CO2 for lactic acid synthesis. As a readily available substrate, several non-catalytic and catalytic attempts were made to use acetaldehyde as a starting material. For instance, via a non-catalytic route, hydrolysis of lactonitrile (obtained by the reaction between acetaldehyde and HCN at high pressures) with the assistance of concentrated acids produced lactic acid but limited by the use of toxic reagents, harsh reaction conditions and downstream purification process.9a Avoiding the need for hazardous reagents, Morooka et al. employed hydrothermal coupling between acetaldehyde and formic acid in acidic (HCl) subcritical water at 225 °C. Despite the impressive yield of lactic acid (68%), their process faced the complications of reactor corrosion and high energy consumption as a result of severe reaction conditions.20 Alternatively, a catalytic route comprised of transition metal salts to catalyze the carbonylation of acetaldehyde for lactic acid synthesis results in a comparatively low yield of lactic acid (∼44%) at 230 °C and 35.5 MPa of CO pressure.21 Moreover, the use of highly toxic and flammable carbon monoxide (CO) gas at high temperatures and pressures is a major drawback of this process. Therefore, it is extremely desirable to develop an accessible and sustainable catalytic method for the synthesis of lactic acid from acetaldehyde.
Herein, we report a simple strategy, which involves compressed CO2 and acetaldehyde to participate in the synthesis of lactic acid over a heterogeneous Ni catalyst at 70 °C in water. The reaction was accelerated in the presence of Lewis acid promoter (Al2O3) and hydrogen. The developed protocol was devoid of any expensive (rare earth metal salts) or hazardous reagents or stoichiometric amounts of bases and/or severe reaction conditions. The extension of the present method to other aldehydes was also explored.
Results and discussion
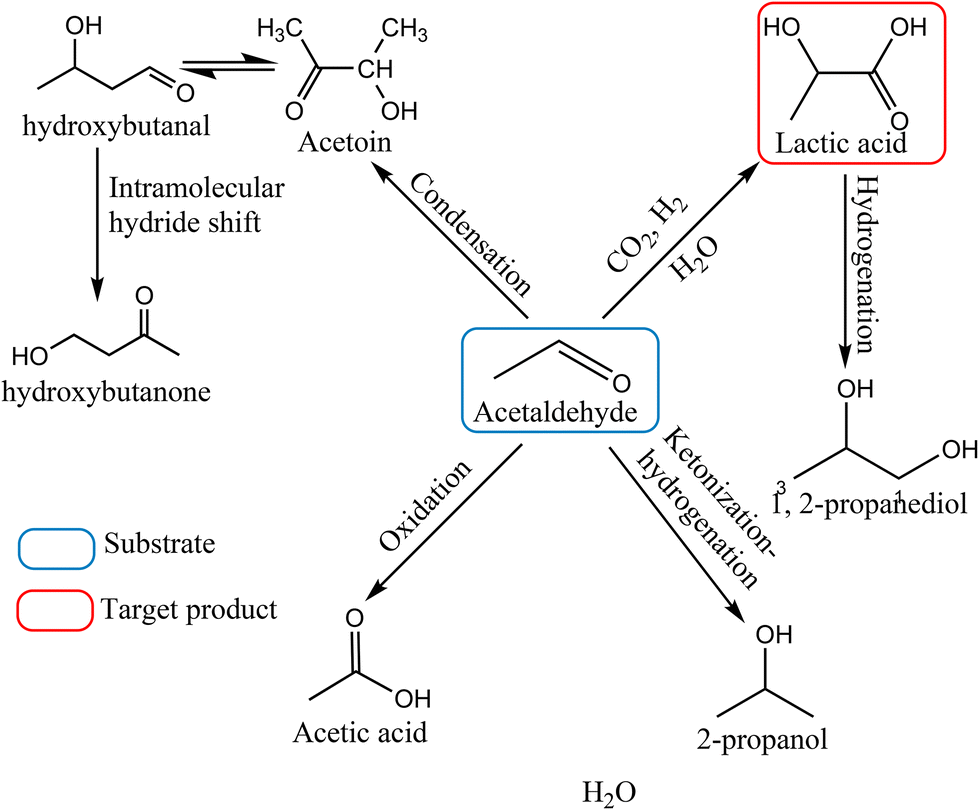
Acetaldehyde is a highly reactive compound, which generally acts as an electrophilic center and possesses an acidic proton in the α-position; thus, it is susceptible to many possible side reactions such as condensation, ketonization, hydrogenation and oxidation under the applied reaction conditions (Scheme 1). For a suitable catalyst, it is indispensable to provide a high yield of the desired product by suppressing those competing side reactions. Furthermore, the sensitivity of each process strongly depends on the reaction conditions; hence, careful modulation of reaction parameters has utmost importance to shift the reaction path towards the preferred direction. We commenced our investigations with the catalyst screening to identify a suitable catalyst followed by the parametric optimization of reaction conditions to achieve the highest yield of lactic acid.
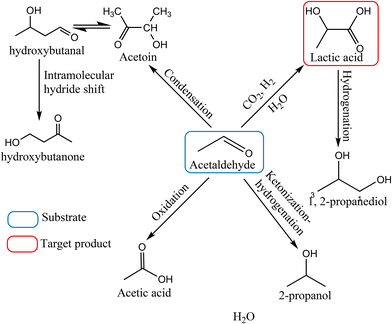 |
| | Scheme 1 Reaction pathway of the conversion of acetaldehyde into lactic acid. | |
Catalyst screening
Our initial experiments began with the exploration of an active catalyst to obtain the maximum yield of lactic acid under mild reaction conditions, which is a challenging task for heterogeneous catalysts, which generally operate under harsh reaction conditions.22 For screening, a series of metals supported on mesoporous MCM-41 were selected as catalysts along with Al2O3; a solid Lewis acid to improve the yield of lactic acid (discussed in the later section). All the metals were chosen based on their expected activity considering CO2 activation and lactic acid synthesis23,24 (Table 1). The result indicates that under the same reaction conditions (PCO2 = 8 MPa, PH2 = 1 MPa, temperature = 70 °C, reaction time = 3 h, water = 1 ml and Al2O3 = 0.0006 mol, and metal content ∼3 wt%), acetaldehyde undergoes different types of reactions depending on the metal used (Table 1; entries 1–8). For instance, instead of lactic acid, Ru produces 2-propanol as the major product with a yield of 65.2% (Table 1; entry 1). However, acetic acid was generated on Pt (yield = 42.1%) and Ir (yield = 24.7%) along with ethanol (Table 1; entries 2 and 3). Interestingly, Au/MCM-41 produces lactic acid with a yield of 10%, which is the highest among the noble metals screened (Table 1; entry 4). Alternatively, non-noble metal catalysts such as Co displayed extremely poor performance (Table 1; entry 5). Among the screened catalysts, Ni/MCM-41 exhibited the best catalytic performance, and lactic acid was obtained with a yield >80% (Table 1; entry 6). In addition to the nature of the metal ion, the catalyst support also plays a crucial role to dictate the activity and product distribution of a specific reaction. Thus, after finding the suitable metal, we studied the activity of Ni catalysts supported on Al2O3 and ZSM-5 as both of the supports are widely used in the synthesis of lactic acid in an aqueous medium without applying alkaline hydrothermal conditions.1,2 Compared to Ni/MCM-41, Al2O3 and ZSM-5-supported Ni catalysts give low yields of lactic acid under the same reaction conditions (Table 1; entries 7 and 8) and follow the trend of MCM-41 (80.2%) > Al2O3 (21.2%) > ZSM-5 (6.3%). It is logical to consider that the highest surface area of MCM-41 facilitated better accessibility of reactants to the active sites of the catalyst and resulted in excellent lactic acid yield. However, despite the larger surface area of ZSM-5 (317.2 m2 g−1) than that of Al2O3 (160 m2 g−1), lactic acid was obtained in poor yields on the former support. Based on this comparison, it can be suggested that the surface area is not necessarily the most decisive factor to obtain the highest yield of lactic acid.
Table 1 Catalyst screening
| Entry |
Catalyst |
Conv. (%) |
Yield (%) |
| Lactic acid |
Cond. productb |
Acetic acid |
Ethanol |
2-Propanol |
Other |
Reaction conditions: catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate = 1:10, Al2O3 = 0.0006 mol, water = 1 ml, temperature = 70 °C, PCO2 = 8 MPa, PH2 = 1 MPa and reaction time = 3 h; MCM-41 supported catalysts were synthesized by the modified method as reported earlier (Catal. Lett., 1999, 61, 199–202); For entries 7 and 8 catalysts were prepared by incipient wetness impregnation method using aqueous solution of Ni-chloride. No additive. 3-Hydroxybutanal, acetoin. Catalysts were chosen based on their efficiency in the utilization of CO2. In each case metal content was ∼3 wt%. All the results presented here are average of 3 measurements. :substrate = 1:10, Al2O3 = 0.0006 mol, water = 1 ml, temperature = 70 °C, PCO2 = 8 MPa, PH2 = 1 MPa and reaction time = 3 h; MCM-41 supported catalysts were synthesized by the modified method as reported earlier (Catal. Lett., 1999, 61, 199–202); For entries 7 and 8 catalysts were prepared by incipient wetness impregnation method using aqueous solution of Ni-chloride. No additive. 3-Hydroxybutanal, acetoin. Catalysts were chosen based on their efficiency in the utilization of CO2. In each case metal content was ∼3 wt%. All the results presented here are average of 3 measurements. |
| 1 |
Ru/MCM-41 |
89.5 |
0.2 |
0 |
20.4 |
0 |
65.2 |
3.8 |
| 2 |
Pt/MCM-41 |
67.4 |
0.3 |
0 |
42.1 |
23.2 |
0 |
1.8 |
| 3 |
Ir/MCM-41 |
47.1 |
0.5 |
0 |
24.7 |
21.3 |
0 |
1.1 |
| 4 |
Au/MCM-41 |
35.0 |
10.5 |
4.9 |
19.5 |
0 |
0 |
0 |
| 5 |
Co/MCM-41 |
26.8 |
0 |
0 |
5.0 |
1.3 |
0 |
6.6 |
| 6 |
Ni/MCM-41 |
92.3 |
80.2 |
0 |
12.1 |
0 |
0 |
0 |
| 7a |
Ni/Al2O3 |
51.9 |
21.2 |
0 |
28.4 |
0 |
0 |
0 |
| 8 |
Ni/ZSM-5 |
35.2 |
6.3 |
7.0 |
10.0 |
10.7 |
0 |
1.2 |
Encouraged by these results, we continued with the Ni/MCM-41 catalyst for the optimization of different reaction parameters involved in the conversion of acetaldehyde. Prior to the investigation of the catalytic activity, structural and morphological characterization of the catalyst was performed by various spectroscopic techniques. Briefly, ordered hexagonal structures of Ni/MCM-41 obtained by a hydrothermal method (details are provided in the Experimental section) can be clearly seen in the XRD pattern of the calcined sample (ESI; Fig. S1†), which was also assessed from the TEM images (ESI; Fig. S2†). Furthermore, mesoporosity was confirmed from the N2 adsorption–desorption analysis (ESI; Fig. S3†). In addition, other spectroscopic techniques are also used to characterize the Ni/MCM-41 catalyst (details are in the ESI†) (Fig. S4 to Fig. S6†). A typical FTIR spectrum was evident for Ni/MCM-41 (Fig. S4†), whereas the existence of Ni in the octahedral coordination and as Ni(II) form was confirmed from the UV-visible spectra (Fig. S5†) and XPS analysis (Fig. S6†), respectively.
Optimization of different reaction parameters
Effect of CO2 pressure.
Fig. 1 evidences the formation of lactic acid after the introduction of CO2 and the yield of lactic acid as a function of CO2 pressure in the range of 4–12 MPa at 70 °C. The result indicates that the yield of lactic acid (presence of lactic acid in the product mixture was confirmed by the 1H NMR spectra; ESI; Fig. S7†) was increased steadily and then reached a maximum of >80% at 8 MPa with the corresponding turnover frequency (TOF) and productivity of 0.98 s−1 (ESI; Fig. S8a†) and 6700 μmol g−1 h−1, respectively. A further increase in CO2 pressure to 12 MPa, substantially reduced the lactic acid yield (4.2%) as well as decreased the TOF (0.37 s−1) and productivity (2544 μmol g−1 h−1). It is worth mentioning that the fluctuation in pressure causes phase separation between gaseous (CO2-hydrogen) and liquid (acetaldehyde-water) phases, consequently determining the existence of acetaldehyde in the water-rich phase or CO2-rich phase.25 As such it is difficult to explain the obtained results; hence, phase behaviour was investigated. Video monitoring of the phase behaviour was conducted in a separate setup (details in the Experimental section) because of the technical constrain related to the reactor used to perform the reaction. After replicating the applied conditions, it is expected to offer an actual scenario on the number of phases present in the system during the reaction. The snapshots of the cell before (ESI; Fig. S9a†) and after the incorporation of acetaldehyde at different CO2 pressures were acquired while investigating the phase behaviour (ESI; Fig. S9b–d†). Naked eye observation indicated a sharp change in the system environment with the alteration of CO2 pressure. At low pressures (4 MPa), CO2 possesses gas-like density; thus, acetaldehyde remains separated as a liquid phase evident below the stirrer bar (Fig. S9b†). Hence, the presence of acetaldehyde is evident in the water-rich phase that shows preference for oxidation to acetic acid,26 which is in line with the observation on the reaction conducted without CO2 (0 MPa); under the applied reaction conditions, acetaldehyde was easily converted into acetic acid in the presence of water (Fig. 1). Interestingly, as the CO2 pressure increased, the liquid phase started to decrease (Fig. S9c†), and at a high pressure of 12 MPa, acetaldehyde-CO2 formed a homogeneous phase (Fig. S9d†). The threshold value of 8 MPa corresponds to the critical pressure at which the substrate existed in a transitional phase with the fluctuation of low-density and high-density regimes was the best pressure (Fig. S9c†) to drive the formation of the intermediate that would allow access to the targeted product, and explain the obtained maximum catalytic efficiency at a particular pressure under the fixed temperature.27 However, at 12 MPa, dense pressurized CO2 starts to extract the reactant from the water-rich phase to the dense CO2-rich phase efficiently,25 which leads to the alteration in the reaction course.28 To explain the maximum rate of reaction (TOF = 0.98 s−1 at 8 MPa), one may invoke in the concept of local concentration.18d In the present system, the local concentration of substrates in the active sites of the catalyst would be very large in a limited pressure range near the critical point, resulting in the promotion of the reaction rate.18d Contrarily, at a higher pressure, increased solubility of acetaldehyde in compressed CO2 diluted the substrate concentration near the catalyst surface, which can explain the reduced catalytic performance (TOF = 0.37 s−1).
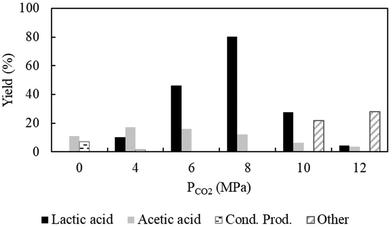 |
| | Fig. 1 Effect of CO2 pressure on product distribution. Reaction conditions: catalyst:substrate = 1:10, Al2O3 = 0.0006 mol, temperature = 70 °C, PH2 = 1 MPa, water = 1 ml and reaction time = 3 h. Cond. Prod. = condensation products. Other = 2-propanol or 1,2-propandiol or unidentified compounds. | |
Effect of temperature.
The results of the assessment of lactic acid yields with the change in temperature from 40 °C to 100 °C are presented in Fig. 2. The significant effect of temperature on the lactic acid yield was prominent; at a low temperature of 40 °C, the yield was as low as 5.9% but reaches a maximum of >80% at 70 °C. However, a further increase in the temperature to 100 °C triggered the secondary reaction of 2-propanol formation via condensation-hydrogenation reactions29 and the yield of lactic acid was significantly dropped to 23.9%. Hence, under the fixed pressure condition, there is every possibility that the change in temperature could disturb the thermodynamic constrain of the system, thus altering the reaction path.28 In addition, the TOF was also accelerated from 0.26 s−1 (productivity = 1787 μmol g−1 h−1) to 1.0 s−1 (productivity = 6876 μmol g−1 h−1) (ESI; Fig. S8b†) induced by the change in temperature from 40 °C to 100 °C at a fixed CO2 pressure of 8 MPa due to kinetics. Notably, the temperature range studied here maintained the vapor state of CO2 and there is a nominal change in the density from 0.14 g ml−1 (40 °C) to 0.09 g ml−1 (100 °C) (NIST chem web book) at a fixed pressure of 8 MPa; thus, no significant changes in the phase behaviour could be observed.
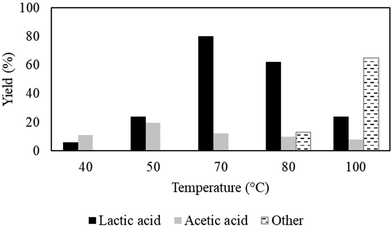 |
| | Fig. 2 Influence of temperature on the catalytic conversion of acetaldehyde to lactic acid. Reaction conditions: catalyst:substrate = 1:10, Al2O3 = 0.0006 mol, PCO2 = 8 MPa, PH2 = 1 MPa, water = 1 ml and reaction time = 3 h. Other = 2-propanol or 1,2-propandiol or unidentified compounds. | |
Variation in hydrogen pressure.
Continued with the optimized CO2 pressure of 8 MPa, we observed that the presence of hydrogen is crucial for the desired reaction to occur. Without hydrogen, the yield of lactic acid was extremely as low as <1% at 70 °C (Fig. 3). Thus, a series of reactions were carried out by varying the hydrogen pressure from 0.2 MPa to 4 MPa at a fixed CO2 pressure and a temperature of 8 MPa and 70 °C, respectively. Fig. 3 shows the promoting effect of hydrogen pressure on the yield of lactic acid, which increased sharply to 18.3% at 0.2 MPa and then reached a maximum of >80% at 1 MPa. With the further increase in hydrogen pressure, the yield of lactic acid started to decrease followed by the generation of 1,2-propanediol. The high solubility of hydrogen in compressed CO2 increased the concentration of the system, consequently facilitating the hydrogenation of lactic acid to 1,2-propanediol;30 hence, a critical control of hydrogen pressure was required to achieve the maximum yield of lactic acid. An optimum hydrogen pressure of 1.0 MPa was chosen throughout the investigation.
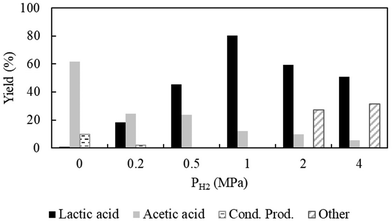 |
| | Fig. 3 Variation in hydrogen pressure. Reaction conditions: catalyst:substrate = 1:10, Al2O3 = 0.0006 mol, temperature = 70 °C, PCO2 = 8 MPa, water = 1 ml and reaction time = 3 h. Cond. Prod. = condensation products. Other = 2-propanol or 1,2-propandiol or unidentified compounds. | |
Variation in the amount of water.
In this study, an aqueous solution of acetaldehyde was used as a reactant to produce lactic acid over Ni/MCM-41. To predict the role of water as a solvent, experiments were conducted in the absence and presence of water (Fig. 4). According to the results, in the absence of water, neat acetaldehyde produces a mixture of condensation products (acetaldol and acetoin) with a very high yield of 86.4%, but lactic acid remains undetectable even in the presence of CO2. In the absence of water, enhanced adsorption of acetaldehyde on the catalyst surface facilitated the bimolecular C–C coupling between the adsorbates, and subsequently, a condensation reaction occurred; but, only after the addition of water, lactic acid was detected. For example, 10% lactic acid was formed with the incorporation of 0.2 ml of water. After confirming the necessity of water, the volume of water varied from 0.2 to 4 ml to check the effect on the catalytic activity (Fig. 4) maintaining the fixed amount of acetaldehyde (1.0 g). As shown in Fig. 4, the yield of lactic acid was increased as a function of the volume of water added and the highest yield of lactic acid (>80%) was obtained using 1 ml of water. Interestingly, the condensation reaction of acetaldehyde was suppressed in the presence of water, which is attributed to the restricted adsorption of acetaldehyde due to the blockage of active sites by water molecules, thus inhibiting the possibility of condensation.31 However, the increased amount of water (4 ml) significantly reduces the yield to only 5.8%, and the TOF of the reaction was also changed from 0.98 s−1 (1 ml) to 0.07 s−1 (4 ml) (ESI; Fig. S8c†) owing to the possible dilution effect (reduced collision between reactants and the catalyst). Hence, water played an important role, which directed the reaction towards the formation of lactic acid and emphasized the requirements of a controlled volume of water to exhibit the best catalytic performance after inhibiting the condensation and minimizing the oxidation of acetaldehyde.
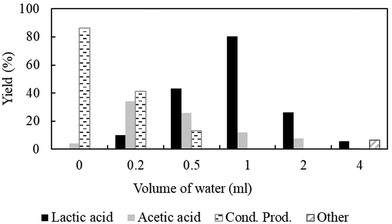 |
| | Fig. 4 Product distribution as a function of the amount of water. Reaction conditions: catalyst:substrate = 1:10, Al2O3 = 0.0006 mol, temperature = 70 °C, PCO2 = 8 MPa, PH2 = 1 MPa and reaction time = 3 h. Cond. Prod. = condensation products. Other = 2-propanol or 1,2-propandiol or unidentified compounds. | |
Effect of the amount of Al2O3.
Even in the presence of water, our investigations on Ni/MCM-41 were detrimental to achieve high yields of lactic acid; only 3.4% desired product was formed (Fig. 5). It has been reported that the presence of solid Lewis acid improves the efficiency of lactic acid formation.32 In analogy to previous studies,32 different solid Lewis acids were tested to obtain high yields of lactic acid. Among them, Al2O3 appeared to be the best choice to augment the catalytic productivity of Ni/MCM-41 towards lactic acid under our standard reaction conditions. Fig. 5 evidences a significant enhancement of lactic acid yield (26.4%) even after the addition of 0.0002 mol of Al2O3, which is almost eight times higher than only Ni/MCM-41 (3.4%) under the same reaction conditions. An increase in the amount of Al2O3 to 0.0006 mol provided the highest yield of lactic acid (>80%). Furthermore, the incorporation of Al2O3 also speeds up the process; TOF was increased from 0.13 s−1 (0 mol) (productivity = 894 μmol g−1 h−1) to 0.98 s−1 (0.0006 mol) (productivity = 6700 μmol g−1 h−1) (ESI; Fig. S8d†). Then, we tested only Al2O3 as a catalyst for the conversion of acetaldehyde into lactic acid by applying the studied condition (Fig. 5). The result indicated an extremely poor lactic acid yield of 0.2% and the significant reduction of TOF (0.029 s−1) (productivity = 138 μmol g−1 h−1) (ESI; Fig. S8d†). The outcome of this investigation confirmed the necessity of the Ni catalyst for lactic acid formation. Moreover, reduced TOF on Al2O3 can be explained by the adsorption of water molecules, which preferentially take up Lewis acid sites (Al+3). This would result in a decrease in the availability of Lewis acid sites for adsorption of acetaldehyde as well as weakens the interaction of CO2 with Al+3 (ref. 32) that inhibits the lactic acid formation. All together, these results inferred a synergism between Ni/MCM-41 and Al2O3, which played a pivotal role to maximize the yield of lactic acid after preventing and/or minimizing side reactions such as condensation and oxidation of acetaldehyde.
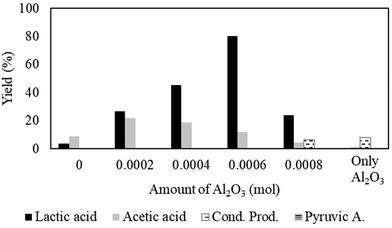 |
| | Fig. 5 Impact of Al2O3 content. Reaction conditions: catalyst:substrate = 1:10, temperature = 70 °C, PCO2 = 8 MPa, PH2 = 1 MPa, water = 1 ml and reaction time = 3 h. Pyruvic A = pyruvic acid, Cond. Prod. = condensation products. | |
Reaction time vs. catalytic activity.
To monitor the progress of the reaction under the optimized condition (temperature = 70 °C, PCO2 = 8 MPa and PH2 = 1 MPa), a time-dependent catalytic performance was studied by varying the reaction time from 0.5 h to 16 h (Fig. 6a) to gain some insights into the reaction path because product distribution of a reaction is closely related to the working conditions of the catalytic system and the stability of the intermediate species on the catalyst surface. There was no detectable product right after the addition of the substrate (0 h). However, after 0.5 h, acetaldehyde was converted into lactic acid and pyruvic acid with yields of 1.6% and 2.3% respectively in addition to acetic acid (yield = 2.1%), which suggested the simultaneous occurrence of CO2-assisted reactions and oxidation of acetaldehyde in an aqueous medium. As a function of reaction time, the yield of lactic acid increased and reached a maximum of >80% within 3 h, but with the further extension of reaction time to 16 h, the yield was reduced to 47.2%. Following the conversion-dependent yield plot (Fig. 6b), a steady increase in lactic acid yield was observed, whereas pyruvic acid disappeared completely after reaching a maximum yield of 5.7%. The observed trend credited pyruvic acid as a possible intermediate of lactic acid formation from acetaldehyde. Therefore, we next examined whether lactic acid could be accomplished using pyruvic acid as a substrate. Only 3.5% of lactic acid was produced from pyruvic acid, which is significantly lower than that obtained from acetaldehyde (>80%) under the same reaction conditions (ESI; Table S1;† entry 1), and discarded the possibility as an intermediate for this reaction. We also tested acetic acid but lactic acid remains undetectable (ESI; Table S1;† entry 2).
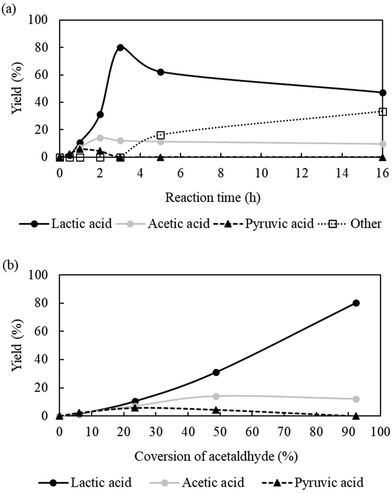 |
| | Fig. 6 (a) Reaction time vs. product distribution profile. (b) Yield plot of lactic acid, acetic acid and pyruvic acid as a function of acetaldehyde conversion. Reaction conditions: catalyst:substrate = 1:10, Al2O3 = 0.0006 mol, temperature = 70 °C, PCO2 = 8 MPa, PH2 = 1 MPa and water = 1 ml. Other = 2-propanol or 1,2-propandiol or unidentified compounds. | |
Substrate scope.
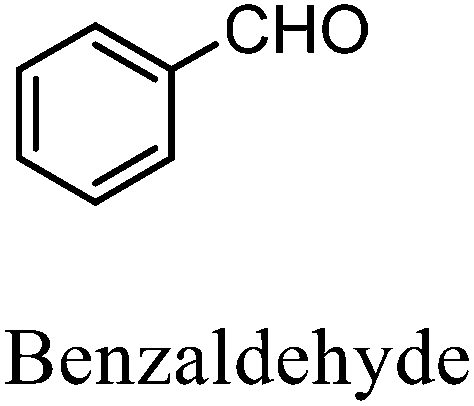
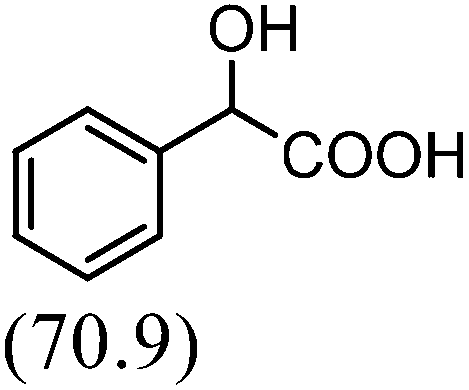
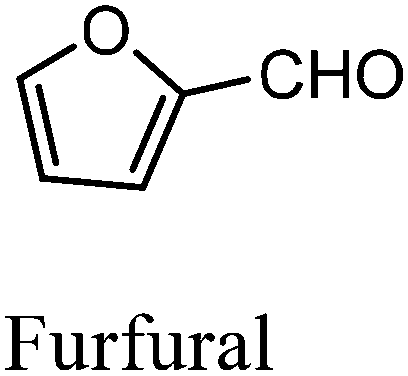
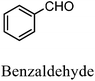
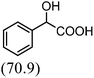
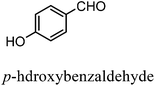
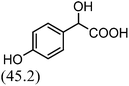
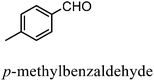
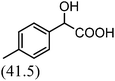
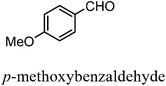
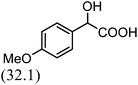
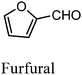
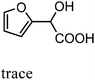
After the successful application of the developed method to the synthesis of lactic acid (hydroxycarboxylic acid), we extended the present protocol on different aldehydes under the optimized reaction conditions, and the results are shown in Table 2. Various hydroxycarboxylic acids can be synthesized through the reaction between CO2 and aldehydes with a high or moderate yield on the Ni catalyst. At first, we evaluated benzaldehyde for the insertion of CO2, which afforded mandelic acid (a biologically important compound and a useful precursor to various drugs) with 70% yield (Table 1; entry 1). Notably, the insertion of CO2 was continued on benzaldehyde with different substitutions. Substituted benzaldehyde was converted into its corresponding hydrocarboxylic acid and the reaction was tolerant to electron-donating and -withdrawing groups but required a longer reaction time of 16 h to obtain satisfactory results. For instance, –OH and –CH3 group-substituted benzaldehyde provided 45.2% and 41.5% yields of their corresponding hydroxycarboxylic acids (Table 2; entries 2 and 3). However, 4-methoxybenzaldehyde underwent a reaction with CO2 to acquire the corresponding product with a yield of 32% (Table 2; entry 3). Next, we turned our attention towards furanic aldehyde such as furfural (Table 2; entry 4). A trace amount of furanglycolic acid was detected in the product mixture after the reaction was conducted for 24 h because furfural is prone to hydrogenation even in the very low hydrogen pressure of 0.1 MPa (Table 2; entry 5) that inhibited the formation of desired hydroxycarboxylic acids.
Table 2 Substrate scope
| Entry |
Substrate |
Product (yield %) |
| Reaction conditions: catalyst:substrate = 1:10, Al2O3 = 0.0006 mol, temperature = 70 °C, PCO2 = 8 MPa, PH2 = 1 MPa and water = 1 ml. Except entry 5, in each case reaction time was 16 h. Entry 5 = 24 h PH2 = 0.1 MPa. |
| 1 |
|
|
| 2 |
|
|
| 3 |
|
|
| 4 |
|
|
| 5 |
|
|
Mechanistic studies.
The elucidation of the reaction mechanism is important and helpful for further implementation of the developed method. On the basis of the aforementioned material characterization and catalytic activity evaluation, a possible mechanism for the synthesis of lactic acid through the insertion of CO2 into acetaldehyde on a Ni catalyst is illustrated in Scheme 2. To prove CO2 as a reaction substrate, further experiments were performed with CO and formic acid (HCOOH), which are well known to be obtained from CO2 by the catalytic hydrogenation as well as carbonate (CO32−) formed by the reaction between CO2 and water. Under the present reaction conditions, when CO was used as a coupling agent, no products except acetic acid were in the detectable range (ESI; Table S1;† entry 3), which confirmed the unsuitability of the reaction conditions. However, lactic acid was formed with an extremely poor yield of 0.6% while using HCOOH as a coupling partner of acetaldehyde (ESI; Table S1;† entry 4), which suggested that the mild reaction condition is ineffective for the C–C bond formation between formic acid and acetaldehyde.20 Since CO2 in a compressed form was present to a significant extent in the system along with water, the generation of carbonic acid/carbonate is possible. To assess the ability of carbonate, the control experiment was conducted using K2CO3 (0.2 mmol), in which the yield of lactic acid was reduced significantly to 2.8% (ESI; Table S1;† entry 5) compared to CO2. In addition, control experiments established that the presence of CO2, hydrogen, water, Ni/MCM-41 and Al2O3 is important to achieve satisfactory yields of lactic acid (ESI; Table S1;† entry 6). Considering the entire scenario, chemical participation of CO2 as a reactant that guided the observed reaction by direct interaction with the substrate catalyzed by the Ni catalyst is proposed. Initially, electron-deficient active Ni(II) (different spectroscopic analyses confirmed the existence of Ni(II) in octahedral coordination; details are provided in the ESI†) facilitated the interaction with the aldehyde. It is worth mentioning that the reduced Ni(0) catalyst was completely inactive for the studied reaction (ESI; Table S1;† entry 7). Thus, the –CHO group of acetaldehyde coordinates with the Ni(II) center in a preferential η2 (C O) configuration resulting in the formation of complex (a).31a In the next step, CO2 would coordinate with the Ni(II) center to form the adduct (b). In this context, Lewis acid is capable of promoting CO2 binding besides increasing the degree of CO2 activation.33 A relevant consideration in this step is the involvement of Al2O3, which could act as a Lewis acid to facilitate the CO2 insertion. This is probably because the coordination of Al(III) to one of the oxygen atoms of CO2 can further polarize CO2 and activate the acceleration of CO2 insertion into the Ni–aldehydic carbon bond,34 which is in line with the experimental observations of dramatic enhancement of the reaction rate from 0.13 s−1 (productivity = 894 μmol g−1 h−1) to 0.98 s−1 (productivity = 6700 μmol g−1 h−1) and the improved yield of lactic acid from 3.4% to >80%, after the addition of Al2O3 along with the Ni catalysts (Fig. 5) under the similar reaction conditions. Next, adduct (b) would form intermediate (c) via the formation of carbon–carbon bond. Finally, protonation provided the desired product (d) and regenerated the active catalyst.
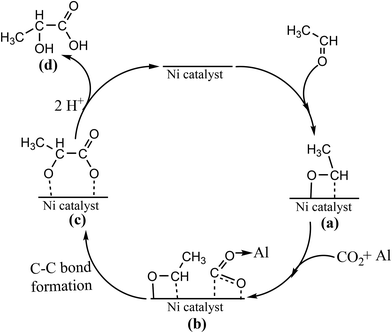 |
| | Scheme 2 A possible reaction mechanism of the transformation of acetaldehyde and CO2 to lactic acid under the studied reaction conditions. | |
DFT calculation.
Although experimental evidences can be correlated with intuitive models to some extent, it is important to identify the transition states, which could have a decisive influence on the reaction path of a particular reaction. In this framework, we carried out calculations using density functional theory (DFT) (details are in the computational method and ESI†) to give the description of the transition states and the reaction mechanism. In view of the reported geometries31a for the adsorption of acetaldehyde, we have considered two different types of geometry for the present case: (i) η1 (O) mode through the oxygen (A) and (ii) η2 (C O) mode via oxygen and aldehydic carbon connecting to the same Ni atom of the Ni–O surface lying parallel to it (B) (Fig. 7). Considering the energy calculations, we observed that the optimized structure B is 7 kcal mol−1 more stable than structure A, which agrees well with the previously reported η2 (C O) mode of adsorption of acetaldehyde on the catalyst surface.31a Furthermore, geometries are also optimized for the interaction of Ni(II)–acetaldehyde adduct (B) with CO2 (Fig. 7; C and D). After the activation by Lewis acid, regarding CO2 insertion, here we considered the options of (i) binding CO2 to the same Ni of the Ni–O surface with which acetaldehyde is bonded (C) and (ii) CO2 may come on the top of aldehydic carbon of acetaldehyde (D) to form a C–C bond. The calculation showed that geometry C was energetically preferred than D by 11.7 kcal mol−1 (energy of C = −33.0 kcal mol−1 and that of D = −21.3 kcal mol−1); thus, D-type geometry was excluded. Continuing with geometry C, the formation of carbon–carbon bonds between CO2 and aldehydic carbon of acetaldehyde affords geometry Evia a transition state of TS-C, and the corresponding activation energy barrier to form E was calculated as 27.8 kcal/mole. In the following step, protonation of E occurs through the transition state of TS-E to produce F with an activation barrier of 25.9 kcal mol−1. Finally, another protonation to F eliminated lactic acid (G) from the catalyst surface after passing the transition state TS-F. The activation barrier for this reaction was obtained as 23.9 kcal mol−1. The energy profile for the total catalytic cycle of Ni-catalyzed conversion of acetaldehyde to lactic acid is also presented (ESI; Fig. S10†).
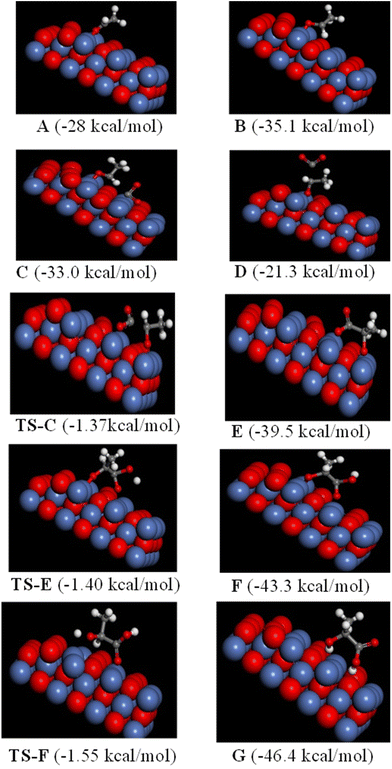 |
| | Fig. 7 Optimized geometries and their corresponding energies related to the step-by step conversion of acetaldehyde to lactic acid. A and B = adsorption mode of acetaldehyde, C and D = insertion of CO2, E = structure of C–C bond formation, F and G = protonation, TS = transition state. Color code: oxygen = red, hydrogen = white, carbon = gray and nickel = blue. | |
Conclusion
In conclusion, we have developed an efficient catalytic strategy rendering sustainable production of lactic acid via the insertion of CO2 into acetaldehyde without the use of any hazardous reagents or stoichiometric amounts of bases or expensive ligands and organic solvents. Some key parameters including CO2 pressure, temperature, volume of water and the presence of Lewis acid that impact the lactic acid yield have been investigated. Indeed, the introduction of the compressed form of CO2 significantly improved the catalytic activity. Neither CO2, nor Ni alone appears as suitable for the reaction, but a combination of both provides high catalytic performance via the proper control of the reaction variables. The optimal temperature (70 °C) and CO2 pressure (8 MPa) were identified to achieve excellent yields, high TOF and production rate in water (1 ml). In addition, the catalytic system provides appreciable tolerance to a series of different types of substrates with various substituents, which generates corresponding hydroxycarboxylic acids in good to excellent yields without affecting the substituent present. A possible reaction mechanism has been proposed from experimental outcome and DFT studies. Overall, this simple straight-forward methodology presented here appears to show great potential to stimulate future research on the further development of clean chemical processes for the incorporation of CO2 into organic frameworks to produce compounds of high interests.
Materials and methods
Acetaldehyde (used as received), Al2O3, sodium hydroxide (NaOH) and Ni(II) chloride were purchased from FUJIFILM Wako Pure Chemicals Corporation. Tetraethyl orthosilicate (TEOS) was used as a silica source obtained from FUJIFILM Wako Pure Chemicals Corporation. Cetyltrimethylammonium bromide (CTAB), a templating agent was obtained from Aldrich. Carbon dioxide (>99.99%) was supplied by Nippon Sanso Co. Ltd.
Catalyst synthesis
Ni/MCM-41 was synthesized in our lab using a modified procedure of the in situ metal introduction method reported previously.35 Briefly, hydrothermal synthesis of Ni containing MCM-41 was accomplished from a gel mixture of molar composition: SiO2:xNi salt:0.14 CTAB:0.56Na2O:120 H2O, where x is the desired amount of metal to be introduced. The gel was prepared by adding 1.30 g of CTAB into 52 g of deionized water containing 0.54 g of NaOH under stirring at ambient temperature for 1 h to obtain a clear solution. To this solution, 5 g of TEOS was added and then stirring was continued for further 1 h. After that, the required amount of metal salt solution was incorporated slowly into the surfactant and silica mixture under continuous stirring until homogeneity was achieved (2 h). The pH of the gel was maintained at 11.3 using NaOH. The resulting gel was then autoclaved at 140 °C for 48 h. After that, the autoclave was quenched with water at room temperature. The obtained solid was separated by filtration and washed thoroughly with deionized water followed by drying at 50 °C for overnight. Prior to use, the template (CTAB) was removed from the synthesized material after calcining in air at 550 °C for 8 h. Catalysts were obtained in 10 different batches with a reproducibility of ±5%. In each case, Si/Ni ratio = 25 was maintained based on the gel composition. After calcination, the final metal content was calculated as 2.67 wt% (≈3 wt%). Prior to the investigation of the catalytic activity, bulk structural characterization and textural analysis were performed using different spectroscopic techniques (XRD, TEM, N2 adsorption–desorption, FTIR spectroscopy, UV-vis spectroscopy and XPS) to confirm the existence of mesoporous structures as well as chemical states of Ni in the calcined materials (details are shown in the ESI; Fig. S1 to Fig. S6†).
Catalytic activity
The catalytic conversion of acetaldehyde into lactic acid was conducted in a 50 ml stainless steel batch reactor. In a typical experiment, 1 g of substrate (acetaldehyde), 0.1 g of Ni/MCM-41 catalyst, 0.0006 mol of Al2O3 and water (1 ml) were charged into the reactor, which was placed in an oven equipped with a magnetic stirrer and a temperature controller. Prior to the temperature stabilization for 1 h without any stirring, the reactor was purged 3 times with nitrogen to remove air. After the required temperature was attained, hydrogen, followed by CO2, was charged into the reactor using a high-pressure liquid pump (JASCO SCF-Get) and then compressed to the desired pressure. The reaction mixture was stirred continuously with a magnetic stirrer during the reaction. After the reaction, the reactor was quenched in an ice bath at 4 °C and then depressurized slowly. The liquid product was separated from the catalyst simply by filtration and then identified by NMR (Bruker Avance III HD 400 MHz) and GC-MS (Varian CP3800 + 1200L), followed by quantitative analysis using a GC (HP 6890) equipped with a capillary column and a flame ionization detector. Detailed analytical methods are given in the ESI.†
Phase observation
In a separate study, the reaction scenario was replicated in a 10 ml high-pressure view-cell fitted with sapphire windows to observe the phase behaviour of CO2-acetaldehyde in the presence of hydrogen. Water and the solid Ni catalyst were omitted for clarity. The cell was placed over a magnetic stirrer for stirring the contents and connected to a pressure controller to regulate the pressure inside the view cell. In addition, a temperature controller was also used to keep the desired temperature of 70 °C ± 0.1 °C. Acetaldehyde was introduced into the view-cell while maintaining the constant hydrogen pressure of 1 MPa and CO2 pressure was varied between 4 and 12 MPa. The content inside the view cell was stirred continuously and the images were recorded in each step (ESI; Fig. S9†).
Computational method
The calculations were formed on a (100) surface model with a 4-layer thickness created from the bulk structure of Ni oxide. The initial geometry of the adsorbed molecules was calculated over the (100) Ni surface using the Adsorption Locator module (Dassault Systemes BIOVIA) that allows finding out the low energy adsorption sites on both periodic and non-periodic substrates as well as investigations on the preferential adsorption of mixtures of adsorbate components. This Adsorption Locator module identifies possible adsorption sites by carrying out Monte Carlo searches of the configurational space of the substrate–adsorbate system as the temperature is slowly decreased within the simulated annealing process of a molecular dynamics run. As described in the ESI,† the minimum energy structures obtained from the Monte Carlo method were then fully optimized by the DFT method.
Conflicts of interest
The authors declare no conflicts of interests.
References
- M. Dusselier, P. V. Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
-
(a) P. Maki-Arvela, I. L. Simakova, T. Salmi and D. Yu. Murzin, Chem. Rev., 2014, 114, 1909–1971 CrossRef CAS PubMed;
(b) P. Maki-Arvela, A. Aho and D. Yu. Murzin, ChemSusChem, 2020, 13, 4833–4855 CrossRef PubMed.
-
(a) M. Dusselier, P. V. Wouwe, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed;
(b) J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. M. Alonso, R. Mariscal and M. L. Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC;
(c) A. Z. Naser, I. Deiaba and B. M. Darras, RSC Adv., 2021, 11, 17151–17196 RSC.
- Y. Wang, H. Meng, D. Cai, B. Wang, P. Qin, Z. Wang and T. Tan, Bioresour. Technol., 2016, 211, 291–297 CrossRef CAS PubMed.
- F. Andres, C. Martinez, E. M. Balciunasa, J. M. Salgado, J. M. D. Gonzalez, A. Convertic and R. P. de Souza Oliveira, Trends Food Sci. Technol., 2013, 30, 70–83 CrossRef.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
-
(a) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed;
(b) Y. Li, S. S. Bhagwat, Y. R. Cortés-Peña, D. Ki, C. V. Rao, Y.-S. Jin and J. S. Guest, ACS Sustainable Chem. Eng., 2021, 9, 1341–1351 CrossRef CAS.
-
(a) M. Finn, J. A. Ridenour, J. Heltzel, C. Cahill and A. Voutchkova-Kostal, Organometallics, 2018, 37, 1400–1409 CrossRef CAS;
(b) L. S. Sharninghausen, J. Campos, M. G. Manas and R. H. Crabtree, Nat. Commun., 2014, 5, 5084 CrossRef CAS PubMed;
(c) P. J. Deuss, K. Barta and J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174–1196 RSC.
-
(a) I. A. Shuklov, N. V. Dubrovina, K. Kghlein and A. Bçrnera, Adv. Synth. Catal., 2016, 358, 3910–3931 CrossRef CAS;
(b) L. Kong, G. Li, H. Wang, W. He and F. Ling, J. Chem. Technol. Biotechnol., 2008, 83, 383–388 CrossRef CAS;
(c) M. Bicker, S. Endres, L. Ott and H. Vogel, J. Mol. Catal. A: Chem., 2005, 239, 151–157 CrossRef CAS;
(d) Y. Sun, L. Shi, H. Wang, G. Miao, L. Kong, S. Li and Y. Sun, Sustainable Energy Fuels, 2019, 3, 1163–1171 RSC.
-
(a) P. Wattanapaphawong, P. Reubroycharoen and A. Yamaguchi, RSC Adv., 2017, 7, 18561–18568 RSC;
(b) M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed;
(c) Y. Wang, W. Deng, B. Wang, Q. Zhang, X. Wan, Z. Tang, Y. Wang, C. Zhu, Z. Cao, G. Wang and H. Wan, Nat. Commun., 2013, 4, 2141 CrossRef PubMed.
-
(a) L. Yang, J. S. S. Carl, J. G. Lynam, X. Yang and H. Lin, Appl. Catal., B, 2015, 162, 149–157 CrossRef CAS;
(b) A. A. Marianou, C. C. Michailof, D. Ipsakis, K. Triantafyllidis and A. A. Lappas, Green Chem., 2019, 21, 6161–6178 RSC;
(c) A. Bayu, A. Yoshida, S. Karnjanakom, K. Kusakabe, X. Hao, T. Prakoso, A. Abudula and G. Guan, Green Chem., 2018, 20, 4112–4119 RSC;
(d) K. Kun-asa, P. Reubroycharoen, K. Yamazaki, N. Mimura, O. Sato and A. Yamaguchi, ChemistryOpen, 2021, 10, 308–315 CrossRef CAS PubMed;
(e) W. Zhang, S. Xu, Y. Xiao, D. Qin, J. Li and C. Hu, Chem. Eng. J., 2021, 421, 130014 CrossRef CAS.
-
(a) Y. Shen, S. Zhang, H. Li, Y. Ren and H. Liu, Chem. – Eur. J., 2010, 16, 7368–7371 CrossRef CAS PubMed;
(b) C. Wang, X. Zhang, J. Li, X. Qi, Z. Guo, H. Wei and H. Chu, ACS Appl. Mater. Interfaces, 2021, 13, 522–530 CrossRef CAS PubMed;
(c) S. Torres, R. Palacio and D. López, Appl. Catal., A, 2021, 621, 118199 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, S. De Smet, R. D. Clercq, L. Verbelen, P. V. Puyvelde, F. E. Du Prez and B. F. Sels, ACS Catal., 2013, 3, 1786–1800 CrossRef CAS.
- Y. Koito, K. Nakajima, M. Kitano and M. Hara, Chem. Lett., 2013, 42, 873–875 CrossRef CAS.
-
(a) G. Innocenti, E. Papadopoulos, G. Fornasari, F. Cavani, A. J. Medford and C. Sievers, ACS Catal., 2020, 10, 11936–11950 CrossRef CAS;
(b) M. S. Rahaman, S. Tulaphol, K. Mills, A. Molley, Md A. Hossain, S. Lalvani, T. Mihom, M. Crocker and N. Sathitsuksanoh, ChemCatChem, 2022, 14, 202101756 CrossRef.
-
P. G. Jessop and R. Noyori, in Green Chemistry using liquid and supercritical carbon dioxide, ed. J. M. DeSimone and W. Tumas; Oxford Press, 2003, ch. 3, pp. 48–61 Search PubMed.
- L. Irwin and Y. Le Moulec, Science, 2017, 356, 805–806 CrossRef CAS PubMed.
-
(a) M. T. Reetz, W. Konen and T. Strack, Chimia, 1993, 47, 493 CrossRef CAS;
(b) P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS;
(c) S. Wesselbaum, U. Hintermair and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8585–8588 CrossRef CAS PubMed;
(d) Z. He, Q. Qian, Z. Zhang, Q. Meng, H. Zhou, Z. Jiang and B. Han, Philos. Trans. R. Soc., A, 2015, 373, 20150006 CrossRef PubMed;
(e) M. Chatterjee, A. Chatterjee, M. Kitta and H. Kawanami, Catal. Sci. Technol., 2021, 11, 4719–4731 RSC.
- H. Zhu, R. Gonzalez and T. A. Bobik, Appl. Environ. Microbiol., 2011, 77, 6441–6450 CrossRef CAS PubMed.
- S. Morooka, N. Matubayasi and M. Nakahara, J. Phys. Chem. A, 2008, 112, 6950–6959 CrossRef CAS PubMed.
- S. K. Bhattacharrya, S. K. Palit and A. R. Das, Ind. Eng. Chem. Prod. Res. Dev., 1970, 9, 92–95 CrossRef.
-
(a) P. Mäki-Arvela, A. Aho and D. Yu. Murzin, ChemSusChem, 2020, 13, 4833–4855 CrossRef PubMed;
(b) P. P. Upare, J. W. Yoon, D. W. Hwang, U.-H. Lee, Y. K. Hwang, D.-Y. Hong, J. C. Kim, J. H. Lee, S. K. Kwak, H. Shin, H. Kim and J.-S. Chang, Green Chem., 2016, 18, 5978–5983 RSC.
-
(a) E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS;
(b) P. Panagiotopoulou, Appl. Catal., A, 2017, 542, 63–70 CrossRef CAS.
- J. Wu, L. Shen, Z.-N. Chen, Q. Zheng, X. Xu and T. Tu, Angew. Chem., Int. Ed., 2020, 59, 10421–10425 CrossRef CAS PubMed.
- M. A. Pigaleva, I. V. Elmanovich, Y. N. Kononevich, M. O. Gallyamov and A. M. Muzafarov, RSC Adv., 2015, 5, 103573–103608 RSC.
- S. Mostrou, A. Nagl, M. Ranocchiari, K. Föttinger and J. A. van Bokhoven, Chem. Commun., 2019, 55, 11833–11836 RSC.
-
(a) E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS;
(b) J. H. Earley, R. A. Bourne, M. J. Watson and M. Poliakoff, Green Chem., 2015, 17, 3018–3025 RSC;
(c) Y. Ikushima, N. Saito, M. Arai and H. W. Blanch, J. Phys. Chem., 1995, 99, 8941–8944 CrossRef CAS;
(d) Y. Ikushima, N. Saito and M. Arai, J. Chem. Eng. Jpn., 1996, 29, 551–553 CrossRef CAS.
- R. Tschan, R. Wandeler, M. S. Schneider, M. M. Schubert and A. Baiker, J. Catal., 2001, 204, 219–229 CrossRef CAS.
- T. Borrmann and P. Swidereka, Eur. Phys. J. D, 2016, 70, 133 CrossRef.
-
(a) K. Liu, X. Huang, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2018, 10, 10810–10817 Search PubMed;
(b) S. Iqbal, S. A. Kondrat, D. R. Jones, D. C. Schoenmakers, J. K. Edwards, L. Lu, B. R. Yeo, P. P. Wells, E. K. Gibson, D. J. Morgan, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 5047–5059 CrossRef CAS.
-
(a) Z. Sun, L. Kong, X. Ding, C. Du, X. Zhao, J. Chen, H. Fu, X. Yang and T. Cheng, Phys. Chem. Chem. Phys., 2016, 18, 9367–9376 RSC;
(b) J. H. Jeong, S. Y. Kim, J. Kim, B. J. Cha, S. W. Han, C. H. Park, T. G. Woo, C. S. Kim and Y. D. Kim, ACS Omega, 2019, 4, 5382–5391 CrossRef CAS PubMed.
-
(a) A. Takagaki, J. Jung and S. Hayashi, RSC Adv., 2014, 4, 43785–43791 RSC;
(b) L. Li, C. Stroobants, K. Lin, P. A. Jacobs, B. F. Sels and P. P. Pescarmona, Green Chem., 2011, 13, 1175–1181 RSC.
- J. A. Buss, D. G. VanderVelde and T. Agapie, J. Am. Chem. Soc., 2018, 140(32), 10121–10125 CrossRef CAS PubMed.
-
(a) F. B. Sayyed and S. Sakaki, Chem. Commun., 2014, 50, 13026–13029 RSC;
(b) X. Lv, L. Zhang, B. Sun, Z. Li, Y.-B. Wu and G. Lu, Catal. Sci. Technol., 2017, 7, 3539–3545 RSC;
(c) D. García-López, L. Pavlovic and K. H. Hopmann, Organometallics, 2020, 39, 1339–1347 CrossRef.
- M. Chatterjee, T. Iwasaki, Y. Onodera and T. Nagase, Catal. Lett., 1999, 61, 199–202 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Abhijit
Chatterjee
b and
Hajime
Kawanami
*a,
Abhijit
Chatterjee
b and
Hajime
Kawanami