
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-driven hydroxylation of testosterone by Synechocystis sp. PCC 6803 expressing the heterologous CYP450 monooxygenase CYP110D1†
Francesco
Mascia
abcd,
Sara B.
Pereira
ab,
Catarina C.
Pacheco
abc,
Paulo
Oliveira
 abc,
Jennifer
Solarczek
e,
Anett
Schallmey
e,
Robert
Kourist
f,
Véronique
Alphand
d and
Paula
Tamagnini
*abc
abc,
Jennifer
Solarczek
e,
Anett
Schallmey
e,
Robert
Kourist
f,
Véronique
Alphand
d and
Paula
Tamagnini
*abc
ai3S – Instituto de Investigação e Inovação em Saúde, Universidade do Porto, R. Alfredo Allen 208, 4200-135 Porto, Portugal. E-mail: pmtamagn@ibmc.up.pt
bIBMC – Instituto de Biologia Celular e Molecular, Universidade do Porto, R. Alfredo Allen 208, 4200-135 Porto, Portugal
cDepartamento de Biologia, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/n, 4169-007 Porto, Portugal
dCNRS, Institut des Sciences Moléculaires de Marseille, UMR7313, Aix Marseille Université, Avenue Escadrille Normandie Niemen 52, 13013 Marseille, France
eInstitut für Biochemie, Biotechnologie & Bioinformatik, Technische Universität Braunschweig, Spielmannstr. 7, 38106 Braunschweig, Germany
fInstitute of Molecular Biotechnology, Graz University of Technology, NAWI Graz, BioTechMed, Petersgasse 14, 8010 Graz, Austria
First published on 6th July 2022
Abstract
The selective hydroxylation of steroids through chemical synthesis is a complex reaction with a high environmental impact. The use of photoautotrophic microorganisms expressing heterologous monooxygenases could overcome this problem by fueling the reaction with electrons and O2 derived from the light-dependent oxidation of water, occurring during photosynthesis. Here, the light-driven selective hydroxylation of testosterone into 15β-hydroxytestosterone was achieved using whole-cells of the unicellular cyanobacterium Synechocystis sp. PCC 6803 expressing the heterologous CYP450 monooxygenase, CYP110D1. Additionally, the reaction conditions including cell density, aeration, and substrate concentration were optimized, leading to a maximum specific activity of 1 U gCDW−1. This value is about 2-fold higher than the one achieved using the model heterotrophic bacterium, E. coli, in which was necessary to express not only CYP110D1 but also its electron partners, and to use glucose as a sacrificial electron donor. Altogether, the results obtained here demonstrate the higher efficiency and sustainability (94% atom economy) of testosterone hydroxylation using our engineered Synechocystis chassis, compared to biocatalysis with heterotrophic microorganisms or chemical synthesis.
Introduction
Cyanobacteria have a great potential to be used as whole-cell biocatalysts for the light-driven oxidation1–4 or reduction of a variety of substrates, with activities close to what is required by industry.5–8 Therefore, cyanobacteria are an appealing solution to perform chemical reactions more sustainably, helping to mitigate the impact that chemical and pharmaceutical industries have on the environment. The potential of these microorganisms as biocatalysts is mainly related to their photoautotrophic metabolism, which allows them to grow using light as energy and CO2 as a carbon source. Moreover, their photosynthetic apparatus can be tapped to provide reducing equivalents resultant from the light-dependent water oxidation reactions to sustain the activity of endogenous or recombinant redox enzymes.9,10 Most of the reports demonstrating the feasibility of this engineering approach used the model unicellular cyanobacterium Synechocystis sp. PCC 6803, owing to its well-known physiology and the availability of established molecular and synthetic biology tools.11–14Among the various classes of redox enzymes, those belonging to the cytochrome P450 (CYP450) monooxygenase superfamily are interesting for several applications, being used as biocatalysts in the production of pharmaceuticals, fine or bulk chemicals, bioremediation agents, flavors, and fragrances.3,15 CYP450s are known to hydroxylate non-activated C–H groups using molecular oxygen as a co-substrate, under mild conditions, and in a regio- and stereo-selective way, with the aid of two electron-carriers, e.g. a NAD(P)H-dependent ferredoxin reductase and a ferredoxin.16,17 CYP450s are particularly suitable to be expressed in cyanobacteria, compared to heterotrophic microorganisms, as the co-expression of the CYP450 electron carriers to sustain the monooxygenase activity may not be required since the photosynthetic apparatus could transfer electrons derived from water oxidation to endogenous ferredoxins, which in turn could provide them to the enzymes.18,19 Consequently, it will not be needed to exogenously supply stoichiometric amounts of sacrificial electron donors (e.g. glucose) for the cofactor regeneration.2 Furthermore, molecular oxygen, the fundamental co-substrate for CYP450 biocatalysis, is produced in situ as a by-product of photosynthesis, thus overcoming air-liquid mass transfer limitations.20
Steroid drugs are an important class of pharmaceutical molecules used as anti-inflammatories, immunomodulators, and physiological regulators, with an estimated market of more than 4 billion USD per year.21 The selective hydroxylation of steroid carbon backbones can alter their physicochemical properties, leading to novel bioactive features, e.g. the hydroxylation in position 15 of the androgenic hormone testosterone was demonstrated to improve its oral bioavailability without affecting liver metabolism.22 This molecule can become a good candidate for the development of oral formulation, overcoming the conventional administration procedure of injecting testosterone undecanoate intramuscularly.23 However, hydroxylation of steroids under synthetic chemistry conditions is often a challenging process, due to the difficulty of selectively oxidizing target non-activated carbon atoms. This may also require several synthetic steps and the extensive use of protecting groups, solvents, and toxic reagents, increasing the process costs and the environmental impact.24 An alternative route to hydroxylate steroids is the use of highly specialized enzymes available in nature, such as a variety of hepatic25 and microbial enzymes26 that are able to selectively hydroxylate testosterone. In a previous study, a library comprised of 213 different bacterial CYP450 genes were expressed in Escherichia coli, and the resulting strains were screened for their ability to use testosterone as a substrate.27 Twenty-four CYP450s were able to mono-hydroxylate testosterone with high selectivity and specificity, and among them, CYP110D1 from the filamentous cyanobacterium Nostoc sp. PCC 7120, was able to convert the substrate into 15β-hydroxytestosterone.27
This study aimed at investigating the potential of cyanobacteria as sustainable whole-cell biocatalysts for the selective hydroxylation of the androgenic steroid testosterone. Since the heterologous expression of CYP450s3,19 was successfully achieved in Synechocystis sp. PCC 6803 and there are a fair number of molecular/synthetic tools available for this model organism, the gene alr4766 (encoding the enzyme CYP110D1 from Nostoc sp. PCC 7120) was introduced into this strain under the control of different regulatory elements and the engineered strains were tested for the conversion of testosterone into 15β-hydroxytestosterone. Subsequently, the conditions promoting this reaction without the addition of sacrificial electron donors were optimized, demonstrating the potential of engineered Synechocystis cells for the selective light-driven hydroxylation of testosterone compared to whole-cell heterotrophic biocatalysts or to chemical synthesis.
Experimental
Organisms and culture conditions
The cyanobacterium Synechocystis sp. PCC 6803 substrain GT-Kazusa28,29 and Synechocystis mutant strains generated in this work were cultivated in BG11 medium,30 supplemented with kanamycin (Km, 50 μg mL−1) in the case of the mutants, at 30 °C and under a 12 hours light (30 μmol photons per m2 per s)/12 hours dark regime. Cultures were routinely maintained in 40 mL of BG11 in 100 mL Erlenmeyer flasks, with orbital shaking at 150 rpm. For cultivation on solid medium, BG11 was supplemented with 1.5% (w/v) noble agar (Difco), 0.3% (w/v) sodium thiosulfate and 10 mM TES-KOH buffer, pH 8.2. For cloning purposes, E. coli strains DH5α (Stratagene) or XL1-Blue (Agilent) were used. For protein expression purposes the E. coli strain C43 (DE3) was used. Cells were grown at 37 °C in LB or TB medium, supplemented with ampicillin (Amp, 100 μg mL−1), kanamycin (Km, 50 μg mL−1), chloramphenicol (25 μg mL−1), or tetracycline (10 μg mL−1).Assembly of the CYP110D1 modules
The open reading frame alr4766 encoding CYP110D1 was amplified by PCR from genomic DNA of Nostoc sp. PCC 7120 (obtained from the Institute Pasteur, France) using primers NS-P-NdeI-FW and NS-P-HindIII-RV (Table S1†). The PCR reaction mixture contained 1 U of PhusionR High-Fidelity DNA Polymerase (NEB), 1× reaction buffer, 200 μM of each deoxyribonucleotide triphosphate, 0.2 μM of each primer, and 150 ng of genomic DNA. The PCR profile was: 5 min at 96 °C, followed by 30 cycles of 1 min at 96 °C, 30 s at 48 °C and 45 s at 72 °C, and a final extension at 72 °C for 10 min. Thereby restriction sites NdeI and HindIII were introduced for subsequent cloning of the gene into vector pIT2-MCS, generating plasmid pIT2::CYP110D1. Generation of vector pIT2-MCS and cloning of vector pACYC::camAB has been described previously.31To assemble the CYP110D1 modules to introduce in Synechocystis, alr4766 was amplified from the vector pIT2 either with primers CYP110D1_RBS0032_SpeI_FW and CYP110D1_PstI_RV, or with primers CYP110D1_His-tag_RBS0032_SpeI_FW and CYP110D1_PstI_RV (Table S1†). The latter pair was designed to introduce a hexahistidine tag at the 5′-end of the sequence; in all cases, the respective open reading frames were preceded by the RBS BBa_B0032 (Registry of Standard Biological Parts, https://parts.igem.org; Table S1†). The PCR reaction mixtures (50 μL) contained 1 U of PhusionR High-Fidelity DNA Polymerase (Thermo-Fisher Scientific), 1× reaction buffer, 200 μM of each deoxyribonucleotide triphosphate, 0.5 μM of each primer, and 10 ng of template DNA. The PCR profile was: 1 min at 98 °C, followed by 10 cycles of 20 s at 98 °C, 20 s at 53 °C and 45 s at 72 °C, 20 cycles of 20 s at 98 °C, 20 s at 65 °C and 45 s at 72 °C and a final extension at 72 °C for 5 min. Each of the amplicons was cloned into pJ201 vectors harboring the synthetic promoters PpsbA2* or Ptrc.x.tetO2,12 between the SpeI and PstI restriction sites. Later, the synthetic modules constituted by the promoter, RBS, and coding sequence were excised with XmaI and PstI restriction enzymes and cloned into the self-replicative vector pSEVA251 (Standard European Vector Architecture repository).32 The correct sequence of the assemblies was confirmed by sequencing (STAB-VIDA).
Generation of the Synechocystis mutants
The synthetic modules generated (see above) were used to transform wild-type Synechocystis as previously described.12 Briefly, cells were grown in BG11 medium until OD730 of ∼0.5. At that point, cells were pelleted at 4190g for 10 minutes and washed three times with 10 mL of 1 mM HEPES buffer, pH 7.5. The cells were finally suspended in 1 mL of 1 mM HEPES and 60 μL aliquots were mixed with 1 μg of purified plasmid DNA (in deionized water), in an electroporation cuvette. The electroporation was performed using a Gene Pulser™ (Bio-Rad), with the capacitor set to 25 μF and the resistor to 400 Ω, for a time constant of 9 ms with an electric field of 12 kV cm−1. After the electric pulse, 400 μL of BG11 were added to the cells, which were then spread onto Immobilon-NC membranes (0.45 μm pore size, 82 mm, Merck Millipore) laying on solid BG11 plates. After incubation at 30 °C under continuous light for 24 hours, the membranes were transferred to solid BG11 plates supplemented with kanamycin and incubated in the same conditions described previously. Colonies were observed after 2–3 weeks. The intracellular presence of the replicative plasmids was confirmed by colony-PCR using specific primers for the gene and the plasmid backbone (Table S1†). To that end, Synechocystis colonies were collected from the BG11 agar plate and suspended in 50 μL of sterile water. The PCR reaction mixture (20 μL) contained 1 U of GoTaq® G2 Flexi DNA Polymerase (Promega), 1× reaction buffer, 2 mM MgCl2, 200 μM of each deoxyribonucleotide triphosphate, 0.5 μM of each primer (Table S1†), and 12 μL of the cell suspension as a template. The PCR profile was: 5 min at 95 °C, followed by 30 cycles of 30 s at 95 °C, 30 s at 55 °C and 70 s at 72 °C, and a final extension at 72 °C for 5 min.Growth measurements (OD), chlorophyll a and dry weight quantification
For growth experiments, Synechocystis harboring the empty pSEVA251 vector or expressing the gene encoding CYP110D1 under the control of either PpsbA2* or Ptrc.x.tetO2 promoters were inoculated in 20 mL of BG11 supplemented with 50 μg mL−1 of kanamycin at OD730 of 0.5 and grown for 7 days under the cultivation conditions described above until an OD730 of ∼2 was reached. Afterward, cells were diluted back to OD730 of 0.5 and 200 μL were transferred to a 96-well microtiter plate that was kept under the same growth conditions. Growth was monitored every day for seven days, using a Synergy™ 2 (Agilent) microplate reader measuring the optical density at 790 nm as previously described.33 For this experiment, three independent biological replicates and four technical replicates were performed. For routine measurements of the growth of Synechocystis cultures, the optical density was measured at 730 nm (ref. 34) using a Shimadzu UVmini-1240 (Shimadzu Corporation) and the chlorophyll a content as described previously.5 The dry weight was also quantified. For that, cells were grown to an OD730 ∼3 and harvested by centrifugation at 4190g, for 10 minutes at RT. After suspending the pellet in BG11 medium to reach an OD730 = 10, 2 mL samples were centrifuged as described above. The resulting pellets were dried overnight at 80 °C and weighed on an analytical scale. The cell dry weight was expressed in g L−1. For this experiment, three independent biological replicates and two technical replicates were performed.SDS-PAGE and western blot
To evaluate the expression of CYP110D1, Synechocystis mutant strains were inoculated in BG11 supplemented with 50 μg mL−1 of kanamycin at OD730 of 0.5 and grown for 7 days under the cultivation conditions described above until an OD730 of ∼2 was reached. Subsequently, these precultures were used to inoculate new cultures to an OD730 of 0.5, which were cultivated under the same conditions for 10 days, until an OD730 of ∼3. At that point, cells were harvested, pelleted by centrifugation, and suspended in lysis buffer (10 mM HEPES pH 8.0, 10 mM EDTA pH 8.0, 0.5% (v/v) Triton X-100, 2 mM DTT) supplemented with EDTA-free protease inhibitor cocktail (Roche), before being disrupted by sonication as described previously.35 After removing cell debris by centrifugation (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 minutes, at 4 °C), proteins in the supernatant were quantified using the BCA protein assay (Thermo Fisher Scientific). For protein analysis, cell extracts were denatured in reducing SDS sample buffer [62.5 Mm Tris-HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 5% (v/v) β-mercaptoethanol, 0.002% (w/v) bromophenol blue] at 95 °C for 5 minutes. 10 or 20 μg of total protein extract were separated by electrophoresis on 12% or 10% (w/v) SDS-polyacrylamide gels.36 The proteins were visualized with PageBlue™ staining solution (Thermo-Fisher Scientific) or blotted onto a nitrocellulose membrane with pore size 0.45 μm (Amersham™ Protan™, GE Healthcare). The transfer of the proteins from the SDS gel to the nitrocellulose membrane was performed within Towbin buffer (25 mM Tris base, 192 mM L-glycine, 20% methanol), applying 100 V for 1 hour. Membranes were probed using a mouse anti-6×-His-Tag (Invitrogen) and an HRP-conjugated anti-mouse IgG a (Sigma) antibodies with a dilution of 1:2000 and 1:5000, respectively. Immunodetection was performed using Clarity™ Western ECL Substrate (Bio-Rad), and images were acquired with a ChemiDoc™ XRS+ Imager (Bio-Rad). The relative signal intensity of the bands obtained was quantified using the Image Lab software (Bio-Rad). Data were statistically analyzed as described below. In this experiment, three independent biological replicates were performed.
000g, 10 minutes, at 4 °C), proteins in the supernatant were quantified using the BCA protein assay (Thermo Fisher Scientific). For protein analysis, cell extracts were denatured in reducing SDS sample buffer [62.5 Mm Tris-HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 5% (v/v) β-mercaptoethanol, 0.002% (w/v) bromophenol blue] at 95 °C for 5 minutes. 10 or 20 μg of total protein extract were separated by electrophoresis on 12% or 10% (w/v) SDS-polyacrylamide gels.36 The proteins were visualized with PageBlue™ staining solution (Thermo-Fisher Scientific) or blotted onto a nitrocellulose membrane with pore size 0.45 μm (Amersham™ Protan™, GE Healthcare). The transfer of the proteins from the SDS gel to the nitrocellulose membrane was performed within Towbin buffer (25 mM Tris base, 192 mM L-glycine, 20% methanol), applying 100 V for 1 hour. Membranes were probed using a mouse anti-6×-His-Tag (Invitrogen) and an HRP-conjugated anti-mouse IgG a (Sigma) antibodies with a dilution of 1:2000 and 1:5000, respectively. Immunodetection was performed using Clarity™ Western ECL Substrate (Bio-Rad), and images were acquired with a ChemiDoc™ XRS+ Imager (Bio-Rad). The relative signal intensity of the bands obtained was quantified using the Image Lab software (Bio-Rad). Data were statistically analyzed as described below. In this experiment, three independent biological replicates were performed.
Light-driven biotransformations with Synechocystis
Cells of Synechocystis mutants were first inoculated in BG11 supplemented with 50 μg mL−1 of kanamycin at OD730 of 0.5 and grown for 7 days under the cultivation conditions described above until an OD730 of ∼2 was reached. Subsequently, these were used to inoculate new cultures to an OD730 of 0.5 and cultivated under the same conditions for 10 days. Afterward, cells were collected by centrifugation (4190g, 10 minutes, at room temperature) and suspended in antibiotic-free BG11 medium to a final OD730 of 5 or 10 (0.9 or 1.8 gCDW L−1). Part of the concentrated cell suspension was used for dry weight and chlorophyll a measurements, and the rest was used for biotransformation experiments. The reaction was initiated by mixing within a 10 mL screw-cap glass vial, 2 mL of cells with 25 μL of testosterone stock solution (40, 80, or 160 mM in DMSO) to reach a final concentration of 0.5, 1 or 2 mM. For each condition tested and every sampling time point, separated reaction vials were used. The reactions were incubated in a photoreactor, providing cool white light (6000–6500 K) with an intensity of 150 μmol photons per m2 per s and magnetic stirring. Reactions in the dark were performed by covering the vials in aluminum foil and keeping them under the same conditions described above. When required, aeration was provided by removing the cap from the vials. Sampling was performed by thoroughly mixing the respective cell suspension with an equal volume of ethyl acetate, drying the resulting organic phase in a vacuum concentrator (SpeedVac), and finally suspending the dried residue in acetonitrile:H2O (60:40). After centrifugation at 14000g for 10 minutes at room temperature, the supernatants were stored at 4 °C until analysis.
HPLC analysis was performed with a Hitachi L-7000 HPLC system (Merck-Hitachi) with a single wavelength UV detector set at 240 nm, equipped with a LiChrospher 100 RP-18 LiChroCART 250 column (250 mm length, 4 mm inner diameter, 5 μm particles size) (Merck). Each sample (10 μL) was chromatographed with a constant acetonitrile concentration in water in a ratio 60:40 (v/v) for 7 min at a flow rate of 1 mL min−1 at 40 °C. For this experiment, three independent biological replicates and two technical replicates were performed.
Expression of CYP110D1 together with Pdx and PdR in E. coli
For activity tests of CYP110D1, the enzyme was co-expressed in E. coli together with redox partners putidaredoxin (Pdx) and putidaredoxin reductase (PdR) from Pseudomonas putida DSM50198.38E. coli C43(DE3)(pACYC::camAB)(pIT2::CYP110D1) cells were grown in 500 mL TB medium containing 25 μg mL−1 chloramphenicol, 10 μg mL−1 tetracycline and 0.1% v/v trace element solution at 37 °C to an optical density at 600 nm (OD600) of 1. Then, expression was induced with isopropyl β-D-1 thiogalactopyranoside (IPTG, 200 μg mL−1), and δ-aminolevulinic acid (84 μg mL−1) was added as a precursor for heme synthesis. After 48 hours of expression at 30 °C, the cells were harvested by centrifugation (4424g, 4 °C for 15 min) and washed once with 40 mM potassium phosphate buffer pH 7.4. Cell pellets were frozen at −20 °C until further use.Testosterone bioconversion with E. coli cells
Frozen cells of E. coli C43 (DE3) (pACYC::camAB) (pIT2::CYP110D1), overexpressing Pdx and PdR together with CYP110D1, were suspended in 100 mM potassium phosphate buffer pH 7.4 to a final OD600 of 40 (corresponding approximately to 13.8 gCDW L−1). Whole-cell reactions were carried out in a 1 mL scale at 30 °C and 200 rpm with 1 mM testosterone [from a 100 mM stock solution in 36% (w/v) hydroxypropyl-β-cyclodextrin in deionized H2O] and 30 mM glucose for cofactor regeneration. After 1 hour, the reaction was extracted with the same volume of ethyl acetate and the solvent was evaporated. The dried residue was dissolved in 200 μL acetonitrile:H2O (60:40) and injected on an HPLC (Prominence, Shimadzu) equipped with a Nucleosil 100-5 C18, 250 × 4.5 mm column (Macherey-Nagel) at 40 °C. Acetonitrile:H2O (60:40) was used as a mobile phase with a flow rate of 1 mL min−1. Detection of substrate and product was performed by UV absorbance at 254 nm. The substrate testosterone and the corresponding hydroxylation product (15β-hydroxytestosterone) were eluted at retention times of 5.9 min and 2.9 min. For this experiment, three independent biological replicates were performed.
Oxygen evolution measurements
The oxygen evolution rates were determined using a Clark-type O2-electrode (Oxygraph; Hansatech Ltd). Calibration was performed using sodium bisulfite and air-saturated water at 30 °C. Cells were grown as described above, harvested, and suspended in fresh BG11 medium to an OD730 of 10 (1.8 gCDW L−1). After incubating 1 mL of concentrated cells in the dark for 1 minute, the assay was started by illuminating the samples for 2 minutes with cool white light (6000–6500 K) with an intensity of 120 μmol photons per m2 per s. After this period 12.5 μL of DMSO or of 80 mM testosterone dissolved in DMSO, were added to the cell suspension, reaching a final substrate concentration of 1 mM. The decrease of the oxygen evolution rate upon the addition of the chemicals was expressed as a percentage of the maximum evolution rate measured during light exposure. For this experiment, three independent biological replicates and two technical replicates were performed.Atom economy calculation
The atom economy of the reaction was calculated as described previously.39Statistical analysis
The statistical analysis was performed using an unpaired t-test, using the software GraphPad Prism 5 (GraphPad Software Inc.).Results and discussion
Expression of the heterologous CYP450 monooxygenase CYP110D1 in Synechocystis
Previously, the selective hydroxylation of testosterone into 15β-hydroxytestosterone mediated by an E. coli co-expressing the CYP450 monooxygenase CYP110D1 (alr4766) from Nostoc sp. PCC 7120, and the electron carriers putidaredoxin (Pdx; camB) and putidaredoxin reductase (PdR; camA) from Pseudomonas putida DSM50198 was reported.27 This reaction was sustained by reducing equivalents originating from the heterotrophic catabolism of glycerol, which was supplemented, in the reaction medium, as a sacrificial electron donor. Here, aiming at performing this hydroxylation using reducing equivalents and molecular oxygen originating from the photosynthetic oxidation of water, the model cyanobacterium Synechocystis sp. PCC 6803 (hereafter Synechocystis) was selected for the heterologous expression of CYP110D1. For this purpose, the gene encoding CYP110D1, in its native form or with the coding sequence of a hexahistidine-tag (His-tag) at its 5′ end, was inserted together with the ribosomal binding site (RBS) BBa_B0032 downstream of the synthetic constitutive promoters PpsbA2* or Ptrc.x.tetO2 (Fig. 1a), since this combination of regulatory parts has been previously successfully used in Synechocystis.12 Each of these synthetic modules was cloned into the self-replicating vector pSEVA251, which was used to transform Synechocystis by electroporation. The presence of the synthetic modules in the mutant strains was confirmed by colony PCR (Fig. S1†). Then, protein extracts were obtained from the different Synechocystis strains, and an SDS-PAGE gel stained with PageBlue revealed the presence of an additional band in Synechocystis expressing CYP110D1 under the control of Ptrc.x.tetO2 promoter, that was not present in the strain harboring the empty pSEVA251 (Fig. 1b–S2†). Under the control of PpsbA2*, it was not possible to observe a distinct band. To further confirm the presence of the heterologous protein CYP110D1, a Western blot analysis using a primary antibody recognizing the His-tag was performed. This method allowed the detection of the tagged CYP110D1 even when under the control of PpsbA2* (Fig. 1c). Moreover, under the regulation of Ptrc.x.tetO2, the level of CYP110D1 was approximately 2-fold higher compared to PpsbA2* (Fig. 1d). This is in agreement with previous studies on the strength of these two promoters that are 6- (PpsbA2*) and 21-fold (Ptrc.x.tetO2) stronger than the cyanobacterial reference PrnpB.12,40 | ||
| Fig. 1 CYP110D1 protein expression in engineered Synechocystis sp. PCC 6803 strains. (a) Schematic representation of the synthetic modules with the coding sequence under the control of the Ppsba2* or Ptrc.x.tetO2 promoter, the BBa_B0032 ribosome binding site, and without or with a sequence coding for a His-tag. Each module was introduced into pSEVA251 upstream the terminator λT0. (b) SDS-PAGE analysis of protein extracts isolated from Synechocystis mutants harboring the empty vector pSEVA251 or the synthetic modules encoding the CYP110D1 His-tag under the control of the Ppsba2* or Ptrc.x.tetO2 promoter. (c) Western blot analysis of protein extracts from Synechocystis mutants harboring the empty vector pSEVA251 or expressing His-tagged CYP110D1 under the control of the Ppsba2* or Ptrc.x.tetO2 promoter. (d) Relative densitometry analysis of CYP110D1 His-tag protein levels. Cells were cultivated in BG11 medium supplemented with 50 μg mL−1 kanamycin, at 30 °C and under a 12 hours light (30 μmol photons per m2 per s)/12 hours dark regimen, until an OD730 ∼3, harvested, suspended in lysis buffer and disrupted by sonication (for details see Experimental section). 20 μg of proteins were loaded in each lane. M, Molecular mass standards are indicated on the left (kDa). Red arrowhead indicates the band corresponding to the heterologous protein CYP110D1. A single representative experiment of three biological replicates is shown. Error bars represent the standard error of the mean (SEM) of 3 biological replicates (*** p-value <0.001). | ||
Light-driven biotransformation of testosterone mediated by Synechocystis expressing CYP110D1
Once the presence of CYP110D1 within the mutant strains was confirmed, a bioconversion reaction was set up to evaluate the efficiency of this enzyme for hydroxylation of testosterone (androst-4-en-17β-ol-3-one). For this purpose, three reaction vials containing either (i) 2 mL of cell-free BG11 medium, (ii) 2 mL of concentrated culture (OD730 = 5; 0.9 gCDW L−1) of Synechocystis harboring the empty pSEVA251, or (iii) 2 mL of concentrated culture (OD730 = 5; 0.9 gCDW L−1) of Synechocystis expressing CYP110D1 under the control of PpsbA2*, plus the substrate (0.5 mM testosterone), were incubated for 21 hours in a homemade photoreactor equipped with white LEDs (150 μmol photons per m2 per s) and a stirring system, placed inside a growth chamber with controlled temperature at 30 °C (Fig. S3a†). The HPLC analysis showed the appearance of a product peak only when Synechocystis cells expressing CYP110D1 were present in the reaction, demonstrating that the conversion was indeed dependent on the enzyme (Fig. S3b†). The product formed was unambiguously identified as 15β-hydroxytestosterone (15-beta,17-beta-dihydroxyandrost-4-en-3-one) by HPLC and GC analysis, having the same mass fragmentation spectra (Fig. S5†) and retention time (Fig. S3b and S4†) as the commercial standard (provided by Steraloids Inc.), and by comparison with 1H and 13C NMR data reported in literature (Tables S3 and S4; Fig. S6–S8†).37 The formation of 15α-hydroxytestosterone was excluded after comparison with the previously described 1H spectrum41,42 of this compound, particularly due to the chemical shifts of H17 and H19 as shown in Table S3.† These results clearly indicate that testosterone was selectively converted by Synechocystis whole-cells harboring the heterologous CYP110D1 into the desired product. Interestingly, and consistently with what was previously observed with two other CYP450s (namely CYP79A1 and CYP77E1), CYP110D1 exhibited its biocatalytic activity without the need to co-express electron carriers and relying only on Synechocystis native ferredoxins, that could be reduced either by photosystem I or by endogenous ferredoxin reductases.19Testosterone hydroxylation by Synechocystis whole-cells harboring different modules for the expression of CYP110D1
After confirming the ability of Synechocystis whole-cells expressing CYP110D1 to hydroxylate testosterone into 15β-hydroxytestosterone, several parameters were evaluated aiming at optimizing this process. To investigate whether the amount of enzyme was a limiting factor, Synechocystis cells expressing the untagged CYP110D1 under the control of the stronger promoter Ptrc.x.tetO2 were tested. As shown in Fig. 2a, the cells of this mutant converted testosterone 3.5-fold more efficiently, achieving 57% substrate conversion, while those with PpsbA2* reached only 16% conversion, after 21 hours. Consistently, the amount of product reached 0.23 mM and 0.06 mM for cells harboring the module under the control of Ptrc.x.tetO2 and PpsbA2*, respectively (Fig. 2b). However, the sum of substrate and product was lower than expected when the biotransformation was catalyzed by cells expressing CYP110D1 under the control of the stronger promoter Ptrc.x.tetO2 (also noticeable in Fig. 3, 4 and Fig. S13†). As incubating the purified product in light with cells harboring the empty pSEVA251 did not cause any change in the product concentration after 21 hours (Fig. S9†), this might be due to the generation of small amounts of side-products or to the product itself being accepted as a substrate by CYP110D1 and be slowly consumed over time. This has been previously reported for other CYP450 monooxygenases, e.g. with Synechocystis expressing the CYP450 CHX from Acidovorax sp. CHX100, the product cyclohexanol was further oxidized to cyclohexanone, but at a slower rate.3 | ||
| Fig. 2 Bioconversion of testosterone into 15β-hydroxytestosterone by Synechocystis whole-cells expressing CYP110D1 under the control of the PpsbA2* or Ptrc.x.tetO2 promoter. (a) % of testosterone converted after 21 hours incubation. (b) Concentration (mM) of testosterone and 15β-hydroxytestosterone in the reaction mixture after 21 hours incubation. Reactions were performed in 2 mL reaction volume, with cells concentrated to OD730 = 5 (0.9 gCDW L−1) in BG11 medium supplemented with 0.5 mM testosterone, in closed vials, at 30 °C, with a light intensity of 150 μmol photons per s per m2. Error bars represent the standard error of the mean (SEM) of 3 biological replicates and 2 technical replicates (*** p-value <0.001). | ||
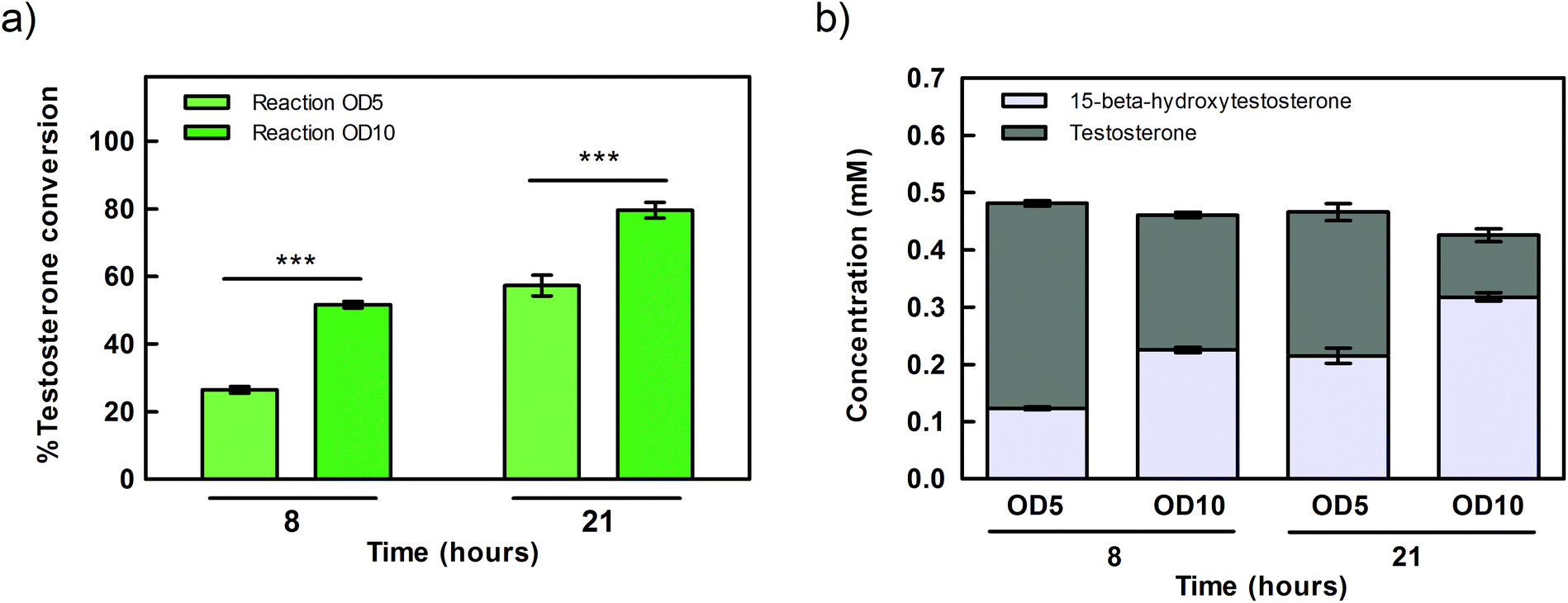 | ||
| Fig. 3 Bioconversion of testosterone into 15β-hydroxytestosterone by Synechocystis whole cells expressing CYP110D1 under the control of Ptrc.x.tetO2 promoter, with cells concentrated to OD730 of 5 or 10 (0.9 or 1.8 gCDW L−1) in the reaction mixture. (a) % of testosterone converted after 8 and 21 hours incubation. (b) Concentration (mM) of testosterone and 15β-hydroxytestosterone in the reaction mixtures after 8 and 21 hours incubation. Reactions were performed by incubating 0.5 mM testosterone in 2 mL reaction volume, in BG11 medium, at 30 °C, in closed vials, with a light intensity of 150 μmol photons per s per m2. Error bars represent the standard error of the mean (SEM) of 3 biological replicates and 2 technical replicates (*** p-value <0.001). | ||
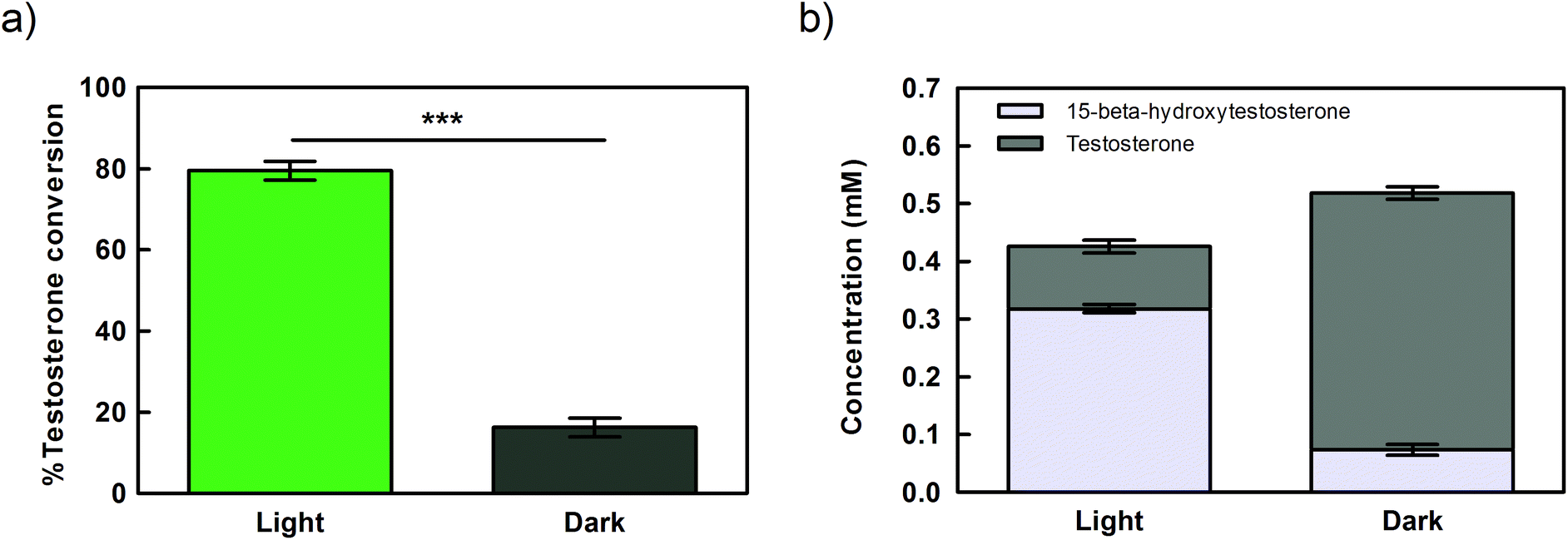 | ||
| Fig. 4 Bioconversion of testosterone into 15β-hydroxytestosterone by Synechocystis whole-cells expressing CYP110D1 under the control of Ptrc.x.tetO2 promoter, under light (150 μmol photons per s per m2) or dark conditions. (a) % of testosterone converted after 21 hours incubation. (b) Concentration of testosterone and 15β-hydroxytestosterone (mM) in the reaction mixtures after 21 hours incubation. Reactions were performed by incubating 0.5 mM testosterone in 2 mL reaction volume, with cells concentrated to OD730 = 10 (1.8 gCDW L−1) in BG11 medium, at 30 °C, in closed vials. Error bars represent the standard error of the mean (SEM) of 3 biological replicates and 2 technical replicates (*** p-value <0.001). | ||
Expressing CYP110D1 under the control of the stronger promoter led to a minor decrease in growth and chlorophyll a content compared to the strains harboring the empty pSEVA251 or expressing the same enzyme under the control of PpsbA2* (Fig. S10 and S11†). This decrease might be due to a small metabolic burden caused by CYP110D1, either directly interfering with cellular processes or generating toxic reactive oxygen species through uncoupling reactions.43
Once we verified that the His-tagged version of CYP110D1 was active (data not shown), the strains expressing this enzyme variant under the control of the two different promoters were also tested and compared. After 21 hours, conversions of 44% and 12% were achieved with cells expressing CYP110D1 under the control of Ptrc.x.tetO2 and PpsbA2*, with the former having a 3.7-fold higher conversion (Fig. S12†). These results are in line with the higher intracellular accumulation of untagged CYP110D1 and His-tag CYP110D1 under the control of Ptrc.x.tetO2 observed previously (Fig. 1b and S2†). Overall, the tagged version had approximately 25% less activity than the native enzyme, therefore Synechocystis expressing the untagged CYP110D1 under the control of Ptrc.x.tetO2 was selected to be used in the next experiments.
Optimization of the cell density in the reaction mixture
Subsequently, the correlation between the reaction mixture cell density and the efficiency of testosterone conversion was investigated. For that, Synechocystis cells expressing CYP110D1 under the control of Ptrc.x.tetO2 were grown under standard conditions and concentrated until OD730 5 or 10 (0.9 or 1.8 gCDW L−1), before being used in the bioconversion assays. The results obtained showed that doubling the cell concentration had a proportional effect on the conversion rate, which raised from 27% to 52% already after 8 hours (Fig. 3a). Consistently with the higher conversion rate, the product formation also increased from 0.12 mM to 0.22 mM (Fig. 3b). After 21 hours of incubation, conversions of 53% and 79% were observed for reaction mixtures at OD730 5 or 10 (0.9 or 1.8 gCDW L−1), respectively (Fig. 3a), corresponding roughly to an increase of about 1.5-fold. This smaller difference could be attributed to the depletion of a reaction component, slowing down the biotransformation after a certain time point. Further increasing cell density was not evaluated, as it was already known that it would lead to a self-shading effect, thus decreasing the overall efficiency of the system.1Light-dependence of testosterone hydroxylation
As the aim of this study was to perform the biotransformation using the reducing power provided by the water oxidation mediated by the photosynthetic electron flow, we evaluated if the reaction was indeed light-dependent. When Synechocystis cells expressing CYP110D1 under the control of Ptrc.x.tetO2 were kept in the dark for 21 hours the bioconversion of testosterone dropped abruptly compared to cells exposed to light (Fig. 4a). This result is corroborated by the analysis of product formation (Fig. 4b), indicating that reducing power and/or the molecular oxygen required to fuel the reaction is mainly provided by the photosynthetic electron flow. Nevertheless, 16% of the substrate was converted under dark conditions (Fig. 4), suggesting that other sources of reducing equivalents, like the storage compounds glycogen or polyhydroxybutyrate, can be catabolized for this purpose.44Influence of aeration and supplementation with inorganic carbon on testosterone hydroxylation
As all monooxygenases, CYP110D1 needs molecular oxygen as a co-substrate to carry out its biocatalytic activity. Although the experiments described above were performed without aeration, with molecular oxygen being generated in situ through photosynthesis, the hypothesis of whether this co-substrate availability may limit CYP110D1 activity was investigated. For this purpose, testosterone was incubated with Synechocystis expressing CYP110D1 under the control of Ptrc.x.tetO2 in either closed or open vials. Higher substrate conversion and increased product formation were obtained with the open vials (Fig. S13†), confirming the importance of aeration for the bioconversion reaction. To verify whether molecular oxygen was indeed the reason for the increased activity observed, cultures of Synechocystis expressing CYP110D1 or harboring the empty pSEVA251 vector were concentrated to OD730 = 10 (1.8 gCDW L−1) and the amount of oxygen dissolved in solution was monitored with an oxygen electrode. Net oxygen evolution rate was measured before and after the addition of testosterone dissolved in DMSO (substrate) or DMSO only (control). The initial oxygen evolution rate decreased by about 25% after the addition of the solvent or, independently of the presence of CYP110D1 (Fig. 5). These results suggest that the decrease is likely due to an effect caused by DMSO. Furthermore, the net oxygen evolution rate detected indicates that O2 is not a limiting factor in the hydroxylation of testosterone under the conditions tested. A possible reason for the increased substrate conversion could be the availability of CO2 for cells with aeration. As previously reported for different cyanobacterial strains, including Synechocystis, in closed systems under light, CO2 can be depleted rather quickly.45–47 CO2 fixation is a major electron sink, and under high-light and carbon limiting conditions an overexcitation of the photosynthetic apparatus may occur, eventually leading to the production of reactive oxygen species and photoinhibition.48 In a previous study, the addition of NaHCO3 in the reaction mixtures resulted in a 25% increase in the specific activity of Synechocystis expressing the operon AlkBGT, encoding an alkane monooxygenase and its electron partners.2 Here, the addition of 50 mM NaHCO3 to closed vials significantly increased testosterone bioconversion and product formation after 8 hours, reaching 83% bioconversion, compared to 57% and 50% in standard BG11 and buffered BG11, respectively (Fig. S14†). Therefore, supplementing inorganic carbon or applying aeration may be ways to prevent photodamage and to maintain the electron flux needed for the activity of heterologous monooxygenases. | ||
| Fig. 5 Net oxygen evolution rate of Synechocystis strains harboring the empty pSEVA251 or expressing CYP110D1 under the control of Ptrc.x.tetO2 promoter, before (t0) or after the addition of 1.25% (v/v) DMSO or 1 mM testosterone dissolved in DMSO (Sub). The measurements were carried out using a Clark-type O2-electrode, in 1 mL volume of concentrated cells OD730 = 10 (1.8 gCDW L−1) in fresh BG11, at 30 °C, with a light intensity of 120 μmol photons per s per m2. Error bars represent the standard error of the mean (SEM) of 3 biological replicates and 2 technical replicates (n.s., not significant, p-value >0.05; ** p-value <0.01; * p-value <0.05). | ||
Influence of substrate concentration on testosterone hydroxylation
The influence of the substrate concentration on the light-driven testosterone hydroxylation catalyzed by Synechocystis expressing CYP110D1 under the control of the Ptrc.x.tetO2 promoter was evaluated. For this purpose, cells concentrated to OD730 = 10 (1.8 gCDW L−1) were incubated in open vials with 0.5, 1, or 2 mM of testosterone. After 21 hours, it was possible to observe 90%, 90%, or 61% conversion of testosterone, respectively (Fig. 6a). Full conversion was never observed, probably due to the substrate hydrophobicity. In fact, a small fraction of it adhered to the vials’ glass as a white precipitate above the reaction mixture, preventing it from getting in contact with the cells. Despite the lower % of conversion observed with 2 mM after 21 hours, it is highly probable that a higher % of conversion will be achieved by extending the reaction time (Fig. 6a). | ||
| Fig. 6 Bioconversion of testosterone into 15β-hydroxytestosterone by Synechocystis whole-cells expressing CYP110D1 under the control of Ptrc.x.tetO2 promoter, incubated with different substrate concentrations. (a) % of testosterone converted at different time points. (b) Concentration of testosterone and 15β-hydroxytestosterone in the reaction mixtures with different substrate concentration, at different time points. (c) Specific activity of the whole-cell biocatalyst expressed as μmol of substrate converted gCDW−1 min−1 after 2 hours of incubation. (d) Biotransformation theoretical productivity after 2 hours of incubation, expressed as μmol of product formed gCDW−1 h−1. Reactions were performed incubating 0.5 mM, 1 mM, or 2 mM testosterone in 2 mL reaction volume, with cells concentrated to OD730 = 10 (1.8 gCDW L−1) in BG11 medium, in open vials, at 30 °C, with a light intensity of 150 μmol photons per s per m2. Error bars represent the standard error of the mean (SEM) of 3 biological replicates and 2 technical replicates (n.s., not significant, p-value >0.05; * p-value <0.05; *** p-value <0.001). When not visible, the error bars are smaller than the size of the symbol. | ||
In order to determine the maximum catalytic capacity of our whole-cell biocatalyst, the initial specific activity was measured. By comparing this value for the substrate concentrations tested, a 21% increase (from 0.85 to 1.03 U gCDW−1) was observed when doubling the testosterone concentration from 0.5 mM to 1 mM. However, a further increase in the substrate concentration to 2 mM did not significantly change this parameter, indicating that the reaction reached a plateau (Fig. 6c). Moreover, the steady specific activity observed with 2 mM excludes the possibility that testosterone had toxic or inhibitory effects on our whole-cell biocatalyst under the conditions tested. An analogous effect was observed with the reaction productivity (Fig. 6d). Nevertheless, the highest product titer was achieved with 2 mM substrate after 21 hours, reaching 0.97 mM, with a 72% yield (Fig. 6b).
As mentioned above, with 0.5 mM substrate it was possible to observe a slight decrease in the product concentration after 21 hours compared to the previous time point (Fig. 6b), supporting our hypothesis that the product itself may be accepted as a substrate by CYP110D1. As shown previously, this problem can be overcome by applying a two-liquid phase system to sequester the product before it could be further converted.3 In this study, the application of a two-liquid phase system was investigated by testing, as an organic overlay, the solvents: di-isononyl phthalate (DINP) or dodecane that were previously shown to be well tolerated by Synechocystis3,49 or ethyl acetate. However, DINP and dodecane failed to efficiently solubilize testosterone and ethyl acetate showed toxic effects on the cyanobacterial cells. Therefore, the possibility to use a two-phase liquid system was not further explored.
Testosterone hydroxylation by photoautotrophic or chemoheterotrophic whole-cell biocatalysts
The hydroxylation of testosterone is generally considered a complex reaction.24 Up to date, only a few examples of whole-cell biocatalysis were reported for the 15β-hydroxylation of this substrate, all carried out using heterotrophic bacteria like Escherichia coli or Pseudomonas putida expressing the enzymes CYP109B1 or CYP106A2.50–52 In these works, although using high cell concentrations and high loads of sacrificial electron donors, low substrate conversions, and sometimes not optimal selectivity were reported (for details see Table 1).50–52| Strains | Enzymes expressed | Testosterone conversion | Cell density | Sacrificial electron donor | Maximum specific activity | Ref. |
|---|---|---|---|---|---|---|
| a Calculated from the original data presented as OD600. b Not determined. | ||||||
| Synechocystis sp. PCC 6803 | CYP110D1 | 67% 1 mM; 86% 0.5 mM; 8 h | ∼1.8 g CDW L −1 | Water | 1.03 U g CDW −1 | This study |
| E. coli C43 (DE3) | CYP110D1 + Pdx + PdR | 42% 1 mM; 1 h | ∼13.8 gCDW L−1 | 5.4 g L−1 glucose (30 mM) | 0.51 U gCDW−1 | This study |
| E. coli BL21 (DE3) | CYP109B1 + Fdx + FdR | 79% 1 mM; 9 h | ∼6.5 gCDW L−1a |
5 g L−1 glucose, 5 g L−1 glycerol | ND | 52 |
| Pseudomonas putida S12 | CYP106A2 + Fdx + FdR | 11% 0.1 mM; 72 h | NDb | 5 g L−1 glucose (27 mM) | ND | 50 |
| E. coli BL21 | CYP106A2 + Adx + AdR + ADH | 40% 0.25 mM; 26 h | ∼4 gCDW L−1a |
400 mM isopropanol | ND | 51 |
In order to compare our engineered Synechocystis expressing CYP110D1 with a heterotrophic whole-cell biocatalyst, an E. coli strain expressing the same CYP450 together with putidaredoxin and putidaredoxin reductase was also generated and tested. For this purpose, 1 mM testosterone was incubated with the aforementioned E. coli strain, concentrated to OD600 = 40 with 30 mM glucose as a sacrificial electron donor. A substrate conversion of 42% was achieved after 1 h (Fig. S15 and S16†), indicating a faster reaction rate compared to Synechocystis. Nonetheless, the maximum specific activity was consistently lower, with 0.51 U gCDW−1 for E. coli and 1.03 U gCDW−1 for Synechocystis (Table 1). Therefore, the higher conversion rate observed with E. coli is most probably due to a higher whole-cell biocatalyst loading, which was 13.8 g L−1 for E. coli and 1.8 g L−1 for Synechocystis. Since the reaction catalyzed by our photoautotrophic strain is light-dependent, higher cell concentrations cannot be reached without addressing the light penetration issue. Therefore, the development of bioreactors with e.g. shorter light pathways such as flat-panel and/or internal illumination systems are mandatory to overcome this challenge.53,54 Moreover, testosterone hydroxylation sustainability was also evaluated according to the principles of green chemistry.39 The use of water as a sacrificial electron donor instead of glucose increased the reaction atom economy55 from 61% to 94%, leading to a minimal waste production and facilitating the downstream processing. Photobiocatalytic testosterone hydroxylation was also considerably more sustainable than the chemical synthesis, not only in terms of waste production and atom economy. The chemical hydroxylation occurs in six synthetic steps, using 3β-androstenedione as a precursor and employing strong acids and bases, and toxic solvents such as dichloromethane. This process also requires derivatization with protecting groups to direct the hydroxylation selectivity, stoichiometric amounts of reagents and high energy inputs.24 In contrast, testosterone hydroxylation with engineered Synechocystis was carried out in a single step, using DMSO (a non-toxic compound) as the only solvent, under mild conditions, with renewable and biodegradable feedstocks, i.e. cyanobacterial whole cells.
Conclusions
In this study, we demonstrated the potential of engineered Synechocystis cells for the selective light-driven hydroxylation of the steroid drug testosterone, using water as the sole sacrificial electron donor. Expressing CYP110D1 under the control of a stronger constitutive promoter (Ptrc.x.tetO2) resulted in higher protein levels and substrate conversion. Reaction parameters were also optimized, and the highest percentage of bioconversion was obtained by adjusting the cell density to OD730 = 10 (1.8 gCDW L−1) and by keeping the vials open (passive aeration). Under these conditions, the maximum specific activity of about 1 U gCDW−1 was reached with 1 mM of substrate. This value is 2-fold higher than the one obtained for E. coli expressing not only the CYP110D1 but also a putidaredoxin, a putidaredoxin reductase, and using glucose as a sacrificial electron donor. Furthermore, the reaction sustainability was improved, being the atom economy close to 100%.Beyond the already promising results obtained with our photoautotrophic biocatalyst, there is still room for improvement. For instance, a more efficient electron supply could be achieved by using other Synechocystis-based chassis (e.g. Δflv1/Δflv3),5 and/or by fusing the CYP450 with a ferredoxin, which will redirect reducing power towards the enzyme in a more efficient way.56 The poor solubility of testosterone in aqueous media can be addressed by using other solvents/co-solvents, or with in situ substrate supply via a two-liquid phase system.3,16,51 Overall, we generated a greener and efficient cyanobacterial whole-cell biocatalyst for the challenging selective hydroxylation of steroid drugs, that could represent a valid alternative to the poorly selective and highly polluting chemical synthesis, or biocatalysis performed by heterotrophic microorganisms.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 764920 MSCA-EJD PhotoBioCat. C. C. Pacheco, P. Oliveira and S. B. Pereira acknowledge FCT – Fundação para a Ciência e a Tecnologia, I.P./Ministério da Ciência, Tecnologia e Ensino Superior for the Assistant Researcher contract CEECIND/00259/2017 (C. C. P.), the FCT investigator grant IF/00256/2015 (P. O.), and Junior Research contract DL57/2016 (S. B. P.).References
- S. Böhmer, K. Köninger, Á. Gómez-Baraibar, S. Bojarra, C. Mügge, S. Schmidt, M. Nowaczyk and R. Kourist, Catalysts, 2017, 7, 240 CrossRef.
- A. Hoschek, B. Buhler and A. Schmid, Biotechnol. Bioeng., 2019, 116, 1887–1900 CrossRef CAS PubMed.
- A. Hoschek, J. Toepel, A. Hochkeppel, R. Karande, B. Buhler and A. Schmid, Biotechnol. J., 2019, 14, 1800724 CrossRef PubMed.
- E. Erdem, L. Malihan-Yap, L. Assil-Companioni, H. Grimm, G. D. Barone, C. Serveau-Avesque, A. Amouric, K. Duquesne, V. de Berardinis, Y. Allahverdiyeva, V. Alphand and R. Kourist, ACS Catal., 2022, 12, 66–72 CrossRef CAS PubMed.
- L. Assil-Companioni, H. C. Büchsenschütz, D. Solymosi, N. G. Dyczmons-Nowaczyk, K. K. F. Bauer, S. Wallner, P. Macheroux, Y. Allahverdiyeva, M. M. Nowaczyk and R. Kourist, ACS Catal., 2020, 10, 11864–11877 CrossRef CAS PubMed.
- K. Koninger, A. Gomez Baraibar, C. Mugge, C. E. Paul, F. Hollmann, M. M. Nowaczyk and R. Kourist, Angew. Chem., Int. Ed., 2016, 55, 5582–5585 CrossRef PubMed.
- V. Jurkaš, C. Winkler, P. Oliveira, S. Poschenrieder, C. Pacheco, E. Ferreira, F. Weissensteiner, P. De Santis, S. Kara, R. Kourist, P. Tamagnini and W. Kroutil, Eng. Microbiol., 2022, 2, 100008 CrossRef.
- H. C. Büchsenschütz, V. Vidimce-Risteski, B. Eggbauer, S. Schmidt, C. K. Winkler, J. H. Schrittwieser, W. Kroutil and R. Kourist, ChemCatChem, 2020, 12, 726–730 CrossRef.
- K. Nakamura and R. Yamanaka, Chem. Commun., 2002, 1782–1783 RSC.
- A. Sengupta, A. V. Sunder, S. V. Sohoni and P. P. Wangikar, J. Biotechnol., 2019, 289, 1–6 CrossRef CAS PubMed.
- E. Englund, F. Liang and P. Lindberg, Sci. Rep., 2016, 6, 36640 CrossRef CAS PubMed.
- E. A. Ferreira, C. C. Pacheco, F. Pinto, J. Pereira, P. Lamosa, P. Oliveira, B. Kirov, A. Jaramillo and P. Tamagnini, Synth. Biol., 2018, 3, ysy014 CrossRef CAS PubMed.
- H.-H. Huang and P. Lindblad, J. Biol. Eng., 2013, 7, 10 CrossRef CAS PubMed.
- C. C. Pacheco, E. A. Ferreira, P. Oliveira and P. Tamagnini, Synthetic biology of cyanobacteria, in The Autotrophic Biorefinery, ed. R. Kourist and S. Schmidt, De Gruyter, 2021, ch. 6, pp. 131–172 Search PubMed.
- Z. Li, Y. Jiang, F. P. Guengerich, L. Ma, S. Li and W. Zhang, J. Biol. Chem., 2020, 295, 833–849 CrossRef PubMed.
- P. Bracco, H. J. Wijma, B. Nicolai, J. A. R. Buitrago, T. Klünemann, A. Vila, P. Schrepfer, W. Blankenfeldt, D. B. Janssen and A. Schallmey, ChemBioChem, 2020, 21, 1–13 CrossRef.
- S. B. Mellor, K. Vavitsas, A. Z. Nielsen and P. E. Jensen, Photosynth. Res., 2017, 134, 329–342 CrossRef CAS PubMed.
- C. Cassier-Chauvat and F. Chauvat, Life, 2014, 4, 666–680 CrossRef PubMed.
- A. Wlodarczyk, T. Gnanasekaran, A. Z. Nielsen, N. N. Zulu, S. B. Mellor, M. Luckner, J. F. B. Thofner, C. E. Olsen, M. S. Mottawie, M. Burow, M. Pribil, I. Feussner, B. L. Moller and P. E. Jensen, Metab. Eng., 2016, 33, 1–11 CrossRef CAS PubMed.
- A. Hoschek, B. Buhler and A. Schmid, Angew. Chem., Int. Ed., 2017, 56, 15146–15149 CrossRef CAS PubMed.
- The Business Research Company, Corticosteroids Global Market Report 2021: COVID 19 Implications And Growth To 2030, https://www.thebusinessresearchcompany.com/report/corticosteroids-therapy-market-global-report-2020-2030-covid-19-implications-and-growth (accessed December 2021).
- E. J. Bunschoten, H. J. T. C. Bennink and R. F. Van Der Linden, US Pat., US7943602B2, 2011 Search PubMed.
- E. Nieschlag and S. Nieschlag, Eur. J. Endocrinol., 2019, 180, R201–R212 CAS.
- I. Černý, J. Fajkoš and V. Pouzar, Steroids, 1996, 61, 58–64 CrossRef.
- T. Niwa, N. Murayama, Y. Imagawa and H. Yamazaki, Drug Metab. Rev., 2015, 47, 89–110 CrossRef CAS PubMed.
- M. Szaleniec, A. M. Wojtkiewicz, R. Bernhardt, T. Borowski and M. Donova, Appl. Microbiol. Biotechnol., 2018, 102, 8153–8171 CrossRef CAS PubMed.
- H. Agematu, N. Matsumoto, Y. Fujii, H. Kabumoto, S. Doi, K. Machida, J. Ishikawa and A. Arisawa, Biosci. Biotechnol. Biochem., 2006, 70, 307–311 CrossRef CAS PubMed.
- Y. Kanesaki, Y. Shiwa, N. Tajima, M. Suzuki, S. Watanabe, N. Sato, M. Ikeuchi and H. Yoshikawa, DNA Res., 2012, 19, 67–79 CrossRef CAS PubMed.
- D. Trautmann, B. Voss, A. Wilde, S. Al-Babili and W. R. Hess, DNA Res., 2012, 19, 435–448 CrossRef CAS PubMed.
- R. Y. Stanier, R. Kunisawa, M. Mandel and G. Cohen-Bazire, Bacteriol. Rev., 1971, 35, 171–205 CrossRef CAS PubMed.
- A. Schallmey, G. den Besten, I. G. P. Teune, R. F. Kembaren and D. B. Janssen, Appl. Microbiol. Biotechnol., 2011, 89, 1475–1485 CrossRef CAS PubMed.
- R. Silva-Rocha, E. Martinez-Garcia, B. Calles, M. Chavarria, A. Arce-Rodriguez, A. de Las Heras, A. D. Paez-Espino, G. Durante-Rodriguez, J. Kim, P. I. Nikel, R. Platero and V. de Lorenzo, Nucleic Acids Res., 2013, 41, D666–D675 CrossRef CAS PubMed.
- F. Opel, N. A. Siebert, S. Klatt, A. Tüllinghoff, J. G. Hantke, J. Toepel, B. Bühler, D. J. Nürnberg and S. Klähn, ACS Synth. Biol., 2022, 11, 1758–1771 CrossRef CAS PubMed.
- S. L. Anderson and L. McIntosh, J. Bacteriol., 1991, 173, 2761–2767 CrossRef CAS PubMed.
- F. Pinto, C. C. Pacheco, P. Oliveira, A. Montagud, A. Landels, N. Couto, P. C. Wright, J. F. Urchueguia and P. Tamagnini, DNA Res., 2015, 22, 425–437 CrossRef CAS PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed.
- N. Fessner, M. Srdič, H. Weber, C. Schmid, D. Schönauer, U. Schwaneberg and A. Glieder, Adv. Synth. Catal., 2020, 362, 2725–2738 CrossRef CAS.
- J. A. Peterson, M. C. Lorence and B. Amarneh, J. Biol. Chem., 1990, 265, 6066–6073 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- H.-H. Huang, D. Camsund, P. Lindblad and T. Heidorn, Nucleic Acids Res., 2010, 38, 2577–2593 CrossRef CAS PubMed.
- N. E. Jacobsen, K. E. Kövér, M. B. Murataliev, R. Feyereisen and F. A. Walker, Magn. Reson. Chem., 2006, 44, 467–474 CrossRef CAS PubMed.
- P. C. Peart, K. P. McCook, F. A. Russell, W. F. Reynolds and P. B. Reese, Steroids, 2011, 76, 1317–1330 CrossRef CAS PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed.
- R. Damrow, I. Maldener and Y. Zilliges, Front. Microbiol., 2016, 7, 966 Search PubMed.
- Y. Allahverdiyeva, H. Mustila, M. Ermakova, L. Bersanini, P. Richaud, G. Ajlani, N. Battchikova, L. Cournac and E.-M. Aro, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4111 CrossRef CAS PubMed.
- P. Lindberg, P. Lindblad and L. Cournac, Appl. Environ. Microbiol., 2004, 70, 2137–2145 CrossRef CAS PubMed.
- J. Zhou, F. Zhang, H. Meng, Y. Zhang and Y. Li, Metab. Eng., 2016, 38, 217–227 CrossRef CAS PubMed.
- N. Murata, S. Takahashi, Y. Nishiyama and S. I. Allakhverdiev, Biochim. Biophys. Acta, 2007, 1767, 414–421 CrossRef CAS PubMed.
- D. Dienst, J. Wichmann, O. Mantovani, J. S. Rodrigues and P. Lindberg, Sci. Rep., 2020, 10, 5932 CrossRef CAS PubMed.
- H. J. Ruijssenaars, E. M. Sperling, P. H. Wiegerinck, F. T. Brands, J. Wery and J. A. de Bont, J. Biotechnol., 2007, 131, 205–208 CrossRef CAS PubMed.
- D. Zehentgruber, F. Hannemann, S. Bleif, R. Bernhardt and S. Lutz, ChemBioChem, 2010, 11, 713–721 CrossRef CAS PubMed.
- X. Zhang, Y. Hu, W. Peng, C. Gao, Q. Xing, B. Wang and A. Li, Front. Chem., 2021, 9, 649000 CrossRef CAS PubMed.
- M. Hobisch, J. Spasic, L. Malihan-Yap, G. D. Barone, K. Castiglione, P. Tamagnini, S. Kara and R. Kourist, ChemSusChem, 2021, 14, 3219–3225 CrossRef CAS PubMed.
- P. Lindblad, D. Fuente, F. Borbe, B. Cicchi, J. A. Conejero, N. Couto, H. Čelešnik, M. M. Diano, M. Dolinar, S. Esposito, C. Evans, E. A. Ferreira, J. Keller, N. Khanna, G. Kind, A. Landels, L. Lemus, J. Noirel, S. Ocklenburg, P. Oliveira, C. C. Pacheco, J. L. Parker, J. Pereira, T. K. Pham, F. Pinto, S. Rexroth, M. Rögner, H.-J. Schmitz, A. M. S. Benavides, M. Siurana, P. Tamagnini, E. Touloupakis, G. Torzillo, J. F. Urchueguía, A. Wegelius, K. Wiegand, P. C. Wright, M. Wutschel and R. Wünschiers, Algal Res., 2019, 41, 101510 CrossRef.
- B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS.
- S. B. Mellor, A. Z. Nielsen, M. Burow, M. S. Motawia, D. Jakubauskas, B. L. Moller and P. E. Jensen, ACS Chem. Biol., 2016, 11, 1862–1869 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1gc04714k |
| This journal is © The Royal Society of Chemistry 2022 |