
Highly efficient Markovnikov hydroaminocarbonylation of alkenes and alkynes catalyzed by a “soluble” heterogeneous Pd catalyst†
Xin
Zhou‡
a,
Zhaozhan
Wang‡
bc,
Bo
Yu
acd,
Shaoping
Kuang
b,
Wei
Sun
 e and
Yong
Yang
*acd
e and
Yong
Yang
*acd
aCAS Key laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology (QIBEBT), Chinese Academy of Sciences, Qingdao 266101, China. E-mail: yangyong@qibebt.ac.cn
bCollege of Environment and Safety Engineering, Qingdao University of Science and Technology, Qingdao 266001, China
cShandong Energy Institute, Qingdao 266101, China
dQingdao New Energy Shandong Laboratory, Qingdao 266101, China
eState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou 730000, China
First published on 8th April 2022
Abstract
Highly efficient and regioselective synthesis of amides from simple starting materials remains a great challenge. Herein, we reported a highly efficient hydroaminocarbonylation of alkenes and alkynes with amines and CO gas catalyzed by a soluble heterogeneous Pd catalyst (Pd@PPOC), in which ultrafine Pd nanoparticles (NPs) were homogeneously dispersed in a well-defined and discrete triphenyl phosphine-built-in porous organic cage (PPOC). The catalyst Pd@PPOC exhibited superior catalytic activity and excellent regioselectivity to Markovnikov adducts, outperforming those previous state-of-the-art Pd catalysts. A diverse set of branched amides and α,β-unsaturated amides were synthesized in high yields, and with broad substrate scopes spanning a range of functional groups that were well tolerated in this synthetic protocol. Remarkably, the soluble catalyst Pd@PPOC demonstrated high stability and could be easily separated and reused up to 10 times with maintenance of the catalytic performance and original structural nature. This study not only provides an efficient and sustainable synthetic method for accessing amides, but also highlights the potential of the MNPs encapsulated in the functional POCs for regioselective catalysis.
Introduction
Hydroaminocarbonylation of alkenes and alkynes represents a most straightforward and atom-economical synthetic approach for accessing amides and α,β-unsaturated amides, which are fundamental structural motifs in natural products, pharmaceuticals, agrochemicals, and functional materials.1,2 Over the past decades, significant advances have been achieved on ligand-controlled Pd-catalyzed hydroaminocarbonylation of readily available alkenes and alkynes as starting materials, and a variety of valuable linear or branched amides and α,β-unsaturated amides have been constructed with strong dependence on phosphine ligands in either a Markovnikov or anti-Markovnikov manner (Scheme 1a).3–13 However, their potential applications, particularly in the pharmaceutical industry, have been severely hampered by the difficulty of separation and recycling of expensive Pd complexes accompanied by purification of the target product. To date, only a few heterogeneous catalysts have been reported for hydroaminocarbonylation, and it remains a formidable challenge to achieve high efficiency and regioselectivity (Scheme 1b). In 2018, Shi and co-workers reported reusable unsupported bulk Pd as a catalyst for ligand-free carbonylation of alkenes with anilines and CO to synthesize diverse amides albeit with poor regioselectivity.14 Later on, the same group developed amide-functionalized porous organic polymer (POP) supported Pd nanoparticles for Markovnikov hydroaminocarbonylation of terminal alkynes to construct α,β-unsaturated amides under phosphine ligand-free conditions, while having a very limited substrate scope.15 Recently, Ding reported a triphenylphosphine-based porous organic polymer supported Pd catalyst (Pd@POPs-PPh3) for hydroaminocarbonylation of aliphatic alkenes with ammonium chloride in the presence of CO, but only a moderate yield and regioselectivity were achieved.16 Therefore, there is a strong need to develop a reusable phosphine-assisted heterogeneous catalyst with high catalytic efficiency to synthesize amides and α,β-unsaturated amides in a regioselective manner.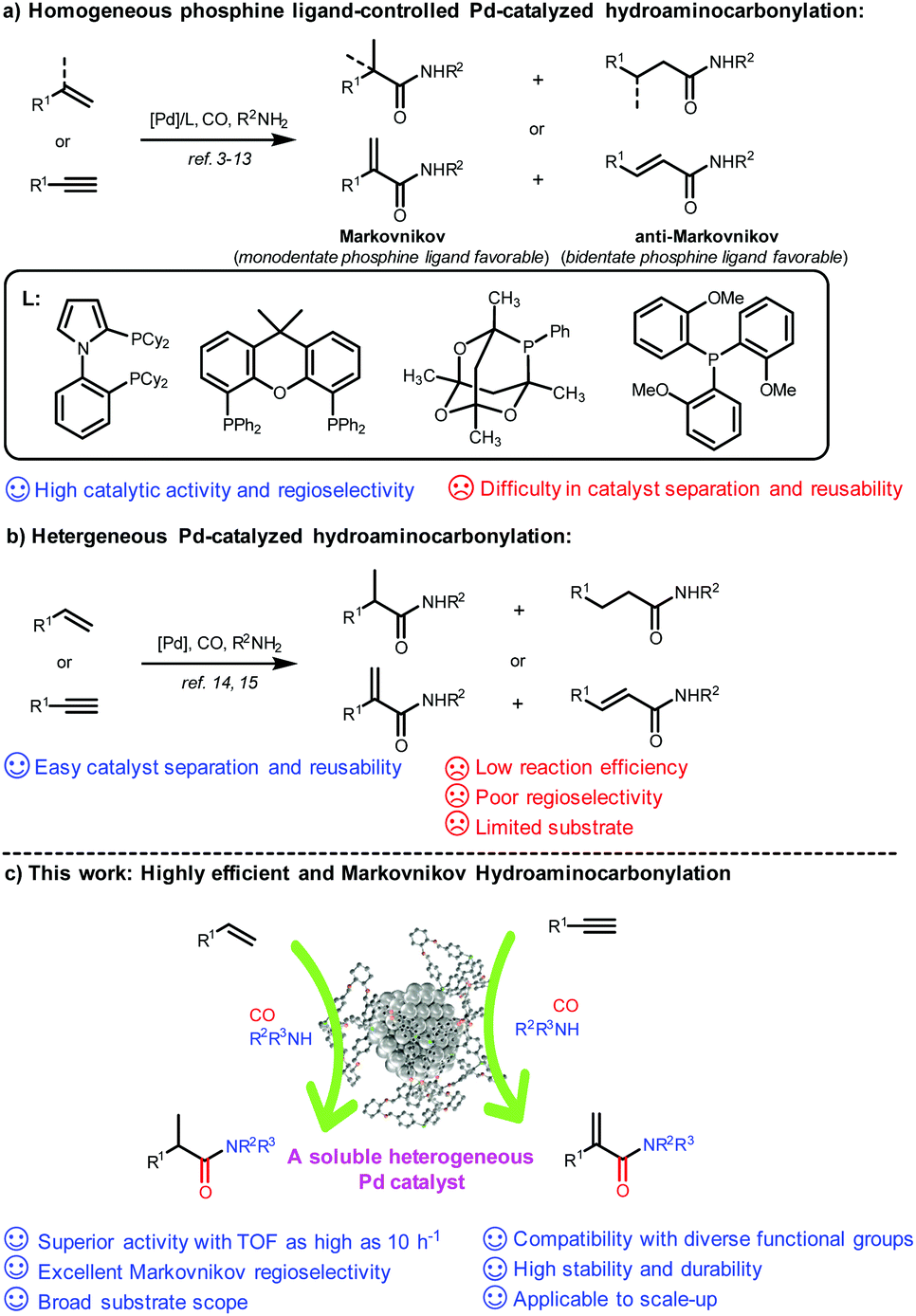 | ||
| Scheme 1 Strategies for Pd-catalyzed hydroaminocarbonylation. | ||
The encapsulation of metal nanoparticles (MNPs) within porous hosts, such as zeolites, metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and dendrimers, represents one of the most significant advances in the field of catalysis and materials sciences.17 However, it still remains a challenge to prepare an encapsulated ultrafine MNP catalyst by simultaneously maintaining superior activity and strong stability and durability.18 In this context, several studies have shown that well-defined and discrete three-dimensional (3D) porous organic cages (POCs) with guest accessible cavities, as a new type of functional molecular open framework,19 could serve as ideal hosts for the size-controlled synthesis of ultrafine MNPs (e.g., Au, Rh, Pd, Pt, etc.) (smaller than 3 nm in size) with high stability by taking full advantage of their confined cage environment and incorporated heteroatoms (N, S, and P) inside the cavity as binding and nucleation sites.20–25 More importantly, POCs have excellent solubility with a well-maintained prefabricated skeleton in certain organic solvents, and thereby could homogenize the heterogeneous ultrafine MNPs to form soluble and stable MNPs with high dispersibility in solution, which could be hardly realized for those insoluble porous hosts.21a,b,22,24b,c,25 As a result, the reactants could rapidly and easily access the encapsulated active MNP sites in the cage as in homogeneous catalysis, thus achieving enhanced catalytic activity and selectivity. In a recent study, we successfully developed a bench-stable triphenyl phosphine-built-in porous organic cage (PPOC) with a large surface area (256 m2 g−1) and hierarchical micro- and mesopores through dynamic imine chemistry, which greatly broadens the functionalities of POCs.26 As a unique porous material, it allows the precisely controlled synthesis of well-dispersed and ultrafine PdNPs with a narrow size distribution (1.7 ± 0.3 nm), labeled as Pd@PPOC, which exhibited superior catalytic activity in a spectrum of cross-coupling reactions of aryl halides due to the positive electronic interaction between ultrafine PdNPs and functional phosphine in the cavities.
Results and discussion
Based on the synergistic effect of the phosphine-built-in POC and ultrafine PdNPs and in combination with our continuous pursuit of facile and sustainable synthetic methods in advanced organic synthesis, we envisioned that this soluble heterogeneous catalyst Pd@PPOC might display a unique catalytic activity and selectivity (including regio- and stereoselectivity) in hydroaminocarbonylation of alkenes and alkynes (Scheme 1c). Hence, we carried out our investigations on hydroaminocarbonylation of styrene with aniline as a benchmark reaction using Pd@PPOC as the catalyst under a CO atmosphere. Firstly, we tested the solubility of the catalyst Pd@PPOC in common organic solvents. As shown in Fig. S1,† it has good solubility and high dispersibility in methanol at room temperature, while it is only partially soluble in N-methylpyrrolidone (NMP). Interestingly, a good solubility with high dispersibility in NMP was observed when the solution was heated to over 60 °C, assuring the formation of a homogeneous phase under relatively higher temperature conditions. Subsequently, several parameters, including catalyst loadings, CO pressures, solvents, and acids, were extensively screened to optimize the reaction conditions, and the results are compiled in Table S1.† It was found that the reaction in the presence of 0.8 mol% of Pd@PPOC, 4.0 MPa pressure of CO, 1.2 equivalents of HCl (with respect to styrene) as an additive in NMP at 110 °C gave the best result, and the Markovnikov adduct 3a was isolated in 96% yield with extremely high regioselectivity (3a/4a = 77/1) within 12 h (Table 1, entry 1). To our surprise, a turnover frequency (TOF) as high as 10 h−1 was achieved, which is the highest value among the previous results for the Pd-catalyzed hydroaminocarbonylation of alkenes (Table S2†). Lowering the catalyst loading to 0.4 mol% also works well but with a slightly longer reaction time for full conversion under otherwise identical conditions (entry 2).|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | NMR yield of 3ab (%) | 3a/4ac |
 a Reaction conditions: Pd source (0.8 mol% Pd), styrene (1a, 0.5 mmol), aniline (2a, 0.6 mmol), HCl (0.6 mmol), CO (4.0 MPa), NMP (3.0 mL), PPh3/Pd molar ratio of 2.
b Determined by NMR.
c Determined by GC-MS of the crude products.
d Isolated yield.
a Reaction conditions: Pd source (0.8 mol% Pd), styrene (1a, 0.5 mmol), aniline (2a, 0.6 mmol), HCl (0.6 mmol), CO (4.0 MPa), NMP (3.0 mL), PPh3/Pd molar ratio of 2.
b Determined by NMR.
c Determined by GC-MS of the crude products.
d Isolated yield.
|
|||
| 1 | None | 98 (96)d | 77 |
| 2 | 0.4 mol% Pd@PPOC instead | 81 | 74 |
| 3 | Pd(PPh3)2Cl2 instead | 91 | 47 |
| 4 | Pd(PPh3)4 instead | 75 | 24 |
| 5 | Pd(OAc)2 plus PPh3 instead | 92 | 15 |
| 6 | Pd(NO3)2 plus PPh3 instead | 88 | 29 |
| 7 | Pd(TFA)2 plus PPh3 instead | 90 | 40 |
| 8 | PdCl2 plus PPh3 instead | 38 | 23 |
| 9 | Pd@CC2 instead | 8 | 17 |
| 10 | Pd@CC3 instead | 4 | 12 |
| 11 | Pd/C instead | 0 | — |
| 12 | Pd@PVP instead | 0 | — |
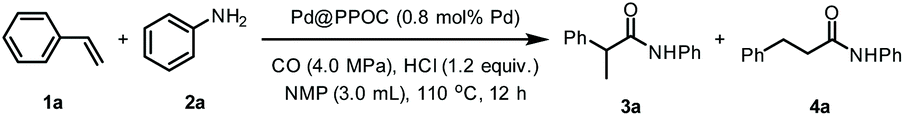
Under the optimized reaction conditions, we employed other commercially available homogeneous Pd catalysts for the reaction, including Pd(PPh3)2Cl2, Pd(PPh3)4, and different Pd salts (e.g., Pd(OAc)2, Pd(NO3)2, Pd(TFA)2 (TFA = trifluoroacetate), and PdCl2) combined with PPh3 as the ligand and maintaining a PPh3/Pd molar ratio of 2 (entries 3–8). It turns out that none of them gave better catalytic activity and regioselectivity than Pd@PPOC. Such an observation shows that the soluble Pd@PPOC with high dispersibility in solution not only outperforms those homogeneous Pd counterparts in catalytic activity but also considerably enhances the regioselectivity to the Markovnikov adduct. This might be ascribable to the electronic effect and confinement of PdNPs in the discrete and well-defined open cavities of PPOC.
To demonstrate the unique advantage of the phosphine-containing POC, we employed two soluble vicinal diamine-containing POCs (CC2 and CC3)19a,23a as the support to encapsulate PdNPs (Schemes S1 and S2, see the details in the ESI†). The resultant catalysts Pd@CC2 and Pd@CC3 with a roughly equal PdNP size (1.9 and 2.1 nm) showed poor reactivity and regioselectivity than Pd@PPOC under otherwise identical conditions, respectively (entries 9 and 10). This finding strongly indicates the critical role of phosphine implanted into the cavity of POC for boosting the catalytic activity and regioselectivity, which is also consistent with our previous results in Pd-catalyzed cross-couplings of aryl halides with the assistance of phosphine ligands.26 In addition, the commercially available heterogeneous Pd/C and the conventional surfactant polyvinylpyrrolidone (PVP) protected PdNPs (Pd@PVP) showed no reactivity (entries 11 and 12), further supporting the necessity of phosphine ligands for achieving superior activity and excellent regioselectivity in the reaction.
We subsequently investigated the stability and recyclability of the catalyst Pd@PPOC using the benchmark reaction under the optimized conditions. After completion of each run, the catalyst was successfully recovered from the reaction solution by centrifugation, washed with water and ethanol, dried at 50 °C for 4 h, and then used in a fresh reaction. As shown in Fig. 1, the catalyst Pd@PPOC could be recovered and recycled for up to 10 times without distinct loss of both activity and regioselectivity, indicating excellent stability and durability. The loading of palladium in Pd@PPOC was determined to be 14.7 wt% after 10 cycles by inductively coupled plasma optical emission spectrometry (ICP-OES), which is very close to the fresh one (15.0 wt%). HR-TEM images (Fig. S2†) for the used catalyst Pd@PPOC show no aggregation in the PdNP size with maintenance of high dispersion as the fresh one, further demonstrating that it has high stability during the catalytic reaction.
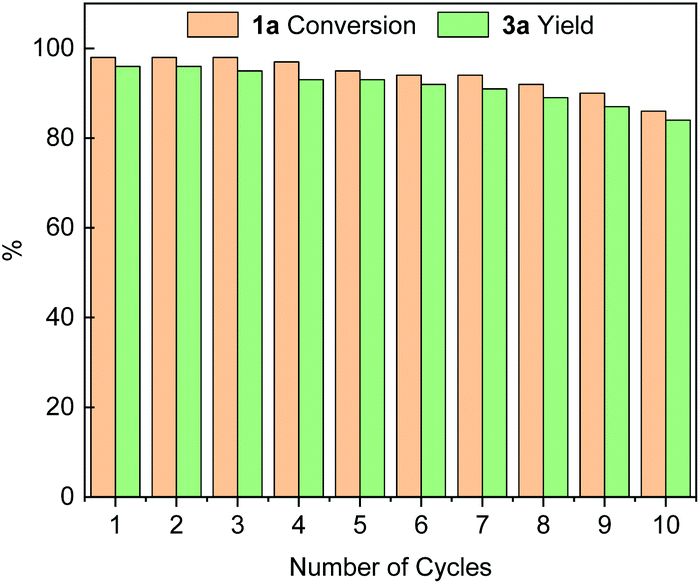 | ||
| Fig. 1 Recycling stability of the catalyst Pd@POC. | ||
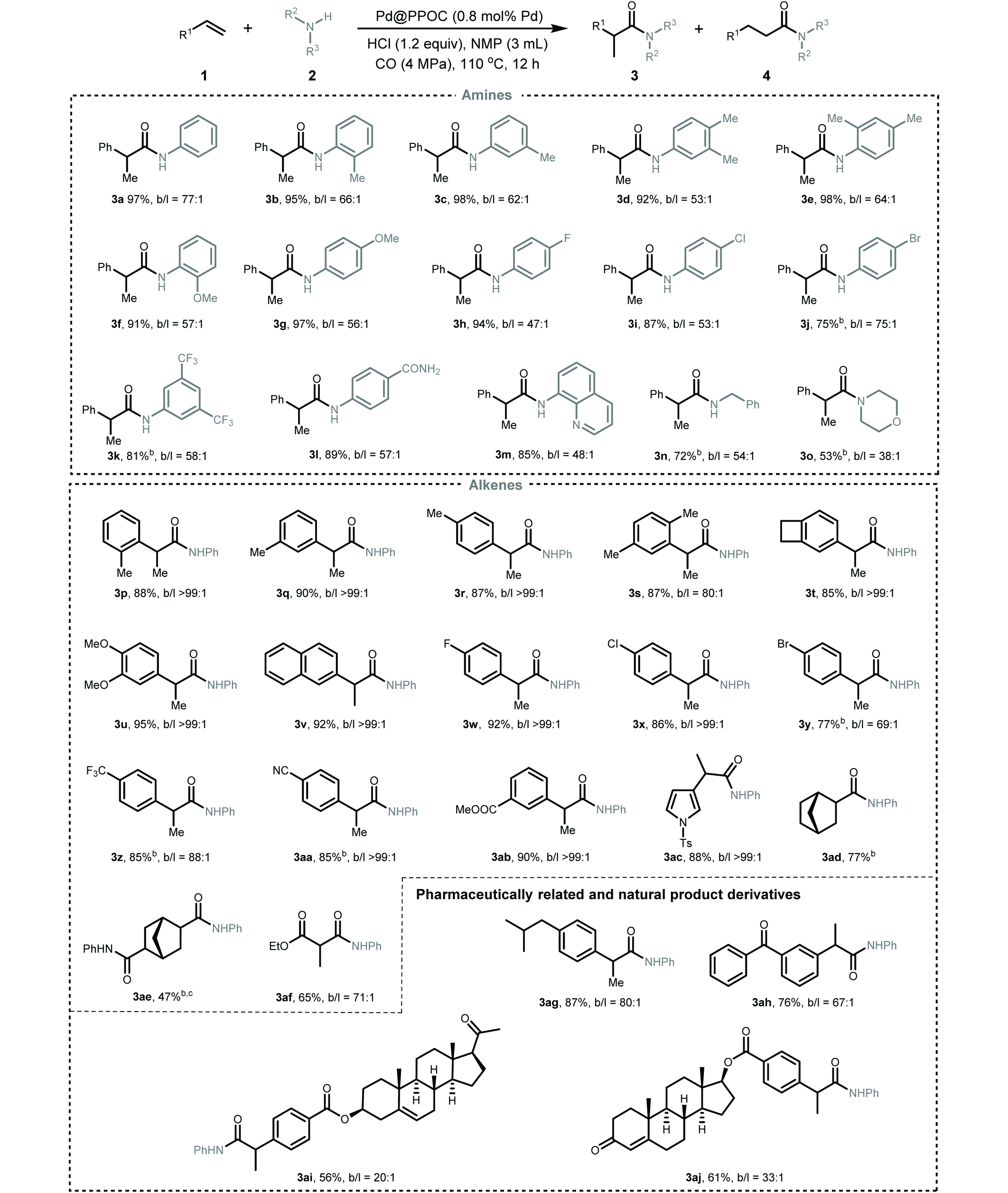
After identifying the optimized reaction conditions, we then explored the scope and limitations of this Pd-catalyzed hydroaminocarbonylation of alkenes for the synthesis of various amides (Table 2). Firstly, the scope of anilines was investigated. A variety of anilines incorporated with electron-neutral, electron-donating, and electron-withdrawing substituents could undergo coupling with styrene and CO to furnish the desired amides in high yields with excellent regioselectivity (3a–3l). ortho-Substituted anilines were also compatible with the present conditions and showed no obvious difference in the catalytic performance compared with meta- and para-substituted anilines (2b, 2e, and 2f), implying a negligible steric effect. Anilines with halide substituents, such as –F (2h), –Cl (2i), and –Br (2j), and strong electron-withdrawing groups, such as –CF3 (2k) and –CONH2 (2l), were tolerable in the reaction to deliver the corresponding amides in a slightly lower yield albeit with excellent regioselectivity. The reaction of styrene with quinolin-8-amine (2m) and CO also proceeded well, regioselectively furnishing the corresponding amide 3m in 85% yield. Pleasingly, the more challenging aliphatic amine (benzylamine 2n) and secondary amine (morpholine 2o) also underwent Markovnikov hydroaminocarbonylation smoothly to afford the corresponding amides in 53 and 72% yields with high regioselectivity, respectively.
| a Reaction conditions: alkene (0.5 mmol), amine (0.6 mmol), HCl (0.6 mmol), Pd@PPOC (0.8 mol% Pd), CO (4.0 MPa), NMP (3.0 mL), 110 °C, 12 h. The ratios of branched/linear amide were determined by GC-MS analysis of the crude products. b 24 h. c Aniline (1.2 mmol), HCl (1.2 mmol). Isolated yields are reported. |
|---|
|
Secondly, the scope of alkenes was examined. A spectrum of styrenes bearing electron-donating and electron-withdrawing substituents at the ortho-, meta-, and para-positions of the phenyl ring could couple with aniline and CO to furnish the corresponding Markovnikov amides (3p–3ab) in high yields with excellent regioselectivity (overall higher than 69). No obvious steric or electronic effect was observed in all investigated styrenes. Heteroaryl alkene, i.e., 1-tosyl-3-vinyl-1H-pyrrole 1ac, also underwent the reaction very well to deliver the desired amide 3ac in 88% yield with excellent regioselectivity. Remarkably, the challenging aliphatic alkenes, including norbornene (1ad), bicyclo[2.2.1]hepta-2,5-diene (1ae), and ethyl acrylate (1af), were also tolerated in the reaction to give the desired amides in moderate to good yields (3ad–3af, 47–77%). To demonstrate the utility of this Pd-catalyzed hydroaminocarbonylation, pharmaceutically related derivatives, i.e., ibuprofen (3ag) and ketoprofen (3ah), and biologically active natural derivatives containing a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, i.e., pregnenolone (1ai) and testosterone (1aj), could be smoothly hydroaminocarbonylated into their respective amides in good to high yields with excellent regioselectivity. In addition, this synthetic protocol could be easily scaled up and the gram-scale synthesis of 3a was successful with maintenance of the activity and regioselectivity under the optimal conditions (Scheme S3†), highlighting its practical potential.
C bond, i.e., pregnenolone (1ai) and testosterone (1aj), could be smoothly hydroaminocarbonylated into their respective amides in good to high yields with excellent regioselectivity. In addition, this synthetic protocol could be easily scaled up and the gram-scale synthesis of 3a was successful with maintenance of the activity and regioselectivity under the optimal conditions (Scheme S3†), highlighting its practical potential.
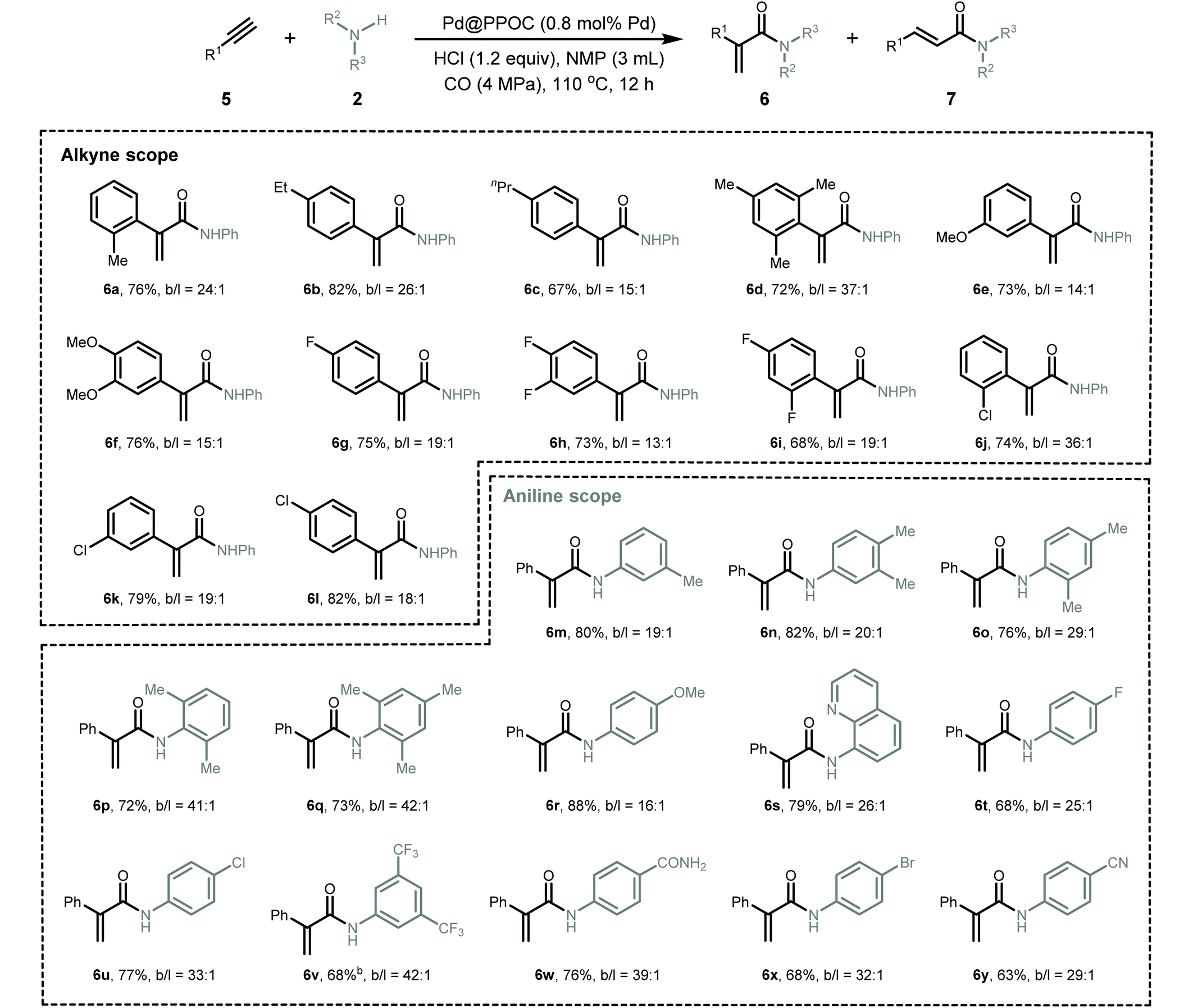
Given the generality of the substrate scope and superior catalytic performance in the Markovnikov hydroaminocarbonylation of alkenes, we next turned our attention to explore terminal alkynes as starting materials for accessing α,β-unsaturated amides. We conducted the reactions using the same conditions as those for hydroaminocarbonylation of alkenes, and the results are presented in Table 3. In general, a diverse set of terminal alkynes smoothly underwent the Markovnikov hydroaminocarbonylation with various anilines and CO pressures to deliver functionalized acrylamides in good to high yields with high Markovnikov regioselectivity. Firstly, phenylacetylenes with substituted groups at the ortho-, meta-, and para-positions of the phenyl ring (5a–5l) coupled with aniline and CO very well, affording their respective α,β-unsaturated amides (6a–6l) in high yields with Markovnikov to anti-Markovnikov regioselectivities varying from 13 to 37. Impressively, halogen-substituted phenylacetylenes (5g–5l) were compatible with the present conditions to deliver their corresponding acrylamides, which allows for their late-stage functionalization to construct more structurally complex and useful compounds via cross-coupling reactions. Secondly, a set of anilines featuring various substituents were examined. Anilines containing electron-donating and electron-withdrawing groups, such as –Me, –OMe, –F, –Cl, –Br, –CN, –CF3, and –CONH2, were well tolerated under the present conditions to give the desired α,β-unsaturated amides (6m–6y) in high yields with excellent regioselectivity.
Our previous study has clearly indicated that the ultrafine Pd NPs confined in PPOC has a strong interaction with PPh3 in the skeleton with the formation of Pd–P coordination bonds,26 which makes this soluble heterogeneous Pd catalyst just like homogeneous phosphine-ligated Pd complexes. According to the above results and previous mechanistic investigation on the Pd-catalyzed hydroaminocarbonylation,3,5–8 we propose that the reaction proceeds via the following pathways as shown in Scheme 2. First, the active [Pd–H] species A is formed in situ from the reaction of Pd@PPOC and HCl, which is similar to the homogeneous system.5 Then, insertion of alkene into [Pd–H] species A affords the alkyl-Pd intermediates B and B′. The alkyl-Pd B and B′ undergo CO insertion to give the corresponding acyl-Pd intermediates C and C′, respectively. Complexes D and D′ are formed through amine insertion, which undergo reductive elimination leading to amides and regenerate the palladium hydride species.
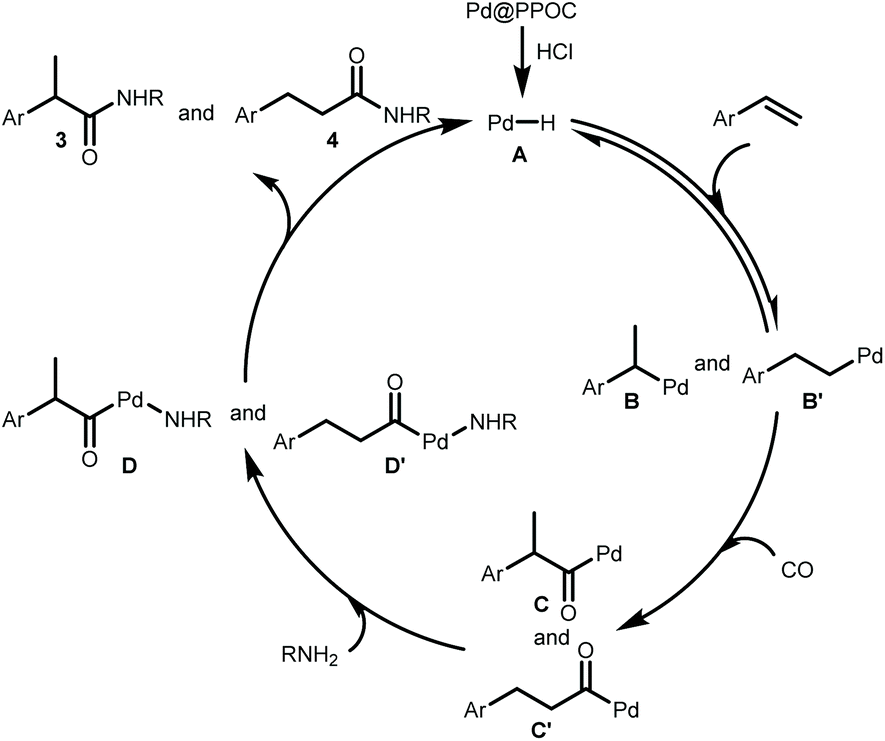 | ||
| Scheme 2 Proposed mechanism. | ||
Conclusions
To sum up, we demonstrated a highly efficient and regioselective hydroaminocarbonylation of alkenes and alkynes over a soluble and stable heterogeneous Pd catalyst consisting of ultrafine PdNPs encapsulated in a phosphine-built-in porous organic cage. A diverse set of amides and α,β-unsaturated amides were synthesized in high yields with excellent Markovnikov regioselectivity. Moreover, this soluble heterogeneous Pd catalyst shows high stability and could be easily recovered for successive reuses without an obvious change in catalytic performance. This work presents an elegant example of soluble porous organic cage-encapsulated ultrafine metal nanoparticles for highly efficient and regioselective catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from QIBEBT, CAS (Grant No. QIBEBT I201902), the Dalian National Laboratory for Clean Energy (DNL) Cooperation Fund, CAS (Grant No. DNL201904), the Shandong Energy Institute Fund (Grant No. SEII202138), the China Postdoctoral Science Foundation (2020M682249), and the Qingdao Postdoctoral Application Research Project is acknowledged. We also gratefully acknowledge the financial support of the National Natural Science Foundation of China (No. 22078305 and 22002178).Notes and references
- (a) R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed; (b) J.-B. Peng and X.-F. Wu, Angew. Chem., Int. Ed., 2018, 57, 1152–1160 CrossRef CAS PubMed; (c) S. Cai, H. Zhang and H. Huang, Trends Chem., 2021, 3, 218–230 CrossRef CAS; (d) H. Wang and F. Shi, Chin. J. Chem., 2021, 39, 1051–1069 CrossRef CAS.
- (a) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed; (b) S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- (a) X. Fang, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14089–14093 CrossRef CAS PubMed; (b) H. Li, K. Dong, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10239–10243 CrossRef CAS PubMed; (c) J. Liu, H. Li, A. Spannenberg, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 13544–13548 CrossRef CAS PubMed.
- C. Jiménez-Rodriguez, A. A. Núñez-Magro, T. Seidensticker, G. R. Eastham, M. R. L. Furst and D. J. Cole-Hamilton, Catal. Sci. Technol., 2014, 4, 2332–2339 RSC.
- (a) G. Zhang, B. Gao and H. Huang, Angew. Chem., Int. Ed., 2015, 54, 7657–7661 CrossRef CAS PubMed; (b) Y. Hu, Z. Shen and H. Huang, ACS Catal., 2016, 6, 6785–6789 CrossRef CAS; (c) J. Zhu, B. Gao and H. Huang, Org. Biomol. Chem., 2017, 15, 2910–2913 RSC; (d) B. Gao, G. Zhang, X. Zhou and H. Huang, Chem. Sci., 2018, 9, 380–386 RSC; (e) X. Zhou, G. Zhang, B. Gao and H. Huang, Org. Lett., 2018, 20, 2208–2212 CrossRef CAS PubMed; (f) J. Li, S. Wang, S. Zou and H. Huang, Commun. Chem., 2019, 20, 14–22 CrossRef; (g) B. Gao and H. Huang, Org. Lett., 2017, 9, 6260–6263 CrossRef PubMed; (h) X. Ji, B. Gao, X. Zhou, Z. Liu and H. Huang, J. Org. Chem., 2018, 83, 10134–10141 CrossRef CAS PubMed.
- (a) T. Xu, F. Sha and H. Alper, J. Am. Chem. Soc., 2016, 138, 6629–6635 CrossRef CAS PubMed; (b) Y. Li, H. Alper and Z. Yu, Org. Lett., 2006, 8, 5199–5201 CrossRef CAS PubMed; (c) S.-M. Lu and H. Alper, J. Am. Chem. Soc., 2008, 130, 6451–6455 CrossRef CAS PubMed; (d) F. Sha and H. Alper, ACS Catal., 2017, 7, 2220–2229 CrossRef CAS.
- (a) H. Liu, N. Yan and P. J. Dyson, Chem. Commun., 2014, 50, 7848–7851 RSC; (b) H. Liu, G. P. S. Lau and P. J. Dyson, J. Org. Chem., 2015, 80, 386–391 CrossRef CAS PubMed.
- (a) Y.-H. Yao, H.-Y. Yang, M. Chen, F. Wu, X.-X. Xu and Z.-H. Guan, J. Am. Chem. Soc., 2021, 143, 85–91 CrossRef CAS PubMed; (b) H.-Y. Yang, Y.-H. Yao, M. Chen, Z.-H. Ren and Z.-H. Guan, J. Am. Chem. Soc., 2021, 143, 7298–7305 CrossRef CAS PubMed.
- S. Torii, H. Okumoto, M. Sadakane and L. H. Xu, Chem. Lett., 1991, 1673–1676 CrossRef CAS.
- (a) B. El Ali, A. El-Ghanam, M. Fettouhi and J. Tijani, Tetrahedron Lett., 2000, 41, 5761–5764 CrossRef CAS; (b) B. El Ali, J. Tijani and A. M. El-Ghanam, Appl. Organomet. Chem., 2002, 16, 369–376 CrossRef CAS; (c) B. El Ali, J. Tijani and A. M. El-Ghanam, J. Mol. Catal. A: Chem., 2002, 187, 17–33 CrossRef CAS; (d) B. El Ali and J. Tijani, Appl. Organomet. Chem., 2003, 17, 921–931 CrossRef CAS; (e) R. Suleiman, J. Tijani and B. El Ali, Appl. Organomet. Chem., 2010, 24, 38–46 CAS.
- U. Matteoli, A. Scrivanti and V. Beghetto, J. Mol. Catal. A: Chem., 2004, 213, 183–186 CrossRef CAS.
- (a) D.-L. Wang, W.-D. Guo, Q. Zhou, L. Liu, Y. Lu and Y. Liu, ChemCatChem, 2018, 10, 4264–4268 CrossRef CAS; (b) D.-L. Wang, W.-D. Guo, L. Liu, Q. Zhou, W.-Y. Liang, Y. Lu and Y. Liu, Catal. Sci. Technol., 2019, 9, 1334–1337 RSC.
- L. Yang, L. Shi, C. Xia and F. Li, Chin. J. Catal., 2020, 41, 1152–1160 CrossRef CAS.
- S. Liu, H. Wang, X. Dai and F. Shi, Green Chem., 2018, 20, 3457–3462 RSC.
- H. Wang, H. Yuan, X. Wang, J. Zhao, D. Wei and F. Shi, Adv. Synth. Catal., 2020, 362, 2348–2353 CrossRef CAS.
- Z. Sun, L. Yan, G. Ji, G. Wang, L. Ma, M. Jiang, C. Li and Y. Ding, Appl. Catal., A, 2021, 614, 118026 CrossRef CAS.
- (a) L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed; (b) A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed; (c) K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed; (d) K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397–1437 CrossRef CAS PubMed.
- (a) D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS; (b) A. Bordet and W. Leitner, Acc. Chem. Res., 2021, 54, 2144–2157 CrossRef CAS PubMed.
- (a) T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed; (b) A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2013, 52, 3746–3749 CrossRef CAS PubMed.
- (a) K. Acharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed; (b) L. Lu, S. Zou and B. Fang, ACS Catal., 2021, 11, 6020–6058 CrossRef CAS.
- (a) J. Sun, W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed; (b) X. Yang, J. Sun, M. Kitta, H. Pang and Q. Xu, Nat. Catal., 2018, 1, 214–220 CrossRef CAS; (c) H. Liu, L. Chen, C.-C. Hou, Y.-S. Wei and Q. Xu, J. Mater. Chem. A, 2021, 9, 13670–13677 RSC.
- Y.-B. Huang, Q. Wang, J. Liang, X. Wang and R. Cao, J. Am. Chem. Soc., 2016, 138, 10104–10107 CrossRef CAS PubMed.
- (a) B. Mondal, K. Acharya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709–1716 CrossRef CAS PubMed; (b) B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS PubMed.
- (a) S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed; (b) L. Qiu, R. McCaffrey, Y. Jin, Y. Gong, Y. Hu, H. Sun, W. Park and W. Zhang, Chem. Sci., 2018, 9, 676–680 RSC; (c) N. Sun, C. Wang, H. Wang, L. Yang, P. Jin, W. Zhang and J. Jiang, Angew. Chem., Int. Ed., 2019, 58, 18011–18016 CrossRef CAS PubMed.
- (a) X.-X. Gou, T. Liu, Y.-Y. Wang and Y.-F. Han, Angew. Chem., Int. Ed., 2020, 59, 16683–16689 CrossRef CAS PubMed; (b) H. Liu, X. Duan, Y.-K. Lv, L. Zhu, Z. Zhang, B. Yu, Y. Jin, Y. Si, Z. Wang, B. Li and P. Peng, Chem. Eng. J., 2021, 426, 130872–130880 CrossRef CAS.
- Z. Wang, C. B. Reddy, X. Zhou, J. J. Ibrahim and Y. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 53141–53149 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc00815g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |