
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical aerobic oxidation of sulfides to sulfoxides: the crucial role of wavelength irradiation†
Elpida
Skolia‡
,
Petros L.
Gkizis‡
,
Nikolaos F.
Nikitas
and
Christoforos G.
Kokotos
 *
*
Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis 15771, Athens, Greece. E-mail: ckokotos@chem.uoa.gr
First published on 5th May 2022
Abstract
The sulfoxide moiety is recognized as one of the most important groups in organic and medicinal chemistry. Many efforts worldwide focus on developing novel and sustainable protocols, accessing sulfoxide-containing molecules. Photochemistry plays a unique role in sulfoxide synthesis, since this new emerging field, employing light as the energy source, is essential in developing novel and eco-friendly protocols. Herein, we report a novel, sustainable, light-driven protocol, where the impact of wavelength irradiation on sulfide aerobic photooxidation was examined. In this work, two different low-catalyst loading (0.05–0.5 mol%) protocols, utilizing anthraquinone as the photocatalyst (under CFL lamps or 427 nm irradiation) were developed and a catalyst-free protocol (under 370 nm irradiation) was also assessed. In addition, a broad scope of substrates was tested and extensive mechanistic studies were performed, in order to distinguish the mechanistic pathways that are followed in the different cases of aryl or alkyl sulfide oxidation under different wavelength irradiation. We also implemented our protocols towards the oxidation of several intermediates en route to Sulforaphane.
Introduction
Sulfoxide-containing compounds display a prominent role in the field of organic synthesis,1 pharmaceuticals,2 agrochemicals3 and polymers.4 Several sulfoxides are known to display a salient pharmacological profile and have been introduced as marketed drugs, including omeprazole, sulindac, sulfinpyrazone and sulforaphane (Scheme 1). Over the past years, scientists have developed numerous protocols for the synthesis of sulfoxides, where a wide variety of oxidants, such as oxone,5m-chloroperbenzoic acid,6 sodium periodate,7 chromic acid,8 chlorine9etc., were employed for the selective oxidation of aryl and/or alkyl sulfides. In all the above-mentioned protocols, the major drawback is the use of more than one equivalent of oxidant, which generates over-stoichiometric amounts of waste. Moreover, in all methodologies presented in literature, another issue is selectivity, highlighted by the formation of the corresponding sulfone, due to over-oxidation of sulfides. Even though the presence of the corresponding sulfone in some cases appears in a very small percentage, in the case of an active pharmaceutical ingredient this is prohibitive. In addition, in most reported methodologies, heating in high temperatures narrows their potential for application in industry. A more green, sustainable, and popular approach is the use of hydrogen peroxide as the oxidant,10 since its main by-product is water. However, the activation of hydrogen peroxide requires either heat11 or metal catalysts.12 From an environmental point of view, metal-free organocatalysts, such as flavins13 or activated ketones14 have been reported.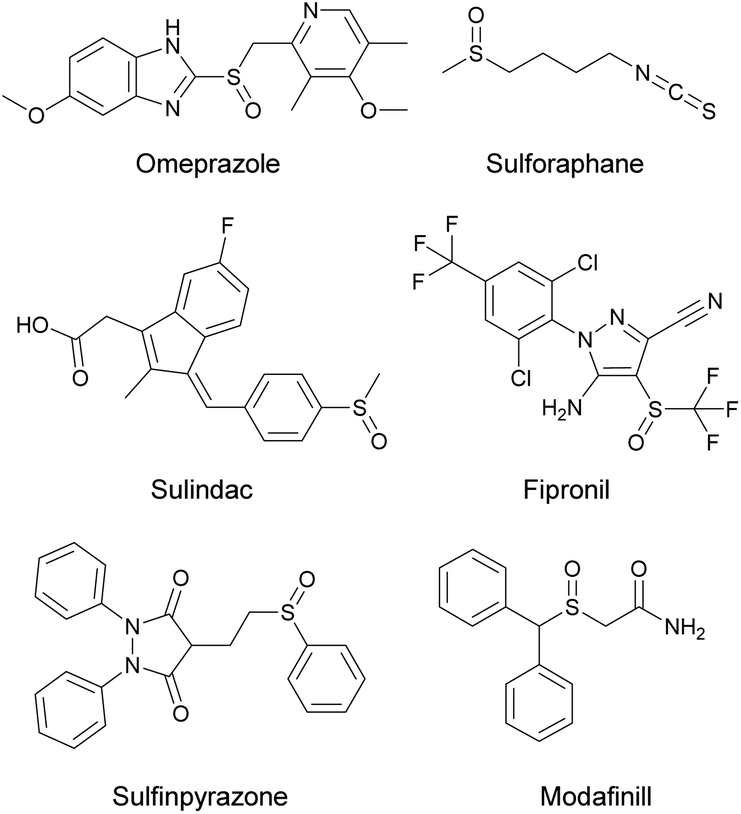 | ||
| Scheme 1 Selected examples of pharmaceuticals’ active ingredients or natural products containing the sulfoxide moiety. | ||
Apart from the environmentally friendly and sustainable hydrogen peroxide, another mild oxidant, widely employed in oxidation protocols is oxygen.15 It is an ambient, abundant, harmless gas, which can be considered the “greenest” approach towards oxidation reactions. The “green” character of molecular oxygen as the oxidant, either deriving from ambient air or being supplied as a gas to the reaction mixture, was very early realized.15 In recent years, different groups have reported metal-catalyzed16 or organocatalyzed17 processes that utilize oxygen as the oxidant in various processes. In a very recent example, in 2020, Petsi and Zografos designed a series of novel proline-based organocatalysts and examined their application in the oxidation of sulfides to sulfoxides, utilizing HFIP as the solvent.18
Photochemistry is an exciting and powerful area of research, which deals with electronically excited molecules and chemical reactions induced by light.19,20 Despite the fact, that it was more than clear that the reactivity of excited molecules differs from those in ground state and light-mediated chemistry has the potential to trigger reactions that are unavailable to ground-state pathways, synthetic photochemistry remained for many years a specialized area of research with limited practical applications. In 2008, seminal publications from MacMillan,20a Stephenson,21 and Yoon22 changed this notion and the synthetic community quickly recognized the new opportunities created by photoredox catalysis and photochemistry.
The inherent advantages of photochemical protocols and aerobic (oxygen)-mediated oxidations were recognized quite early and the merger of these two areas was envisaged in the oxidation of sulfides to sulfoxides from the 1960s.23,24 Throughout the years, many synthetic efforts were made for the photochemical aerobic oxidation of sulfides, however most of them focused on the use of specialized apparatus for irradiation or the use of heterogeneous and in some cases homogeneous catalysts (metal-based or organocatalysts).24 With the advent of Photoorganocatalysis, the use of small organic molecules for photochemical reactions,25 the renewal of research interest in this photochemical aerobic protocols occurred. In 2019, Liu and co-workers, following their interest in developing a new protocol using a mild oxidant, proposed the use of molecular oxygen and the non-photochemical reaction was held in the presence of an ether, which promoted the reaction.26 In that case, the use of oxygen in the presence of a flammable organic solvent limited their industrial application. To overcome these disadvantages, in 2021, they exploited molecular oxygen as the sole oxidant in a visible-light-induced sulfide oxidation using CF3SO2Na (25 mol%) as the photocatalyst (Scheme 2A).27 However, a high catalyst loading for such a process had to be employed. Since 2017, a number of protocols have been reported for the photochemical aerobic oxidation of sulfides, such as the selective oxidation of sulfides induced by visible light in the presence of uranyl salts (Scheme 2B)28 or porphyrin derivatives as the photocatalyst (Scheme 2C).29 In addition, a variety of visible-light induced oxidations, employing either naturally-occurring30b,c or commercially available molecules30a as the organic photocatalyst were also reported recently in literature (Scheme 2D). However, expensive uranyl salts, expensive and hazardous solvents or/and the synthesis of the photoorganocatalysts, not only increases the manufactory cost, but poses difficulties in the final purification of the product. Furthermore, in all these cases where organic photocatalysts were employed (Scheme 2D), the two generic mechanisms of aerobic oxidations (singlet oxygen or single electron transfer) were proposed to be operating, without proving if one of them is dominant and if the mechanism of action changes, depending on the nature of the employed substrate.
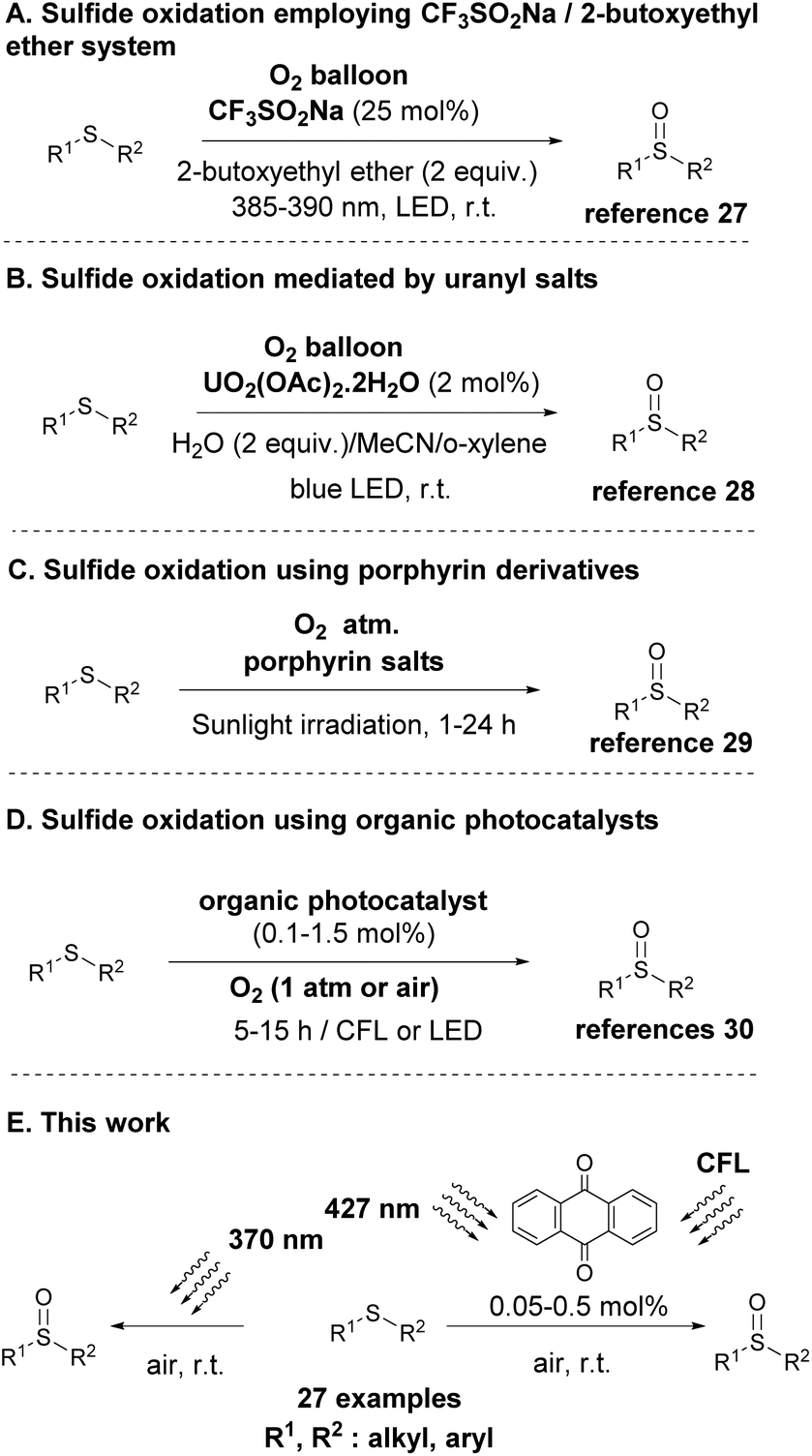 | ||
| Scheme 2 Photochemical aerobic oxidation of sulfides. | ||
Apart from the use of several catalysts for either oxygen or substrate activation, many efforts have targeted the development of catalyst-free protocols. Since 1990s, a series of publications were focused on studying the behaviour of sulfides under visible light irradiation,24,31 however, the major drawback was the formation of undesired by-products, since the C–S fragmentation pathway prevailed the oxidation one. Lately, Sun and coworkers,32 while testing the use of a benzothiadiazole-based conjugated microporous polymer as the photocatalyst to trigger sulfide oxidation, discovered that under blue LED irradiation (100 Watt), the oxidation proceeded in the absence of a catalyst. This novel catalyst-free approach of sulfide oxidation suffers from limited substrate scope (only aryl aryl or alkyl aryl sulfides were successfully employed) and prolonged reaction times, which in some cases is crucial for the formation of several by-products. Also, in their proposed mechanism, although they employ DFT calculations, they proposed a thiadioxirane intermediate as the active intermediate,32 however, this intermediate has been discarded by researchers as a possible intermediate in the recent past.24
Following our continuous interest in developing novel light-driven methodologies,33 herein, we report a mild, green and chemoselective direct photochemical aerobic oxidation of sulfides to the corresponding sulfoxides (Scheme 2E). From an environmental and economical point of view, this novel, green and sustainable protocol for the oxidation of sulfides using a mild oxidant, such as molecular oxygen, in the presence of a low-cost commercially available photocatalyst serves the basic principles of green chemistry, which is highly desirable on the chemical and pharmaceutical industry. We, furthermore, point out the crucial role of the wavelength irradiation source, towards the selectivity and the mechanism of the oxidation process. In this sense, we have developed a fast and green catalyst-free protocol for the photochemical aerobic oxidation of sulfides under 370 nm irradiation, while two low-catalyst loading (0.05 or 0.5 mol%) protocols (under CFL lamps or 427 nm irradiation) were also developed. Finally, in our attempt to shed light on the reaction mechanism, we examined the role and application of several known quenchers into the investigation of the reaction mechanism, and we propose a chart flow, researchers could follow in the future.
Results and discussion
We initially investigated the visible light-mediated aerobic oxidation of thioanisole (1a), as the model reaction to optimize the reaction parameters (Table 1). In the beginning, we studied the influence of the wavelength irradiation source in the outcome of the photochemical aerobic oxidation (Table 1). The self-excitation of sulfides is well established,31 however, usually harsh reaction conditions are employed (Xe or Hg lamps for the irradiation). For this purpose, the reaction was conducted in the absence of a photocatalyst, under various wavelengths of LEDs or CFL household lamps (Table 1). More specifically, under the irradiation of compact fluorescence lamps (CFL), the reaction appears to be sluggish (Table 1, entry 1). In this case, the use of an appropriate photocatalyst to facilitate the reaction is required.34 On the contrary, the use of LEDs, as the irradiation source, facilitates the reaction, leading not only to the desired product, but also to the corresponding sulfone, due to over-oxidation (Table 1, entries 2–8). Also, it is obvious that upon decrease of the wavelength irradiation of the LED, the ratio of the products alters and the sulfone by-product increases significantly (Table 1, entries 2 and 3 vs. 4–8). Next, we turned our attention into optimizing the reaction conditions, employing a suitable photocatalyst when needed, to shorten the reaction time and suppress the formation of the overoxidized byproduct.34 A thorough investigation on the reaction outcome based on different irradiation wavelength and reaction conditions can be found in the ESI.†34 From these studies, we concluded that the irradiation wavelength and reaction conditions are crucial for product distribution (sulfide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sulfoxide:sulfone), and as will be described below, CFL lamp irradiation, as well as 370 nm or 427 nm, were chosen as optimum for the development of catalyst and catalyst-free protocols.34
:sulfoxide:sulfone), and as will be described below, CFL lamp irradiation, as well as 370 nm or 427 nm, were chosen as optimum for the development of catalyst and catalyst-free protocols.34
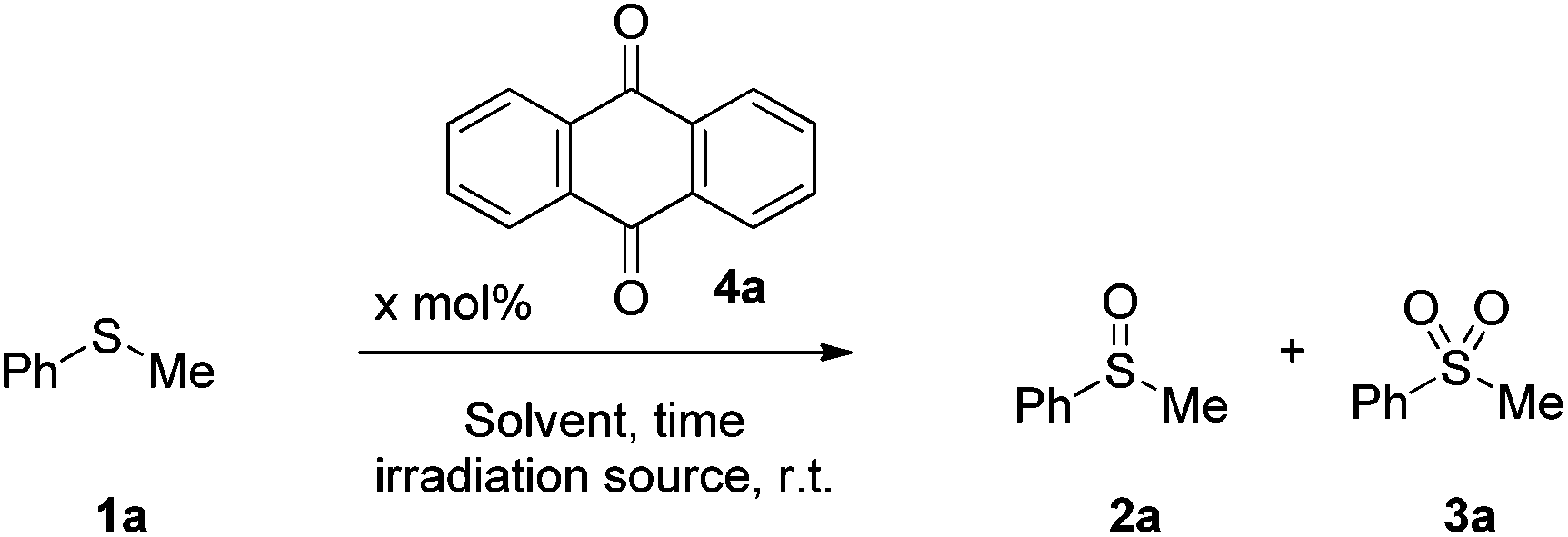
Initially, we investigated the sulfide oxidation under household lamp irradiation, employing a wide range of photocatalysts and we concluded that anthraquinone (4a) was the one that outperformed the others.34 Then, we turned our attention to the nature of the reaction medium (Table 2). Protic solvents, i.e. methanol, ethanol afforded the best results (Table 2, entries 2–5), while in aprotic solvents, the reaction proved to be sluggish (Table 2, entries 7 and 8).34 Addition of 10 equiv. of H2O was essential in accelerating the reaction rate and suppressing the formation of the sulfone by-product (Table 2, entry 4 vs. entry 3).34 Interestingly, when the reaction was conducted using water as the solvent, no formation of the desired product was observed, due to the insolubility of anthraquinone in water (Table 2, entry 6). Under LED irradiation (427 nm), sulfide oxidation led to a selective formation of the corresponding sulfoxide vs. the sulfone, after 5 h of irradiation in the presence of 0.05 mmol% of 4a (Table 2, entry 10 vs. entry 9). When the reaction was performed in methanol, the desired product was obtained in quantitative conversion, suppressing the undesired sulfone by-product (Table 2, entry 11). In aprotic solvents, the reaction was incomplete (Table 2, entries 12–15). The optimum reaction conditions were also tested under sunlight irradiation, leading to a quantitative yield of 2a.34 In both cases (CFL lamps and 427 nm), low-catalyst loadings were employed, outperforming most of the known procedures highlighted in Scheme 1, while in the 427 nm protocol, the reaction required short reaction time.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Lamp (nm) | Solvent, time (h) | 4a (mol%) | 1a (%) | 2a/3ab (%) |
| a The reaction was performed with 1a (0.20 mmol) in solvent (1 mL) and H2O (40 μL), under open air irradiation for 18 h. b Conversion was determined by 1H-NMR. c The reaction was performed without the addition of H2O. d 0.5 mL of solvent instead of 1 mL. | |||||
| 1 | CFL | MeCN, 18 | 0.1 | 71 | 29/0 |
| 2 | CFL | MeOH, 18 | 0.1 | 61 | 39/0 |
| 3c | CFL | MeOH, 18 | 0.5 | 0 | 95/5 |
| 4 | CFL | MeOH, 18 | 0.5 | 0 | 100/0 |
| 5 | CFL | EtOH, 18 | 0.1 | 69 | 31/0 |
| 6 | CFL | H2O, 18 | 0.1 | 100 | 0/0 |
| 7 | CFL | CH2Cl2, 18 | 0.1 | 96 | 4/0 |
| 8 | CFL | DMF, 18 | 0.1 | 67 | 26/0 |
| 9c | 427 | MeCN, 18 | 0.05 | 0 | 81/19 |
| 10c | 427 | MeCN, 5 | 0.05 | 36 | 64/0 |
| 11c,d | 427 | MeOH, 5 | 0.05 | 0 | 100/0 |
| 12c,d | 427 | EtOAc, 5 | 0.05 | 11 | 89/0 |
| 13c,d | 427 | CH2Cl2, 5 | 0.05 | 20 | 80/0 |
| 14c,d | 427 | Benzene, 5 | 0.05 | 72 | 28/0 |
| 15c,d | 427 | DMF, 5 | 0.05 | 50 | 50/0 |
We also targeted in identifying a catalyst-free protocol for the photochemical aerobic oxidation of 1a (Table 3). To our delight, when the reaction was performed under UVA-LED irradiation (370 nm), quantitative formation of the desired product was obtained after 3 h, under catalyst-free conditions (Table 3, entry 3). Prolonged reaction time and the presence of a photocatalyst led to the formation of sulfone 3a (Table 3, entry 1). Decreasing the reaction time and omitting the presence of the photocatalyst, led to a better yield of 2a (Table 3, entry 2). Adding of 10 equiv. of H2O led to an accelerated reaction, while suppressing the formation of the overoxidation by-product (Table 3, entry 3). Due to hydrogen bond formation, water inhibits the conversion of sulfoxide to sulfones.35 Again, solvent optimization did not reveal any medium leading to better yields (Table 3, entries 4–8). It is important to note that this fast and mild reaction protocol (3 h and 45 W 370 nm) provides a better alternative to literature (11 h and 100 W Blue LED).32 A major drawback of the literature process32 is that it cannot be applied for dialkyl sulfides. Our desire to develop a photochemical protocol with broad scope of substrates led us to investigate the optimized reactions conditions on the less reactive dialkyl sulfides. Dodecyl methyl sulfide (1q) was chosen as a model substrate to apply the optimum reaction conditions (Table 3). We were pleased to observe that our optimum conditions were successfully implemented. The major difference was observed in the reaction time and in order to obtain the desired product 2q, the reaction time was prolonged from 3 h to 18 h (Table 3, entry 9). For the cases of sensitive substrates, the presence of 0.5 mol% of anthraquinone (4a) was used and the desired product was formed quantitatively after 3 h of irradiation at 370 nm (Table 3, entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Sulfide | Solvent, time (h) | 1 (%) | 2/3b (%) |
| a The reaction was performed with 1a or 1r (0.20 mmol) in solvent (1 mL), open air under 370 nm irradiation. b Conversion was determined by 1H-NMR. c The reaction was performed with 0.5 mol% anthraquinone (4a). d 40 μL of H2O were added. | ||||
| 1c | 1a | MeCN, 18 | 0 | 29/71 |
| 2 | 1a | MeCN, 2 | 11 | 89/0 |
| 3d | 1a | MeCN, 3 | 0 | 99/1 |
| 4 | 1a | MeOH, 2 | 20 | 80/0 |
| 5 | 1a | EtOH, 2 | 11 | 89/0 |
| 6 | 1a | iPrOH, 2 | 17 | 83/0 |
| 7 | 1a | Benzene, 2 | 93 | 17/0 |
| 8 | 1a | DMF, 2 | 72 | 28/0 |
| 9 | 1q | MeCN, 18 | 0 | 100/0 |
| 10c | 1q | MeCN, 3 | 0 | 100/0 |
With the optimized conditions in hand, we turned our attention in examining the substrate scope (Scheme 3). We decided to follow three different approaches towards the identification of broad and easily-replicated photochemical protocols, using air as the sole oxidant. The first protocol is the catalyst-free approach, using irradiation at 370 nm (45 W, protocol c). In the same time, we opted for an anthraquinone-mediated photochemical reaction, using a low catalyst loading (0.05 mol%) under 427 nm irradiation (45 W, protocol b). Finally, if one cannot have access to LED irradiation source, a CFL-mediated approach was developed, using anthraquinone at 0.5 mol% (protocol a). A variety of sulfides were tested, affording the corresponding sulfoxides in good to excellent yields (Scheme 3). Initially, the easier type of sulfoxides, according to literature, was probed, using alkyl aryl sulfides (Scheme 3A). Same as thioanisole (1a), other phenyl-substituted sulfides bearing an alkyl group afforded the corresponding sulfoxides in high to quantitative yields, when the reaction took place under household lamp irradiation for 18 h (Scheme 3, protocol a, 2a–e). Furthermore, phenyl-substituted sulfides with an alkyl group decorated with different functional groups, i.e. double or triple bonds, hydroxy or carboxyl moiety, afforded in all cases the desired products in good to excellent yields (Scheme 3, protocol a, 2f–k). Similarly, benzyl phenyl sulfides afforded the corresponding sulfoxides in excellent yields (Scheme 3, protocol a, 2l). Naphthyl or heteroaryl methyl sulfides were also employed successfully, although prolonged reaction time was required in some cases (Scheme 3, protocol a, 2m and 2n). In the case of aryl aryl sulfides, both diphenyl sulfide (1o) and fused-aryl sulfide 1p were tested, leading to high yields of the desired product (Scheme 3, protocol a, 2o and 2p). Finally, a broad scope of dialkyl sulfides were employed under the optimum conditions leading to the dialkyl sulfoxides in good to excellent yields (Scheme 3, protocol a, 2q–y). The obtained lower yields in the cases of dialkyl sulfoxides 2s, 2t and 2u are attributed to the difficulties posed during the purification process and not to the formation of undesired byproducts (Scheme 3, protocol a).
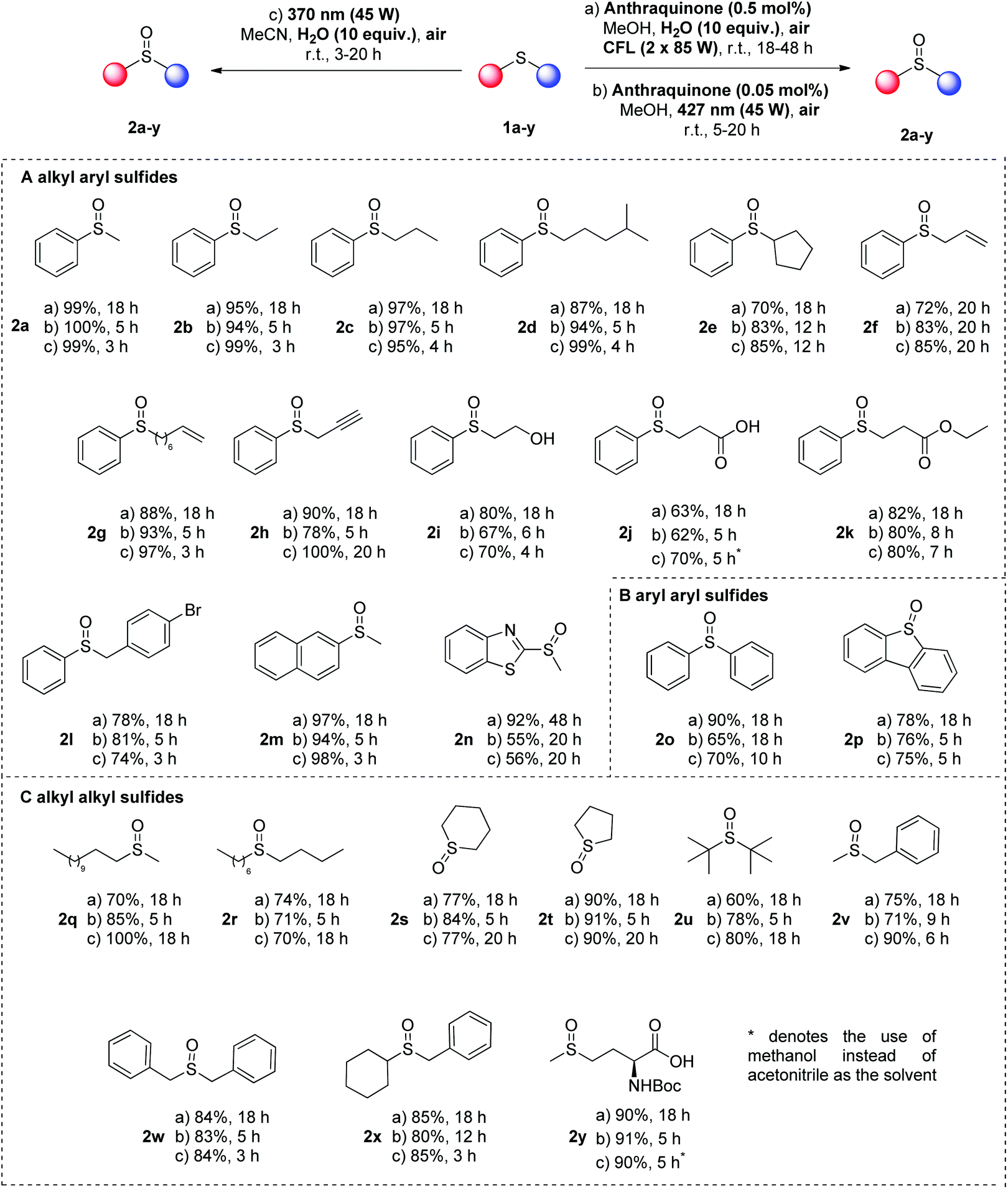 | ||
| Scheme 3 Substrate scope. | ||
When the same substrates were employed under LED irradiation (427 nm), the catalyst loading of 4a can be reduced to 0.05 mol% and the reaction time varied from 5–20 h, while similar results were obtained (Scheme 3, protocol b). In the cases of alkyl aryl sulfides, the reaction time was usually 5–6 h, leading to excellent yields in all cases (Scheme 3, protocol b, 2a–n). Only in the cases of phenyl secondary alkyl sulfide 1e, allyl phenyl 1f and heteroaryl methyl 1n, an extended reaction time was required (up to 20 h), in order to achieve similar high yields. In the same vein, aryl aryl sulfides led to high yields, under these conditions, as well (Scheme 3, protocol b, 2o and 2p). Finally, dialkyl sulfide 1q-y led to excellent yields in short reaction time (Scheme 3, protocol b, 2q–y).
The substrate scope was also examined under catalyst-free conditions upon irradiation at 370 nm (Scheme 3, protocol c). Sulfides bearing an aryl group afforded the corresponding sulfoxides in good to excellent yields in short reaction times (Scheme 3, protocol c, 2a–e). Furthermore, functional groups, i.e. double or triple bonds, hydroxy and carboxyl moiety, were well tolerated, as well as aryl aryl sulfides (Scheme 3, protocol c, 2f–p). Interestingly, the less reactive dialkyl sulfides under the catalyst-free conditions were converted into sulfoxides in good to excellent yields (Scheme 3, protocol c, 2q–y). In this case, prolonged reaction times were required, due to the reactivity of the substrates. Furthermore, in all cases, NMR monitoring of the crude reaction mixture did not reveal any byproduct, derived either from overoxidation or from the C–S fragmentation pathway. In the last case of the catalyst-free protocol (Scheme 3, protocol c), for the cases of 3-phenyl-thio-propionic acid (1j) and Boc-protected methionine 1y, the solvent medium was switched to methanol, instead of acetonitrile, due to the insolubility of the substrates in acetonitrile.
In addition, to demonstrate the synthetic utility of our protocol in a real-life application, we attempted to use our optimized reaction conditions into the oxidation of sulfide intermediates that appear in the synthetic preparation of sulforaphane 8 (Scheme 4). Sulforaphane [4-(methylsulfinyl)butylisothiocyanate] 8 was isolated in 1992 by Talalay and coworkers.36 Sulforaphane is the product of the enzymatic hydrolysis of glucoraphanin by the enzyme myrosinase, which is most represented in broccoli. Recently, it has been found to exhibit excellent anti-cancer properties.37 In literature, sulforaphane is synthesized from 5 and in two steps, isothiocyanate 6 is obtained (Scheme 4, top).38 Then, a m-CPBA-mediated oxidation leads to sulforaphane 8.38 Alternatively, sulfoxidation of 5 leads to 7, which can be converted into sulforaphane 8 (Scheme 4).39 Our photochemical aerobic oxidation protocol was successfully implemented in the oxidation of sulfides 5 or 6 to afford the corresponding sulfoxides 7 or 8, respectively, in good to excellent yields (Scheme 4). In the case of 5, photochemical aerobic oxidation under all three different protocols led to excellent yields of sulfoxide 7. Similar good yields were obtained in the case of the oxidation of 6 to sulforaphane 8 (Scheme 4). One of the major issues in the preparation of sulforaphane is its decomposition under open air conditions. We came across this problem, when the photooxidation took place under household lamp irradiation, when we attained sulforaphane in a moderate yield. To avoid this issue in our catalyst-free conditions, we added 0.5 mol% of anthraquinone (4a) to decrease the reaction time, increasing the yield up to 87%.
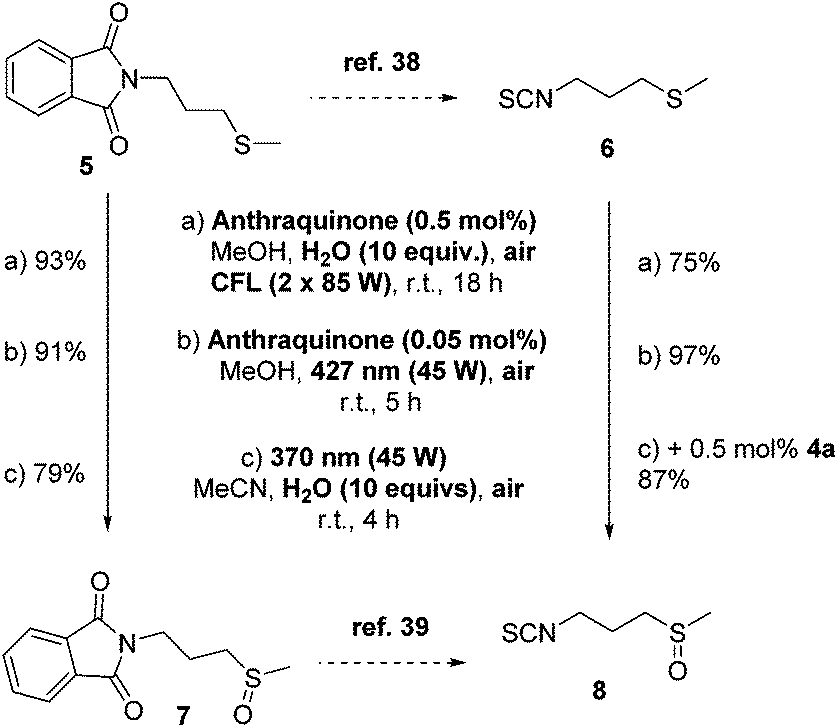 | ||
| Scheme 4 Photochemical aerobic synthesis of sulforaphane 8. | ||
Mechanistic studies
One of the most challenging tasks in photooxidation protocols is the complete understanding of the reaction mechanism. In general, two different mechanistic pathways for the photochemical aerobic sulfide oxidation are reported in literature.24 The first mechanism is promoted by singlet oxygen (1O2, I), which is generated in the presence of a photosensitizer (Scheme 5A). The generated singlet oxygen mediates the oxygenation of the sulfide to afford persulfoxide intermediate II (Scheme 5A). The latter reacts with a second molecule of sulfide to afford two molecules of the desired sulfoxide. In the second mechanistic pathway, a single electron transfer (SET) event occurs between the excited photocatalyst and the sulfide, affording a sulfide radical cation (like IV in Scheme 7A). Reaction with either the superoxide radical anion or triplet state oxygen 3O2 leads to the desired sulfoxide.24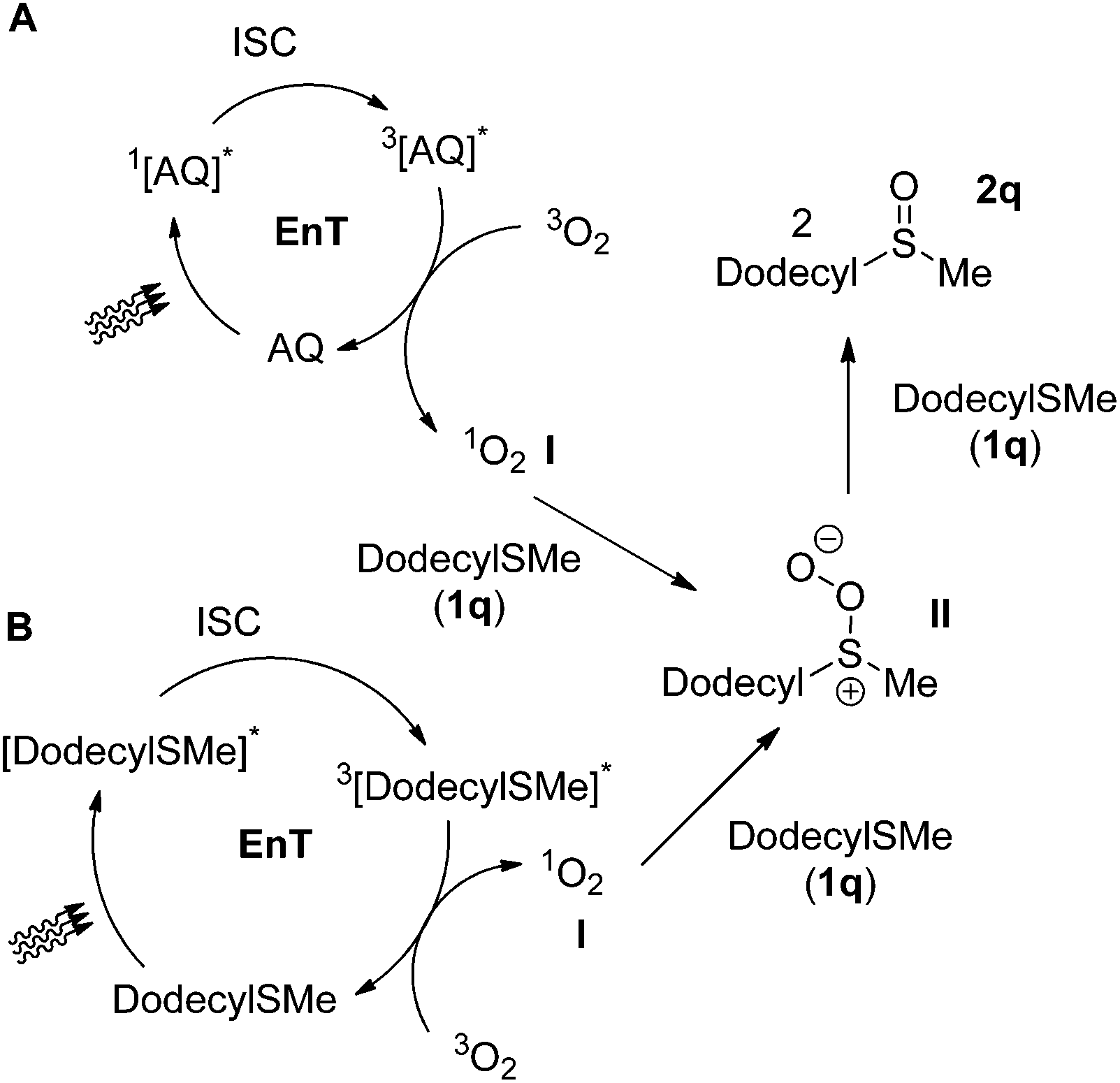 | ||
| Scheme 5 Proposed mechanistic pathways for photochemical aerobic oxidation of dodecyl methyl sulfide (1q). | ||
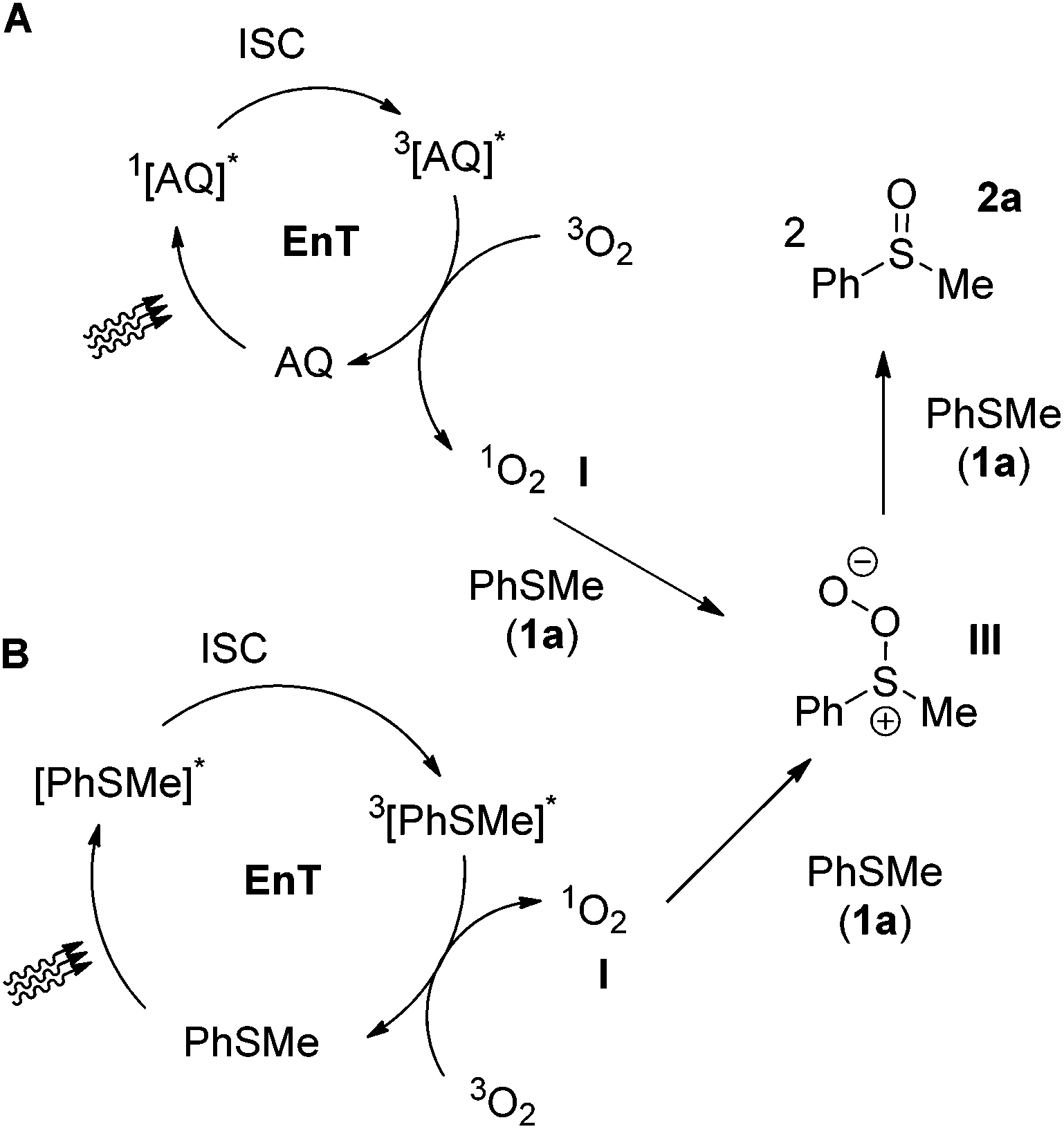 | ||
| Scheme 6 Proposed mechanistic pathways for the photochemical aerobic oxidation of thioanisole (1a). | ||
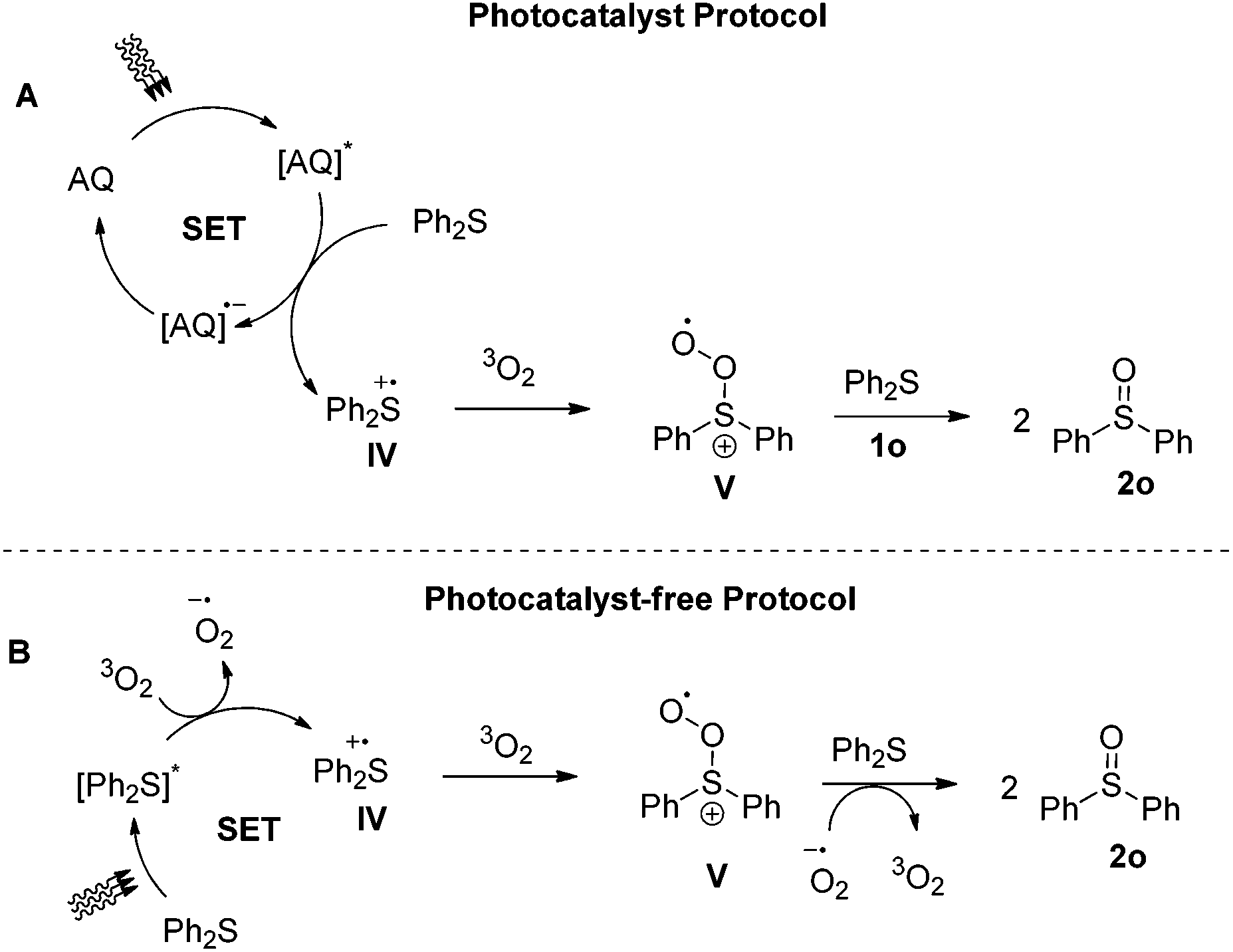 | ||
| Scheme 7 Proposed mechanistic pathways for the photochemical aerobic oxidation of diphenyl sulfide (1o). | ||
In many literature reports, both mechanisms are proposed to be involved.24 The use of different irradiation sources in our protocols, along with the use of alkyl aryl, aryl aryl or dialkyl sulfides may suggest that different reaction mechanism may be in place in each case. In general, anthraquinone (4a) is known in literature that can generate singlet oxygen via energy transfer.40 Furthermore, upon excitation, anthraquinone (4a) possesses the appropriate potential (Ered = 1.77 V vs. SCE)41 to oxidize sulfides (Eox = 1.34 V vs. SCE, Eox = 1.43 V vs. SCE and Eox = 1.63 V vs. SCE for thioanisole, diphenyl sulfide and dibutyl sulfide, respectively).42 However, fluorescence quenching experiments of excited anthraquinone did not show any quenching either by thioanisole (1a) (alkyl aryl sulfide) or dodecyl methyl sulfide (1q) (dialkyl sulfide).34 Not following the same trend, diphenyl sulfide (1o) (aryl aryl sulfide) does quench excited anthraquinone.34 In addition, it is also reported that sulfides, without the use of an external photocatalyst, upon irradiation, may either generate singlet oxygen or mediate an electron transfer event.31
To clarify the accurate mechanistic pathway in a photooxidation protocol, we examined the behavior of three representative examples of sulfides. Thioanisole (1a), dodecyl methyl sulfide (1q) and diphenyl sulfide (1o) were chosen as representative examples of an alkyl aryl sulfide, a dialkyl sulfide and a diaryl sulfide, respectively. First of all, in the absence of oxygen (reaction performed under argon), no oxidation took place.34 Then, a variety of known additives, able to quench either mechanistic pathways, have been reported in literature.24 Initially, to commence our investigations in establishing the exact mechanistic pathway of our protocols, we conducted a series of fluorescence quenching studies.34 For this purpose, we examined the impact of singlet oxygen quenchers, such as sodium azide, DABCO or Co(acac)3 on the fluorescence of anthraquinone (4a).34 Similar experiments were performed, using SET quenchers, such as 1,4-dimethoxybenzene (DMB) or benzoquinone.34 Our studies revealed that among all, benzoquinone and Co(acac)3 appears to decrease greatly the anthraquinone fluorescence and thus, their use is not indicated for distinguishing the sulfide photooxygenation mechanism, when anthraquinone (4a) is employed as the photocatalyst.34 In the same vein, it was found that benzoquinone also quenches the fluorescence of thioanisole (1a).34 Thus, the remarkable suppression of the oxidation processes, both in the case of the anthraquinone-mediated oxidation protocols or the catalyst-free conditions by benzoquinone can be attributed to the fluorescence quenching of the catalyst or 1a and by no means can be assigned to electron transfer events.34 Thus, it is clearly supported that benzoquinone is not a suitable additive for the investigation of the anthraquinone-mediated or the catalyst-free thioanisole photooxygenation.34 Similarly, Co(acac)3is not a suitable additive for the investigation of the anthraquinone-mediated photooxidations.34 On the other hand, DABCO, sodium azide and 1,4-dimethoxybenzene do not interact with anthraquinone.34
We begin with the case of dodecyl methyl sulfide (1q), supporting that dialkyl sulfide quenching studies indicate that in both type of conditions (photocatalyst-free and anthraquinone-mediated), the reaction follows the singlet oxygen mechanistic pathway (Scheme 5). Photochemical aerobic oxidation of 1q, in the presence of DABCO, sodium azide or Co(acac)3, was totally suppressed (0–5%), while the presence of DMB decreased slightly the yield of the sulfoxide (88%).34 These results indicate that under the photocatalyst-free conditions, the singlet oxygen mechanism is in place (Scheme 5B). Upon irradiation, triplet excited 3[1q]* generates singlet oxygen I, via an energy transfer event (Scheme 5B). Singlet oxygen reacts with a ground state 1q, leading to II, which leads to 2q. In the case of the anthraquinone-mediated photooxygenations, upon 427 nm irradiation, the photochemical aerobic oxidation of 1q, in the presence of DABCO or sodium azide, was also suppressed, while the presence of DMB, no change in the yield of the sulfoxide was observed (88%).34 These results indicate that under 427 nm irradiation in the presence of anthraquinone, the singlet oxygen mechanism is in place (Scheme 5A). Upon irradiation, triplet excited 3[AQ]* generates singlet oxygen I, via an energy transfer event (Scheme 5A). Singlet oxygen reacts with a ground state 1q, leading to II, which leads to 2q. Upon household lamps irradiation, similar results with DABCO, sodium azide or DMB were obtained.34 Similarly as before, in this case, the energy transfer mechanism (Scheme 5A) is the major pathway.
We then moved to the cases of alkyl aryl sulfide, using thioanisole as the benchmark study. When the reaction is performed under the photocatalyst-free conditions (370 nm), the impact of the substrate into the mechanism of the reaction is more easily distinguished. Photochemical aerobic oxidation of 1a, in the presence of DABCO, sodium azide or Co(acac)3, led to diminished yields (12–28%), while the presence of DMB decreased slightly the yield of the sulfoxide (84%).34 These results indicate that under the photocatalyst-free conditions, the singlet oxygen route is dominant (Scheme 6B). In conclusion, thioanisole (1a), upon excitation, leads to triplet 3[1a]*, which generates singlet oxygen I, through an energy transfer event. Singlet oxygen reacts with a ground state 1a, leading to III, which leads to 2a. This appears to be the major mechanistic pathway of the photocatalyst-free aerobic oxidation of aryl alkyl sulfides at 370 nm (Scheme 6B). This is in accordance with the results of Bonesi and coworkers for the reaction of thioanisoles at 310 nm.31 In the case of the anthraquinone-mediated photooxygenations, except from benzoquinone, Co(acac)3 cannot be employed as a reliable quencher, since it decreases the fluorescence of anthraquinone (4a).34 When the anthraquinone-mediated processes were performed in the presence of DABCO or sodium azide (singlet oxygen quenchers), decreased yields (23–31%) were obtained, thus singlet oxygen is involved. Similarly, when the anthraquinone-mediated processes were performed in the presence of DMB (SET quencher), high yields (75–89%) were also obtained, indicating that the singlet oxygen pathway is again the dominant mechanism of action (Scheme 6A).
The investigation of the mechanistic pathway when diphenyl sulfide is employed, proved to be rather intricate, since fluorescence quenching studies reveals an interaction between diphenyl sulfide and excited anthraquinone.34 Considering the triplet energy of anthraquinone (ET 260 kJ mol−1)43 with the ET of diphenyl sulfide (280 kJ mol−1),44 an energy transfer event is unlikely to occur. However, the energy transfer mechanism from anthraquinone to oxygen is viable.40 On the other hand, based on the potentials of excited anthraquinone and diphenyl sulfide, (1.77 V vs. SCE and 1.43 V vs. SCE, respectively),42 an electron transfer event is more likely to occur. Photocatalyst-free aerobic oxidation of 1o, in the presence of DABCO, sodium azide or Co(acac)3, suppressed the reaction (0–8%), while the presence of DMB also suppressed the generation of sulfoxide (6%).34 These results could indicate that under the photocatalyst-free conditions, both mechanistic pathways (singlet oxygen and electron transfer) are involved. However, careful consideration of a variety of parameters is required. The mechanism of the reaction at 370 nm involves excitation of diphenyl sulfide. An electron transfer event takes place, generating sulfide radical cation IV (Scheme 7B), which reacts with ground state oxygen to afford the corresponding sulfoxide, through intermediate V (Scheme 7B). The alternative well-accepted mechanism that leads to the desired sulfoxide involves the reaction of sulfide radical cation IV with superoxide radical ion,24 however, for the specific case of diphenyl sulfide, Bonesi and coworkers have proven through kinetic studies that superoxide radical anion does not react directly with IV, but plays a secondary role in the process, more likely mediating the conversion of V to 2o.31 Also, the energy transfer mechanism from excited 1o to oxygen, in order to generate singlet oxygen is also not operative, as Bonesi and coworkers has also proven for the case of 1o.31 However, our quenching experiments with DABCO or sodium azide delivered minimum yields of the reaction.34 Herein, it is necessary to highlight the need to be fully be aware of the oxidation potentials of the compounds involved in the process. Taking into consideration the potential of diphenyl sulfide (Eox = 1.43 V vs. SCE),42 as well as the potentials of DABCO (Eox = 0.57 V vs. SCE)45 and sodium azide (Eox = 1.29 V vs. NHE,46 ∼approx. 1.05 V vs. SCE), then it can easily be deducted that the reduced yields observed in the presence of DABCO or sodium azide are due to redox reactions between sulfide radical cation IV with either DABCO or NaN3 and not due to quenching of singlet oxygen. The singlet oxygen pathway is also not viable, since it is known that product 2o constitutes a quencher of persulfoxide intermediates (like intermediates II or III of Schemes 5 and 6).24 In the photocatalyst-mediated protocols, the reaction in the presence of DABCO or sodium azide or DMB, was suppressed (0–19%).34 As before though, upon anthraquinone irradiation, an electron transfer event occurs, generating sulfide radical cation IV, which reacts with oxygen to afford the corresponding sulfoxide, through intermediate V (Scheme 7A). All other potential mechanisms are not viable, due to the same reasons.
Based on the literature, Cismesia and Yoon employed the quantum yield as a mechanistic tool.47 Using potassium ferrioxalate as the actinometer, we calculated the quantum yield of the thioanisole photooxidation under the anthraquinone-mediated conditions.34 The achieved quantum yields (Φ = 2.0 and 2.1 for 427 nm and CFL irradiation) are impressive, since the maximum value based on the proposed mechanism is 2.0, regardless the pathway followed.
Conclusions
Photochemical aerobic oxidations merge two interesting fields, photochemistry, and air-mediated processes. Although for many years, both areas received limited attention, with the uprise of modern synthetic photochemistry and increasing number of literature reports on oxygen-mediated oxidations, the last five years, the photochemical aerobic oxidation of sulfides to sulfoxides has attracted exponential attention. Herein, we provide a general, fast, mild, green and easy-to-operate procedure to perform these reactions. Initially, we explored the role of wavelength irradiation towards sulfides aerobic photooxygenation to the corresponding sulfoxides. Compared to all the previous methods presented in the literature, two low-catalyst loading (0.05–0.5 mol%) anthraquinone-mediated protocols (CFL lamps or 427 nm) and one photocatalyst-free aerobic protocol (370 nm) are reported, combining short reaction time and overcome the unwanted overoxidation reaction. These conditions outperform most known literature procedures as far as reaction time and catalyst loading are concerned. Furthermore, expensive photocatalysts or complicated non-natural occurring catalysts, which are commonly employed in sulfide oxidation, are replaced by a cheap and commercially available photocatalyst, like anthraquinone. A broad scope of substrates was successfully tested, under the optimum conditions. We also applied our photooxidation protocols towards the synthesis of Sulforaphane, a promising anti-cancer agent. The mechanism of the reaction was extensively studied and for this purpose a series of quenching studies were performed, and we herein provide a flowchart for future researchers. Firstly, after UV-Vis studies, the potential of developing a photocatalyst-free protocol should be defined. When the appropriate photocatalyst is identified, via literature reports, the potential mechanistic scenarios should be identified. Utilizing fluorescence quenching studies (photocatalyst with substrates, photocatalyst with quenchers and sulfides with quenchers), the appropriate probes (quenchers) can be identified, in order to correctly recognize which probes can be used and which cannot, since they provide false positive or negative results. In this case, for the photocatalyst-free process, via the appropriate quenchers, we identified that dodecyl methyl sulfide (dialkyl sulfides) are oxidized via singlet oxygen, while when anthraquinone is employed, the singlet oxygen pathway is the dominant. In the case of thioanisole (alkyl aryl sulfides), in the photocatalyst-free protocol and under anthraquinone-mediated protocols, the singlet oxygen mechanism is the major pathway. Finally, in the case of diphenyl sulfide (aryl aryl sulfides), in the photocatalyst-free protocol and under the use of anthraquinone, the sulfide radical cation mechanism is operative.Author contributions
C. G. K conceived the study, E. S., P. L. G., N. F. N. and C. G. K. designed the experiments and analyzed the data. E. S., P. L. G. and N. F. N. performed the experiments. P. L. G. and C. G. K. prepared the draft of the manuscript and C. G. K. performed the final editing. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Hellenic Foundation for Research and Innovation (HFRI), since this research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “1st Call for H.F.R.I. Research Projects to support Faculty Members & Researchers and the Procurement of High-and the procurement of high-cost research equipment grant” (grant number 655). The authors would like to thank the Chemical Laboratories of Department of Food Science and Human Nutrition of the Agricultural University of Athens for providing access to their fluorescence instrumentation.Notes and references
- (a) M. C. Carreno, Chem. Rev., 1995, 95, 1717–1760 CrossRef CAS; (b) E. Block, Angew. Chem., Int. Ed. Engl., 1992, 31, 1135–1178 CrossRef; (c) B. Ferber and H. B. Kagan, Adv. Synth. Catal., 2007, 349, 493–507 CrossRef CAS.
- (a) E. A. Ilardi, E. Vidaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (b) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed; (c) R. A. Hartz, A. G. Arvanitis, C. Arnold, J. P. Rescinito, K. L. Hung, G. Zhang, H. Wong, D. R. Langley, P. J. Gilligan and G. L. Trainor, Bioorg. Med. Chem. Lett., 2006, 16, 934–937 CrossRef CAS PubMed.
- (a) P. Devendar and G.-F. Yang, Top. Curr. Chem., 2017, 375, 1–44 CrossRef CAS PubMed; (b) C. Lamberth, J. Sulfur Chem., 2004, 25, 39–62 CrossRef CAS.
- (a) X. Xu, C. Fu, X. Huang, Y. Chang, Y. Yu, J. Zhao, N. Isahak, J. Teng, R. Qiao, H. Peng, C.-X. Zhao, T. P. Davis, C. Fu and A. K. Whittaker, Biomacromolecules, 2021, 22, 330–339 CrossRef CAS PubMed; (b) Y. Yu, W. Xu, X. Huang, X. Xu, R. Qiao, Y. Li, F. Han, H. Peng, T. P. Davis, C. Fu and A. K. Whittaker, ACS Macro Lett., 2020, 9, 799–805 CrossRef CAS.
- (a) H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3317 CrossRef CAS PubMed; (b) B. Yu, A. H. Liu, L. N. He, B. Li, Z. F. Diao and Y. N. Li, Green Chem., 2012, 34, 957–962 RSC.
- (a) N. K. Jana and J. G. Verkade, Org. Lett., 2003, 5, 3787–3790 CrossRef CAS PubMed; (b) R. J. Griffin, A. Henderson, N. J. Curtin, A. Echalier, J. A. Endicott, I. R. Hardcastle, D. R. Newell, N. E. Noble, L. Z. Wang and T. B. Golding, J. Am. Chem. Soc., 2006, 128, 6012–6013 CrossRef CAS PubMed.
- R. S. Varma, R. K. Saini and H. M. Meshram, Tetrahedron Lett., 1997, 38, 6525–6528 CrossRef CAS.
- D. Edwards and J. B. Stenlake, J. Chem. Soc., 1954, 3272–3274 RSC.
- N. Fukuda and T. Ikemoto, J. Org. Chem., 2010, 75, 4629–4631 CrossRef CAS PubMed.
- (a) K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS; (b) I. Triandafillidi, D. I. Tzaras and C. G. Kokotos, ChemCatChem, 2018, 10, 2521–2535 CrossRef CAS.
- F. Shi, M. K. Tse, H. M. Kaiser and M. Beller, Adv. Synth. Catal., 2007, 349, 2425–2430 CrossRef CAS.
- (a) K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS; (b) J. Brinshma, R. La Crois, B. L. Feringa, M. I. Donnoli and C. Rosini, Tetrahedron Lett., 2001, 42, 4049–4052 CrossRef; (c) F. Hosseinpoor and H. Golchoubian, Tetrahedron Lett., 2006, 47, 5195–5197 CrossRef CAS; (d) S. Velusamy, A. V. Kumar, R. Saini and T. Punniyamurthy, Tetrahedron Lett., 2005, 46, 3819–3822 CrossRef CAS; (e) Y. Mekmouche, H. Hummel, R. Y. N. Ho, L. Que, V. Schünemann, F. Thomas, A. X. Trautwein, C. Lebrun, K. Gorgy, J.-C. Lepêtre, M.-N. Collomb, A. Deronzier, M. Fontecave and S. Ménage, Chem. – Eur. J., 2002, 8, 1196–1204 CrossRef CAS PubMed; (f) E. Baciocchi, M. F. Gerini and A. Lapi, J. Org. Chem., 2004, 69, 3586–3589 CrossRef CAS PubMed; (g) K. Jeyakumar and D. K. Chand, Tetrahedron Lett., 2006, 47, 4573–4576 CrossRef CAS; (h) C. A. Gamelas, T. Lourenço, A. P. da Costa, A. L. Simplício, B. Royo and C. C. Romáo, Tetrahedron Lett., 2008, 49, 4708–4712 CrossRef CAS; (i) C. Yang, Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. Deng, Green Chem., 2009, 11, 1401–1405 RSC; (j) R. D. Chakravarthy, V. Ramkumar and D. K. A. Chand, Green Chem., 2014, 16, 2190–2196 RSC; (k) Y. Yuan and Y. Bian, Tetrahedron Lett., 2007, 48, 8518–8520 CrossRef CAS.
- (a) A. A. Lindén, L. Krüger and J.-E. Bäckvall, J. Org. Chem., 2003, 68, 5890–5896 CrossRef PubMed; (b) A. Lindén, M. Johansson, N. Hermanns and J.-E. Bäckvall, J. Org. Chem., 2006, 71, 3849–3853 CrossRef PubMed.
- E. Voutyritsa, I. Triandafillidi and C. G. Kokotos, Synthesis, 2017, 49, 917–924 CAS.
- X. Zhang, K. P. Rakesh, L. Ravindar and H.-L. Qin, Green Chem., 2018, 20, 4790–4833 RSC.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- (a) R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Russ. Chem. Rev., 2018, 87, 821–830 CrossRef CAS; (b) S. P. Brown, M. P. Brochu, C. J. Sinz and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 10808–10809 CrossRef CAS PubMed.
- M. Petsi and A. L. Zografos, ACS Catal., 2020, 10, 7093–7099 CrossRef CAS.
- For selected reviews, see: (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (d) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed; (e) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (f) Visible Light Photocatalysis in Organic Chemistry, ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley, Weinheim, 2018 Search PubMed; (g) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC; (h) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS PubMed; (i) S. Reischauer and B. Pieber, iScience, 2021, 24, 102209–102224 CrossRef CAS PubMed.
- For selected examples, see: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nature, 2010, 2, 527–532 CAS; (c) R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS PubMed; (d) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed; (e) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (f) A. Trowbridge, D. Reich and M. J. Gaunt, Nature, 2018, 561, 522–527 CrossRef CAS PubMed; (g) E. Speckmeier, T. Fische and K. A. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed; (h) S. P. Morcillo, E. D. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945–12949 CrossRef CAS PubMed; (i) J. Wu, P. S. Grant, X. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 5697–5701 CrossRef CAS PubMed; (j) T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 CrossRef CAS PubMed; (k) A. Ruffoni, F. Julia, T. D. Svejstrup, A. J. McMillan, J. J. Douglas and D. Leonori, Nat. Chem., 2019, 11, 426–433 CrossRef CAS PubMed.
- J. M. R. Narayanan, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- For the first examples on photochemical aerobic oxidation of sulfides, see: (a) G. O. Schenck and C. H. Krauch, Chem. Ber., 1963, 96, 517–519 CrossRef CAS; (b) K. Gollnick, in Advances in Photochemistry, Vol. 6, ed. W. A. Noyes Jr., G. S. Hammond and J. N. Pitts Jr., John Wiley & Sons, Inc., 1968, pp. 109–110 Search PubMed; (c) C. S. Foote and J. W. Peters, J. Am. Chem. Soc., 1971, 93, 3795–3796 CrossRef CAS.
- For a recent review, see: E. Skolia, P. L. Gkizis and C. G. Kokotos, ChemPlusChem, 2022, 87, e202200008 CrossRef CAS PubMed.
- For selected reviews on photoorganocatalysis, see: (a) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (b) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (c) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC; (d) M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833–857 CrossRef CAS PubMed; (e) N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Org. Biomol. Chem., 2021, 19, 5237–5253 RSC; (f) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- Z. Cheng, P. Sun, A. Tang, W. Jin and C. Liu, Org. Lett., 2019, 21, 8925–8929 CrossRef CAS PubMed.
- K.-J. Liu, Z. Wang, L.-H. Lu, J.-Y. Chen, F. Zeng, Y.-W. Lin, Z. Cao, X. Yu and W.-M. He, Green Chem., 2021, 23, 496–500 RSC.
- Y. Li, S. A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499–13506 CrossRef CAS PubMed.
- A. G. Mojarrad and S. Zakavi, Catal. Sci. Technol., 2018, 8, 768–781 RSC.
- (a) C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 2205–2210 CrossRef PubMed; (b) J. Li, W. Bao, Z. Tang, B. Guo, S. Zhang, H. Liu, S. Huang, Y. Zhang and Y. Rao, Green Chem., 2019, 21, 6073–6081 RSC; (c) Y. Zhang, J. Lou, M. Li, Z. Yuan and Y. Rao, RSC Adv., 2020, 10, 19747–19750 RSC.
- S. M. Bonesi, S. Crespi, D. Merli, I. Manet and A. Albini, J. Org. Chem., 2017, 82, 9054–9065 CrossRef CAS PubMed.
- Q. Fan, L. Zhu, X. Li, H. Ren, G. Wu, H. Zhu and W. Sun, Green Chem., 2021, 23, 7945–7949 RSC.
- For contributions employing metal complexes in photoredox reactions, see: (a) I. Triandafillidi, M. G. Kokotou and C. G. Kokotos, Org. Lett., 2018, 20, 36–39 CrossRef CAS PubMed For contributions employing organic molecules in photochemical reactions, see: (b) G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2014, 20, 13811–13814 CrossRef CAS PubMed; (c) N. Kaplaneris, A. Bisticha, G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Green Chem., 2017, 19, 4451–4456 RSC; (d) N. F. Nikitas, I. Triandafillidi and C. G. Kokotos, Green Chem., 2019, 21, 669–674 RSC; (e) E. Voutyritsa and C. G. Kokotos, Angew. Chem., Int. Ed., 2020, 59, 1735–1741 CrossRef CAS PubMed; (f) G. N. Papadopoulos, M. G. Kokotou, N. Spiliopoulou, N. F. Nikitas, E. Voutyritsa, D. I. Tzaras, N. Kaplaneris and C. G. Kokotos, ChemSusChem, 2020, 13, 5934–5944 CrossRef CAS PubMed; (g) N. F. Nikitas, D. I. Tzaras, I. Triandafillidi and C. G. Kokotos, Green Chem., 2020, 22, 471–477 RSC; (h) N. Spiliopoulou and C. G. Kokotos, Green Chem., 2021, 23, 546–551 RSC; (i) C. S. Batsika, C. Mantzourani, D. Gkikas, M. G. Kokotou, O. G. Mountanea, C. G. Kokotos, P. K. Politis and G. Kokotos, J. Med. Chem., 2021, 64, 5654–5666 CrossRef CAS PubMed; (j) N. F. Nikitas, M. K. Apostolopoulou, E. Skolia, A. Tsoukaki and C. G. Kokotos, Chem. – Eur. J., 2021, 27, 7915–7922 CrossRef CAS PubMed; (k) I. Triandafillidi, N. F. Nikitas, P. L. Gkizis, N. Spiliopoulou and C. G. Kokotos, ChemSusChem, 2022, 15, e202102441 CrossRef CAS PubMed.
- For detailed experimental procedures, optimization studies and mechanistic experiments, see ESI.†.
- P. Pitchen, E. Dunach, M. N. Deshmukh and H. B. Kagan, J. Am. Chem. Soc., 1984, 106, 8188–8193 CrossRef CAS.
- Y. Zhang, P. Talalay, C. Cho and G. H. Pooner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2399–2403 CrossRef CAS PubMed.
- (a) N. Juge, R. F. Mithen and M. Traka, Cell. Mol. Life Sci., 2007, 64, 1105–1127 CrossRef CAS PubMed; (b) M. G. Kokotou, P. K. Revelou, C. Pappas and V. Constantinou-Kokotou, Food Chem., 2017, 237, 566–573 CrossRef CAS PubMed.
- X. Chen, Z. Li, X. Sun, H. Ma, X. Chen, J. Ren and K. Hu, Synthesis, 2011, 3991–3996 CrossRef CAS.
- N. Kuhnert and Y. Lu, J. Labelled Compd. Radiopharm., 2004, 47, 501–507 CrossRef CAS.
- (a) S. C. Nunez Montoya, L. R. Comini, M. Sarmiento, C. Becerra, I. Albesa, G. A. Arguello and J. L. Cabera, J. Photochem. Photobiol., B, 2003, 78, 77–83 CrossRef PubMed; (b) N. Liu and G. Sun, Ind. Eng. Chem. Res., 2011, 50, 5326–5333 CrossRef CAS.
- W. Lee, S. Jung, M. Kim and S. Hong, J. Am. Chem. Soc., 2021, 143, 3003–3012 CrossRef CAS PubMed.
- S. Fukuzumi, K. Shimoosako, T. Suenobu and Y. Watanabe, J. Am. Chem. Soc., 2003, 125, 9074–9082 CrossRef CAS PubMed.
- C. W. Kee, K. F. Chin, M. W. Wong and C.-H. Tan, Chem. Commun., 2014, 50, 8211–8214 RSC.
- Y. Shiraishi, H. Koizumi and T. Hirai, J. Phys. Chem. B, 2005, 109, 8580–8586 CrossRef CAS PubMed.
- S. F. Nelsen and P. J. Hintz, J. Am. Chem. Soc., 1972, 94, 7114–7117 CrossRef CAS.
- Z. B. Alfassi, A. Harriman, R. E. Huie, S. Mosseri and P. Neta, J. Phys. Chem., 1987, 91, 2120–2122 CrossRef CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data, 1H and 13C NMR data, UV-Vis, optimization and mechanistic studies. See DOI: https://doi.org/10.1039/d2gc00799a |
| ‡ Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |