
Selective catalysis for the reductive amination of furfural toward furfurylamine by graphene-co-shelled cobalt nanoparticles†
Xiuzheng
Zhuang‡
a,
Jianguo
Liu‡
 *b,
Shurong
Zhong
a and
Longlong
Ma
*b
*b,
Shurong
Zhong
a and
Longlong
Ma
*b
aKey Laboratory of Renewable Energy, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, People's Republic of China
bKey Laboratory of Energy Thermal Conversion and Control of Ministry of Education, School of Energy and Environment, Southeast University, Nanjing 210096, People's Republic of China. E-mail: liujg@ms.giec.ac.cn; mall@ms.giec.ac.cn
First published on 9th November 2021
Abstract
Amines with functional groups are widely used in the manufacture of pharmaceuticals, agricultural chemicals, and polymers but most of them are still prepared through petrochemical routes. The sustainable production of amines from renewable resources, such as biomass, is thus necessary. For this reason, we developed an eco-friendly, simplified, and highly effective procedure for the preparation of a non-toxic heterogeneous catalyst based on earth-abundant metals, whose catalytic activity on the reductive amination of furfural or other derivatives (more than 24 examples) proved to be broadly available. More surprisingly, the cobalt-supported catalyst was found to be magnetically recoverable and reusable up to eight times with an excellent catalytic activity; on the other hand, the gram-scale tests catalyzed by the same catalyst exhibited the similar yield of the target products in comparison to its smaller scale, which was comparable to the commercial noble-based catalysts. Further results from a series of analytical technologies involving XRD, XPS, TEM/mapping, and in situ FTIR revealed that the structural features of the catalyst are closely in relation to its catalytic mechanisms. In simple terms, the outer graphitic shell is activated by the electronic interaction as well as the induced charge redistribution, enabling the easy substitution of the –NH2 moiety toward functionalized and structurally diverse molecules, even under very mild industrially viable and scalable conditions. Overall, this newly developed catalyst introduces the synthesis of amines from biomass-derived platforms with satisfactory selectivity and carbon balance, providing cost-effective and sustainable access to the wide applications of reductive amination.
1. Introduction
Given the expected shortage of fossil-based resources, the interest in the development of sustainable chemistry is growing to synthesize valuable chemicals and fuels from renewable materials.1,2 In this regard, biomass stands out as a promising carbon source for the chemical industry because of its advantages of abundant reserves, eco-friendliness, and worldwide distribution.3–5 As we already know, a variety of platform molecules, including but not limited to furfural, 5-hydroxymethyfurfural (5-HMF), levulic acid, and phenol (or phenol derivatives), can be easily obtained from lignocellulosic biomass,6 which are substitutes for fossil-based feedstock in the manufacture of valuable compounds, especially in the field of fine chemicals.7–9 One of the recent strategies to synthesize high-value chemicals is related to the reductive amination of biomass-derived platforms, which is superior in terms of its high selectivity toward the targeted N-containing components, its availability under very mild conditions, and its diversity in the product compounds.10 Among them, furfural is easily accessible from the acid-catalyzed tandem hydrolysis dehydration of hemicellulose or acid-facilitated transformation of cellulose-derived hexoses, exhibiting an attractive economic benefit when compared to other biomass-derived platforms.11,12 In addition, furfural can react with ammonia or primary amines to synthesize valuable amines, such as furfurylamine, which have a large extent of applications in the manufacture of pharmaceuticals, pesticides, synthetic resins, and useful agrochemicals. As reported by Mariscal et al.,13 the reductive amination of furfural with amines or ammonia is a kind of cascade reaction consisting of the formation of imines and the sequential hydrogenation of furfurylamine or its derivatives, and the key step during this process is the selective hydrogenation of the in situ formed imines toward amines owing to the presence of the competitive side reaction.14 In the reaction network depicted in Fig. 1, furfural can be hydrogenated to alcohol (step A), react with ammonia to form the reactive intermediates, or react with the target product (i.e., furfurylamine) to form the secondary imine (step B). It is worthwhile to mention that there are still minor mechanistic details under debate surrounding the similar reaction in step B, which was discussed in a thorough review by Gomez et al.15 Once the secondary imine is formed, it is easily hydrogenated to the secondary amine, which can be further condensed with furfurylamine as step B, or can be decomposed to the primary imine (or amine) in the presence of ammonia (step C). Also, the primary imine can also be hydrogenated to the target amine in step D. Unfavorable side reactions involve the formation of difurfurylamine and furfuryl alcohol, competing with the formation of furfurylamine as well as rendering the selective synthesis of furfurylamine challenging.6 This was further corroborated by Hara et al.16 through the density functional theory (DFT) calculations, suggesting that the formation of difurfurylamine and furfuryl alcohol were more thermodynamically favorable in comparison to the formation of furfurylamine. As a result, the competition between the main and side reactions absolutely induced the carbon loss of the target product. Thus, seeking suitable catalysts to regulate the reaction balance is of importance for the sustainable synthesis of functional amines from biomass-derived platforms.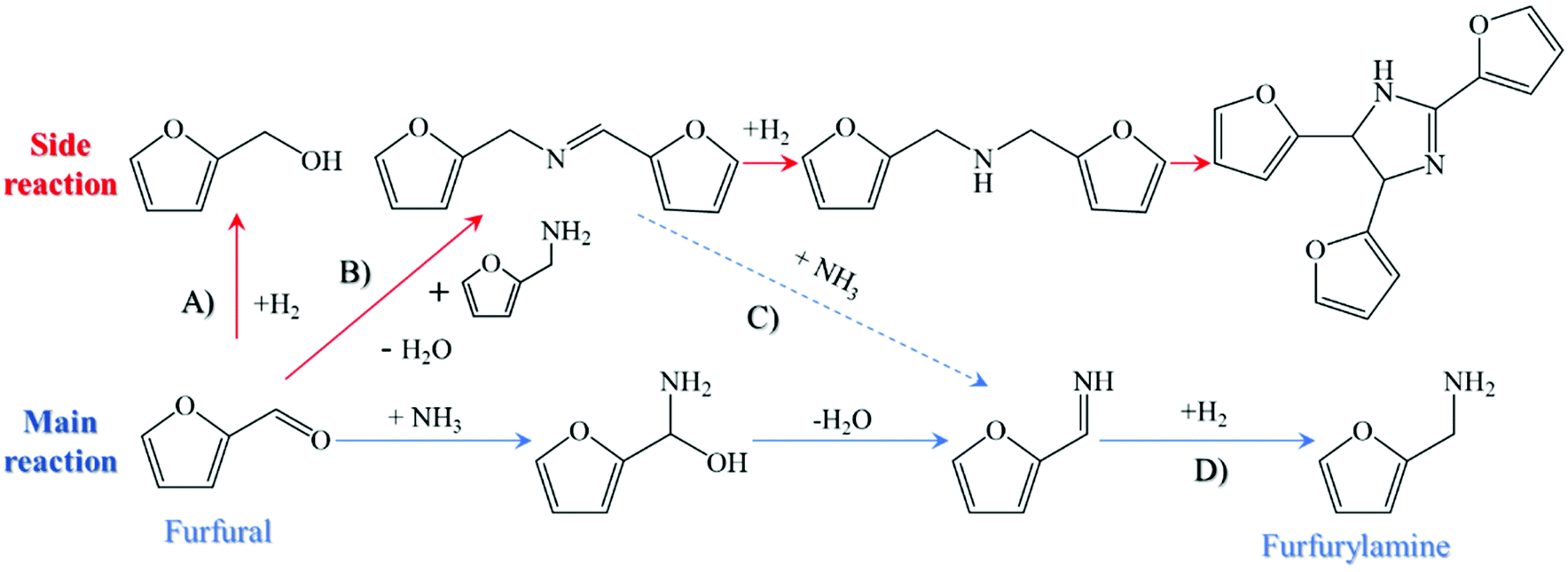 | ||
| Fig. 1 Reductive pathway for the reductive amination of furfural with ammonia. | ||
A series of noble metal-based catalysts have been developed for the reductive amination of furfural using ammonia as the nitrogen source and H2 as the reducing agent, and the details are given in Table 1. For instance, with the help of homogeneous RuCl2(PPh3)3 as a catalyst, Jagadeesh et al.17,18 reported 85% furfurylamine from the reductive amination of furfural at 130 °C, 24 h, and 4 MPa H2 but the difficulties in the separation and reuse limited its practical applications. The exploitation of heterogeneous catalysts has thus become the focus of relevant research. Ebitani and co-workers19 indicated that the Ru-based catalyst supported by hydroxyapatite (Ru-PVP/HAP) facilitated the reductive amination of furfural but only afforded a relatively lower yield of furfurylamine at 60%, similar to other Ru-based oxide-supported catalysts reported by Chatterjee et al.14 and Komanoya et al.16 The unfavorable side reactions are the main reason that challenge the reductive amination of furfural, while Martinez et al.20 found that screening and adjusting the structural features of the supports is beneficial for improving the catalytic selectivity owing to the synergistic effect of the acid sites and metal sites. On the other hand, the scarcity of noble metal-based catalysts has negative effects on its cost and large-scale application, which makes the low-cost and abundant non-noble metal-based catalysts favorable for the reductive amination of furfural.6 Xu and co-workers21 indicated that both RANEY® Ni and RANEY® Co enabled the reductive amination of furfural with ammonia to proceed well but the latter was proved to be superior in terms of its higher selectivity as metallic cobalt facilitates the hydrogenolysis of N-furfurylidenefurfurylamine. In addition, Varma et al.22 prepared a magnetic Fe3O4@SiO2-Ni catalyst with a core–shell structure for the reductive amination of furfural in ammonia solution at 115 °C, 2 h, and 2 MPa H2, which yielded 73% furfurylamine but exhibited the advantages of high stability, facile separation, and long-term reusability at the same time. The reductive amination of furfural with other N-containing compounds (e.g., aminopropanol, glycine, and β-alanine) also proceeded well according to the type of heterogeneous catalysts, giving 50–90% yields of the corresponding target amines. Based on the above findings concerning the reductive amination of furfural, it can be summarized that the current state of relevant works commonly has one or several of the following issues:3,4,9,11–14,19–21 (1) noble metal-based homogeneous catalysts need complex or even toxic ligands, tedious product/catalyst separation process, difficult catalyst recycling and reusability; (2) heterogeneous catalysts with earth-abundant transition metal can induce reductive amination but commonly require harsh conditions; (3) the structural features of heterogeneous supports affect the catalytic activity and selectivity, which requires a more simplified method for catalyst preparation. In an attempt to overcome these problems, especially from the perspectives of sustainability and environmental goals, several main factors are necessary for providing a relatively brand-new methodology of reductive amination, i.e., a green and renewable reagent, a full atom-economic process without any byproduct generation, fairly cheap and possible widely applied industrial scalable heterogeneous catalysts, relatively mild conditions, easy and convenient procedures of product separation, and downstream processing.
| Type | Catalysts | Nitrogen source | Reaction conditions | Conv./%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Yield/%c |
Ref. |
|---|---|---|---|---|---|---|
| a Molar equivalent relative to furfural. b Conversion of furfural. c Yield of furfurylamine. | ||||||
| Noble metals | RuCl2(PPh3)3 | NH3 (0.5–0.7 MPa) | 130 °C, 24 h, 4 MPa H2 | — | 85 | 17 |
| Ru–PVP/HAP | NH3 solution (25%) | 100 °C, 2 h, 0.4 MPa H2 | — | 60 | 19 | |
| Ru/Nb2O5 | NH3 (16 equiv.)a | 90 °C, 4 h, 4 MPa H2 | >99 | 89 | 19 | |
| Ru/SiO2 | NH3 (16 equiv.)a | 90 °C, 4 h, 4 MPa H2 | — | 84 | ||
| Ru/TiO2 | NH3 (16 equiv.)a | 90 °C, 4 h, 4 MPa H2 | — | 72 | ||
| Ru/Al2O3 | NH3 (33 equiv.)a | 80 °C, 2 h, 2 MPa H2 | >99 | 91.5 | 14 | |
| Non-noble metals | RANEY® Ni | NH3 (0.1 MPa) | 120 °C, 2 h, 1 MPa H | >99 | 65.1 | 21 |
| RANEY® Co | NH3 (0.1 MPa) | 120 °C, 2 h, 1 MPa H | >99 | 98.9 | ||
| Fe3O4@SiO2·Ni | NH3 solution (25%) | 120 °C, 2 h, 2 MPa H | >99 | 73 | 22 | |
Herein, we describe an environment friendly and simplified procedure for the preparation of porously graphitic spheres that encapsulate uniform cobalt nanoparticles (Co@C) by adopting cobalt acetate as the metallic precursor. This strategy originates from the “chainmail for catalyst” provided by the group of Bao et al.,23,24 who elaborated the unique electron penetration through the graphene layer from the encapsulated metals to promote the relevant catalytic reaction on the outermost surface of graphene. We avoid the use of hazardous solvents and meanwhile reduce the number of synthetic steps, as described in the Experimental section. The structural features of the catalysts in relation to their catalytic activity were characterized by multiple techniques, and the selected catalysts were next applied to the reductive amination of furfural under varied conditions (70–120 °C, 1–5 MPa H2, and 1–5 h in this study) to investigate the effects of the operational factors. Subsequently, using the optimized conditions and starting from inexpensive, readily available reactants and molecular ammonia, we conducted the synthesis of more than 24 biomass-derived platforms for a series of primary amines with varied functional groups. Last but not least, we have also demonstrated the scale-up of the heterogeneous amination protocol to the gram-scale synthesis as well as the lifecycle performance in the batch reactor to prove its value in industrial application, which can enable the preparation of simple but highly efficient catalysts for the reductive amination of renewable sources in the near future.
2. Experimental section
2.1 Chemical materials
The chemicals and solvents used in this study were purchased from certified companies registered in China Academy Science On-line market systems, such as Aladdin Chemicals Co. Ltd (Beijing, China), Sigma-Aldrich Chemicals Co. Ltd (Shanghai, China), and Macklin Chemical Reagent Co., Ltd (Shanghai, China). All of them were used without any further purification but the purity of the substrates was determined prior to the experiments, such as furfural (≥98.5%), 5-HMF (≥99%), and furfurylamine (≥99%).2.2 Preparation of the heterogeneous catalyst
The cobalt-based catalyst with porous graphitic structures was synthesized through a simplified three-step procedure described in our previous work.25 Initially, citric acid (as the carbon source, 0.03 mol) and cobalt acetate (as the Co source, 0.03 mol) were assembled in H2O at approximately 70 °C to form an aqueous mixture, where the solvent was very slowly evaporated in a vacuum environment within 72 h to enable the good distribution of metallic atoms. A sparking jelly was finally obtained once the aqueous phase was fully evaporated from the mixture systems, which served as the carbonaceous precursor coated with cobalt acetate. It is noteworthy that since the choice of the solvent and the cobalt source affects the catalytic properties to a certain extent, relatively safer solvents (e.g., EtOH) and other cobalt salts (e.g., cobalt nitrate and cobalt carbonate) were also studied as a reference. The preliminary results indicated that the influence of the solvent was more important than that of cobalt salts. Afterward, the carbonaceous precursor was pyrolyzed at 500–700 °C for 3 h under N2 atmosphere at a constant flow of 40 ml L−1 and then rinsed with 1 M H2SO4 and H2O to remove the unloaded metals and readjust the pH of the catalyst back to 7, respectively. Cobalt encapsulated in graphene layer (Co@C) nanoparticles catalyst could be obtained after vacuum freeze-drying at −41 °C overnight. Some of the Co@C catalysts were further oxidized under O2/Ar (5% O2 in Ar, 99.999% purity) atmosphere at 200 °C for 3 h to provide the Co/CoO@C catalyst, which is helpful in distinguishing the catalytic properties between metallic atoms and oxides. All these materials were labeled as Co@C-x–y or Co/CoO@C-x–y, where x suggests the pyrolysis temperature while y denotes the solvent. These controllable factors associated with the preparation of the cobalt-based catalyst were discussed in detail from its structural features as well as the catalytic activity to establish the correlation between the processing conditions and the catalyst properties.2.3 Analysis of the catalyst properties
In general, the performance of heterogeneous catalysts is characterized by two important aspects: one is relevant to the structural features of the carbonaceous support, while another associated with the catalytic activity is closely related to the metallic nanoparticles. To explore the comprehensive information, both of them were analyzed with the help of multiple techniques, as described below.As for the structural features of the carbonaceous support: (1) the morphological information was measured using high resolution transmission electron microscopy (HRTEM), and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, JEM-2100F, Japan) was coupled with energy dispersive X-ray spectrometry (EDS, Thermo Scientific, Waltham, MA) to measure the distribution of the metallic atoms. Meanwhile, the surface appearance of the sample was observed by scanning electron microscopy (SEM, S-4800, HITACHI, Japan). The surface area and pore structure of the samples were also analyzed through a gas-adsorption analyzer (Gemini VII 2390, USA) according to the N2 isothermal adsorption/desorption at −196 °C in the relative pressure (P/P0) between 0.01 and 0.99; (2) the crystal structure was explored with the help of XRD (PANalytical, X'Pert PRO, Netherlands) by employing Cu Kα radiation (λ = 0.15406 nm), and the scan conditions were adjusted from 20° to 80° in the 2θ range with 0.0167° step interval. In addition, given the correlation of the Raman spectra with the parameters calculated from XRD, a Raman spectrometer (LabRAM HR800-LS55, France) was applied for the specific microstructure information of the cobalt-based catalysts. The light source was provided by Nd-YAG at 532 nm, and the scanning region was selected between 1800 cm−1 and 1000 cm−1. For FTIR analysis, 10 mg sample and 200 mg KBr were ground together and pressed into slices for each run. Afterward, these slices were directly measured on a Bruker Vertex 70 spectrophotometer in the range from 4000 to 400 cm−1 with 64 accumulated scans at a resolution of 4 cm−1. XPS analysis was adopted to characterize the surface chemistry, especially for cobalt and carbon functionalities, of the solid samples to a depth of about 0.1–1 nm. The XPS spectra were obtained on a ThermoScientific ESCALAB 250Xi spectrometer, which was equipped with an Al (Kα) X-ray radiation source (hν = 1486.6 eV) at 20 eV pass energy, 0.1 eV energy step, and 0.1 s dwelling time.
As for the catalytic activity of the metallic nanoparticles: (1) the acid strength distribution was determined by the temperature-programmed desorption of ammonia (NH3-TPD) with the help of an ASIQACIV200-2 automatic physical/chemical adsorption analyzer (Quantachrome, USA). The sample (150 mg) was loaded into a U-tube quartz reactor and heated to 200 °C for 30 min (heating ramp of 10 °C min−1) under 30 mL min−1 of He flow. The system was cooled to 80 °C, 8% NH3/He mixed gas was adsorbed onto the catalyst for 60 min until the saturation state was obtained, and then the catalyst was purged with a He flow of 30 mL min−1 to remove the physically adsorbed NH3. Next, the system was heated to 700 °C with a heating ramp of 10 °C min−1 under the same He flow. (2) The distribution of Brønsted (B) and Lewis (L) acidity was examined by pyridine-absorbed Fourier transform infrared spectroscopy (Py-FTIR; Nicolet 6700, Orlando, FL). For each run, a 100 mg sample was vacuum-activated (1 × 10−4 mmHg) at 350 °C for 60 min. The background spectrum was recorded after cooling the sample to 50 °C, and the sample was then exposed to pyridine (Aldrich, GC, purity ≥99.5%) vapor for 15 min. Excess pyridine was subsequently removed by a vacuum pump at room temperature for 30 min, followed by measuring the pyridine-adsorption spectrum of the sample.
2.4 General procedure of reductive amination
The process of reductive amination was performed in a stainless-steel autoclave reactor (MS100-P5-T3-HC1-SV, Anhui Kemi Machinery Technology Co., Ltd, China) coupled with six independent channels (each of them is 10 mL). In a typical run, 0.5 mmol furfural, 10 mg catalyst, and 5 ml 7 M NH3 (in MeOH) were accurately weighed into one of the channels; afterward, the reactor was flushed with H2 several times to remove air. The reactor was then heated up to the target temperature for a given period and H2 pressure, and the magnetic stirrer was rotated at a constant speed of 300 rpm throughout the whole period to ensure a homogeneous reaction. Subsequently, the products in the mixture were filtered and primarily detected by gas chromatography using 1,3,5-trimethoxybenzene as the internal standard. The products were also identified by GC/MS (ThermoTrace 1300-ISQ QD, USA) and 1H NMR (Avance III 400 MHz NMR, Bruker, Germany) to calculate the corresponding conversion and selectivity. Further studies on the influence of the reaction conditions include the temperature range of 60–120 °C, the reaction time of 0–8 h, the catalyst amount of 5–15 mg, aqueous NH3 concentration of 2–7 M, and H2 pressure of 1–5 MPa. The scope of the substrates focuses on the typical biomass-derived platforms, involving 5-HMF, substituted furfural, and phenol derivatives.3. Results and discussion
3.1 Structural and catalytic features of the prepared catalyst
First of all, SEM images were taken to primarily investigate the morphological information of the catalysts. As shown in Fig. 2(a)–(c), the magnified images exhibit that anomalous sponge-like structures were observed for these catalysts, and this trend was more serious with increasing pyrolysis temperatures because that of Co@C-700-EtOH started to collapse into smaller particles with large pores. Such sponge-like structures were mainly caused by the volatilization of organic matter during the pyrolysis stage to form a heterogeneous material,26 which could assemble the metallic nanoparticles depending on its pore size, as reported by Lu et al.27 in their studies. Interestingly, an extra step of oxidation at 200 °C for 3 h might result in the formation of crystal cubes or rods as a part of the metallic nanoparticles were aggregated or oxidized into a relatively large size (displayed in Fig. 2(e)). The corresponding EDS result of Co/CoO@C-600-EtOH revealed the coexistence of C, O, and Co with a mass fraction of 76.3%, 18.0%, and 5.7%, respectively, where the ratio of C to O was superior to that of Co@C-600-EtOH owing to the superficial oxidation of cobalt nanoparticles in air, coinciding well with the similar observations reported previously.18,28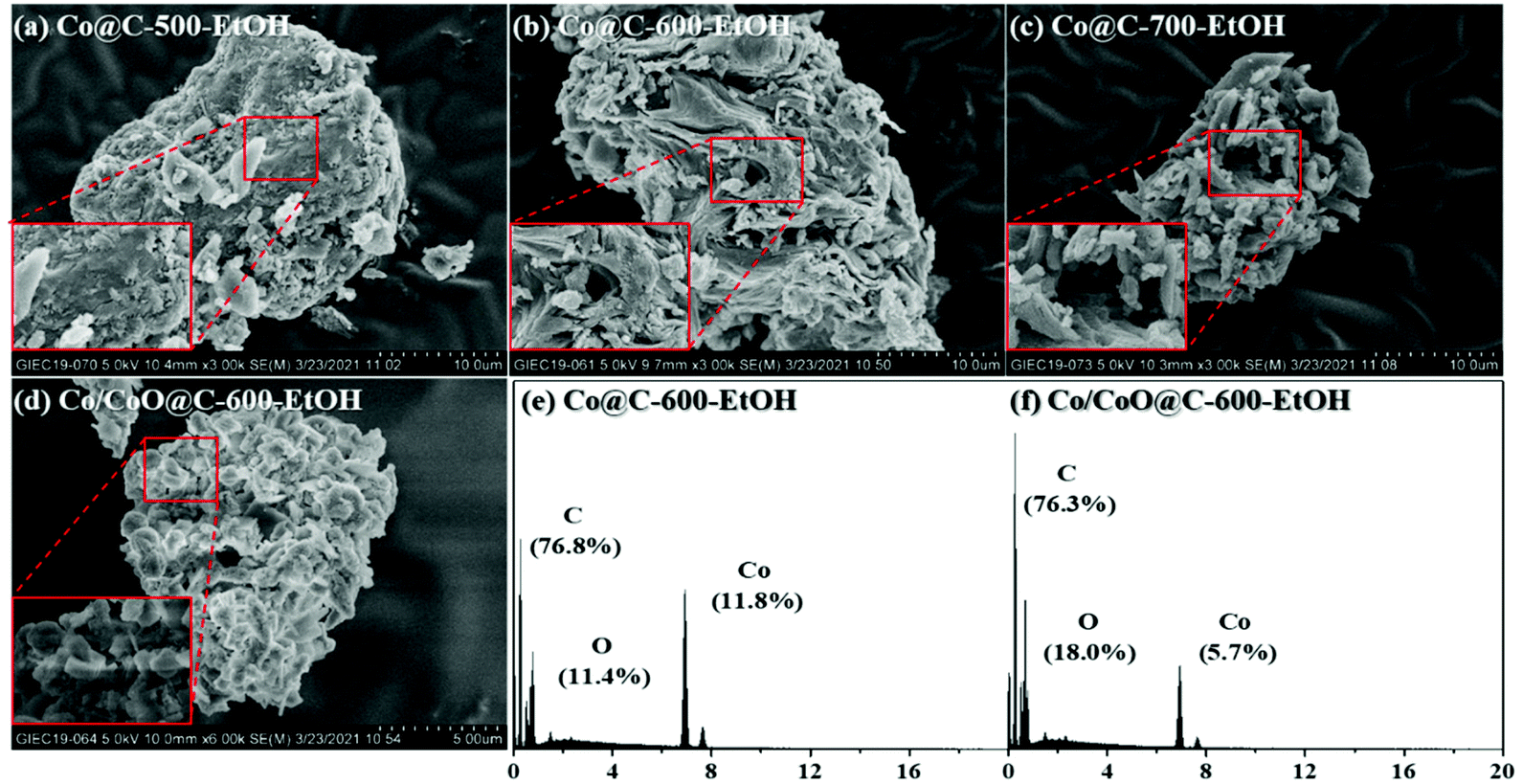 | ||
| Fig. 2 SEM image of Co@C-500-EtOH (a), Co@C-600-EtOH (b), Co@C-700-EtOH (c), and Co/CoO@C-600-EtOH (d), together with the corresponding EDS results of Co@C-600-EtOH (e) and Co/CoO@C-600-EtOH (f). | ||
In addition, the prepared catalysts were subsequently analyzed using TEM to observe the distribution of the supported metal on the nanoparticle surface and its average size, as shown in Fig. 3(a) and (b). It is obvious that the metallic nanoparticles were dispersed uniformly on the surface of the carbonaceous matrix and completely coated by the graphene shells without any significant aggregation of the clusters. The use of ethanol instead of H2O as the solvent provided a better distribution of the metallic phase. According to the statistical data, the average diameters of these metallic nanoparticles were 5.56 ± 0.35 nm (Co@C-500-EtOH), 6.82 ± 0.42 nm (Co@C-600-EtOH), 7.31 ± 0.28 nm (Co@C-700-EtOH), and 8.32 ± 0.24 nm (Co/CoO@C-600-EtOH), indicating: (1) at a temperature over 500 °C; the metallic nanoparticles were finally obtained by the encapsulation of graphene layers on the surface, and the layer number could be controlled by the pyrolysis temperature;29 (2) the extra step of oxidation could promote the growth and agglomeration of cobalt oxides, thus causing the poor dispersity.30 Well-resolved fringes with an interplanar spacing of 0.20 nm can also be observed in Co@C-600-EtOH, which is attributed to the (111) plane d-spacing of the Co metal (i.e., Co0) in Co@C-600-EtOH. Given these results, it can be proposed that Co2+ in the structure of the carbonaceous precursor was reduced during the pyrolysis process to Co0, and the final size of the formed Co nanoparticles can be controlled by varying the pyrolysis temperature.28 In contrast, the metallic nanoparticles in the Co/CoO@C-600-EtOH samples not only had the (111) plane of the Co metal but also exhibited the (111) plane of the CoO alloy with a d-spacing of approximately 0.25 nm.31 These crystal planes exactly agree with the number of diaphragms shown in their corresponding diffraction pattern, which are discussed together with the Raman spectra in the following section. Moreover, the corresponding elemental mappings in Fig. 3(c)–(f) also suggested the homogeneous distribution of Co atoms over the particle surface of Co@C-600-EtOH, whereas that of Co/CoO@C-600-EtOH indicated a higher concentration of the oxygen species, which coexist aqueous with Co atoms as cobalt oxide (CoO), in good agreement with the TEM images. By and large, most of these metallic atoms are surrounded by a combination of some graphitic layers and short-range ordered graphitic shells, functioning as chainmail for the catalyst, which is accessible to reactants and acts as the active center in the catalytic reaction.
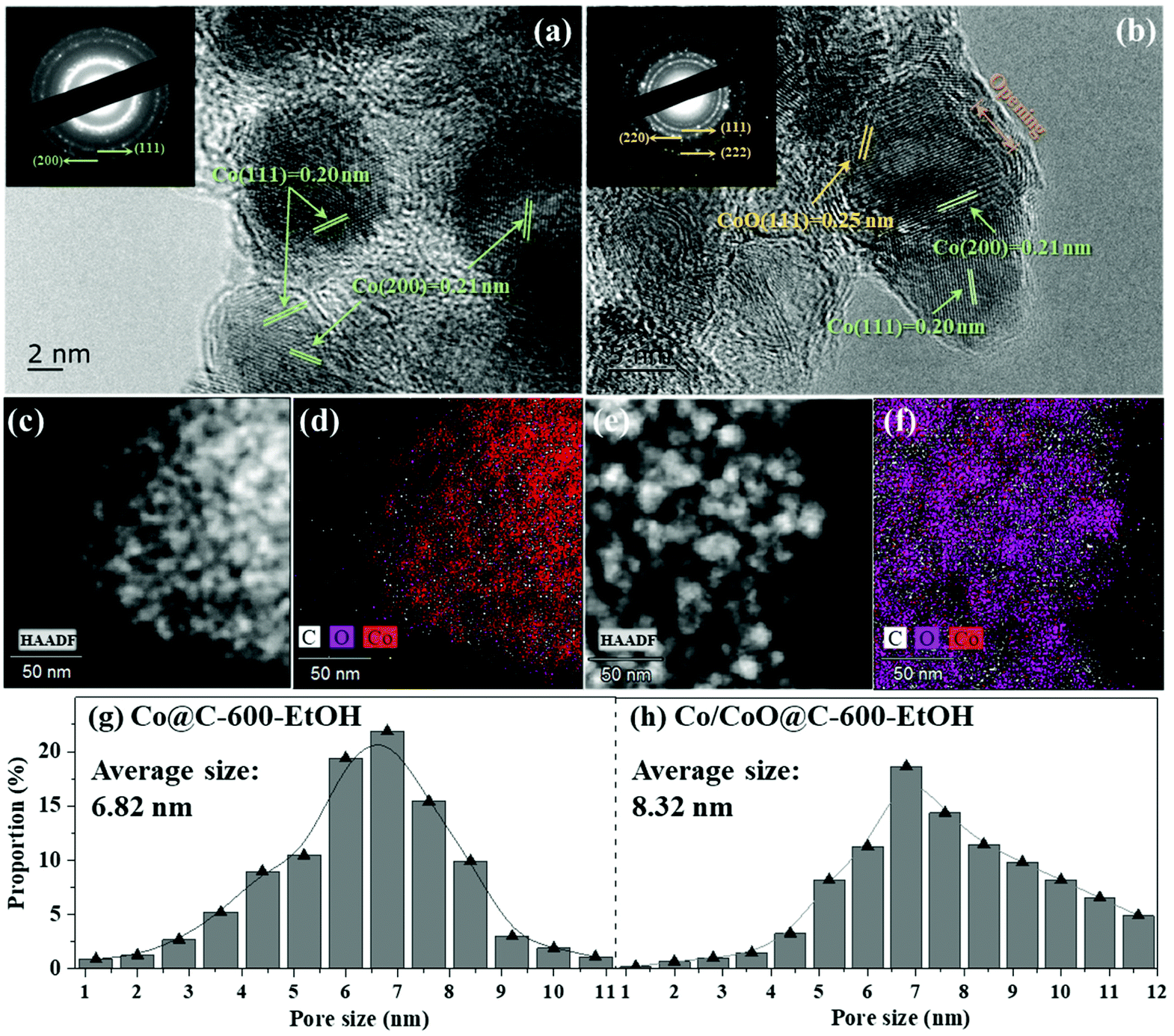 | ||
| Fig. 3 TEM image of Co@C-600-EtOH (a) and Co/CoO@C-600-EtOH (b), and the insets here show the corresponding diffraction pattern; HAADF-TEM images of a single particle in Co@C-600-EtOH (c) and Co/CoO@C-600-EtOH (e) with their elemental mapping of C, O, and Co in (d) and (f), respectively; the statistical results of the size distribution for Co@C-600-EtOH (g) and Co/CoO@C-600-EtOH (h). | ||
The N2 adsorption–desorption isotherms of the prepared catalyst at −196 °C were adopted to analyze the specific surface area through the Brunauer–Emmett–Teller (BET) method, while the pore structures were calculated using the Barrett–Joyner–Halenda (BJH) model to identify the proportion of microporous to mesoporous distribution (0.35–40 nm).28,31,32 As shown in Fig. 4(a), all the prepared catalysts presented the typical type IV isotherm (i.e., Langmuir adsorption–desorption isotherms), corresponding to the well-developed microporous and mesoporous structures as a large amount of N2 was mainly adsorbed in the entire relative pressure range.32 Herein, the hysteresis loop suggested that capillary condensation occurred at the desorption stage and led to a lower P/P0 than that at the adsorption process with the same quantity.28 With increasing pyrolysis temperatures, it can be observed that the specific surface area of the catalyst was gradually increased from 297.4 m2 g−1 (at 500 °C) to 407.8 m2 g−1 (at 600 °C) due to the devolatilization process when organic matter escaped in the form of CO2, leading to the opening of the pore structure, as confirmed by the SEM images. However, a slight decrease was detected for Co@C-700-EtOH (375.2 m2 g−1) because of the collapse of the pores at severe conditions. In addition, the Co/CoO@C-600-EtOH catalyst prepared with an extra oxidation step had a similar but relatively lower specific area (359.2 m2 g−1) than that of Co@C-600-EtOH, which implies that the introduction of oxygen assembles more metallic oxides, thereby slightly blocking the pore structure. More importantly, such hierarchical porosity of the Co@C catalyst is of great importance for the catalytic application as the heterogeneous porous property facilitates the chemisorption and dispersion of metal particles according to the relevant literature.31 It was also noted in Fig. 4(b) that all the samples displayed a peak centered at about 6.5 nm, indicating that heterogeneous materials are dominated by a mesoporous structure that can enhance the catalytic activity and provide efficient diffusion.33 The average pore diameters of Co@C-600-EtOH and Co/CoO@C-600-EtOH were calculated to be 5.6 and 4.9 nm, while the corresponding total pore volumes were 0.801 and 0.687 cm3 g−1, respectively. Interestingly, some early studies demonstrated that catalysts containing large pores could lead to the rapid transportation of products and reactants, thus influencing the selectivity for reduction amination.32,33
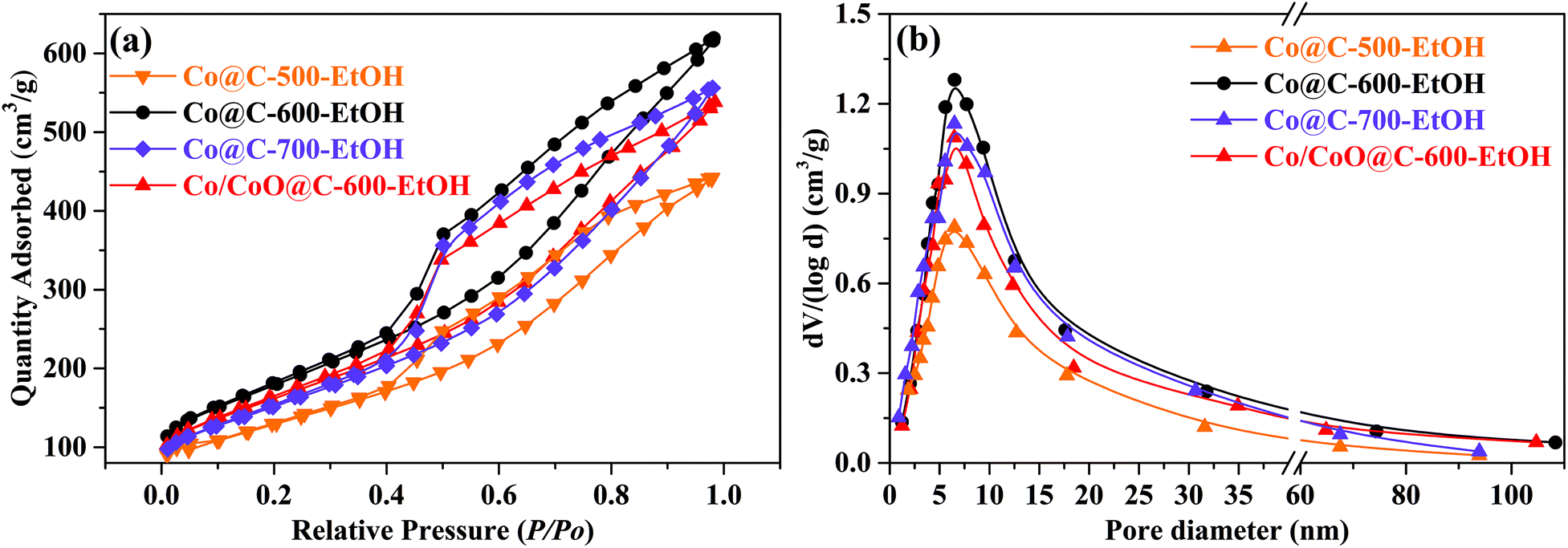 | ||
| Fig. 4 N2 adsorption–desorption isotherms and pore size distribution of the prepared catalysts. | ||
To further explore the structure–activity relationship, several techniques were adopted for a comprehensive understanding, whose results are provided in Fig. 5. From the XRD patterns depicted in Fig. 5(a), it can be found that only three characteristic peaks corresponding to the planes of Co metal (i.e., 43° (111), 52° (200), and 75° (220)) were observed in Co@C-600-EtOH when compared to Co/CoO@C-600-EtOH, and all of them were relatively narrow and sharp in the metallic states. This indicates that the Co@C-600-EtOH sample has high purity levels, single crystal phases, and high structural regularities.28,34 After being oxidized under 5% O2/N2 atmosphere at 200 °C, the reflected planes of the Co metal were weakened or even eliminated but the newly developed peaks at 37° (111), 65° (220), and 78° (222) can be ascribed to the incorporation of O into the lattice of the cobalt metal to form cobalt oxides.31 The broad reflection of Co/CoO@C-600-EtOH implied an amorphous structure, which might limit the active sites, thus affecting the catalytic activity during reduction amination.35,36 This can also be inferred from the Raman spectra shown in Fig. 5(b), where the spectra of heterogeneous materials exhibited a D band at about 1350 cm−1 and a G band at about 1580 cm−1, corresponding to the disordered arrangement of graphite lattice structures and the stretching of the sp2 atomic pairs on the carbon ring or long carbon-chain in graphite, respectively.36 The value of La was calculated through the strength of the D and G bands, as previously reported (Co@C-600-EtOH: 27.5; Co/CoO@C-600-EtOH: 23.0), which confirms that the oxidation step reduced the graphitization degree of carbon-supported catalysts.37 Acid functional groups were detected on the catalyst surface by means of FTIR spectroscopy, as shown in Fig. 5(c), where the wide peak of v-O–H (between 3600 cm−1 and 3200 cm−1) and the peak of v-C–O (between 1100 cm−1 and 1000 cm−1) were attributed to the hydroxyl group, while the peak of v-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (between 1760 cm−1 and 1680 cm−1), v-C–O (between 1320 cm−1 and 1210 cm−1), and δ-O–H (between 955 cm−1 and 915 cm−1) reflected the existence of the carboxyl group.35 Both the prepared catalysts exhibited a sufficient amount of acid sites, which can probably catalyze the reductive amination. In addition, the characteristic peaks associated with aromatic structures (v-CC and v–C–C) were found to be strong in the range from 1650 cm−1 to 1450 cm−1. The chemical states of the surface functionalities were also studied by XPS spectroscopy to understand the interaction between the carbon and the cobalt species, as shown in Fig. 5(d)–(e). In the C 1s spectra, the wide peak ranging from 282 eV to 295 eV can be resolved into individual peaks at approximately 283.9 eV, 284.8 eV, 286.2 eV, 287.6 eV, and 289.1 eV, which are in accordance with the -C–H bonds, the –C–(C, H)/CC bonds in the graphitic structures, –C–(O, N) graphene bonds, –CO bonds, and O–CO bonds, respectively;26,30,36 among them, the bond of –C–(C, H)/CC was dominant but suffered from a slight decrease after oxidation. Further, it is also noted that the chemical state of Co 2p was mainly in the metallic form with the binding energy of about 778 eV and 793 eV, while the peaks at 782 eV and 796 eV belonged to the oxidized state of the metallic atom.28,30 A strong Co 2p3/2 satellite peak at 786 eV and Co 2p1/2 satellite peak at 802 eV were found, which has been used for the identification of the cobalt species.31 The varied intensities of the specific peaks from different catalysts were in good agreement with these trends, for instance, the oxidation step strengthened the peak of Co 2p1/2 as well as weakened that of Co 2p3/2, which concludes that the functions of Co0 and Co2+ in the catalysts might be mutually affected by the preparation conditions.
O (between 1760 cm−1 and 1680 cm−1), v-C–O (between 1320 cm−1 and 1210 cm−1), and δ-O–H (between 955 cm−1 and 915 cm−1) reflected the existence of the carboxyl group.35 Both the prepared catalysts exhibited a sufficient amount of acid sites, which can probably catalyze the reductive amination. In addition, the characteristic peaks associated with aromatic structures (v-CC and v–C–C) were found to be strong in the range from 1650 cm−1 to 1450 cm−1. The chemical states of the surface functionalities were also studied by XPS spectroscopy to understand the interaction between the carbon and the cobalt species, as shown in Fig. 5(d)–(e). In the C 1s spectra, the wide peak ranging from 282 eV to 295 eV can be resolved into individual peaks at approximately 283.9 eV, 284.8 eV, 286.2 eV, 287.6 eV, and 289.1 eV, which are in accordance with the -C–H bonds, the –C–(C, H)/CC bonds in the graphitic structures, –C–(O, N) graphene bonds, –CO bonds, and O–CO bonds, respectively;26,30,36 among them, the bond of –C–(C, H)/CC was dominant but suffered from a slight decrease after oxidation. Further, it is also noted that the chemical state of Co 2p was mainly in the metallic form with the binding energy of about 778 eV and 793 eV, while the peaks at 782 eV and 796 eV belonged to the oxidized state of the metallic atom.28,30 A strong Co 2p3/2 satellite peak at 786 eV and Co 2p1/2 satellite peak at 802 eV were found, which has been used for the identification of the cobalt species.31 The varied intensities of the specific peaks from different catalysts were in good agreement with these trends, for instance, the oxidation step strengthened the peak of Co 2p1/2 as well as weakened that of Co 2p3/2, which concludes that the functions of Co0 and Co2+ in the catalysts might be mutually affected by the preparation conditions.
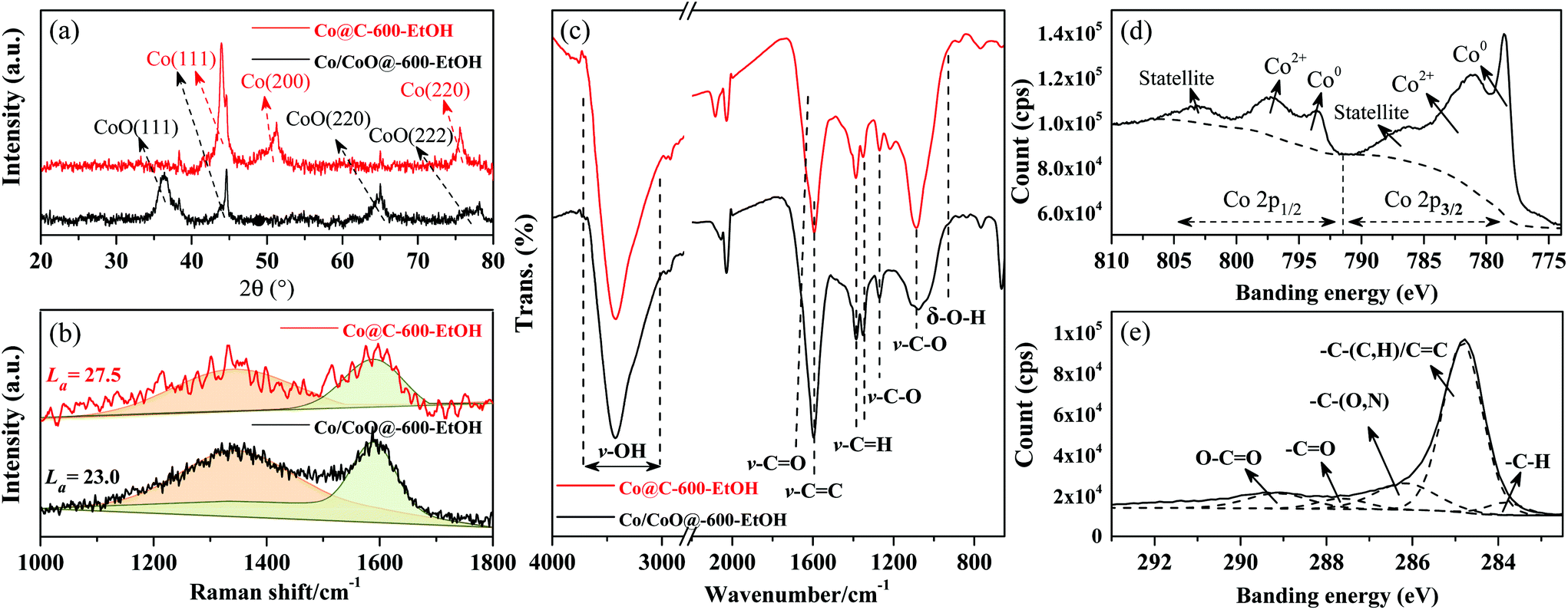 | ||
| Fig. 5 Structural features of the prepared catalyst. XRD results of Co@C-600-EtOH and Co/CoO@C-600-EtOH (a); Raman spectra of Co@C-600-EtOH and Co/CoO@C-600-EtOH (b); FTIR spectra of Co@C-600-EtOH and Co/CoO@C-600-EtOH (c); XPS image of Co@C-600-EtOH related with its spectrum (d) and Co spectrum (e). | ||
In an attempt to identify the effect of the acidity on the catalytic activity, its strength distribution and relevant type were collaboratively examined by Py-FTIR adsorption and NH3-TPD, respectively, whose profiles were depicted in the ESI.† The NH3 desorption peaks positioned at the temperature of 200 °C were rarely observed for both Co@C-600-EtOH and Co/CoO@C-600-EtOH, and this might explain that no weak acidity was formed on the particle surface.31 At higher temperatures, two desorption peaks at about 331 °C and 382 °C were observed for Co@C-600-EtOH, suggesting the existence of medium acidity. In contrast, it is found that the NH3 desorption peak of Co/CoO@C-600-EtOH was stronger (i.e., 470 °C), which indicates that the number of strong acid sites was much more than that of Co@C-600-EtOH. As for the type of acid sites, the Py-FTIR spectrum was further obtained at about 1650 cm−1 and 1400 cm−1, where the reflection bonds at 1450 cm−1 and 1590 cm−1 were all ascribed to the Lewis acid sites; meanwhile, a tiny reflection of the Brønsted acid site was also observed at 1540 cm−1. Both of them are similar to the catalysts of Co@C-600-EtOH and Co/CoO@C-600-EtOH. In general, the Brønsted acid sites can be attributed to the hydroxyl groups, which were easily dissociated at a high temperature,38 while the introduced Co species can act as Lewis acid sites or electrophilic sites to polarize and facilitate the cleavage of the C–O bond.37
3.2 Catalyst application on the reductive amination reaction
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalysts | Conv./% | Yield of 1a/% | Entry | Catalyst | Conv./% | Yield of 1a/% |
| Note: Screening of the prepared and commercial catalysts are based on the below conditions: 90 °C, 2 h, 10 mg catalyst, 5 ml ammonia solution (7 M NH3 in MeOH), 2 MPa H2, and 0.5 mmol furfural; the best catalyst Co@C-600-EtOH was selected to further optimize the reaction conditions. Yields and conversion were determined by GC-MS and 1H NMR spectroscopy using 1,3,5-trimethoxybenzenen as the internal standard. | |||||||
| 1 | Co@C-500-EtOH | >99 | 80.4 | 6 | 5% Ru/C | >99 | 0.0 |
| 2 | Co@C-600-EtOH | >99 | 86.9 | 7 | 5% Pt/C | >99 | 81.1 |
| 3 | Co@C-700-EtOH | >99 | 71.5 | 8 | 5% Rh/C | >99 | 68.5 |
| 4 | Co@C-600-H2O | >99 | 47.2 | 9 | 5% Pd/C | >99 | 14.7 |
| 5 | Co/CoO@C-600-EtOH | >99 | 8.5 | 10 | RANEY® Co | >99 | 83.7 |
| — | — | — | — | 11 | RANEY® Ni | >99 | 67.8 |
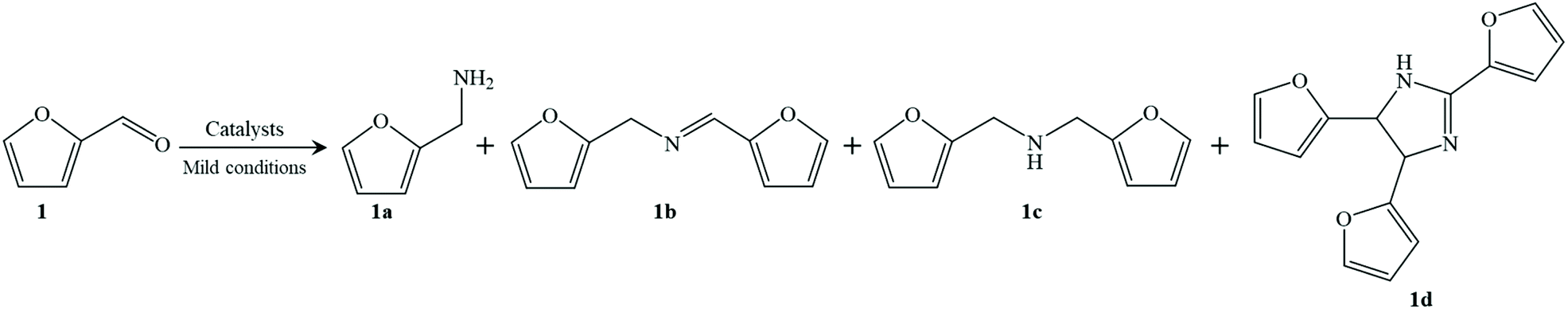
From these results, reductive amination can be successfully done using the Co@C-600-EtOH sample under very mild reaction conditions but several side reactions are one of the main reasons for carbon loss according to the reaction formula of reductive amination illustrated in Table 2. In addition to furfurylamine, another condensation product of N-furfurylidenefurfurylamine (i.e., 1b) was identified to be one of the secondary intermediates. The hydrogenation of N-furfurylidenefurfurylamine could easily produce the byproduct difurfurylamine (i.e., 1c), while the subsequent thermal cyclization of 1c toward furfurin (i.e., 1d) has also been noted in the previous works.39 Interestingly, the hydrogenation of furfural into furfuryl alcohol was not detected in our catalyst system, which is commonly observed in other catalytic systems, especially using noble metal catalysts.6 This may be caused because the cobalt-based catalyst was inactive to the hydrogenation of furfuryl alcohol, in accordance with the higher selectivity for furfurylamine in the RANEY® Co system when compared to that of RANEY® Ni (entries 10 and 11). Xu et al.21 also confirmed that the cobalt nanoparticles were identified as the active sites for reductive amination, which is very meaningful to get a satisfactory yield of the target product.
With the optimal reaction temperature in hand, the time course of the distributions of processing the product was recorded for the reductive amination of furfural with NH3 and H2, as shown in Fig. 6(b). For all the cases, the conversion of furfural was high to quantitative (>99.5%); as far as the target product is concerned, the molar percentage of furfurylamine greatly increased from 2.1% at 0 h to 44.3% at 1 h, 78.8% at 2 h, and 86.2% at 4 h, followed by a constant level even prolonging the reaction period up to 8 h. The variation of the byproducts through the side reactions was also observed and suffered from a similar tendency as that of the reaction temperatures.32 With regard to the influence of catalyst addition (shown in Fig. 6(c)), 5 mg amount was sufficient to fully convert furfural but only yielded 10.2% furfurylamine, which might be owing to the fact that a large amount of furfural tends to react with the produced furfurylamine for N-furfurylidenefurfurylamine without the help of the Co@C-600-EtOH catalyst. Accordingly, adding more catalysts could improve the catalytic selectivity for the target products by up to 87.6%. It was also noted that the 10 mg was the threshold value as a further increase in the catalyst showed slight effects on the catalytic selectivity. Moreover, the effects of NH3 concentration were also illustrated, as depicted in Fig. 6(d). In contrast to the aforementioned factors, the NH3 concentration exhibited relatively weak effects on the distribution of the products, and the yield of furfurylamine could be accumulated with the increasing concentration of NH3 to some extent. For example, the selectivity of furfural toward furfurylamine was calculated to be 76.9% by the use of 2 M concentration of NH3 in MeOH, while they were 83.3% and 86.2% in the presence of MeOH solution with 5 M and 7 M NH3 concentration, respectively. Last but not least, the H2 pressure also reflected a negligible effect on the conversion of furfural and its selectivity toward the target product, as shown in Fig. 6(e). All cases with varied H2 pressure in the range of 1–5 MPa could provide the full conversion and the high selectivity of furfurylamine over 99% and 85%, respectively. The byproduct of difurfurylamine was not observed under 1–2 MPa H2 pressure but it was produced under 3–5 MPa H2 pressure with an increasing trend, whose selectivity was determined to be 3.4%, 5.0%, and 4.2% under the H2 pressure of 3, 4, and 5 MPa, respectively. These results suggest that both the transformation of furfural into furfurylamine as well as the side product of difurfurylamine were simultaneously promoted by the increase in the H2 pressure. Consequently, the optimal H2 pressure of 2 MPa was adopted for the reductive amination of furfural, which provides not only the high conversion but also the high selectivity of furfurylamine.
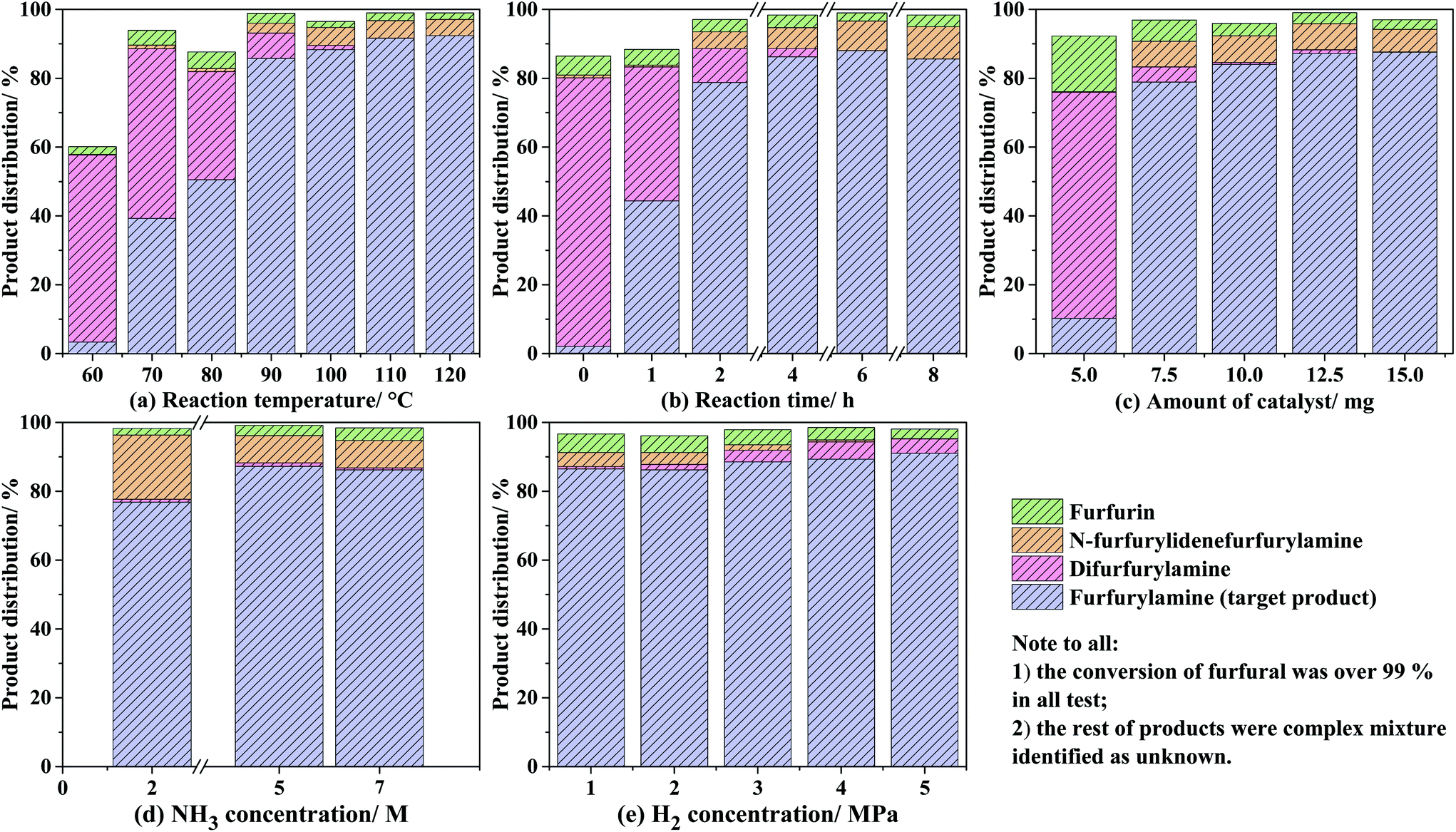 | ||
| Fig. 6 Influences of the reaction conditions on the reductive amination of furfural. Reaction temperature (entries 12–18), reaction time (entries 19–24), amount of catalyst (entries 25–29), NH3 concentration (entries 30–33), and H2 pressure (entries 34–38). Yields and conversion were determined by GC-MS and 1H NMR spectroscopy using 1,3,5-trimethoxybenzenen as the internal standard. | ||
During the process of reductive amination, N-furfurylidenefurfurylamine was identified to be the sole intermediate by gas chromatography, which was formed from the condensation of furfural with furfurylamine via the direct pathway according to the previous literature;13,39 herein, methylfurimidate formed by the condensation of furfural with NH3 was unstable to be detected by gas chromatography. The molar percentage of difurfurylamine was mostly eliminated after 4 h while the formation of furfurylamine remained almost unchanged, which suggests that the transformation of N-furfurylidenefurfurylamine into furfurylamine might be the rate-determining step.14 The addition of one NH3 molecule to N-furfurylidenefurfurylamine gave rise to the unstable germinal diamine, which was subsequently hydrogenated toward furfurylamine. This trend is confirmed by the increased selectivity of furfurylamine by adding more NH3 into the reaction systems discussed above. In summary, the reaction conditions of 90 °C, 4 h, 2 MPa H2, 10 mg catalyst, and 7 M NH3 solution have been considered as the optimal conditions.
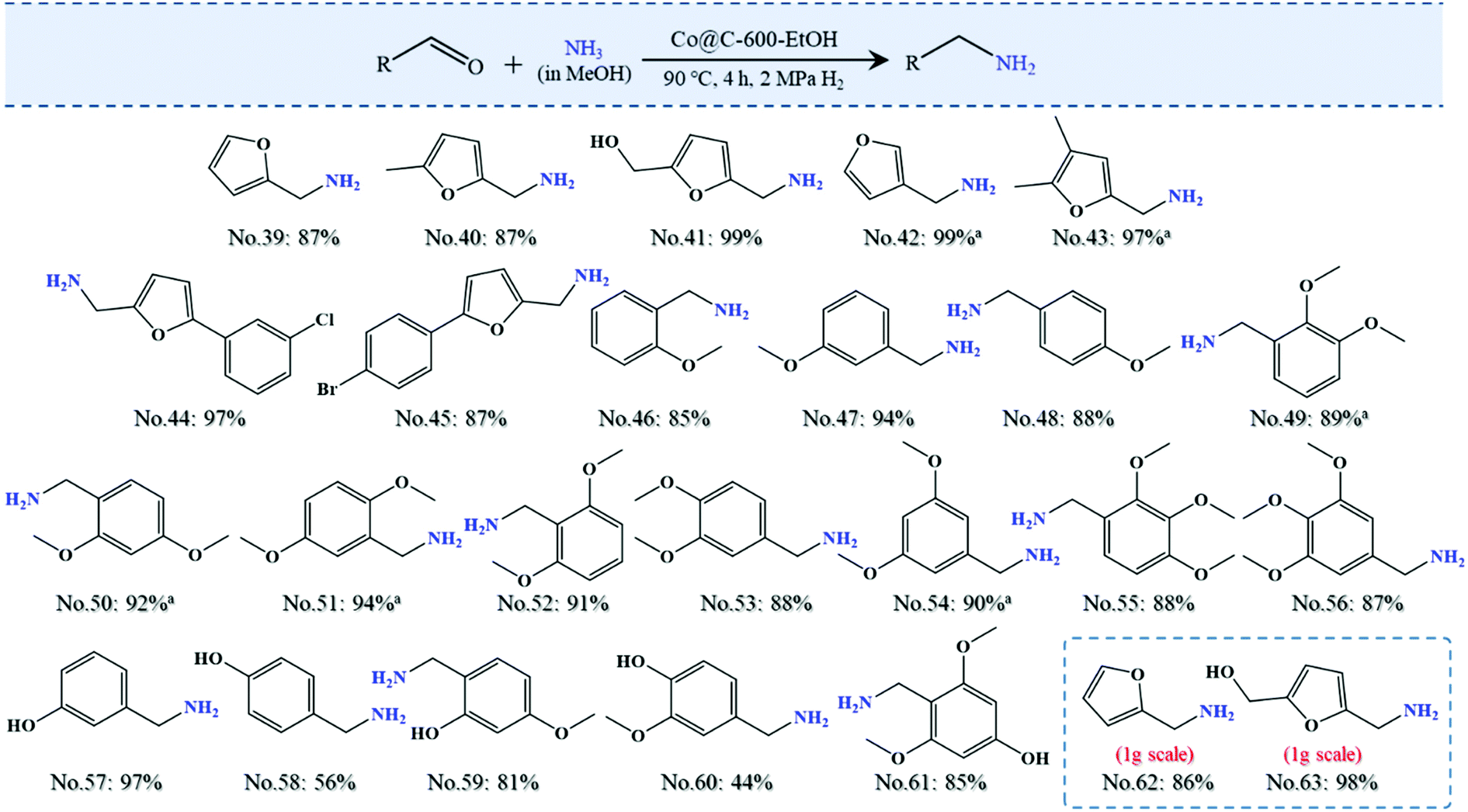 | ||
| Fig. 7 The result of reductive amination based on biomass-derived platforms for the synthesis of primary amines. Reaction conditions: reactant (0.05 mmol), 90 °C, 4 h reaction time, 10 mg catalyst, 7 M NH3 solution, and 2 MPa H2 pressure. Note: aThe reaction temperature was increased to 100 °C. Yields and conversion were determined by GC-MS and 1H NMR spectroscopy using 1,3,5-trimethoxybenzenen as the internal standard. | ||
According to our studies and controlled experiments, we proposed the mechanism of Co@C-600-EtOH for catalyzing the reductive amination of biomass-derived platform with NH3 solution as the N source. In this reaction process depicted in Fig. 8, furfural reacted with NH3 first to form the intermediate methanol adsorbed on the catalyst surface, whose outer graphitic shell was activated by the electronic interaction between the inner metallic nanoparticles and the carbon layer as well as the induced charge redistribution.23,24 This characteristic facilitated the delivery of electrons catalyzed by metallic cobalt, thereby forming imines after the dehydration reaction. In the following step, the H–H bond was activated on the surface of Co@C-600-EtOH, and meanwhile, the imine was reduced to furfurylamine by the introduction of H+ under a very mild environment eventually.39 The most accepted reductive amination pathway refers to the nucleophilic addition of the amine and carbonyl groups to yield an imine, and the imine is then hydrogenated to the desired amine.25 However, as for the second amines, the corresponding imine cannot be formed according to the above pathway. An in situ Raman experiment was previously reported to record the presence of the intermediate species during the reaction under 80 °C,43 whose results illustrated that metallic Co promotes imine formation by activating the CO bond in the aldehyde groups rather than the interaction between furfural and furfurylamine, while the acid sites on the particle surface are responsible for the hydrogenation of primary imines to the corresponding amines.
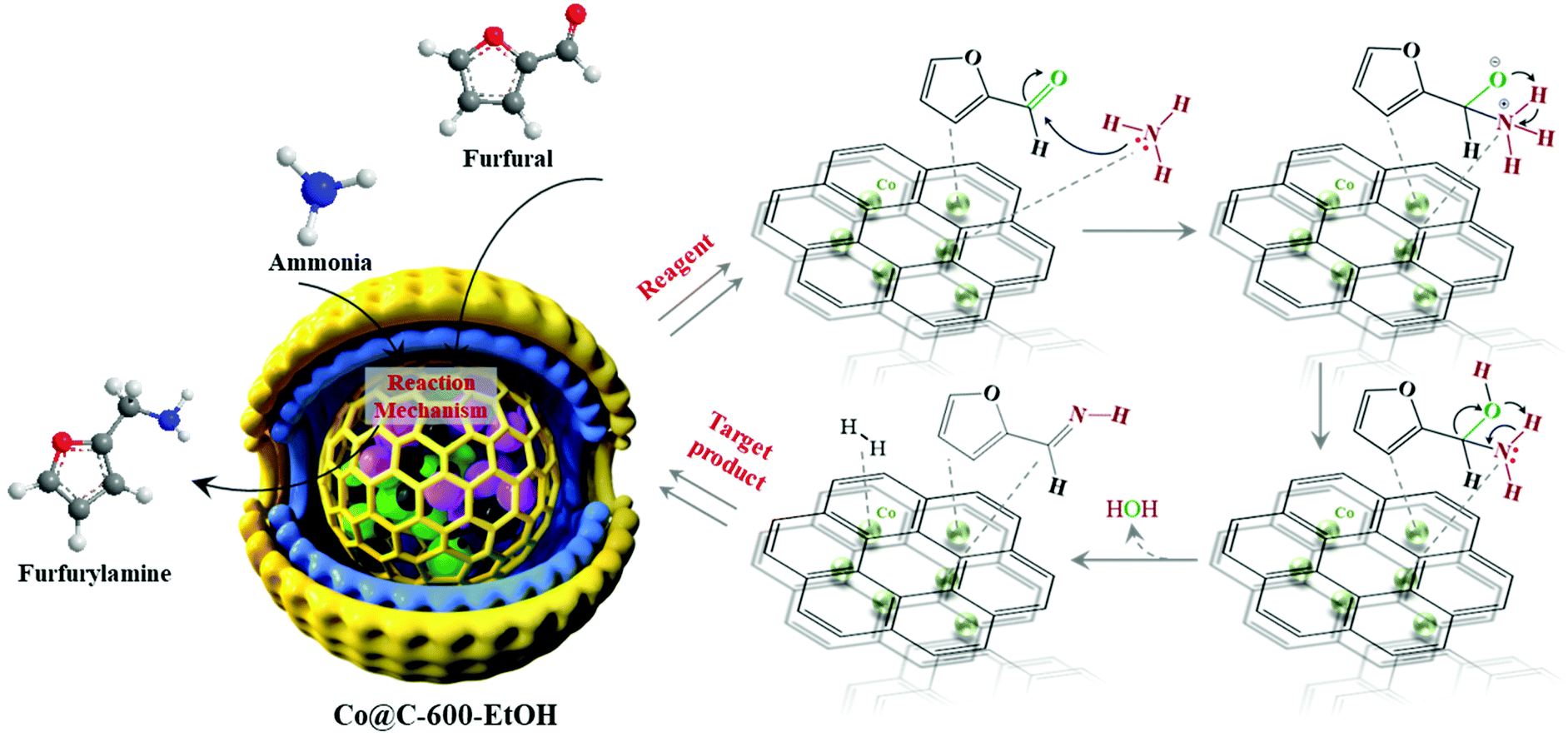 | ||
| Fig. 8 A possible pathway for Co@C-600-EtOH to catalyze the reduction amination of furfural with ammonia and H2. | ||
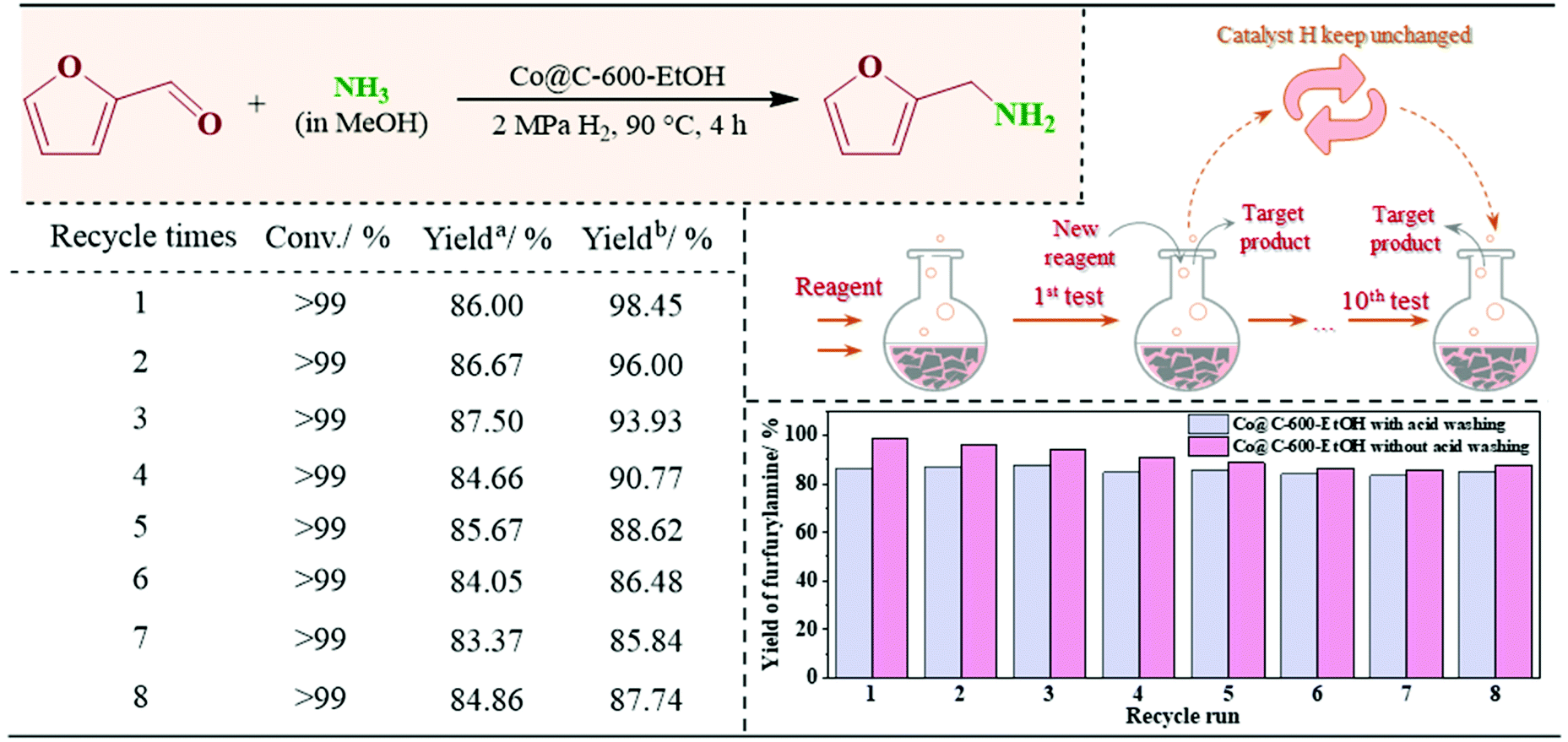 | ||
| Fig. 9 The recycling experiment of the Co@C-600-EtOH catalyst on the reductive amination of furfural. Reaction conditions: furfural (0.05 mmol), 90 °C h reaction time, 4 h reaction time, 10 mg catalyst, 7 M NH3 solution, and 2 MPa H2 pressure. Yielda and Yieldb were calculated based on the Co@C-600-EtOH with and without acid washing, respectively. | ||
4. Conclusions
In summary, uniform nanoparticle catalysts encapsulated in the multilayer graphene structure using cobalt acetate as the metallic precursor are synthesized successfully, followed with a simple and environment friendly method. According to its structural features associated with the catalytic activity and selectivity, we found that the metallic nanoparticles are dispersed uniformly on the surface of the carbonaceous matrix and completely coated by the graphene shells without any significant aggregation of the clusters, and the sponge-like structures of heterogeneous materials are the reason for the good distribution of metallic nanoparticles depending on its pore size. Moreover, the introduced Co species can act as Lewis acid sites or electrophilic sites to polarize and facilitate the reductive amination but an extra step of oxidation will significantly weaken its catalytic selectivity. The processing conditions (i.e., temperature, time, H2 pressure, NH3 concentration, and amount of catalyst) affect the distribution of the reaction products to different extents, and the optimal conditions were found to be 90 °C, 4 h, 2 MPa H2 pressure, 7 M NH3 solution, and 10 mg catalyst. More importantly, under the mild industrially viable and scalable conditions, the catalysts couple easily to a wide range of biomass-derived platforms containing aldehyde groups; thus, a series of functionalized structurally diverse branched benzylic and heterocyclic amines (more than 24 samples) were synthesized in good to excellent yield. The reaction pathways reflect that metallic Co promotes imine formation by the activating CO group of the aldehyde groups, while the acid sites on the particle surface are responsible for the hydrogenation of imine to amine. Also, this protocol exhibits a similar reactivity during the gram-scale test as well as a longer run of the lifecycle as it can be easily recycled and reused up to eight times without any significant loss of the catalytic activity and selectivity. The above findings strongly confirm the available and feasible preparation of simple but highly efficient catalysts toward the reductive amination of renewable sources. By and large, the advantages of this newly developed method include operational simplicity, high stability, easily recyclable, cost-effective catalyst, and good functional group compatibility for the synthesis of functional amines, as well as the highly efficient and industrial applicable synthetic process.
Author contributions
J. G. L. and L. L. M. supervised and designed the research. X. Z. Z. performed most of the experiments and wrote the original paper. S. R. Z. performed substrates scope experiments. J. G. L. reviewed and corrected the original manuscript. All authors discussed the results and assisted during manuscript preparation.Data availability
Data supporting the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported financially by the National Key R&D Program of China (2018YFB1501500), National Natural Science Foundation of China (51976225).References
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu, M. Heggen, L. Lin, C. Ma, M. Wang, F. Fan, S. Jin and F. Wang, Nat. Energy, 2019, 4, 575–584 CrossRef CAS.
- E. Hayashi, Y. Yamaguchi, K. Kamata, N. Tsunoda, Y. Kumagai, F. Oba and M. Hara, J. Am. Chem. Soc., 2019, 141, 890–900 CrossRef CAS PubMed.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS.
- H. Li, S. Yang, S. Saravanamurugan and A. Riisager, ACS Catal., 2017, 7, 3010–3029 CrossRef CAS.
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Green Chem., 2020, 22, 6714–6747 RSC.
- I. Bodachivskyi, U. Kuzhiumparambil and D. B. G. Williams, ChemSusChem, 2018, 11, 642–660 CrossRef CAS PubMed.
- E. C. Gaudino, G. Cravotto, M. Manzoli and S. Tabasso, Green Chem., 2019, 21, 1202–1235 RSC.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS.
- H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021–1049 CrossRef CAS.
- D. Carnevali, O. Guevremont, M. G. Rigamonti, M. Stucchi, F. Cavani and G. S. Patience, ACS Sustainable Chem. Eng., 2018, 6, 5580–5587 CrossRef CAS.
- Y. Wang, X. Yang, H. Zheng, X. Li, Y. Zhu and Y. Li, Mol. Catal., 2019, 463, 130–139 CrossRef CAS.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba and M. Lopez Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC.
- S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS.
- T. Komanoya, T. Kinemura, Y. kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef CAS PubMed.
- T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumann, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Nat. Commun., 2018, 9, 4123 CrossRef PubMed.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- S. Nishimura, K. Mizuhori and K. Ebitani, Res. Chem. Intermed., 2016, 42, 19–30 CrossRef CAS.
- J. J. Martinez, E. Nope, H. Rojas, M. H. Brijaldo, F. Passos and G. Romanelli, J. Mol. Catal. A: Chem., 2014, 392, 235–240 CrossRef CAS.
- K. Zhou, B. Chen, X. Zhou, S. Kang, Y. Xu and J. Wei, ChemCatChem, 2019, 11, 5562–5569 CrossRef CAS.
- M. Manzoli, E. C. Gaudino, G. Cravotto, S. Tabasso, R. B. N. Baig, E. Colacino and R. S. Varma, ACS Sustainable Chem. Eng., 2019, 7, 5963–5974 CrossRef CAS.
- L. Yu, D. H. Deng and X. H. Bao, Angew. Chem., Int. Ed., 2020, 59, 15294–15297 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- J. Liu, Y. Zhu, C. Wang, T. Singh, N. Wang, Q. Liu, Z. Cui and L. Ma, Green Chem., 2020, 22, 7387–7397 RSC.
- W. Lv, Y. T. Zhu, J. Liu, C. G. Wang, Y. Xu, Q. Zhang, G. Y. Chen and L. L. Ma, ACS Sustainable Chem. Eng., 2019, 7, 5751–5763 CrossRef CAS.
- C. G. Lu and J. Liu, J. Phys. Chem. B, 2006, 110, 20254–20257 CrossRef CAS PubMed.
- X. Y. Li, C. M. Zeng, J. Jiang and L. H. Ai, J. Mater. Chem. A, 2016, 4, 7476–7482 RSC.
- X. Sun, A. I. Olivos-Suarez, L. Oar-Arteta, E. Rozhko, D. Osadchii, A. Bavykina, F. Kapteijn and J. Gascon, ChemCatChem, 2017, 9, 1854–1862 CrossRef CAS.
- K. Murugesan, T. Senthamarai, M. Sohail, A. S. Alshammari, M. M. Pohl, M. Beller and R. V. Jagadeesh, Chem. Sci., 2018, 9, 8553–8560 RSC.
- D. Li, Q. Y. Liu, C. H. Zhu, H. Y. Wang, C. H. Cui, C. G. Wang and L. L. Ma, J. Energy Chem., 2019, 30, 34–41 CrossRef.
- Z. L. Yuan, B. Liu, P. Zhou, Z. H. Zhang and Q. Chi, J. Catal., 2019, 370, 347–356 CrossRef CAS.
- Y. L. Zhang, X. Z. Lin, X. J. Li, C. G. Wang, Q. Long and L. L. Ma, New J. Chem., 2018, 42, 15968–15973 RSC.
- X. H. Liu, C. G. Wang, Y. Zhang, Y. Qiao, Y. Pan and L. L. Ma, ChemSusChem, 2019, 12, 4791–4798 CrossRef CAS PubMed.
- X. Z. Zhuang, H. Zhan, Y. Q. Huang, Y. P. Song, X. L. Yin and C. Z. Wu, Bioresour. Technol., 2018, 267, 17–29 CrossRef CAS.
- X. Z. Zhuang, H. Zhan, Y. P. Song, C. He, Y. Q. Huang, X. L. Yin and C. Z. Wu, Fuel, 2019, 236, 960–974 CrossRef CAS.
- L. Wang, Y. Weng, P. Duan, X. Liu, X. Wang, Y. Zhang, C. Wang, Q. Liu and L. Ma, SN Appl. Sci., 2019, 1, 404 CrossRef CAS.
- C. H. Zhu, H. Y. Wang, H. Li, B. J. Cai, W. Lv, C. L. Cai, C. G. Wang, L. Yan, Q. Y. Liu and L. L. Ma, ACS Sustainable Chem. Eng., 2019, 7, 19556–19569 CrossRef CAS.
- N. S. Gould, H. Landfield, B. Dinkelacker, C. Brady, X. Yang and B. J. Xu, ChemCatChem, 2020, 12, 2106–2115 CrossRef CAS.
- W. Chen, Y. Sun, J. Du, Z. Si, X. Tang, X. Zeng, L. Lin, S. Liu and T. Lei, J. Chem. Technol. Biotechnol., 2018, 93, 3028–3034 CrossRef CAS.
- W. Wu, W. Zhang, Y. Long, J. H. Qin and J. T. Ma, Mol. Catal., 2020, 497, 10 Search PubMed.
- Y. L. Cao, K. K. Liu, C. Wu, H. P. Zhang and Q. Y. Zhang, Appl. Catal., A, 2020, 592, 9 CrossRef.
- G. F. Liang, A. Q. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050–3054 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03578a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |