
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic conversion of glycerol and co-feeds (fatty acids, alcohols, and alkanes) to bio-based aromatics: remarkable and unprecedented synergetic effects on catalyst performance†
Songbo
He
a,
Thomas Sjouke
Kramer
a,
Dian Sukmayanda
Santosa
 a,
Andre
Heeres
b and
Hero Jan
Heeres
*a
a,
Andre
Heeres
b and
Hero Jan
Heeres
*a
aGreen Chemical Reaction Engineering, Engineering and Technology Institute Groningen, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: h.j.heeres@rug.nl
bHanze University of Applied Sciences, Zernikeplein 11, 9747 AS Groningen, The Netherlands
First published on 18th December 2021
Abstract
Glycerol is an attractive bio-based platform chemical that can be converted to a variety of bio-based chemicals. We here report a catalytic co-conversion strategy where glycerol in combination with a second (bio-)feed (fatty acids, alcohols, alkanes) is used for the production of bio-based aromatics (BTX). Experiments were performed in a fixed bed reactor (10 g catalyst loading and WHSV of (co-)feed of 1 h−1) at 550 °C using a technical H-ZSM-5/Al2O3 catalyst. Synergistic effects of the co-feeding on the peak BTX carbon yield, product selectivity, total BTX productivity, catalyst life-time, and catalyst regenerability were observed and quantified. Best results were obtained for the co-conversion of glycerol and oleic acid (45/55 wt%), showing a peak BTX carbon yield of 26.7 C%. The distribution of C and H of the individual co-feeds in the BTX product was investigated using an integrated fast pyrolysis-GC-Orbitrap MS unit, showing that the aromatics are formed from both glycerol and the co-feed. The results of this study may be used to develop optimized co-feeding strategies for BTX formation.
Introduction
Crude glycerol is an abundantly available co-product of the biodiesel industry (global amount of ca. 41.9 billion liters in 20201). It is considered a platform chemical with a large potential for further catalytic conversions to value-added bio-based chemicals, either as such2 or after purification3. The production of bio-aromatics such as benzene, toluene, and xylenes (abbreviated as bio-BTX) from glycerol using e.g., H-ZSM-5-based catalysts4,5 is an attractive option to green up the aromatics industry and thus to enhance the sustainability of the petrochemical industry.Several studies on the catalytic conversion of glycerol over H-ZSM-5-based catalysts in continuous fixed-bed reactors6–13 have shown a BTX carbon yield of at max 28.1 ± 0.2 C% when using an un-modified H-ZSM-5 zeolite catalyst.6 Such catalysts often show a rather short catalyst life-time (5–19 h) due to coke formation6,7,14,15 and thus require frequent catalyst regeneration by an oxidative treatment. However, a few studies on reaction-regeneration cycles6,9,10,12,15,16 have shown that also irreversible deactivation of the catalyst occurs, lowering the catalyst performance for the regenerated catalysts.
Co-feeding of glycerol with alcohols (e.g., methanol, ethanol, i-propanol, and i-butanol15,17–22), methanol/aromatics (e.g., benzene and toluene23), alkanes (e.g., n-hexane,24 dodecane and hexadecane,25 has also been studied, next to the use of pure glycerol.6,13,14,18,25–30 Based on literature data, it can be summarized that upon co-feeding, (i) the aromatics yield is increased, e.g., from 3.0 wt% for glycerol to 10.8 wt% for glycerol/methanol (55/45 wt%)18 and from 4.8 wt% for hexane to 12.0 wt% for glycerol/hexane (50/50 wt%);24 (ii) the selectivity of aromatics changes, e.g., a higher alcohol content leads to higher selectivities to benzene and toluene and lower xylenes and trimethyl benzenes selectivity;19 and (iii) the rate of catalyst deactivation is reduced leading to prolonged catalyst life-times, e.g., from 3 h for glycerol/methanol (40/60 wt%) to 8 h for glycerol/methanol (10/90 wt%).20
Literature studies have shown that improved catalyst activity and stability when using co-feeds is correlated with higher hydrogen to carbon effective ratio (abbreviated as H/Ceff, H/Ceff = (H-2O)/C) of the feed.31 Besides, an early study on catalytic co-conversion of glycerol and 13C-labeled methanol (5/95 wt%) showed that the 13C content in the aromatics was higher than that in the feed, indicating that carbon atoms in the co-feed are involved in pathways leading to the formation of aromatics.32
However, most of the above-mentioned co-feeding studies only applied glycerol in combination with one specific co-feed at a single mixing ratio and did not consider synergetic effects. We here report a systematic study with a broad range of co-feeds at similar and well-defined conditions, allowing proper comparison of data. We aimed to determine the synergetic effects of co-feeds on catalyst performance (e.g., BTX yield and selectivity, and catalyst life-time), and to determine the origin of the carbon atoms in the formed BTX (glycerol and/or the co-feed). In addition, the catalyst consumption (kgcat tonprod−1, or catalyst productivity, tonprod kgcat−1), which is one of the important performance metrics for industrial implementation33 has been determined and will be reported, and this is also an absolute novelty of this paper. This metric is particularly important for the catalytic conversion of glycerol to BTX using H-ZSM-5 based catalysts considering the occurrence of irreversible catalyst deactivation (ca. 10–15% decrease in BTX production after each regeneration6,10). Irreversible deactivation was shown to be most likely due to dealumination of the H-ZSM-5 framework by steam34 generated in a large amount 6 by dehydration reactions.35 As such, the effect of co-feeds on catalyst stability and particularly irreversible deactivation is also of interest.
In this contribution, we report a comprehensive study on the catalytic conversion of glycerol in combination with a co-feed for BTX production. Three types of co-feeds were used, including alcohols (methanol and ethanol), alkanes (dodecane and hexadecane), and free fatty acids (oleic acid). The latter is of particular interest as crude glycerol may contain considerable amounts of such free fatty acids. Previous studies have shown that fatty acids and vegetable oils are good feeds for bio-BTX production, examples are the use of oleic acid36 and glycerol blended with canola oil.37 In total, 13 different glycerol – co-feeds at different blending ratios were tested in this study using a technical H-ZSM-5/Al2O3 catalyst27,36 in a fixed bed reactor at times on stream (TOS) between 8.5–12 h. We particularly aimed to determine synergetic effects between glycerol and the co-feed on catalyst performance including peak BTX yields, catalyst productivity, and regenerability. Besides, using catalytic pyrolysis integrated with GC-Orbitrap MS, we also show the distribution of the C and H of the co-feeds in the products.
Experimental
Materials
The granular H-ZSM-5/Al2O3 catalyst (60/40 wt%, ϕ 1.2–1.8 mm) used in this study was supplied by Yangzhou Baisheng Catalyst Co., Ltd, PR China. The as-received catalyst was pretreated by calcination at 600 °C under air for 8 h in a muffle furnace (LT 9/11/P330, Nabertherm GmbH) and was stored in a vacuum desiccator (Bel-Art™ F42400-2141, BEL-ART – SP Scienceware & HB Instruments) filled with silica gel (Fisher Scientific Nederlands) at reduced pressure.Glycerol (>99.5% purity) was supplied by Boom BV, The Netherlands. Oleic acid (>95% purity, Product No. O/0200/17) was supplied by Fischer Scientific Netherlands. All the other chemicals such as methanol, ethanol, dodecane, hexadecane, tetrahydrofuran (THF), n-nonane, C3D8O3 (≥98 atom % D, ≥98% CP, Product No. 447498-1G), and 13C3H8O3 (≥99 atom % 13C, ≥99% CP, Product No. 489476-1G), are of analytical grade and were supplied by Sigma-Aldrich. N2 gas (99.999% purity) was supplied by Linde.
Catalytic (co-)pyrolysis of the (co-)feed(s) to aromatics in a fixed-bed reactor unit
The catalytic conversion of glycerol with various co-feeds was performed in a bench-scale fixed-bed reactor setup (Fig. S1†) loaded with 10 g of the H-ZSM-5/Al2O3 catalyst. The reactor was heated to 550 °C with a heating rate of 5 °C min−1 under an N2 flow of 50 ml min−1. The liquid feedstocks (weight hourly space velocity, WHSV of 1 h−1) were pumped into a preheater (350 °C) by four syringe pumps (NE-1010, Prosense B.V.) using four 100 ml gastight syringes (Part No. 86020, Hamilton). The vaporized feedstock(s) mixed with N2 gas were then passed over the catalyst bed. After the reaction, the pyrolysis vapor was cooled in two parallel condensation-separation units (switched every 30 min) and transferred to a sampling unit (−40 to 1 °C), equipped with automated pneumatic switches (V11, V12, and V15, Fig. S1†). The liquid samples were collected in 20 ml glass vials, the gases in 5-L FlexFoil Plus sample bags (Part No. 207104, SKC Ltd).After the reaction, the reactor was cooled to room temperature under N2 (50 ml min−1). The used catalyst was removed from the reactor followed by an oxidative regeneration in a muffle furnace (LT 9/11/P330, Nabertherm GmbH). The used catalyst was placed in a 90 ml Haldenwanger porcelain crucible (Product No. 10493081, Fisher Scientific Nederlands) and was oxidized at 680 °C for 12 h. The regenerated catalyst was then loaded to the reactor to determine the performance by following the same protocol for testing the fresh catalyst. In total, 5 reaction-regeneration cycles were performed.
The liquid products were diluted approximately 7 times with a stock solution containing ca. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm of n-nonane in a solvent mixture (ethanol/THF, ca. 10/90 vol%). The samples were analyzed by GC-MS (HP 6890/5973, Hewlett-Packard) and GC-FID (HP 5890, Hewlett-Packard) equipped with a capillary column (Rtx-1701, 30 m × 0.25 mm × 0.25 μm, Restek). The relative response factors of the individual aromatics to n-nonane were applied for their quantification. The gaseous products were analyzed by a GC-TCD (HP 5890, Hewlett-Packard) equipped with a CP-PoraBOND Q column (50 m × 0.53 mm × 10 μm, Varian) and a HP-Molsieve column (30 m × 0.53 mm × 50 μm, Agilent). Before and after sample analyses, the GC-TCD was calibrated with a standard reference gas mixture containing C1–C3, CO, CO2, and N2 (Product No. G322243, Westfalen AG). The coke content on the used catalyst was analyzed using an elemental analyzer (EuroEA3000, Eurovector), which was calibrated using sulfanilamide as the standard.
000 ppm of n-nonane in a solvent mixture (ethanol/THF, ca. 10/90 vol%). The samples were analyzed by GC-MS (HP 6890/5973, Hewlett-Packard) and GC-FID (HP 5890, Hewlett-Packard) equipped with a capillary column (Rtx-1701, 30 m × 0.25 mm × 0.25 μm, Restek). The relative response factors of the individual aromatics to n-nonane were applied for their quantification. The gaseous products were analyzed by a GC-TCD (HP 5890, Hewlett-Packard) equipped with a CP-PoraBOND Q column (50 m × 0.53 mm × 10 μm, Varian) and a HP-Molsieve column (30 m × 0.53 mm × 50 μm, Agilent). Before and after sample analyses, the GC-TCD was calibrated with a standard reference gas mixture containing C1–C3, CO, CO2, and N2 (Product No. G322243, Westfalen AG). The coke content on the used catalyst was analyzed using an elemental analyzer (EuroEA3000, Eurovector), which was calibrated using sulfanilamide as the standard.
The carbon yields and selectivities of the products and the total BTX productivity were calculated using eqn (1)–(3). The catalyst life-time is defined as the TOS at which the yield of BTX was negligible (<1 C%).
 | (1) |
 | (2) |
 | (3) |
Catalytic (co-)conversion of 13C- and D-labeled glycerol mixed with oleic acid in an integrated fast pyrolysis-GC-Orbitrap MS unit
Catalytic fast pyrolysis-GC-Orbitrap MS was performed using an EGA/PY-3030D Multishot pyrolyzer equipped with an AS-1020E Autoshot autosampler (Frontier Laboratories). The pyrolyzer was coupled to an inline GC (Trace 1300, Thermo Scientific), which was equipped with an Rtx-1701 column (30 m × 0.25 mm × 0.25 μm, Restek) and an Exactive series EI/VeV/CI quadrupole-Orbitrap mass spectrometer (Thermo Scientific).38 The fine catalyst powder (ca. 50 μg) was mixed with the liquid feed containing glycerol and oleic acid (ca. 50 μg) in a crucible (Frontier Laboratories), which was loaded into the autosampler and automatically injected into the pyrolyzer operated at 550 °C. The detailed GC-Orbitrap MS operation parameters, standard calibration, and data processing are described in a previous publication.39 The molecular weight used for the identification of the BTX products with various labeled D and 13C atom(s) is shown in Table S1† and an example of GC-Orbitrap MS analysis is shown in Fig. S15.† All the experiments were performed in triplicate and the averaged results are reported.Results and discussion
Catalytic conversion of glycerol with various co-feeds
The catalytic conversion of glycerol with various co-feeds (WHSV of 1 h−1) was continuously performed in a fixed-bed reactor loaded with 10 g of the H-ZSM-5/Al2O3 catalyst at 550 °C for TOS of 12 h. An overview of experiments including the abbreviations used for the experiments is given in Table 1.| Entry | (co-)Feed | Abbreviations | H/Ceff | Weigh ratio (wt/wt) | Molar ratio (mol/mol) | Carbon ratio (mol/mol) |
|---|---|---|---|---|---|---|
| 1 | Glycerol | G | 0.67 | — | — | — |
| 2 | Methanol | M | 2.00 | — | — | — |
| 3 | Ethanol | E | 2.00 | — | — | — |
| 4 | Dodecane | D | 2.17 | — | — | — |
| 5 | Hexadecane | H | 2.13 | — | — | — |
| 6 | Oleic acid | O | 1.67 | — | — | — |
| 7 | Glycerol/methanol | G/M | 1.41 | 43/57 | 21/79 | 44/56 |
| 8 | Glycerol/ethanol | G/E | 1.37 | 54/46 | 37/63 | 47/53 |
| 9 | Glycerol/dodecane | G/D | 1.37 | 71/29 | 82/18 | 53/47 |
| 10 | Glycerol /hexadecane | G/H | 1.37 | 70/30 | 85/15 | 52/48 |
| 11 | Glycerol/oleic acid | G/O | 1.37 | 45/55 | 72/28 | 30/70 |
| 12 | Glycerol/oleic acid | G/O | 1.00 | 80/20 | 82/18 | 43/57 |
| 13 | Glycerol/oleic acid | G/O | 1.55 | 20/80 | 53/47 | 16/84 |
The carbon yields for the various aromatics (BTX and others) versus the TOS for a representative experiment (glycerol and oleic acid, entry 11 in Table 1) is given in Fig. 1. The carbon yields of the gas (CO, CO2, and C1–C3 hydrocarbons) and liquid products (BTX, ethylbenzene, naphthalene, and methyl naphthalenes), and the carbon selectivity's of the individual BTX components versus TOS for all other experiments are shown in Fig. S2–S11.† In general, the aromatics yield curves (e.g., Fig. 1) show a volcano-type shape,6,15,19,20 showing that the BTX yield increases to a maximum value (termed as peak BTX carbon yield) after TOS of ca. 1.5 h (associated with the start-up of the reaction system6,15). Then it decreases gradually with TOS (associated with catalyst deactivation, e.g., by coking26,40,41) to a negligible level. The catalyst life-time is defined as the TOS when the BTX yield is <1 C%.
 | ||
| Fig. 1 Carbon yields of aromatics (BTX and others) versus TOS for a representative experiment (glycerol/oleic acid, 45/55 wt%, entry 11 in Table 1). Reaction conditions: H-ZSM-5/Al2O3 (60/40 wt%) catalyst of 10 g, WHSV of the (co-)feeds of 1 h−1, N2 flow of 50 ml min−1, reactor temperature of 550 °C, and atmospheric pressure. | ||
| (co-)Feeda | G | M | G/M | E | G/E | D | G/D | H | G/H | O | G/O |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a (Co-)feed: G – glycerol, M – methanol, E – ethanol, D – dodecane, H – hexadecane, O – oleic acid, G/M (43/57 wt%), G/E (54/46 wt%), G/D (71/29 wt%), G/H (70/30 wt%), and G/O (45/55 wt%). b Total BTX productivity for catalyst life-time or for a TOS of 12 h. c Cumulative carbon yield of all the products analyzed (including BTX, the other aromatics, CO & CO2, C1C3, and coke, but excluding glycerol and oxygenates). d Overall carbon selectivity of the cumulative product collected during a run. (The numbers in brackets are calculated according to the feed ratios of the individual feeds (Table 1, entries 7–11) and their corresponding performance data. See the ESI† for a calculation example.). | |||||||||||
| Peak BTX carbon yield (C%) | 19.5 | 4.3 | 22.6 (11.0) | 25.0 | 25.6 (22.4) | 18.1 | 21.7 (18.8) | 18.3 | 22.3 (18.9) | 22.0 | 26.7 (21.3) |
| Total BTX productivityb (mgBTX gcat−1) | 426 | 155 | 911 (274) | 1379 | 972 (931) | 2428 | 798 (1367) | 2619 | 748 (1479) | 739 | 834 (645) |
| Catalyst life-time (h) | 8.5 | 5.5 | >12 | >12 | >12 | >12 | 8 | >12 | 9 | 6.5 | 11 |
| Total carbon yieldc (%) | 34.2 | 79.4 | 59.8 | 67.6 | 71.1 | 66.8 | 52.0 | 51.7 | 45.3 | 48.1 | 38.4 |
| Overall carbon selectivityd (%) | |||||||||||
| BTX | 35.0 | 3.8 | 32.3 (17.5) | 27.5 | 24.2 (31.0) | 25.5 | 26.9 (30.5) | 33.5 | 29.5 (34.3) | 25.9 | 32.6 (28.6) |
| Benzene | 6.7 | 0.3 | 2.1 | 5.4 | 4.4 | 6.1 | 5.3 | 7.6 | 5.6 | 7.0 | 7.8 |
| Toluene | 16.9 | 1.4 | 10.3 | 13.8 | 11.7 | 12.4 | 13.1 | 16.3 | 14.2 | 12.2 | 15.8 |
| m,p-Xylene | 9.3 | 1.7 | 15.7 | 6.4 | 6.4 | 5.2 | 6.9 | 7.2 | 7.8 | 5.2 | 7.2 |
| o-Xylene | 2.1 | 0.4 | 4.2 | 1.9 | 1.7 | 1.7 | 1.6 | 2.4 | 1.9 | 1.6 | 1.8 |
| Other aromatics | 3.9 | 0.1 | 1.3 (1.8) | 1.2 | 1.7 (2.5) | 2.8 | 2.4 (3.4) | 3.4 | 2.7 (3.7) | 4.0 | 3.5 (4.0) |
| Ethylbenzene | 0.7 | 0.0 | 0.3 | 0.4 | 0.5 | 0.4 | 0.6 | 0.5 | 0.7 | 0.5 | 0.6 |
| Naphthalene | 1.2 | 0.0 | 0.3 | 0.4 | 0.5 | 1.0 | 0.8 | 1.2 | 0.8 | 1.5 | 1.3 |
| 2-Methyl naphthalene | 1.5 | 0.1 | 0.6 | 0.3 | 0.6 | 1.0 | 0.8 | 1.2 | 0.9 | 1.4 | 1.2 |
| 1-Methyl naphthalene | 0.5 | 0.0 | 0.2 | 0.1 | 0.2 | 0.5 | 0.3 | 0.5 | 0.3 | 0.6 | 0.4 |
| CO & CO2 | 7.8 | 33.2 | 12.3 (22.0) | 1.8 | 9.7 (4.6) | 0.0 | 13.0 (4.1) | 0.0 | 11.8 (4.1) | 9.6 | 8.4 (9.1) |
| CO | 0.8 | 31.0 | 9.8 | 1.1 | 6.4 | 0.0 | 9.8 | 0.0 | 8.8 | 6.7 | 5.1 |
| CO2 | 7.0 | 2.3 | 2.5 | 0.6 | 3.3 | 0.0 | 3.2 | 0.0 | 3.0 | 2.9 | 3.3 |
| C1–C3 | 33.1 | 58.7 | 39.3 (47.4) | 67.7 | 54.4 (51.4) | 69.4 | 45.4 (50.2) | 59.9 | 42.1 (46.0) | 53.0 | 42.9 (47.0) |
| Methane | 0.9 | 38.5 | 8.7 | 5.2 | 3.2 | 9.9 | 5.1 | 8.5 | 4.4 | 6.7 | 6.1 |
| Ethane | 3.3 | 1.8 | 7.2 | 10.0 | 11.0 | 13.9 | 5.8 | 10.8 | 7.1 | 9.3 | 5.9 |
| Ethene | 13.5 | 8.5 | 11.5 | 18.9 | 23.8 | 6.7 | 11.7 | 6.3 | 10.5 | 10.1 | 10.6 |
| Propane | 10.6 | 4.2 | 4.8 | 20.1 | 5.7 | 33.1 | 10.4 | 28.8 | 8.4 | 14.4 | 9.3 |
| Propene | 4.8 | 5.7 | 7.1 | 13.5 | 10.8 | 5.7 | 12.4 | 5.6 | 11.7 | 12.4 | 11.0 |
| Coke on the spent catalyst | 20.2 | 4.1 | 14.8 (11.2) | 1.8 | 10.1 (10.4) | 2.3 | 12.3 (11.8) | 3.1 | 13.9 (12.0) | 7.5 | 12.6 (11.3) |
Nevertheless, it is of high interest to see (Table 2 and Fig. S12†) that the peak BTX carbon yields for the co-feeding experiments are higher than the calculated ones based on the feed ratios of the individual feeds (Table 1, entries 7–11) and their corresponding peak BTX carbon yields. This is clearly illustrated for glycerol/oleic acid in Fig. 2.
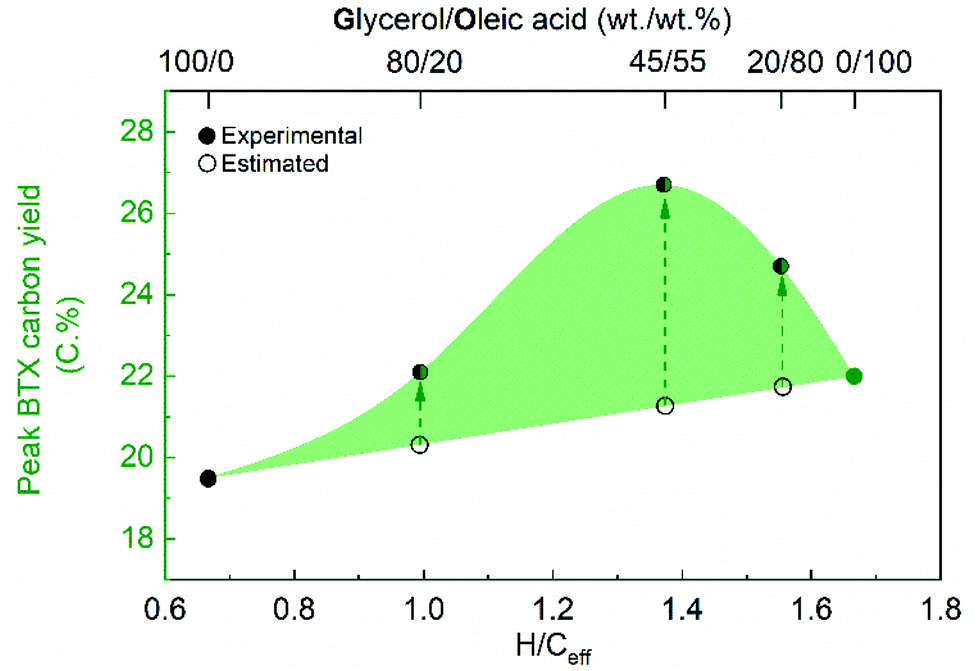 | ||
| Fig. 2 Peak BTX carbon yield versus the H/Ceff of the (co-) feed. Reaction conditions: H-ZSM-5/Al2O3 (60/40 wt%) catalyst of 10 g, WHSV of the (co-)feeds of 1 h−1, N2 flow of 50 ml min−1, reactor temperature of 550 °C, and atmospheric pressure. (The estimated peak BTX carbon yields for the co-feeds with various H/Ceff ratio's (Table 1, entries 11–13) are based on plots of the peak BTX carbon yield versus the H/Ceff. See the ESI† for a calculation example). | ||
For those co-feeds having a H/Ceff of ca. 1.4 (Table 1, entries 7–11), the highest peak BTX carbon yield (26.7 C%, Table 2 and Fig. S12†) was obtained from the co-conversion of glycerol/oleic acid (45/55 wt%). These results indicate the presence of a synergetic effect between glycerol and the other feedstock and this leads to a higher peak BTX carbon yield than anticipated based on results for individual feeds.
For the co-feeding experiments of glycerol with alkanes or alcohols, the overall selectivity to COx (CO and CO2) is considerably higher than calculated (the numbers in the brackets in Table 2) based on feed ratios of the individual feeds (Table 1, entries 7–11) and their corresponding selectivities (Table 2). This leads to a reduction in the selectivity to aromatics and low-molecular-weight hydrocarbons (C1–C3) upon co-feeding. However, the reverse was found for the co-conversion of glycerol with oleic acid.
When considering coke formation for the pure feeds, glycerol gives the highest amount of coke (20% selectivity, Table 2), while the other three types of feedstocks show rather low coke selectivity (2–7%, Table 2). However, the coke selectivity for the co-feeding experiments is the same as or only slightly higher than expected according to the feed ratio of the individual feeds (Table 1, entries 7–11) and their corresponding coke selectivity (Table 2).
Synergetic effects for glycerol/oleic acid
The experimental and calculated carbon yields of the major gaseous and liquid products from co-feeding experiment of glycerol/oleic acid (45/55 wt%) versus TOS are provided in Fig. S14,† and a representative result is shown in Fig. 3. Compared to the calculated carbon yields (Fig. 3-black curves) based on the feed ratio of the individual feeds (Table 1, entry 11) and their corresponding carbon yields (Fig. 3), the experimental yields for the liquid aromatics products (BTX in particular) are considerably higher (Fig. 3-red curve). Furthermore, the yields for the gas phase components like COx (CO2 in particular) and C1–C3 are decreased (Fig. 3-green curves). Apparently, upon co-feeding of glycerol with oleic acid, more of the carbon is ending up in the BTX components than in the gas phase. The observation that COx yields are reduced upon co-feeding imply that the rates of deoxygenation by decarboxylation/decarbonylation reactions are affected in a different manner for individual and co-feeds and that these are less favoured when co-feeding. The reduction in the yields of C1–C3 suggest that either the hydrocarbon pool on the catalyst surface is more tailored towards BTX formation and/or that the rate of cracking reactions to smaller hydrocarbons like propane is retarded during co-feeding. Most likely, intermediates formed during pyrolysis from both glycerol and oleic acid have a higher tendency to react with each other resulting in increased BTX formation.
 | ||
| Fig. 3 Experimental and calculated carbon yields of a representative liquid BTX component (m,p-xylene, top), gaseous hydrocarbon (propane, bottom) and CO2 (middle) versus TOS for the co-feeding of glycerol/oleic acid (45/55 wt%). Reaction conditions: H-ZSM-5/Al2O3 (60/40 wt%) catalyst of 10 g, WHSV of the (co-)feed of 1 h−1, N2 flow of 50 ml min−1, reactor temperature of 550 °C, and atmospheric pressure. (The calculated carbon yields are based on the carbon yields of the individual feeds (Table 1, entry 11) and the feed ratio. See the ESI† for a calculation example.). | ||
 | ||
| Fig. 4 Total BTX productivity (a), catalyst life-time (b), and average coking rate (c) versus the hydrogen to carbon effective ratio of the (co-)feed. Reaction conditions: H-ZSM-5/Al2O3 (60/40 wt%) catalyst of 10 g, WHSV of the (co-)feeds of 1 h−1, N2 flow of 50 ml min−1, reactor temperature of 550 °C, and atmospheric pressure. (The estimated total BTX productivities, catalyst life-times, and average coking rates for the co-feeds with various H/Ceff (Table 1, entries 11–13) are based on the linear fittings of their plots versus H/Ceff. See the ESI† for a calculation example.). | ||
Catalyst deactivation for zeolites used for the conversion of alcohols and organic acids is typically associated with rapid coke formation in the time frame of hours.6,36 Catalyst regeneration by coke removal using oxidative methods has proven to be well possible.6,14,27 In addition, irreversible changes in the zeolite framework may occur, like dealumination and associated loss in acidity,6,10 though this typically is only visible after a number of regeneration cycles. As such, differences in catalyst life-time when only performing single experiments without catalyst regeneration are likely related to coke built-up phenomena. When considering coke formation on the catalyst after reaction between glycerol (0.25 g C per h, Fig. 4c) and oleic acid (0.34 g C per h), it is clear that the coking rate is higher when using oleic acid as the feed. This is also expressed by the higher catalyst life-time when using glycerol (8.5 h) compared to oleic acid (6.5 h, Table 2). However, the total BTX productivity (mgBTX gcat−1) is higher when using oleic acid as the feed. Apparently, a higher amount of coke on the catalyst is not necessarily leading to a lower BTX productivity and other factors like the molecular composition and associated properties of the coke may play a role as well. This may lead to differences in the micro-environment at an acidic center in the zeolite structure between experiments with oleic acid and glycerol and impact the relative rates of aromatization and coke forming reactions.43 Further detailed studies on relevant zeolite properties after reaction including coke characteristics as well as the specific surface area, pore volume (total pore volume and micropore volume), relative crystallinity, acidity (total acidity, Brønsted acidity, and Lewis acidity) to substantiate this hypothesis are in progress and will be reported in due course.44 However, this hypothesis only provides a possible explanation for the differences in catalyst performance between oleic acid and glycerol, and not in the synergetic effects observed when using mixtures of the two. Apparently, a micro-environment at the acidic center with both oleic acid and glycerol derived (hydrocarbon) products complemented with a specific type of coke is beneficial and prolongs catalyst life-time. Further experimental studies among others detailed characterization of the catalysts, supported by theoretical calculations, will be required to substantiate this hypothesis.
In total, 5 reaction-regeneration cycles were performed for glycerol/oleic acid and the total BTX productivity over the fresh and regenerated catalysts is shown in Fig. 5. For comparison, the total BTX productivity for individual glycerol and oleic acid is also plotted in Fig. 5. Irreversible catalyst deactivation is reflected by a decrease in the total BTX productivity after 2 reaction-regeneration cycles for glycerol27 and after 3 reaction-regeneration cycles for oleic acid conversion36 (Fig. 5). It is interesting to see that upon co-feeding, the regenerated catalysts show considerably higher total BTX productivity compared to the calculated values based on the feed ratio of the individual feeds (Table 1, entry 11) and their corresponding total BTX productivities of the regenerated catalysts for the individual feeds (Fig. 5). A negligible decrease of the total BTX productivity after 5 reaction-regeneration cycles for the co-conversion of glycerol/oleic acid (45/55 wt%) is observed. These results indicate that co-feeding also has a positive effect on catalyst regenerability. Irreversible catalyst deactivation is known to be mainly due to dealumination, most likely caused by the high amount of water formed during the reaction due to dehydration reactions. Speculatively, it is possible that upon co-feeding oleic acid, the long hydrocarbon chain or fragments thereof lead to a reduction of the hydrophobicity on the catalyst surface and as such reduces the rate of dealumination.
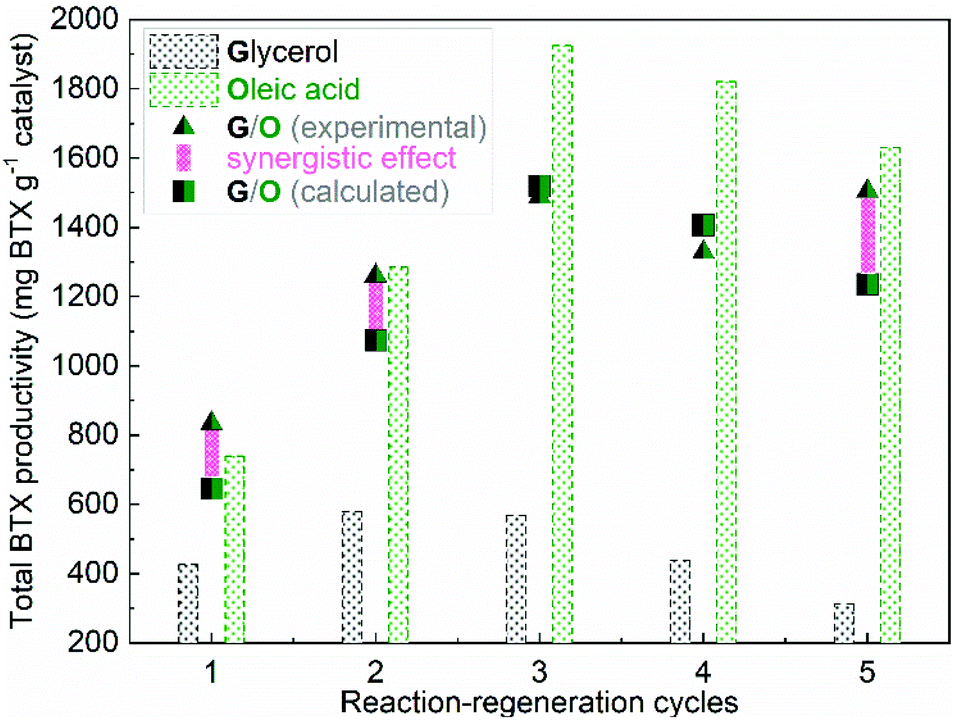 | ||
| Fig. 5 The total BTX productivity over the fresh and regenerated catalysts versus the reaction-regeneration cycles. Reaction conditions: H-ZSM-5/Al2O3 (60/40 wt%) catalyst of 10 g, WHSV of the (co-)feed (glycerol, oleic acid, and glycerol/oleic acid (45/55 wt%)) of 1 h−1, N2 flow of 50 ml min−1, reactor temperature of 550 °C, atmospheric pressure, and TOS of 12 h. (The calculated total BTX productivities are based on the feed ratio of the individual feeds (Table 1, entry 11) and their corresponding total BTX productivities. See the ESI† for a calculation example.). | ||
Labeling studies
To determine the extent of incorporation of both feeds into the final BTX products, isotopic-labeling experiments have been performed with D- and 13C-labeled glycerol in combination with unlabeled oleic acid using a dedicated Pyrolysis GC-Orbitrap MS unit operated at 550 °C. Experiments were carried using a mixture of a powdered catalyst and glycerol alone or a glycerol/oleic acid mixture (catalyst to liquid weight ratio of ca. 1). The hydrogen and carbon selectivity of the individual BTX for D- and 13C-labeled glycerol and non-labeled oleic acid are shown in Fig. 6. | ||
| Fig. 6 Hydrogen (top) and carbon (bottom) selectivity's of the individual BTX for the catalytic conversion of (a) C3D8O3, (b) the mixed C3D8O3 and C18H34O2, (c)13C3H8O3, and (d) the mixed 13C3H8O3 and C18H34O2 over the H-ZSM-5/Al2O3 catalyst performed on the Pyrolysis-GC-Orbitrap-MS. (The calculated data are based on the feed ratios of the labeled feeds and their corresponding performance data. See the ESI† for a calculation example.). | ||
When using D-labeled C3D8O3 (>98% isotopic purity) in the absence of oleic acid, the BTX products contain not only D but also H atoms (Fig. 6a). The latter may be from the H containing impurities present in the C3D8O3 (D purity is ≥98 atom % according to the specification). Besides, this rather surprising finding may also be explained by the presence of Brønsted acid sites on the zeolite catalyst.45 H/D exchange between deuterated hydrocarbons on the catalyst surface (hydrocarbon pool) and the acid sites (H+) may take place and lead to H incorportion in the BTX during the reaction. Both H/D exchange pathways hamper the interpretation of the results for co-feeding experiments with oleic acid. Nevertheless, upon co-feeding C3D8O3 with oleic acid (Fig. 6b) the experimental H incorporation level in benzene is lower than the calculated value (78 vs. 85%, Fig. 6, see the ESI† for the calculation procedure). These results indicate that oleic acid (and its pyrolysis intermediates) are indeed involved in BTX formation. However, due to the presence of H/D exchange reactions it is not possible to exactly quantify the extent of carbon incorporation of glycerol and oleic acid into the final BTX products.
Additional labeling experiments were performed using fully 13C-labeled glycerol. When using 13C3H8O3 (>99% purity) only, the BTX components contain a high amount of the 13C-label (Fig. 6c). Upon co-feeding 13C3H8O3 with the non-labeled oleic acid, the amount of labeled carbon in BTX is significantly reduced, indicating that carbon atoms from oleic acid are indeed involved in the reaction network and participate in the hydrocarbon pool15,46 on the catalyst surface. The experimentally observed amounts of C in all BTX formed is on average equal to the calculated value (see Fig. 6d), suggesting that the 13C label is randomly distributed over the catalyst surface. For the xylenes, the experimental values are very close to the calculated ones. Remarkably, the experimental 13C fraction in benzene (12%) is lower than the calculated one (23%) while the opposite trend is true for toluene (Fig. 6). So far, we do not have a sound explanation for the latter observation.
Conclusions
We have discovered an unprecedented synergetic effect between glycerol and various co-feeds including alkanes, alcohols, and free fatty acids for the catalytic conversion to bio-based aromatics. A detailed investigation on the co-feeding of glycerol and oleic acid revealed that (i) the peak BTX carbon yield is increased, (ii) the catalyst life-time is enhanced, (iii), the total BTX productivity is higher, and (iv) the level of irreversible deactivation is reduced when compared to experiments with the pure feeds. Best results were obtained for the co-conversion of glycerol/oleic acid (45/55 wt%), showing a peak BTX carbon yield of 26.7 C.%, a total BTX productivity of 834 mgBTX gcatalyst−1, and negligible irreversible catalyst deactivation after 5 cycles of reaction-regeneration. These studies reveal that co-feeding strategies can lead to improved catalyst performance. It also indicates that (expensive) separation processes to obtain pure feeds can be avoided. This is particularly relevant when considering the glycerol case. Crude glycerol from the biodiesel industry is typically contaminated with fatty acids and methanol, of which both are shown to be excellent co-feeds. As such, it is not necessary and even undesired to purify the crude glycerol by e.g., distillation before using it as a feed for BTX manufacture. Labeling studies show that the BTX components formed originate from both glycerol and oleic acid. Studies to elucidate the origin of this remarkable synergistic effect are in progress and will be reported in due course.Author contributions
Songbo He: Conceptualization, validation, investigation, data curation, writing – original draft, writing – review & editing. Thomas Sjouke Kramer: Validation, investigation. Dian Sukmayanda Santosa: Validation, investigation. Andre Heeres: Conceptualization, writing – review & editing, funding acquisition. Hero Jan Heeres: Conceptualization, data curation, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO, LIFT programme, Grant No. 731.016.401) is greatly acknowledged. We are also very grateful for BIOBTX B.V. for the collaboration and valuable discussions. Peter de Gijsel and Dr Gijs van Erven from the Laboratory of Food Chemistry at Wageningen University & Research are acknowledged for their help with the experiments with the Pyrolysis-GC-Orbitrap MS unit.References
- M. R. Monteiro, C. L. Kugelmeier, R. S. Pinheiro, M. O. Batalha and A. D. Cesar, Renewable Sustainable Energy Rev., 2018, 88, 109–122 CrossRef CAS.
- X. Luo, X. Ge, S. Cui and Y. Li, Bioresour. Technol., 2016, 215, 144–154 CrossRef CAS PubMed.
- C. H. C. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. M. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- J. Zhang, L. Wang, Y. Y. Ji, F. Chen and F. S. Xiao, Front. Chem. Sci. Eng., 2018, 12, 132–144 CrossRef CAS.
- O. Muraza, Front. Chem., 2019, 7, 233 CrossRef CAS PubMed.
- S. He, K. Zuur, D. S. Santosa, A. Heeres, C. Liu, E. Pidko and H. J. Heeres, Appl. Catal., B, 2021, 281, 119467 CrossRef CAS.
- F. Wang, X. Z. Chu, P. S. Zhao, F. X. Zhu, Q. Q. Li, F. Y. Wu and G. M. Xiao, Fuel, 2020, 262, 116538 CrossRef CAS.
- D. H. Pan, S. Q. Xu, Y. A. Miao, N. N. Xu, H. Z. Wang, X. H. Song, L. J. Gao and G. M. Xiao, Catal. Sci. Technol., 2019, 9, 739–752 RSC.
- N. N. Xu, D. H. Pan, Y. F. Wu, S. Q. Xu, L. J. Gao, J. Zhang and G. M. Xiao, React. Kinet., Mech. Catal., 2019, 127, 449–467 CrossRef CAS.
- S. He, I. Muizebelt, A. Heeres, N. J. Schenk, R. Blees and H. J. Heeres, Appl. Catal., B, 2018, 235, 45–55 CrossRef CAS.
- F. Wang, M.-x. Zhou, X.-h. Yang, L.-j. Gao and G.-m. Xiao, Mol. Catal., 2017, 432, 144–154 CrossRef CAS.
- F. Wang, X. Kang, M. X. Zhou, X. H. Yang, L. J. Gao and G. M. Xiao, Appl. Catal., A, 2017, 539, 80–89 CrossRef CAS.
- T. Q. Hoang, X. L. Zhu, T. Danuthai, L. L. Lobban, D. E. Resasco and R. G. Mallinson, Energy Fuels, 2010, 24, 3804–3809 CrossRef CAS.
- S. He, H. R. Goldhoorn, Z. Tegudeer, A. Chandel, A. Heeres, C. Liu, E. Pidko and H. J. Heeres, Fuel Process. Technol., 2021, 221, 106944 CrossRef CAS.
- F. Wang, W. Y. Xiao, L. J. Gao and G. M. Xiao, RSC Adv., 2016, 6, 42984–42993 RSC.
- W. Y. Xiao, F. Wang and G. M. Xiao, RSC Adv., 2015, 5, 63697–63704 RSC.
- Y. W. Suh, H. S. Jang and K. B. Bae, United States Pat, US2015336856A1, 2015 Search PubMed.
- L. H. Dao, M. Haniff, A. Houle and D. Lamothe, ACS Symp. Ser., 1988, 376, 328–341 CrossRef CAS.
- H. S. Jang, K. Bae, M. Shin, S. M. Kim, C. U. Kim and Y. W. Suh, Fuel, 2014, 134, 439–447 CrossRef CAS.
- G. Q. Luo and A. G. McDonald, Energy Fuels, 2014, 28, 600–606 CrossRef CAS.
- Y.-W. Suh, Proceeding of Annual/Fall Meetings of the Japan Petroleum Institute, 2014, 2014f, 119.
- D. Kumar, N. Anand and K. K. Pant, Clean Technol. Environ. Policy, 2018, 20, 751–757 CrossRef CAS.
- Y. W. Suh, H. S. Jang and K. B. Bae, United States Pat, US9834489B2, 2016 Search PubMed.
- R. Le Van Mao, H. Yan, A. Muntasar and N. Al-Yassir, in New and Future Developments in Catalysis, ed. S. L. Suib, Elsevier, Amsterdam, 2013, pp. 143–173, DOI:10.1016/B978-0-444-53876-5.00007-6.
- A. Shahnazari, Master, The University of New Brunswick, 2016.
- S. He, H. R. Goldhoorn, Z. Tegudeer, A. Chandel, A. Heeres, M. C. A. Stuart and H. J. Heeres, A time- and space-resolved catalyst deactivation study on the conversion of glycerol to aromatics using H-ZSM-5, 2022, Submitted Search PubMed.
- S. He, T. S. Kramer, F. G. H. Klein, A. Chandel, Z. Tegudeer, A. Heeres, C. Liu, E. Pidko and H. J. Heeres, Appl. Catal., A, 2022, 629, 118393 CrossRef CAS.
- Y. Xiao and A. Varma, ACS Energy Lett., 2016, 1, 963–968 CrossRef CAS.
- S. Tamiyakul, W. Ubolcharoen, D. N. Tungasmita and S. Jongpatiwut, Catal. Today, 2015, 256, 325–335 CrossRef CAS.
- A. Corma, G. W. Huber, L. Sauvanaud and P. O'Connor, J. Catal., 2007, 247, 307–327 CrossRef CAS.
- H. Y. Zhang, Y. T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 RSC.
- U. V. Mentzel and M. S. Holm, Appl. Catal., A, 2011, 396, 59–67 CrossRef CAS.
- J.-P. Lange, Nat. Catal., 2021, 4, 186–192 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, J. Catal., 2013, 307, 194–203 CrossRef CAS.
- F. Fantozzi, A. Frassoldati, P. Bartocci, G. Cinti, F. Quagliarini, G. Bidini and E. M. Ranzi, Appl. Energy, 2016, 184, 68–76 CrossRef CAS.
- S. He, F. G. H. Klein, T. S. Kramer, A. Chandel, Z. Tegudeer, A. Heeres and H. J. Heeres, ACS Sustainable Chem. Eng., 2021, 9, 1128–1141 CrossRef CAS PubMed.
- M. Buchele, M. Swoboda, A. Reichhold and W. Hofer, Chem. Eng. Process., 2019, 142, 107553 CrossRef.
- Pyrolysis-GC-Orbitrap MS, https://www.wur.nl/en/Value-Creation-Cooperation/Collaborating-with-WUR-1/show-srf/Pyrolysis-GC-Orbitrap-MS-and-GC-Orbitrap-MS.htm, (accessed 20th June, 2021).
- G. van Erven, R. de Visser, P. de Waard, W. J. H. van Berkel and M. A. Kabel, ACS Sustainable Chem. Eng., 2019, 7, 20070–20076 CrossRef CAS.
- A. Errekatxo, A. Ibarra, A. Gutierrez, J. Bilbao, J. M. Arandes and P. Castano, Chem. Eng. J., 2017, 307, 955–965 CrossRef CAS.
- M. Shin, T. Kim and Y. W. Suh, Top. Catal., 2017, 60, 658–665 CrossRef CAS.
- G. J. S. Dawes, E. L. Scott, J. Le Notre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231–3250 RSC.
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41–48 CrossRef CAS.
- S. He, F. G. H. Klein, T. S. Kramer, A. Chandel, Z. Tegudeer, A. Heeres and H. J. Heeres, Catalytic co-conversion of glycerol and oleic acid to bio-aromatics: catalyst deactivation studies for a technical H-ZSM-5/Al2O3 catalyst, 2022, Submitted Search PubMed.
- Y. Ono, Catal. Rev.: Sci. Eng., 1992, 34, 179–226 CrossRef CAS.
- S. Wang, Y. Y. Chen, Z. F. Qin, T. S. Zhao, S. B. Fan, M. Dong, J. F. Li, W. B. Fan and J. G. Wang, J. Catal., 2019, 369, 382–395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03531b |
| This journal is © The Royal Society of Chemistry 2022 |