
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHesperidin protects against cisplatin-induced cardiotoxicity in mice by regulating the p62–Keap1–Nrf2 pathway†
Yuxin
Jia‡
a,
Hui
Guo‡
a,
Xizhen
Cheng
a,
Yuling
Zhang
a,
Mingdong
Si
a,
Jing
Shi
*b and
Donglai
Ma
 *ac
*ac
aSchool of Pharmacy, Hebei University of Chinese Medicine, Shijiazhuang, 050200, Hebei, China. E-mail: mdl_hebei@aliyun.com
bDepartment of Scientific Research Management, the Fourth Hospital of Hebei Medical University, Shijiazhuang, 050011, Hebei, China. E-mail: 13931143718@139.com
cHebei Technology Innovation Center of TCM Formula Preparations, Shijiazhuang, 050200, Hebei, China
First published on 21st March 2022
Abstract
Hesperidin (HES) is an abundant and economical dietary bioflavonoid, and it has several pharmacological properties such as antioxidant activity and powerful cardiac protection. However, HES protection against cisplatin (CP)-induced cardiotoxicity and its mechanism have not been fully clarified. The current study was performed to further elucidate the mechanism of HES against CP-induced cardiotoxicity. Mice were orally administered HES (100 or 300 mg kg−1 day−1) for 7 consecutive days and then injected intraperitoneally (i.p.) with CP (5 mg kg−1) on days 3 and 6. On day 8, mice were anaesthetised with sodium pentobarbital (50 mg kg−1, i.p.), and blood and heart samples were collected for analysis. HES treatment reduced CP-induced cardiac pathologic damage and leakage of the myocardial markers cardiac troponin I (cTnI), creatine kinase (CK), and lactate dehydrogenase (LDH). HES treatment reduced levels of reactive oxygen species (ROS) and malondialdehyde (MDA), which is an oxidative product, and increased antioxidant marker levels including superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH). HES also reduced the CP-induced release of the inflammatory factors tumour necrosis factor (TNF)-α and interleukin (IL)-6. Additionally, HES treatment up-regulated the expression of anti-apoptotic protein Bcl-2 and down-regulated the expression of pro-apoptotic proteins Bax and Caspase-3. HES treatment also improved the expression of pathway proteins p62 and Nrf2 and inhibited the increase in CP-induced Keap1 expression. Thus, HES may provide protection against CP cardiotoxicity through inhibiting oxidative stress, inflammation, and apoptosis, which may contribute to activation of the p62–Keap1–Nrf2 signalling pathway. These findings suggest that HES may be a promising protective agent against CP cardiotoxicity in future anticancer clinical practice.
1. Introduction
Cisplatin (CP) kills tumour cells by inducing senescence or triggering innate apoptotic pathways that cause irreparable DNA damage, and it is the most commonly used first-line drug for cancer treatment.1 However, its clinical use is often limited by dose-related cardiotoxicity,2 which includes arrhythmias,3 angina pectoris,4 and cardiac failure.5 These events may increase the prevalence of cardiovascular disease in tumour patients who have received CP. Although the mechanism of CP-induced cardiotoxicity has not been fully defined, oxidative stress, inflammation, and apoptosis caused by CP are considered to be involved in inducing cardiotoxicity.6,7Oxidative stress is primarily identified as a main mechanism of CP-induced cardiotoxicity, which is characterised by the imbalance between reactive oxygen species (ROS) and the antioxidant defence system.8–10 Additionally, oxidative stress has been shown to contribute to various biological processes, such as inflammation and apoptosis.11 Excessive ROS levels induce nuclear transcription factor activation and increase pro-inflammatory cytokine release, which aggravates inflammatory damage. An increasing amount of evidence indicates that the cytotoxic effects of CP can be exacerbated by pro-inflammatory cytokine release.12 Furthermore, ROS overproduction can target mitochondria and lead to mitochondrial dysfunction, which in turn induces Caspase cascade apoptosis.7,13 Nuclear factor erythroid-related factor 2 (Nrf2) plays a crucial role in protecting cells against oxidative stress.14 Its hyperactivation often follows excessive build-up of p62 that sequesters Kelch-like ECH-associated protein 1 (Keap1), which is an adaptor of the E3-ubiquitin ligase that binds to Nrf2. Nrf2 activation could protect cells from free radical damage and apoptosis, thus promoting cell survival.15
Hesperidin (HES; C28H34O15) (Fig. 1) is a natural compound that is abundantly found in citrus fruits, and its concentration appears to be high in Citrus sinensis (15.25 ± 8.21 mg per 100 g fresh fruit weight) and C. reticulate (19.26 ± 11.56 mg per 100 g fresh fruit weight).16 In addition, HES is one of the primary components of Chenpi, a traditional Chinese medicine made from Citrus unshiu peel with the content of 6.25%,17 and it possesses multiple medicinal benefits including antioxidant and anti-inflammatory activities.18 Previous studies have demonstrated that HES could significantly attenuate myocardial ischaemia/reperfusion injury,19 myocardial infarction,20 and CO-induced cardiotoxicity,21 suggesting powerful protection of myocardial tissue by HES. Furthermore, Oguzturk et al.,22 suggested that HES could play a protective role against CP-induced cardiotoxicity in rats, but the underlying mechanism remains unclear.
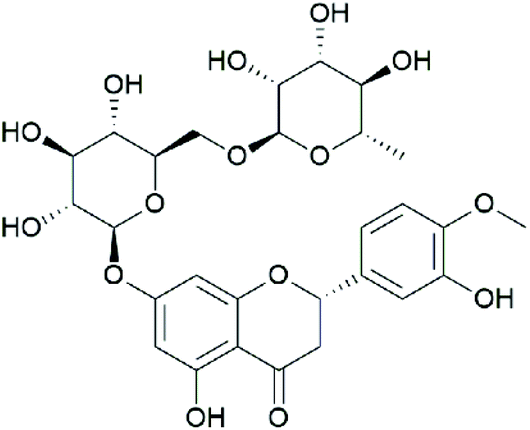 | ||
| Fig. 1 Chemical structure of HES. | ||
In the present study, we evaluated the protective effect of HES on CP-induced cardiotoxicity by observing changes in cardiac histopathology and myocardial marker levels in mice. Additionally, the underlying mechanisms were explored by measuring indicators related to oxidative stress, inflammation, apoptosis, and the p62–Keap1–Nrf2 signalling pathway.
2. Methods
2.1 Drugs
CP was purchased from Qilu Pharmaceutical Co., Ltd (Shandong, China). HES (purity >90.0%) was obtained from Tokyo Chemical Industry (Tokyo, Japan). Sodium carboxymethyl cellulose was dissolved in ionic water at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 to prepare homogeneous HES suspension. It was prepared as high and low doses using a magnetic stirrer.23
:200 to prepare homogeneous HES suspension. It was prepared as high and low doses using a magnetic stirrer.23
2.2 Animals
Fifty male Kunming mice (18–22 g) were obtained from Hebei Medical University (Certificate No. SCXK [Hebei] 2018-004), and they were housed in a room with a standard environmental, with a constant temperature of 22–25 °C and a relative humidity of 40%–60%. Mice had free access to a normal diet and purified water for 1 week before the experiment. Ethics committee approval was obtained from Hebei University of Chinese Medicine Animal Experiments Ethics Committee (DWLL2018015).2.3 Experimental groups and administration methods
Fifty mice were randomly divided into the following five groups (ten mice per group): Control (CONT), CP, low-dose HES + CP (L-HES), high-dose HES + CP (H-HES), and HES. The CP group received CP (5 mg kg−1, intraperitoneal injection [i.p.]). The L-HES and H-HES groups received HES (100 mg kg−1 day−1 or 300 mg kg−1 day−1, orally) + CP (5 mg kg−1, i.p.). The HES group received HES (300 mg kg−1 day−1, orally). The CP and HES doses were selected on the basis of previous studies.24–26 Drug administration continued for 7 days; CP was injected on days 3 and 6, and HES was administered orally once a day. Twenty-four hours after the last oral HES dose, mice were anaesthetised with sodium pentobarbital (50 mg kg−1, i.p.), and blood and tissue samples were quickly collected for the analyses described below.2.4 Histopathology analysis
Heart samples were collected immediately after blood samples were obtained, and they were fixed in 4% paraformaldehyde. The fixed tissues were dehydrated and embedded with conventional paraffin to prepare paraffin blocks. Then, the blocks were cut into 4 μm sections and stained with haematoxylin and eosin (H&E) to evaluate the histopathological changes under an optical microscope (Leica DM4000B, Solms, Germany). The degree of myocardial injury was quantified using Image Pro Plus 6.0 software.2.5 Colorimetric analysis
2.6 Fluorescence microscopy
Fresh heart tissue was embedded at the optimum cutting temperature and sectioned with a frozen slicer. To detect ROS content in cardiomyocytes, heart tissue was stained with dihydroethidium (Sigma Chemical Co., St Louis, MO, USA; Catalogue: D7008) for 30 min at 37 °C in a dark incubator and then stained with DAPI solution (Servicebio, Wuhan, China; Catalogue: G1012) for another 10 min at room temperature in dark. Nuclei were labelled blue using DAPI, and ROS-positive cells were labelled red by fluorescein. The staining results were observed. The images were collected under a fluorescence microscope (Leica DM4000B, Solms, Germany), and Image-Pro Plus 6.0 software (Media Cybernetics, Inc.) was used to determine the ROS content in the images.2.7 ELISA analysis
Cardiac troponin I (cTnI) (Sigma Chemical Company; Catalogue: SEKM-0153) levels in the serum and tumour necrosis factor-(TNF)-α (Thermo Fisher Scientific, Waltham, MA, USA; Catalogue: 88-7324) and interleukin (IL)-6 (Multi Sciences, Hangzhou, China; Catalogue: EK206/3-01) levels in the heart tissue were determined using the respective commercially available ELISA kits, according to the kit instructions.2.8 Western blot analysis
Heart tissue was homogenised in RIPA lysis buffer (Servicebio; Catalogue: G2002) and centrifuged at 12000 rpm for 10 min at 4 °C to obtain the protein. Equal amounts of protein were separated using 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride membranes that were placed in blocking buffer containing 5% non-fat milk for 2 h at room temperature. Subsequently, the membranes were incubated overnight at 4 °C in primary antibodies (dilution 1:1000) of anti-B-cell lymphoma-2 (Bcl-2) (Abways, Shanghai, China; Catalogue: CY5032), anti-Bcl-2-associated X protein (Bax) (Abways; Catalogue: CY5059), anti-Caspase-3 (Abways; Catalogue: CY5051), anti-p62 (Servicebio; Catalogue: GB11531), anti-Keap1 (Proteintech, Wuhan, China; Catalogue: 10503-2-AP), and anti-Nrf2 (Proteintech; Catalogue: 16396-1-AP). Anti-β-actin (ABclonal, Wuhan, China; Catalogue: AC026, dilution 1:10000) was used as the internal standard. The next day, membranes were washed three times with TBST. The membranes were incubated at room temperature for 1 h in the dark with secondary antibodies (Bioeasy, Beijing, China; Catalogue number: BE0101, dilution 1:10000). After washing three more times, the films were scanned in the dark, and Image Pro Plus 6.0 software (Media Cybernetics, Inc.) was used to analyse the grey values of the target bands.
2.9 Statistical analysis
Statistical analyses were conducted using SPSS v.21.0 software (IBM Corp., Armonk, NY, USA). The data results were recorded as the mean ± standard deviation (SD). A one-way analysis of variance (ANOVA) followed by the Tukey's test were used to analyse the differences between the measurements among groups. Statistical significance was set at p < 0.05.3. Results
3.1 Effects of HES on histopathology
Fig. 2 shows histopathology results for heart tissue, which had a normal histological appearance in the CONT and HES groups. However, heart tissue in the CP group showed inflammatory cells (arrow 1) and apoptotic (arrow 2) and myocardial oedema cells (arrow 3). These histological changes were alleviated in the L-HES and H-HES groups in a dose-dependent manner compared with the CP group (p < 0.01), which suggested that HES significantly prevented CP-induced inflammatory cell infiltration and myocardial cell oedema and apoptosis.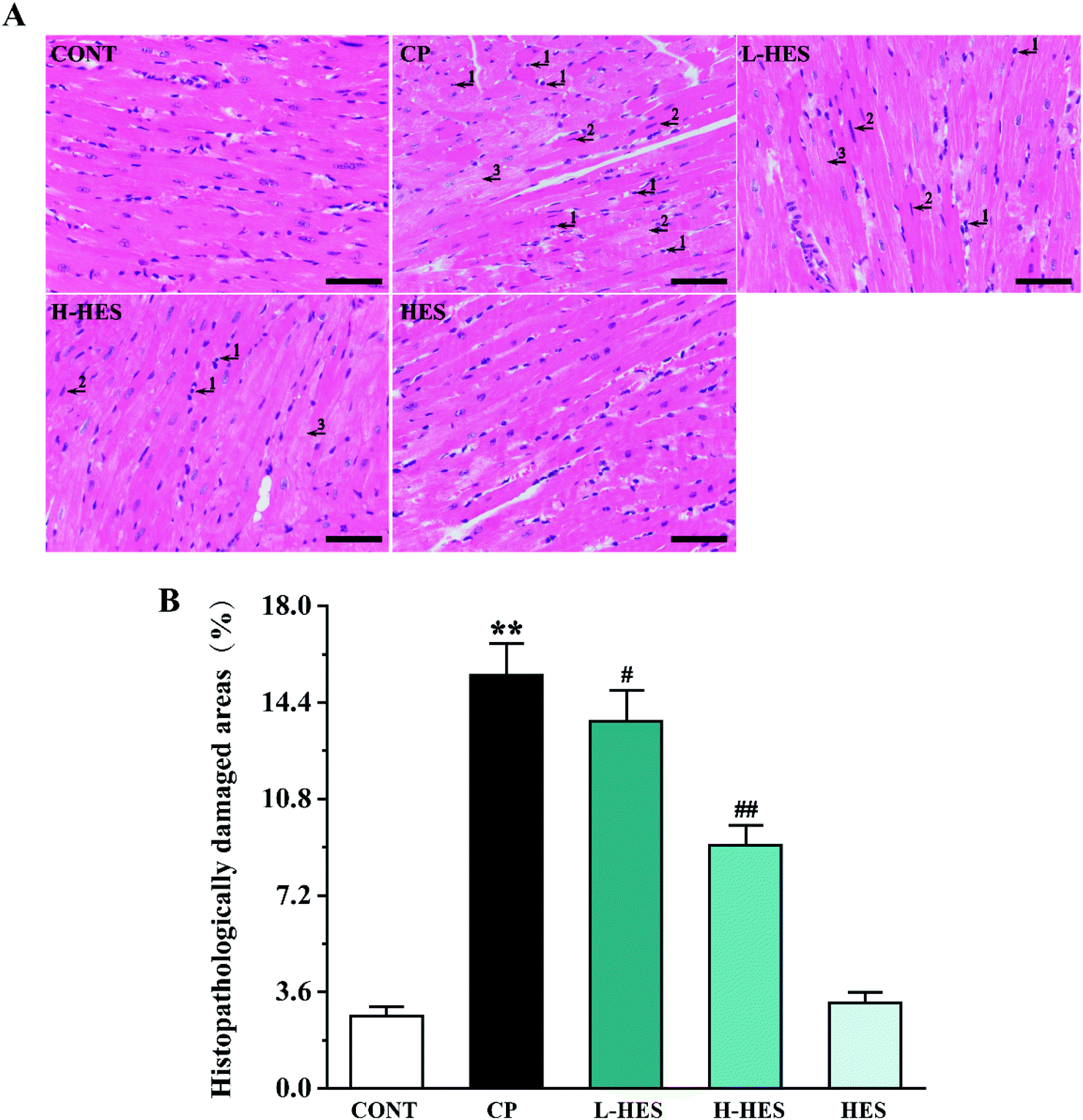 | ||
| Fig. 2 Effects of HES on cardiac histopathological changes in CP-induced mice (A) H&E stain images from the CONT, CP, L-HES, H-HES, and HES groups (scale bar: 50 μm, magnification: × 400). (B) The area of myocardial injury in each group was calculated. Arrows 1 to 3 represent inflammatory cells, apoptotic cells, and myocardial oedema cells, respectively. Data are presented as the mean ± SD for each group (n = 3). ** p < 0.01 versus the CONT group; #p < 0.05 and ##p < 0.01 versus the CP group. | ||
3.2 Effects of HES on myocardial markers
Serological analysis in Fig. 3 shows that the levels of the myocardial makers cTnI (Fig. 3A), CK (Fig. 3B), and LDH (Fig. 3C) were markedly higher in the CP group than that in the CONT group (p < 0.01). However, cTnI, CK, and LDH activity in the L-HES and H-HES groups were obviously reduced compared to the CP group (p < 0.05 or p < 0.01). Additionally, the HES group showed non-significant differences compared to the CONT group (p > 0.05).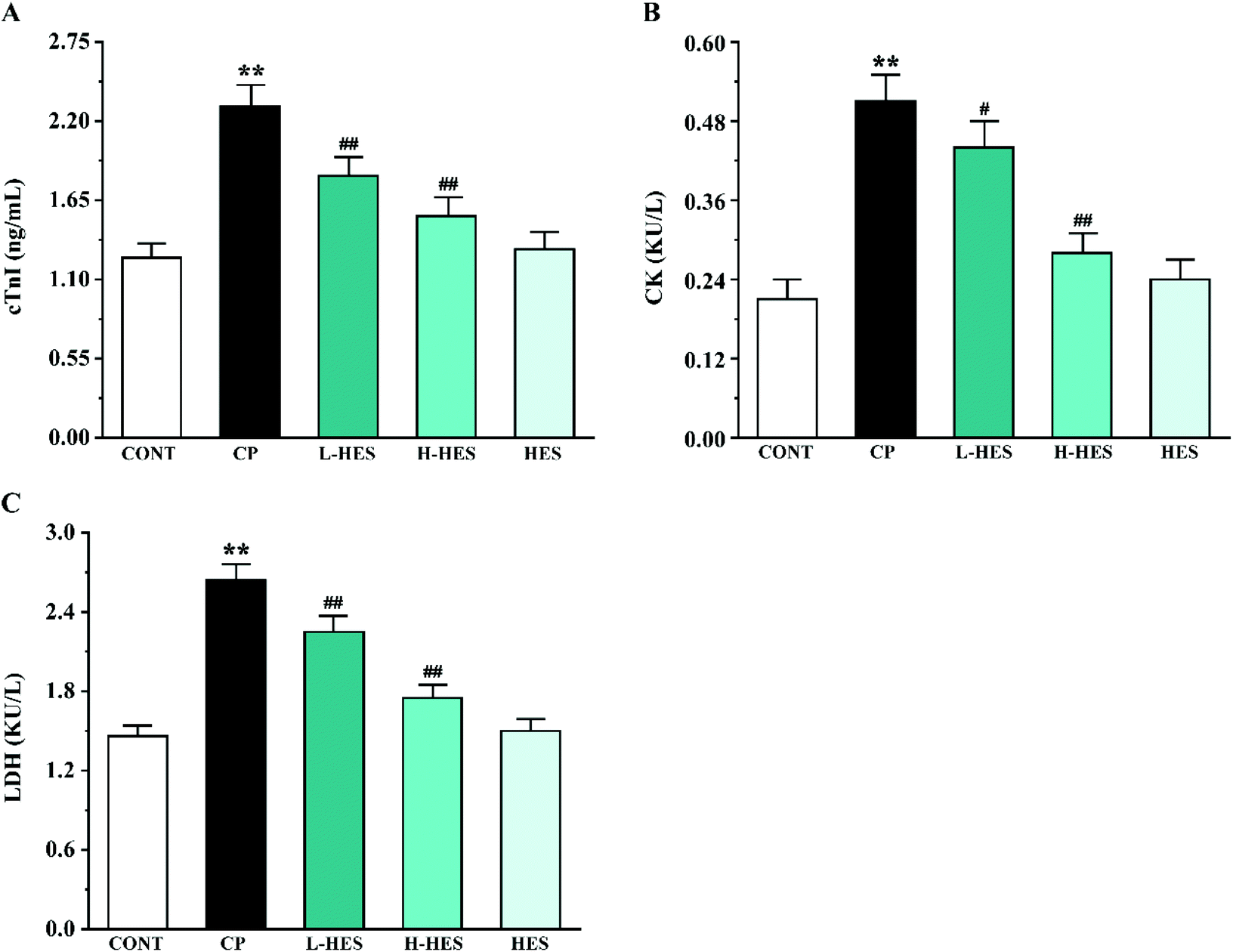 | ||
| Fig. 3 Effects of HES on serum levels of myocardial markers cTnI (A), LDH (B), and CK (C) in CP-induced mice. Data are presented as the mean ± SD for each group (n = 6). ** p < 0.01 versus the CONT group; #p < 0.05 and ##p < 0.01 versus the CP group. | ||
3.3 Effects of HES on oxidative stress
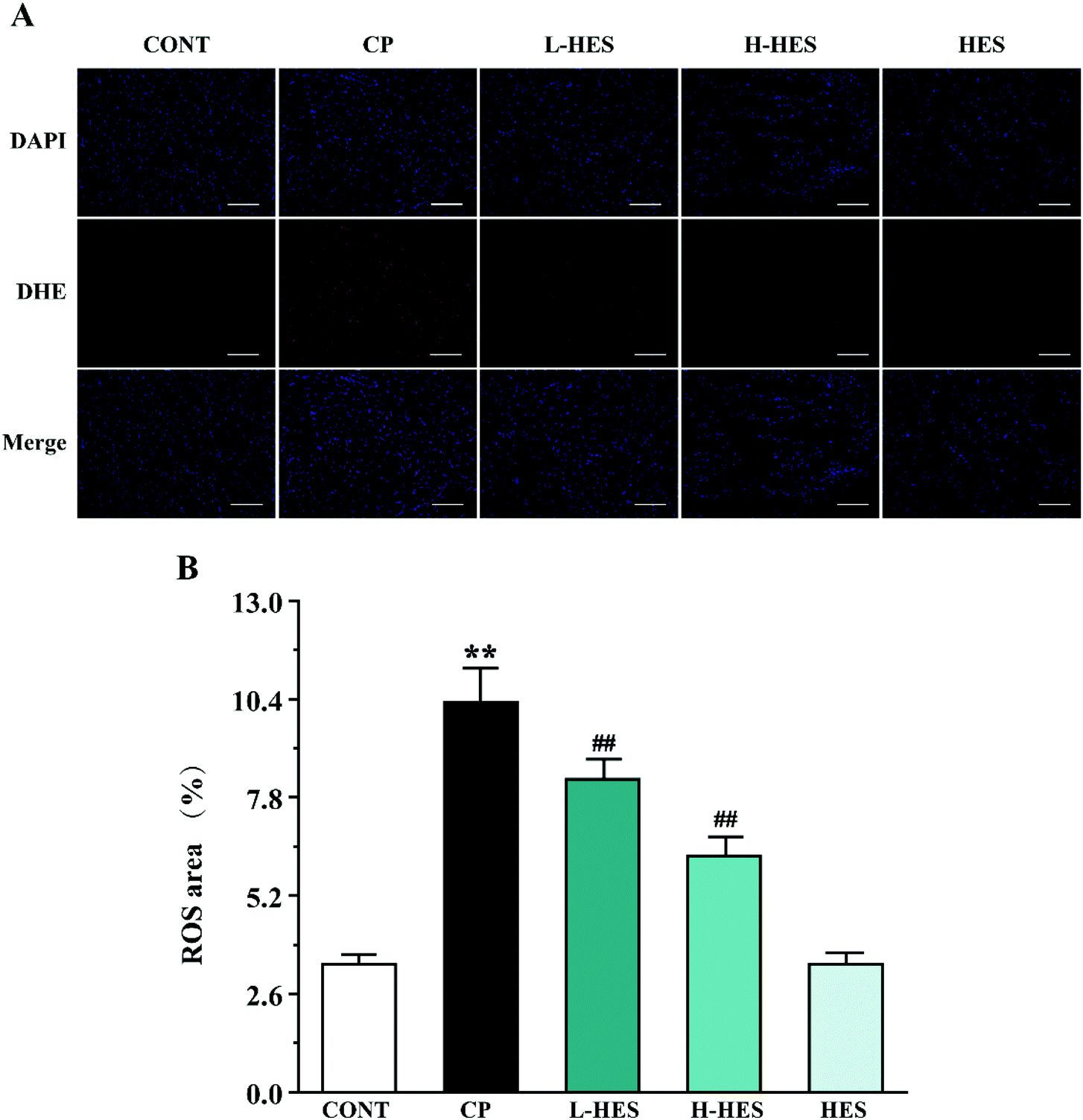 | ||
| Fig. 4 Effects of HES on the ROS level in CP-induced mice. (A) Representative ROS expression from CONT, CP, L-HES, H-HES, and HES groups (scale bar = 50 μm). (B) ROS fluorescent area. Data are presented as the mean ± SD for each group (n = 3). ** p < 0.01 versus the CONT group; ##p < 0.01 versus the CP group. | ||
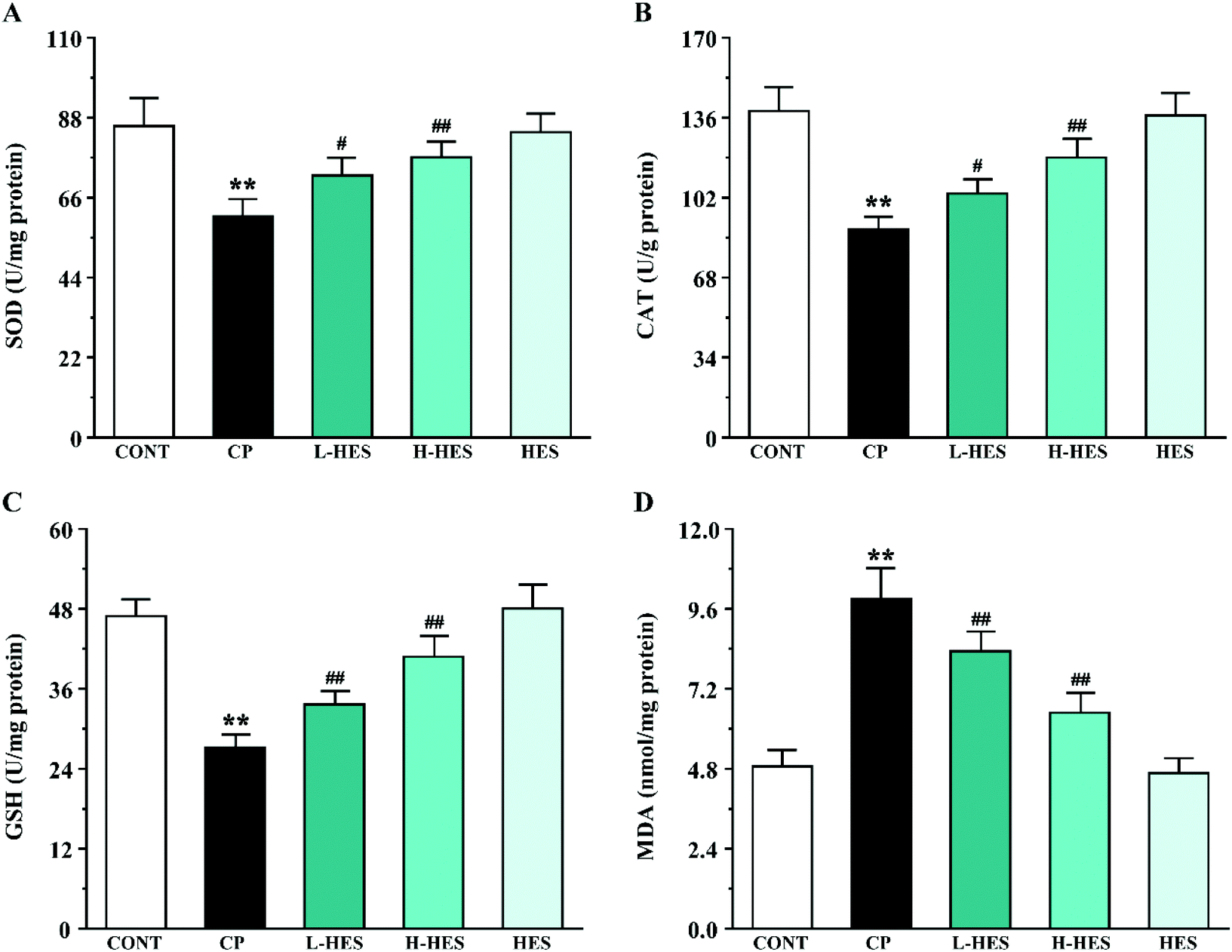 | ||
| Fig. 5 Effects of HES on tissue levels of the oxidative stress markers SOD (A), CAT (B), GSH (C), and MDA (D) in CP-induced mice. Data are presented as the mean ± SD for each group (n = 6). ** p < 0.01 versus the CONT group; #p < 0.05 and ##p < 0.01 versus the CP group. | ||
3.4 Effects of HES on inflammatory cytokines
ELISA analysis showed a marked elevation in TNF-α and IL-6 levels in the CP group compared to the CONT group (p < 0.01; Fig. 6). However, these parameters in the L-HES and H-HES groups were distinctly lower than those in the CP group (p < 0.01). Additionally, the HES group showed no significant differences compared to the CONT group (p > 0.05).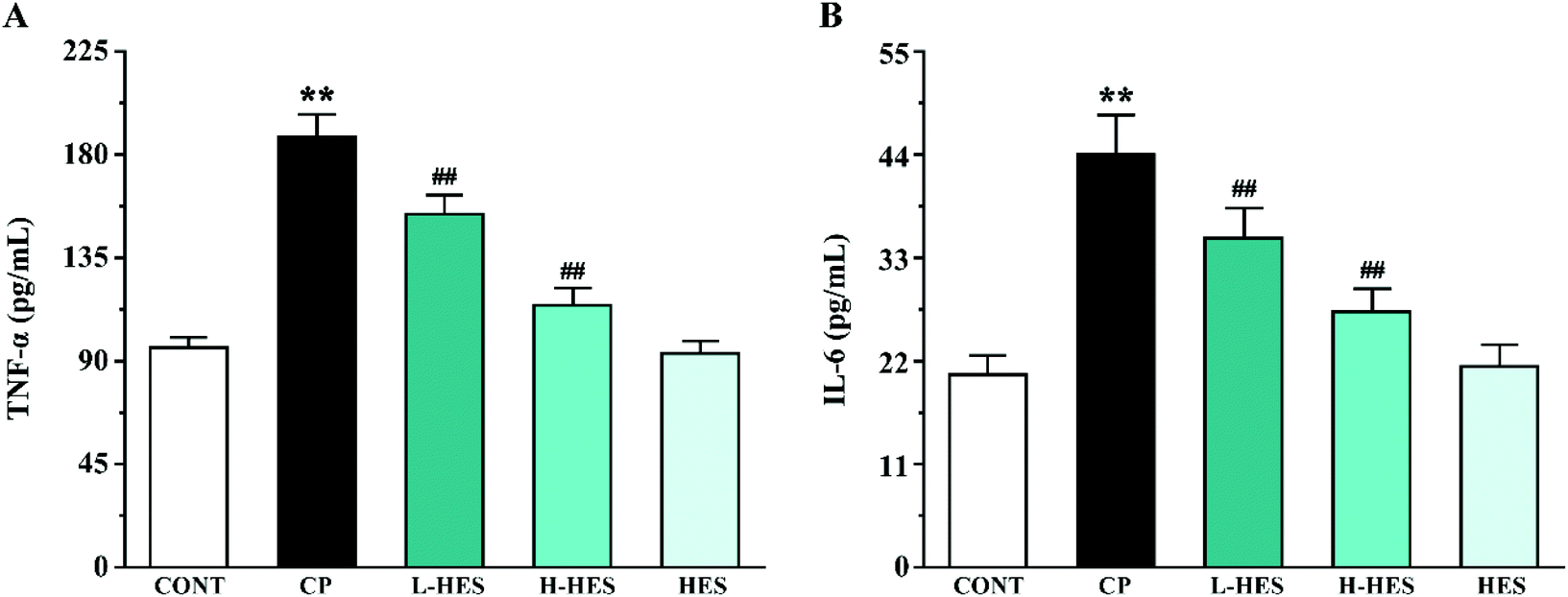 | ||
| Fig. 6 Effects of HES on levels of the inflammatory cytokines TNF-α (A) and IL-6 (B) in CP-induced mice. Data are presented as the mean ± SD for each group (n = 6). ** p < 0.01 versus the CONT group; ##p < 0.01 versus the CP group. | ||
3.5 Effects of HES on apoptosis
Western blot analysis for apoptosis factors showed that Bax and Caspase-3 expression was significantly upregulated in the CP group compared to the CONT group (p < 0.01), while Bcl-2 expression was lower than that in the CONT group (p < 0.01; Fig. 7). Compared with the CP group, Bax and Caspase-3 expression in the L-HES and H-HES groups was significantly downregulated (p < 0.05 or p < 0.01) and Bcl-2 expression was higher than that in the CP group (p < 0.05 or p < 0.01). There was no significant difference in each protein expression between the HES and CONT groups (p > 0.05).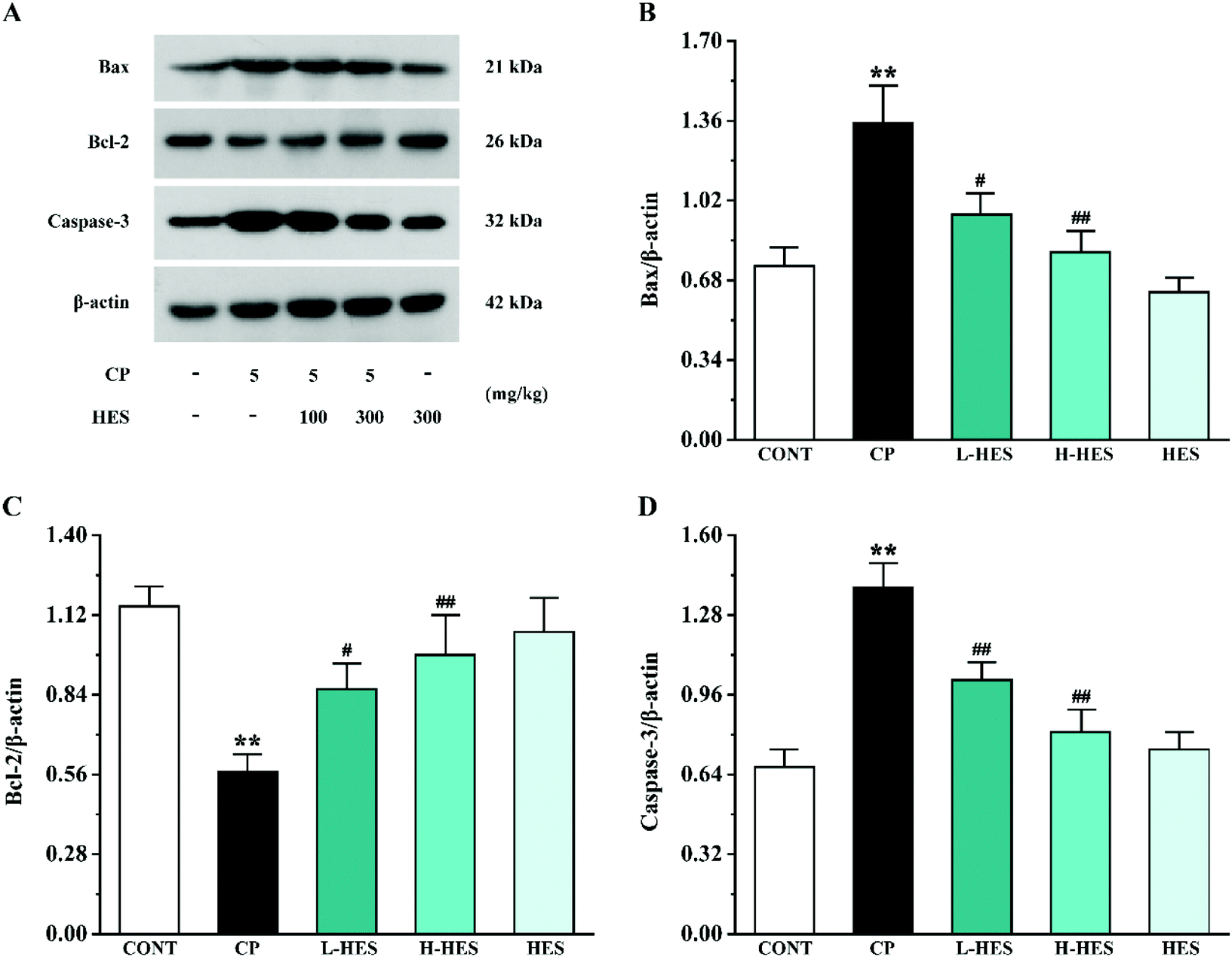 | ||
| Fig. 7 Effects of HES on levels of the apoptosis factors Bax (B), Bcl-2 (C), and Caspase-3 (D) in CP-induced mice (A). Data are presented as the mean ± SD for each group (n = 3). ** p < 0.01 versus CONT group; #p < 0.05 and ##p < 0.01 versus CP group. | ||
3.6 Effects of HES on signal pathway proteins
Western blot analysis for signal pathway proteins is shown in Fig. 8. p62 and Nrf2 expression was significantly decreased (p < 0.01) while the Keap1 expression level was significantly increased (p < 0.01) in the CP group compared to the CONT group. However, compared with the CP group, increased p62 and Nrf2 expression (p < 0.05 or p < 0.01) and decreased Keap1 levels (p < 0.01) were observed in the L-HES and H-HES groups compared with the CP group. Additionally, there was no significant difference in the expression of each protein between the HES and CONT groups (p > 0.05).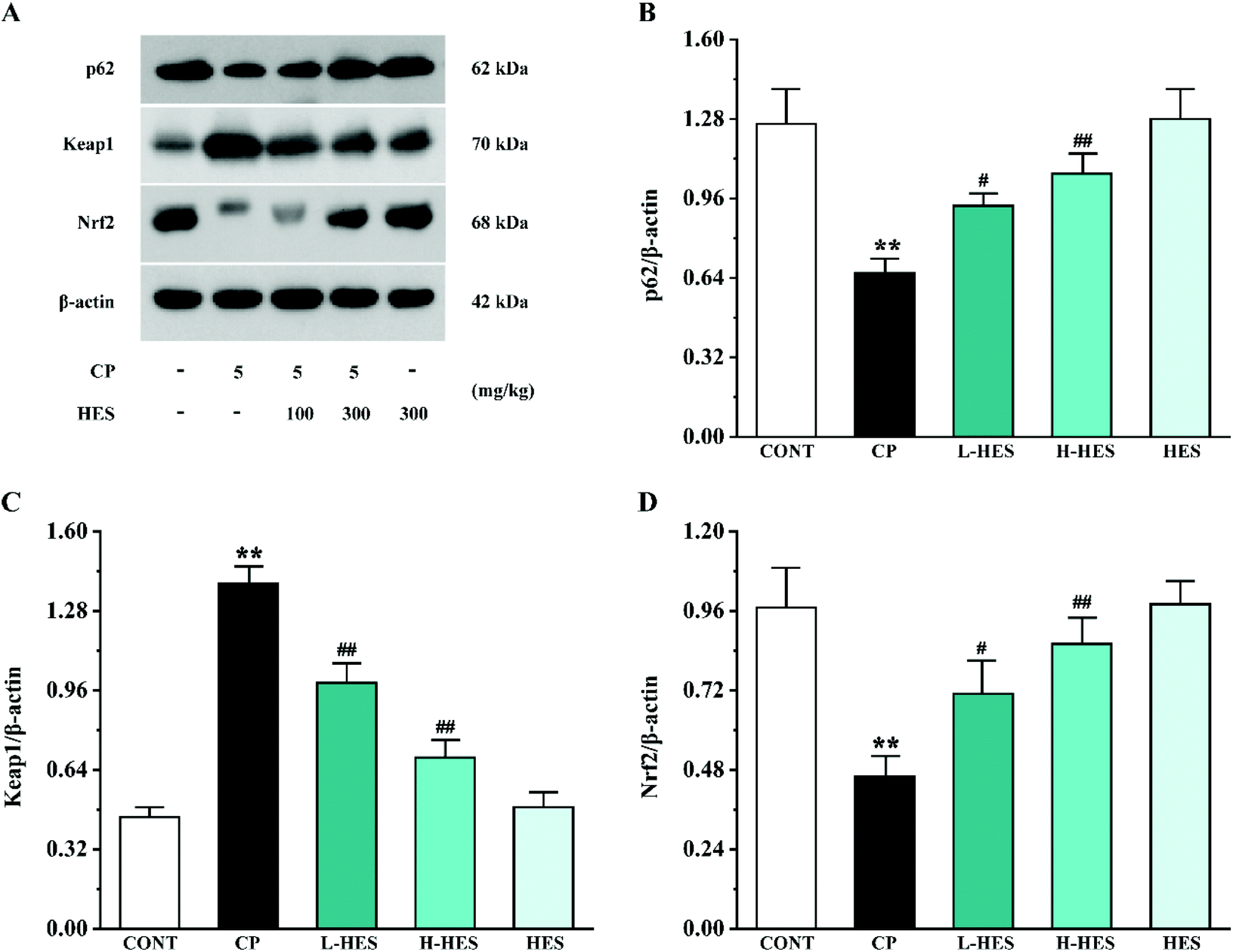 | ||
| Fig. 8 Effects of HES on levels of the signal pathway proteins p62 (B), Keap1 (C), and Nrf2 (D) in CP-induced mice (A). Data are presented as the mean ± SD for each group (n = 3). ** p < 0.01 versus the CONT group; #p < 0.05 and ##p < 0.01 versus the CP group. | ||
4. Discussion
CP has serious cardiotoxicity and other side effects.27–29 As a result, many tumour patients have to stop taking CP early, and the desired therapeutic effect is not achieved. Therefore, new therapeutic agents with better cardiac protection and safety to attenuate CP-induced cardiotoxicity are required. HES is an abundant and economical dietary bioflavonoid, and it has a variety of pharmacological properties and powerful cardiac protection.19,20,30 A recent study showed that HES could ameliorate CP-induced cardiac damage in rats on histomorphology and biochemistry.22 However, the specific mechanism by which HES protects against CP-induced cardiotoxicity remains unknown.Histopathological observation is crucial for evaluation of myocardial injury. In our present study, we found that CP triggered a series of pathological changes in heart tissue, including inflammatory cell infiltration and myocardial cell oedema and apoptosis. HES alleviated the myocardial lesions caused by CP, especially the high dose of HES, which initially confirms that HES may have a protective effect against CP-induced cardiotoxicity (Fig. 2).
CTnI, is a reliable marker of cardiac dysfunction, and CK and LDH are serum marker enzymes commonly used to detect cardiotoxicity. Consistent with previous studies,10,31 we found that cTnI, CK, and LDH levels were increased after CP administration compared to the CONT group. However, these parameters were significantly reduced after HES treatment, which confirms that HES may have a protective effect against CP-induced cardiotoxicity. This could be interpreted as an irreversible modification of the membrane structure and function resulting from increased lipid peroxidation of the myocardial membrane, which leads to leakage of myocardial enzymes and cTnI.32 This suggests that HES may protect the heart by reducing lipid peroxidation levels (Fig. 3).
CP can cause ROS production,12,33 and ROS are involved in a variety of cellular pathways. Excessive ROS can lead to the disruption or depletion of antioxidant defences. SOD and CAT, the main enzymes in the endogenous antioxidant system, are the first line of defence against oxidative damage. Excessive ROS can reduce SOD and CAT activity and subsequent oxidative stress. GSH is a small molecular-weight endogenous antioxidant, and its depletion intensifies ROS activity that triggers a lipid peroxidation cascade leading to a loss of membrane integrity. MDA, a degradation product of lipid hydroperoxide, is an index of lipid peroxidation in biological tissues. Increased MDA levels can cause serious damage to the cell membrane and aggravate oxidative stress.34 In the current research, CP markedly increased ROS and MDA production and decreased SOD, CAT, and GSH activity, which is consistent with previous studies.8,12 However, antioxidant marker activity was significantly increased and ROS and MDA levels were significantly decreased after HES treatment. Thus, the cardioprotective effect provided by HES in this study could be attributed to its antioxidant property through reducing lipid peroxidation and oxidative stress (Fig. 4 and 5).
Inflammation is considered to be another important determinant of CP-induced cardiotoxicity.35 High ROS concentrations induce activation of redox-sensitive transcription factors, which is followed by the release of pro-inflammatory mediators such as TNF-α and IL-6.36 TNF-α is an important inflammatory cytokine that can induce the release of other inflammatory mediators to promote the inflammatory cascade, and it can also induce myocardial apoptosis and participate in ventricular remodeling.37 IL-6 is also associated with cardiac depression.38 Studies have shown that CP increased the release of TNF-α and IL-6 in heart tissue,39–41 which are directly involved in tissue damage.42 Our results are consistent with these previous findings.26,43,44 CP administration resulted in a significant increase in TNF-α and IL-6 levels, but HES can significantly decrease TNF-α and IL-6 levels, indicating its potential anti-inflammatory role. Therefore, the cardioprotective effect of HES may also depend on its anti-inflammatory effect (Fig. 6).
Additionally, an increasing amount of evidence suggests that CP can induce serious cardiotoxicity by triggering mitochondria-mediated apoptosis.45 CP-induced ROS accumulation and overproduction often lead to myocardial mitochondrial dysfunction, which results in the release of pro-apoptotic factors.13 The Bcl-2 protein family, which plays an important regulatory role in the progress of apoptosis, is mainly mediated by the mitochondria.46 Both the anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax belong to this family. Bcl-2 plays a crucial role in maintaining mitochondrial structure and function. However, CP induces ROS production, leading to Bax translocation to the outer mitochondrial membrane and ultimately induces Caspase-3 activation. Caspase-3 is a key mediator of the apoptosis signalling pathway, and its activation eventually leads to apoptosis.47 We also detected apoptosis-related protein expression by western blot, which showed a significant increase in Bax and Caspase-3 expression levels and a decrease in Bcl-2 expression in the CP group. These results are consistent with previous research results.26 However, Bax and Caspase-3 expression was significantly decreased, while Bcl-2 was significantly increased after HES treatment. Thus, it can be speculated that HES can ameliorate cardiotoxicity by inhibiting apoptosis (Fig. 7).
Different factors have been shown to be involved in CP-induced cardiotoxicity, including oxidative stress, inflammation, and apoptosis. Among them, oxidative stress is the primary factor, and it is a key determinant of CP cardiotoxicity. Oxidative stress was shown to be a state of imbalance between oxidation and anti-oxidation in vivo, and overproduction of ROS causes oxidative stress.48 Under pathological conditions, oxidative stress can activate the transcription of various pro-inflammatory cytokines, and the release of cytokines during the inflammatory process can also induce systemic changes including cardiac damage.49,50 Inflammatory responses can, in turn, increase oxidative stress.51 Oxidative stress is also an important inducer of programmed cell death. Most apoptotic signals originate from the mitochondria, and the endogenous apoptotic pathways mediated by mitochondria are activated by numerous stimuli, including oxidative stress.32 In the mechanism of regulating oxidative stress, the Keap1–Nrf2 pathway is considered to be a well-characterised antioxidant defence mechanism.52,53 Nrf2 is usually maintained in an inactive cytoplasmic form by binding with its negative regulator Keap1.54 Under stress conditions, the Keap1–Nrf2 complex separates and releases the inhibitory effect of Keap1 on Nrf2, which is then activated and transported to the nucleus.54 Nrf2 is the core regulator of downstream antioxidant enzymes, and it is responsible for SOD and CAT activation, which helps prevent oxidative damage to myocardial cells.55,56 A recent study showed that p62 is involved in regulating oxidative stress through the Keap1–Nrf2 pathway,57 and p62–Keap1 interaction promotes Nrf2 nuclear translocation and its downstream signalling cascade.54 In the present study, CP administration significantly reduced p62 and Nrf2 and increased Keap1 expression. However, HES treatment results in enhanced p62–Keap1 interaction and Nrf2 nuclear translocation, which is demonstrated as increased p62 and Nrf2 and inhibited Keap1 expression. Up-regulation of Nrf2 promoted the increase of antioxidant enzyme expression and subsequent ROS reduction, thus alleviating CP-induced cardiotoxicity (Fig. 8).
5. Conclusion
In summary, HES has a significant protective effect against CP-induced cardiotoxicity, which may be achieved by inhibiting oxidative stress, inflammation, and apoptosis, and the mechanism may be attributed to activation of the p62–Keap1–Nrf2 signalling pathway. These findings suggest that HES may be a promising protective agent against CP cardiotoxicity in future anticancer clinical practice (Fig. 9).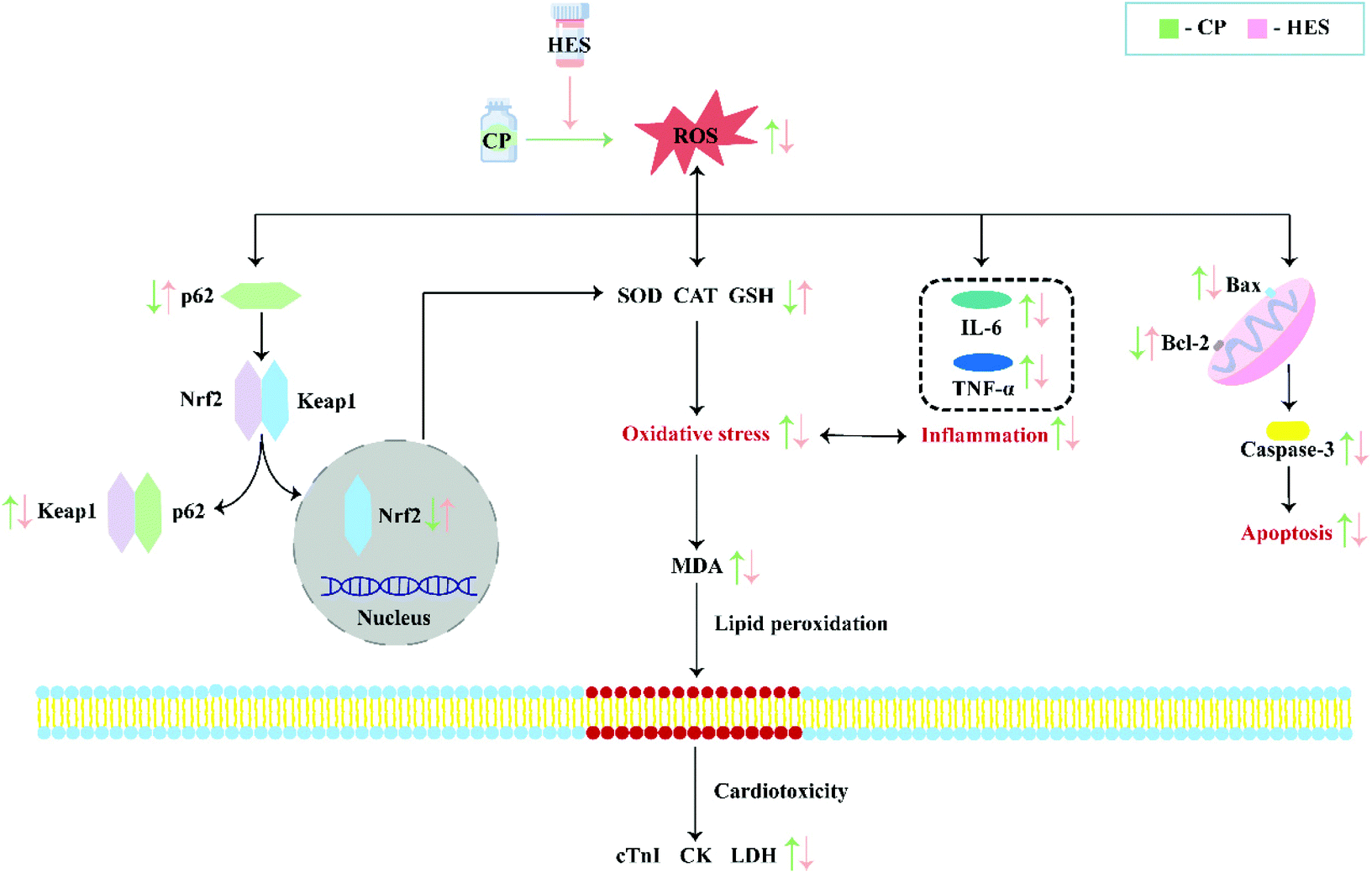 | ||
| Fig. 9 Mechanism for the protective effects of HES against CP-induced cardiotoxicity. | ||
Author contributions
Yuxin Jia: writing-original draft, investigation. Hui Guo: methodology, data curation. Xizhen Cheng: formal analysis. Yuling Zhang: investigation. Mingdong Si: software. Jing Shi: investigation, writing-review & editing. Donglai Ma: conceptualization, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Medical Science Research project of the Office of Hebei Provincial of China (No. 20222869), and Innovation Ability for Postgraduate of Hebei province of China (No. CXZZSS2022102).References
- N. Devarajan, R. Manjunathan and S. Ganesan, Tumor hypoxia: The major culprit behind cisplatin resistance in cancer patients, Crit. Rev. Oncol. Hematol., 2021, 162, 103327 Search PubMed
.
- K. J. M. Schimmel, D. J. Richel, R. B. A. v. d. Brink and H.-J. Guchelaar, Cardiotoxicity of cytotoxic drugs, Cancer Treat. Rev., 2004, 30, 181–191 Search PubMed
- J. Alexandre, J. J. Moslehi, K. R. Bersell, C. Funck-Brentano, D. M. Roden and J.-E. Salem, Anticancer drug-induced cardiac rhythm disorders: current knowledge and basic underlying mechanisms, Pharmacol. Ther., 2018, 189, 89–103 Search PubMed
- S. Khan, C. L. Chen, M. S. Brady, R. Parameswaran, R. Moore, H. Hassoun and R. D. Carvajal, Unstable angina associated with cisplatin and carboplatin in a patient with advanced melanoma, J. Clin. Oncol., 2012, 30, e163–e164 Search PubMed
- H. Ma, K. R. Jones, R. Guo, P. Xu, Y. Shen and J. Ren, Cisplatin compromises myocardial contractile function and mitochondrial ultrastructure: role of endoplasmic reticulum stress, Clin. Exp. Pharmacol. Physiol., 2010, 37, 460–465 Search PubMed
- K. Kalam and T. H. Marwick, Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: a systematic review and meta-analysis, Eur. J. Cancer, 2013, 49, 2900–2909 Search PubMed
- D. Shaloam and T. P. Bernard, Cisplatin in cancer therapy: molecular mechanisms of action, Eur. J. Pharmacol., 2014, 740, 364–378 Search PubMed
- B. Anzel, C. Ayşe, Ö. G. Özlem, Y. Betül, Ü. Menekşe, Ö. T. Merve and Y. Arzu, Protective effects of curcumin and beta-carotene on cisplatin-induced cardiotoxicity: An experimental rat model, Anatolian J. Cardiol., 2018, 19, 213–221 Search PubMed
- G. J. Dugbartey, L. J. Peppone and I. A. M. d. Graaf, An integrative view of cisplatin-induced renal and cardiac toxicities: Molecular mechanisms, current treatment challenges and potential protective measures, Toxicology, 2016, 371, 58–66 Search PubMed
- T. İsmail, Ö. B. Aslı, K. Ç. Ferda, K. Nezahat, S. Zeynep, B. Yasin, Ö. Adalet and A. Durdu, The effect of rutin on cisplatin-induced oxidative cardiac damage in rats, Anatolian J. Cardiol., 2018, 20, 136–142 Search PubMed
- H. Xue, H. Danfeng, P. Zhuma, L. Gaoxing and D. Jun, Reactive-oxygen-species-scavenging nanomaterials for resolving inflammation, Mater. Today Bio, 2021, 11, 100124 Search PubMed
- A. F. Soliman, L. M. Anees and D. M. Ibrahim, Cardioprotective effect of zingerone against oxidative stress, inflammation, and apoptosis induced by cisplatin or gamma radiation in rats, Naunyn-Schmiedeberg's Arch. Pharmacol., 2018, 391, 819–832 Search PubMed
- Q. Peng, Y. Jie, L. Qiang, Y. Tao, D. Yan, W. Tao, F. Wei and W. Ling, Cyanidin ameliorates cisplatin-induced cardiotoxicity via inhibition of ROS-mediated apoptosis, Exp. Ther. Med., 2018, 15, 1959–1965 Search PubMed
- C. T. Tan, H.-C. Chang, Q. Zhou, C. Yu, N. Y. Fu, K. Sabapathy and V. C. Yu, MOAP-1-mediated dissociation of p62/SQSTM1 bodies releases Keap1 and suppresses Nrf2 signaling, EMBO Rep., 2021, 22, e50854 Search PubMed
- J. W. Kaspar, S. K. Niture and A. K. Jaiswal, Nrf2:INrf2 (Keap1) signaling in oxidative stress, Free Radicals Biol. Med., 2009, 47, 1304–1309 Search PubMed
- C. Li and H. Schluesener, Health-promoting effects of the citrus flavanone hesperidin, Crit. Rev. Food Sci. Nutr., 2017, 57, 613–631 Search PubMed
- Y. Lu, C. Zhang, P. Bucheli and D. Wei, Citrus flavonoids in fruit and traditional Chinese medicinal food ingredients in China, Plant Foods Hum. Nutr, 2006, 61, 57–65 Search PubMed
- A. M. Ali, M. A. Gabbar, S. M. Abdel-Twab, E. M. Fahmy, H. Ebaid, I. M. Alhazza, O. M. Ahmed and A. Lloret, Citrus reticulataantidiabetic potency, antioxidant effects, and mode of actions of fruit peel hydroethanolic extract, hesperidin, and quercetin in nicotinamide/streptozotocin-induced wistar diabetic rats, Oxid. Med. Cell. Longevity, 2020, 2020, 1730492 Search PubMed
- S. He, X. Wang, Y. Zhong, L. Tang, Y. Zhang, Y. Ling, Z. Tan, P. Yang and A. Chen, Hesperetin post-treatment prevents rat cardiomyocytes from hypoxia/reoxygenation injury in vitro via activating PI3 K/Akt signaling pathway, Biomed. Pharmacother., 2017, 91, 1106–1112 Search PubMed
- S. S. Rekha, J. A. Pradeepkiran and M. Bhaskar, Bioflavonoid hesperidin possesses the anti-hyperglycemic and hypolipidemic property in STZ induced diabetic myocardial infarction (DMI) in male Wister rats, J. Nutr. Intermed. Metab., 2019, 15, 58–64 Search PubMed
- R. Ramin, S. Alireza, J. Saeedeh, E. Sarvenaz, B. Somayeh, K. Zahra, D. Madjid, O. D. Anca, T. Konstantinos, A. S. Dimosthenis, K. Spyros, T. Aristidis, K. Leda and H. Mahmoud, Cardioprotective effects of hesperidin on carbon monoxide poisoned in rats, Drug Chem. Toxicol., 2019, 1–6, DOI:10.1080/01480545.2019.1650753
- H. Oguzturk, O. Ciftci, A. Cetin, K. Kaya, O. M. Disli, M. G. Turtay, S. Gürbüz and N. Basak, Beneficial effects of hesperidin following cis-diamminedichloroplatinum-induced damage in heart of rats, Niger. J. Clin. Pract., 2016, 19, 99–103 Search PubMed
- J. Li, S. Wang, J. Duan, P. Le, C. Li, Y. Ding, R. Wang and Y. Gao, The protective mechanism of resveratrol against hepatic injury induced by iron overload in mice, Toxicol. Appl. Pharmacol., 2021, 424, 115596 Search PubMed
- S. Di, Y. Ma and L. Zhao, Clinical application and dosage of dried tangerine peel, Jilin J. Chin. Med., 2019, 39, 733–736 Search PubMed
- E. Turk, F. Kandemir, S. Yildirim, C. Caglayan, S. Kucukler and M. Kuzu, Protective Effect of Hesperidin on Sodium Arsenite-Induced Nephrotoxicity and Hepatotoxicity in Rats, Biol. Trace Elem. Res., 2019, 189, 95–108 Search PubMed
- J. Xing, J. Hou, Y. Liu, R. Zhang, S. Jiang, S. Ren, Y. Wang, Q. Shen, W. Li, X. Li and Z. Wang, Panax quinquefoliussupplementation of saponins from leaves of mitigates cisplatin-evoked cardiotoxicity via inhibiting oxidative stress-associated inflammation and apoptosis in mice, Antioxidants, 2019, 8, 347 Search PubMed
- A. A. El-Hawwary and N. M. Omar, The influence of ginger administration on cisplatin-induced cardiotoxicity in rat: Light and electron microscopic study, Acta histochemica, 2019, 121, 553–562 Search PubMed
- E.-S. E. El-Awady, Y. M. Moustafa, D. M. Abo-Elmatty and A. Radwan, Cisplatin-induced cardiotoxicity: Mechanisms and cardioprotective strategies, Eur. J. Pharmacol., 2011, 650, 335–341 Search PubMed
- E. E. Gunturk, B. Yucel, I. Gunturk, C. Yazici, A. Yay and K. Kose, The effects of N-acetylcysteine on cisplatin induced cardiotoxicity, Bratisl. Lek. Listy, 2019, 120, 423–428 Search PubMed
- P. Pratibha and K. Fahad, A mechanistic review of the anticancer potential of hesperidin, a natural flavonoid from citrus fruits, Nutr. Res., 2021, 92, 21–31 Search PubMed
- Y. A. Khadrawy, E. N. Hosny, M. M. El-Gizawy, H. G. Sawie and H. S. Aboul Ezz, The effect of curcumin nanoparticles on cisplatin-induced cardiotoxicity in male wistar albino rats, Cardiovasc. Toxicol., 2021, 21, 433–443 Search PubMed
- B. Han, S. Li, Y. Lv, D. Yang, J. Li, Q. Yang, P. Wu, Z. Lv and Z. Zhang, Dietary melatonin attenuates chromium-induced lung injury via activating the Sirt1/Pgc-1α/Nrf2 pathway, Food Funct., 2019, 10, 5555–5565 Search PubMed
- L. Antunes, J. Darin and M. Bianchi, Protective effects of vitamin c against cisplatin-induced nephrotoxicity and lipid peroxidation in adult rats: a dose-dependent study, Pharmacol. Res., 2000, 41, 405–411 Search PubMed
- V. Vineetha and K. Raghu, An Overview on Arsenic Trioxide-Induced Cardiotoxicity, Cardiovasc. Toxicol., 2019, 19, 105–119 Search PubMed
- N. Kou, M. Xue, L. Yang, M. Zang, H. Qu, M. Wang, Y. Miao, B. Yang and D. Shi, Panax quinquefolius saponins combined with dual antiplatelet drug therapy alleviate gastric mucosal injury and thrombogenesis through the COX/PG pathway in a rat model of acute myocardial infarction, PLoS One, 2018, 13, e0194082 Search PubMed
- J. Liu, G. Chang, J. Huang, Y. Wang, N. Ma, A. Roy and X. Shen, Sodium Butyrate Inhibits the Inflammation of Lipopolysaccharide-Induced Acute Lung Injury in Mice by Regulating the Toll-Like Receptor 4/Nuclear Factor κB Signaling Pathway, J. Agric. Food Chem., 2019, 67, 1674–1682 Search PubMed
- J. Li, C. Xie, J. Zhuang, H. Li, Y. Yao, C. Shao and H. Wang, Resveratrol attenuates inflammation in the rat heart subjected to ischemia-reperfusion: Role of the TLR4/NF-κB signaling pathway, Mol. Med. Rep., 2015, 11, 1120–1126 Search PubMed
- I. Mikaelian, D. Coluccio, K. Morgan, T. Johnson, A. Ryan, E. Rasmussen, R. Nicklaus, C. Kanwal, H. Hilton, K. Frank, L. Fritzky and E. Wheeldon, Temporal gene expression profiling indicates early up-regulation of interleukin-6 in isoproterenol-induced myocardial necrosis in rat, Toxicol. Pathol., 2008, 36, 256–264 Search PubMed
- A. A. Al-Majed, M. M. Sayed-Ahmed, A. A. Al-Yahya, A. M. Aleisa, S. S. Al-Rejaie and O. A. Al-Shabanah, Propionyl-L-carnitine prevents the progression of cisplatin-induced cardiomyopathy in a carnitine-depleted rat model, Pharmacol. Res., 2006, 53, 278–286 Search PubMed
- S. A. Abdellatief, A. A. A. Galal, S. M. Farouk and M. M. Abdel-Daim, Ameliorative effect of parsley oil on cisplatin-induced hepato-cardiotoxicity: A biochemical, histopathological, and immunohistochemical study, Biomed. Pharmacother., 2017, 86, 482–491 Search PubMed
- P. Kumar, K. Sulakhiya, C. C. Barua and N. Mundhe, TNF-α, IL-6 and IL-10 expressions, responsible for disparity in action of curcumin against cisplatin-induced nephrotoxicity in rats, Mol. Cell. Biochem., 2017, 431, 113–122 Search PubMed
- G. Schuller-Levis, M. Quinn, C. Wright and E. Park, Taurine protects against oxidant-induced lung injury: possible mechanism(s) of action, Adv. Exp. Med. Biol., 1994, 359, 31–39 Search PubMed
- Y. Liang, B. Zheng, J. Li, J. Shi, L. Chu, X. Han, X. Chu, X. Zhang and J. Zhang, Crocin ameliorates arsenic trioxide-induced cardiotoxicity via Keap1-Nrf2/HO-1 pathway: Reducing oxidative stress, inflammation, and apoptosis, Biomed. Pharmacother., 2020, 131, 110713 Search PubMed
- A. A. K. El-Sheikh and Z. Khired, Morphine deteriorates cisplatin-induced cardiotoxicity in rats and induces dose-dependent cisplatin chemoresistance in mcf-7 human breast cancer cells, Cardiovasc. Toxicol., 2021, 21, 553–562 Search PubMed
- Z. V. Varga, P. Ferdinandy, L. Liaudet and P. Pacher, Drug-induced mitochondrial dysfunction and cardiotoxicity, Am. J. Physiol.: Heart Circ. Physiol., 2015, 309, H1453–H1467 Search PubMed
- A. Cagla, T. Charles and G. Yair, Arsenic trioxide and paclitaxel induce apoptosis by different mechanisms, Cell cycle, 2004, 3, 324–334 Search PubMed
- E. A. Kosenko, I. N. Solomadin, L. A. Tikhonova, V. P. Reddy, G. Aliev and Y. G. Kaminsky, Pathogenesis of Alzheimer disease: role of oxidative stress, amyloid-β peptides, systemic ammonia and erythrocyte energy metabolism, CNS Neurol. Disord.: Drug Targets, 2014, 13, 112–119 Search PubMed
- X. Sun, X. Wang, Q. He, M. Zhang, L. Chu, Y. Zhao, Y. Wu, J. Zhang, X. Han, X. Chu, Z. Wu and S. Guan, Investigation of the ameliorative effects of baicalin against arsenic trioxide-induced cardiac toxicity in mice, Int. Immunopharmacol., 2021, 99, 108024 Search PubMed
- Z. Zhang, C. Guo, H. Jiang, B. Han, X. Wang, S. Li, Y. Lv, Z. Lv and Y. Zhu, Inflammation response after the cessation of chronic arsenic exposure and post-treatment of natural astaxanthin in liver: potential role of cytokine-mediated cell-cell interactions, Food Funct., 2020, 11, 9252–9262 Search PubMed
- C. Campana, R. Dariolli, M. Boutjdir and E. Sobie, Inflammation as a Risk Factor in Cardiotoxicity: An Important Consideration for Screening During Drug Development, Front. Pharmacol., 2021, 12, 598549 Search PubMed
- X. Xu, A. Grijalva, A. Skowronski, M. van Eijk, M. Serlie and A. Ferrante, Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation, Cell Metab., 2013, 18, 816–830 Search PubMed
- P. Canning, F. J. Sorrell and A. N. Bullock, Structural basis of Keap1 interactions with Nrf2, Free Radicals Biol. Med., 2015, 88, 101–107 Search PubMed
- Q. M. Chen and A. J. Maltagliati, Nrf2 at the heart of oxidative stress and cardiac protection, Physiol. Genomics, 2018, 50, 77–97 Search PubMed
- C. A. Silva-Islas and P. D. Maldonado, Canonical and non-canonical mechanisms of Nrf2 activation, Pharmacol. Res., 2018, 134, 92–99 Search PubMed
- A. K. Jaiswal, Nrf2 signaling in coordinated activation of antioxidant gene expression, Free Radicals Biol. Med., 2004, 36, 1199–1207 Search PubMed
- Z. Li, Protective effects of trimetazidine and coenzyme Q10 on cisplatin-induced cardiotoxicity by alleviating oxidative stress and mitochondrial dysfunction, Anatolian J. Cardiol., 2019, 22, 232–239 Search PubMed
- B. Desirée, D. Katiuscia, T. Pierangelo, P. Marta and G. Francesco, Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma, Transl. Res., 2018, 193, 54–71 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2fo00298a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |