 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resonant X-ray photoelectron spectroscopy: identification of atomic contributions to valence states†
J. M.
Seymour
 a,
E.
Gousseva
a,
R. A.
Bennett
a,
A. I.
Large
b,
G.
Held
b,
D.
Hein
c,
G.
Wartner
c,
W.
Quevedo
c,
R.
Seidel
c,
C.
Kolbeck
d,
C. J.
Clarke
e,
R. M.
Fogarty
f,
R. A.
Bourne
g,
R. G.
Palgrave
h,
P. A.
Hunt
i and
K. R. J.
Lovelock
*a
a,
E.
Gousseva
a,
R. A.
Bennett
a,
A. I.
Large
b,
G.
Held
b,
D.
Hein
c,
G.
Wartner
c,
W.
Quevedo
c,
R.
Seidel
c,
C.
Kolbeck
d,
C. J.
Clarke
e,
R. M.
Fogarty
f,
R. A.
Bourne
g,
R. G.
Palgrave
h,
P. A.
Hunt
i and
K. R. J.
Lovelock
*a
aDepartment of Chemistry, University of Reading, Reading, UK. E-mail: k.r.j.lovelock@reading.ac.uk
bDiamond Light Source, Didcot, Oxfordshire, UK
cHelmholtz-Zentrum Berlin für Materialien und Energie (HZB), Berlin, Germany
dFritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin, Germany
eSchool of Chemistry, University of Nottingham, Nottingham, UK
fDepartment of Materials, Imperial College London, London, UK
gInstitute of Process Research and Development, Schools of Chemistry and Chemical and Process Engineering, University of Leeds, Leeds, UK
hDepartment of Chemistry, University College London, London, UK
iSchool of Chemical and Physical Sciences, Victoria University of Wellington, Wellington, New Zealand
First published on 16th February 2022
Abstract
Valence electronic structure is crucial for understanding and predicting reactivity. Valence non-resonant X-ray photoelectron spectroscopy (NRXPS) provides a direct method for probing the overall valence electronic structure. However, it is often difficult to separate the varying contributions to NRXPS; for example, contributions of solutes in solvents or functional groups in complex molecules. In this work we show that valence resonant X-ray photoelectron spectroscopy (RXPS) is a vital tool for obtaining atomic contributions to valence states. We combine RXPS with NRXPS and density functional theory calculations to demonstrate the validity of using RXPS to identify atomic contributions for a range of solutes (both neutral and ionic) and solvents (both molecular solvents and ionic liquids). Furthermore, the one-electron picture of RXPS holds for all of the closed shell molecules/ions studied, although the situation for an open-shell metal complex is more complicated. The factors needed to obtain a strong RXPS signal are investigated in order to predict the types of systems RXPS will work best for; a balance of element electronegativity and bonding type is found to be important. Additionally, the dependence of RXPS spectra on both varying solvation environment and varying local-covalent bonding is probed. We find that RXPS is a promising fingerprint method for identifying species in solution, due to the spectral shape having a strong dependence on local-covalency but a weak dependence on the solvation environment.
1. Introduction
Theories linking electronic structure to reactivity are well-established.1–5 For many applications which revolve around electron donation in the liquid phase, e.g. electrochemical energy storage and catalysis,6–10 knowledge of the occupied electronic states is vital for understanding and predicting reactivity. Experimental measures of the occupied valence states (OVSs)11 and calculations of the occupied molecular orbitals (OMOs)1–5,12 for gas phase molecules and for solids/adsorbates on solids are widely available. The highest OVS (HOVS) from experiments and highest OMO (HOMO) from calculations are often found to be important for explaining reactivity, but deeper OVSs/OMOs at larger binding energy (EB) have also been found to be critical.13–15 Therefore, knowledge of both EB and the atomic contributions to the OVSs/OMOs is crucial to understanding and predicting reactivity for the liquid phase. Ideally, this question would be answered using experimental data, but measuring every solute/solvent combination is not possible; calculations offer a potential for screening solute/solvent combinations. EB(HOMO) calculated on gas phase species has been suggested as a suitable descriptor for choosing solutes (e.g. redox mediators) and solvents for battery electrolytes,6,7,16,17 although gas phase calculations do not include solvation effects.18 Calculations on models of the liquid phase8,19 or where solvent effects are accounted for,20 have been published, but capturing solvation with calculations is very challenging, both in terms of the methods and the system size. Therefore, benchmark experimental data is required for validation of these calculations.Information about the electronic structure can be obtained from valence non-resonant X-ray photoelectron spectroscopy (NRXPS) (Fig. 1b). Currently there is little liquid phase experimental data for OVSs compared to the amount of experimental data available for the gas phase and solids, especially for solvents other than water, as solutions pose significant challenges. Given the volatile nature of most solutions, liquid jet sample set-ups are usually required,21–24 although UHV-compatible ionic liquids (ILs, liquids containing only ions and no neutral molecules) have also been studied.25 A further challenge is when contributions from the solute and solvent overlap; contributions from the solvent dominate, making OVSs from the solute particularly hard to identify. There are a small number of solutions where the solute HOVS was clearly the sample HOVS, meaning that NRXPS could be used to measure EB for the solute HOVS, e.g. NaI in water.26 For certain solutes, the solute HOVS has been found to overlap with the solvent HOVS; therefore, the solute EB(HOVS) can be measured with very careful valence NRXPS measurements and subtraction of the solvent valence NRXPS contribution. However, EB for most other solute OVSs cannot be readily measured, e.g. imidazole in water.27 Little valence NRXPS exists for solutes with large HOVS EB due to the significant experimental challenges.
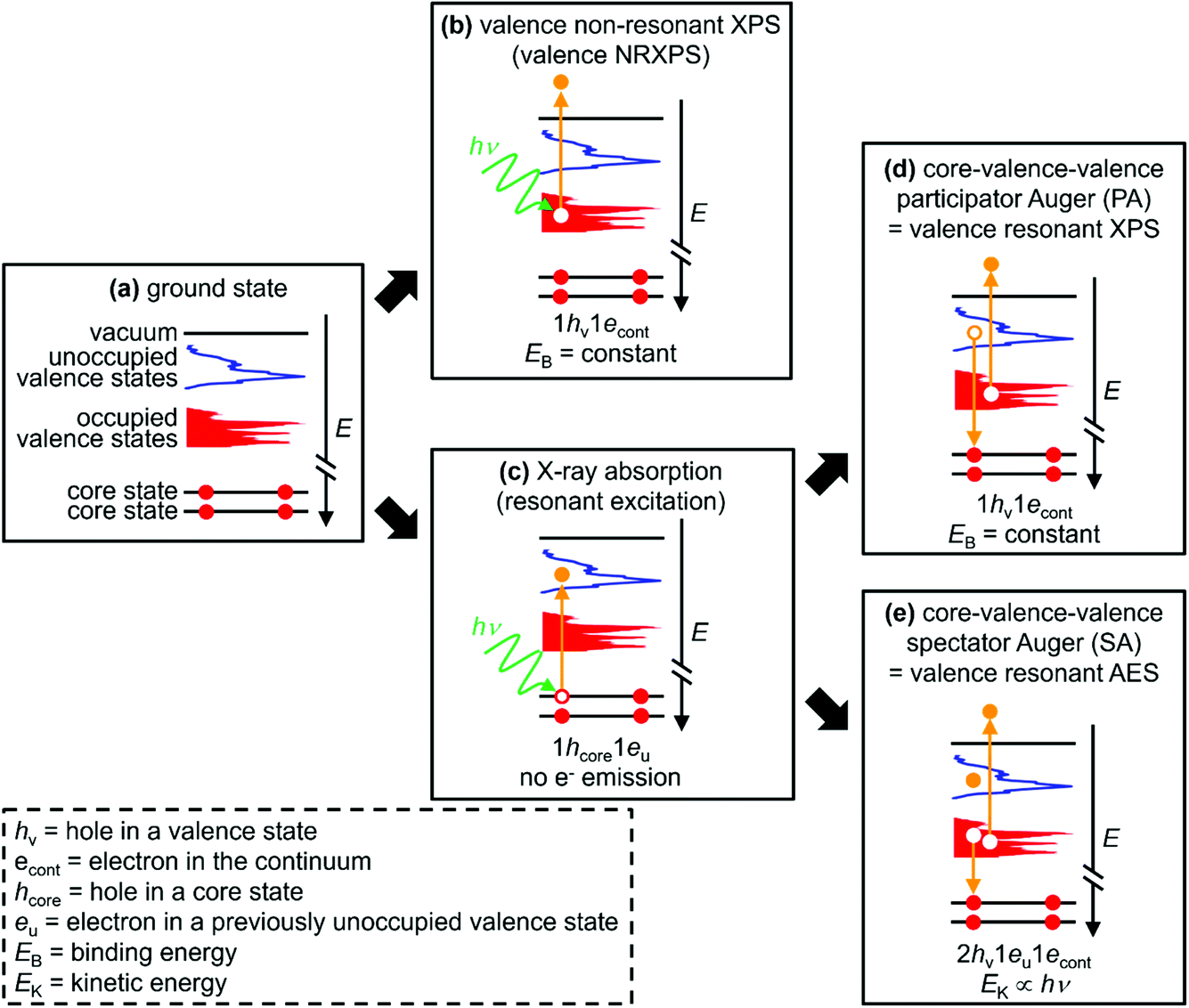 | ||
| Fig. 1 Sketches explaining the different electron dynamics events featured in this article. (a) Ground state. (b) Valence non-resonant XPS (NRXPS). (c) X-ray absorption (resonant excitation). (d) Core–valence–valence participator Auger (PA) transition; X-ray absorption followed by a valence PA transition = valence resonant XPS (RXPS). (e) Core–valence–valence spectator Auger (SA) transition; X-ray absorption followed by a valence SA transition = valence resonant Auger electron spectroscopy (RAES). The text at the bottom of each sketch gives the final state of each process and the approximate electron kinetic energy (EK) or binding energy (EB). 1econt = electron in the continuum. 1eu = electron in a previously unoccupied valence state. Element- and orbital-specificity are achieved through studying different element core state X-ray absorptions followed by RXPS for the same samples; site-specificity can be achieved for certain samples by studying two or more different X-ray absorptions followed by RXPS for the same element of the same sample. | ||
A comprehensive description of liquid-phase electronic structure can potentially be obtained from valence resonant XPS (RXPS, Fig. 1c and d), which is element-specific and orbital-specific, and at times site-specific.28–78 In particular, contributions from overlapping OVSs can be identified using valence RXPS, which is very important for complex systems such as solutions. RXPS has been used for many years to study readily vaporised small molecules, e.g. water, N2.28–32 In the condensed phase, RXPS has been used to study many solids: metal salts/complexes,33–35 inorganic salts,36–45 small organic molecules,46–52 protein,53 polymers,54–59 organic perovskites.60,61 Furthermore, RXPS has been used at the solid–adsorbate interface.62–66 More recently, RXPS has been used to study liquids, both for the solvent (water,67 ILs68,69) and more commonly for solutes in water (H2O2,67 metal complexes70–75 such as Fe3+ in aqueous solution70 and Ti3+ in aqueous solution,71 and nanoparticles76,77).
Valence RXPS is best pictured as a two-step process. (i) X-ray absorption (XA) by a core state to give a 1hcore1eu state (1hcore represents the core-hole and 1eu represents an electron in a previously unoccupied valence state, Fig. 1c), where the absorption cross-section is large for dipole allowed transitions (e.g. N 1s → N 2p π* or Co 2p3/2 → Co 3d). (ii) A core–valence–valence participator Auger (PA) transition as the de-excitation step; the excited valence electron refills the core-hole (i.e. participating in the process through direct recombination) and an electron from an OVS is ejected (Fig. 1d). The final state for valence PA emission is one hole in the valence, 1hv (Fig. 1d), 1+ relative to the ground state (Fig. 1a), the same final state as for valence NRXPS (Fig. 1b). Hence, XA followed by valence PA emission is known as valence RXPS and can be interpreted in terms of a one-electron picture, i.e. the frozen orbital approximation.
The selection rules are different for valence NRXPS and valence RXPS, with the selection rules well-known for valence NRXPS (explained by the Gelius method79,80) but unclear for valence RXPS.31,52,55 One of the requirements for a PA transition is that the OVS (in the ground state) involved in the PA process has good overlap with the core-hole. Therefore, features due to RXPS appear as resonant enhancements in the intensity of valence NRXPS contributions and normally appear at constant binding energy, EB; the same EB as for valence NRXPS from the same OVS.34,56 Using a combination of results from RXPS (i.e. PA transitions), valence NRXPS and ground state calculations to obtain the partial density-of-states (pDoS), it has been shown that RXPS can be used to identify atomic contributions to OVSs for a number of relatively simple systems, e.g. pyridine,46 pyrazine,46 s-triazine,46 butadiene,51 furan,49 pyrrole,49 benzene,48 and also the polymer poly(4-hydroxystyrene) (PHS),55i.e. the one-electron picture held. Leading on from these findings, RXPS has been used to identify the HOVS as originating from: (a) DNA bases and not in the backbone of DNA duplexes,57 and (b) the cation (and not the anion) for nine ILs (out of 60 ILs studied).68,69 However, most RXPS studies have been limited to organic molecules containing π-bonding.
To probe the OVSs, intense RXPS transitions are ideal. Therefore, it is important to determine what structures give intense RXPS transitions. At EB > ∼20 eV (depending on the sample and element edge studied) transitions are detected, e.g. spectator Auger (SA) transitions, where the final state is 2hv1eu with one-electron ejected to the continuum (Fig. 1e). For these SA transitions the one-electron picture does not hold.49 SA transitions compete with PA transitions to fill the core-hole; large PA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SA intensity ratios are best for probing OVSs. In general, valence SA features are more intense than valence PA features.31,36,43–45,49,50,52,54,55 For X-ray absorption for core state → π* versus X-ray absorption for core state → σ*, organic molecules with π* showed much larger PA:SA intensity ratios than organic molecules with no π* and only σ* (which generally showed no PA intensity),50,54e.g. ethylene versus cyclohexane.50 For different elements, much larger PA:SA intensity ratios were obtained for the B 1s → π* absorption for solid BN and solid B2O3 than for the N 1s → π* absorption for solid BN.43–45 For solid pyrrole the PA:SA intensity ratios were 1:10 for the C 1s → π* absorption and 1:50 for the N 1s → π* absorption.49 Furthermore, the PA:SA intensity ratios were 1:10 for the C 1s → π* absorption for solid furan, polystyrene and poly(4-hydroxystyrene).49,54,55 For the gas phase of four simple O-containing organic molecules (formic acid, acetaldehyde, acetic acid and methyl formate), the PA:SA intensity ratio was small even for O 1s → π* absorption.31 Lastly, no PA intensity was observed at the F 1s absorption edge for solid CaF2 and fluorinated nanotubes.36,78
:SA intensity ratios are best for probing OVSs. In general, valence SA features are more intense than valence PA features.31,36,43–45,49,50,52,54,55 For X-ray absorption for core state → π* versus X-ray absorption for core state → σ*, organic molecules with π* showed much larger PA:SA intensity ratios than organic molecules with no π* and only σ* (which generally showed no PA intensity),50,54e.g. ethylene versus cyclohexane.50 For different elements, much larger PA:SA intensity ratios were obtained for the B 1s → π* absorption for solid BN and solid B2O3 than for the N 1s → π* absorption for solid BN.43–45 For solid pyrrole the PA:SA intensity ratios were 1:10 for the C 1s → π* absorption and 1:50 for the N 1s → π* absorption.49 Furthermore, the PA:SA intensity ratios were 1:10 for the C 1s → π* absorption for solid furan, polystyrene and poly(4-hydroxystyrene).49,54,55 For the gas phase of four simple O-containing organic molecules (formic acid, acetaldehyde, acetic acid and methyl formate), the PA:SA intensity ratio was small even for O 1s → π* absorption.31 Lastly, no PA intensity was observed at the F 1s absorption edge for solid CaF2 and fluorinated nanotubes.36,78
Studies have investigated whether valence RXPS spectra gave fingerprint contributions that represent functional groups within a sample. For solid M[NO3] (where M = Li+, Na+) the [NO3]− anion was essentially quasi-isolated, as the RXPS spectra were broadly independent of the counterion identity.41,42 Conversely, for four gas phase small C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O containing organic molecules (formic acid, acetaldehyde, acetic acid and methyl formate), the RXPS spectra did not match; RXPS was deemed too sensitive to the chemical bonding to enable identification of specific functional groups in these molecules.31
O containing organic molecules (formic acid, acetaldehyde, acetic acid and methyl formate), the RXPS spectra did not match; RXPS was deemed too sensitive to the chemical bonding to enable identification of specific functional groups in these molecules.31
In this article, we investigate the liquid phase electronic structure of both solutes and solvents using valence RXPS and DFT calculations. The key molecules and ions studied are given in Fig. 2. A full list of samples studied using valence RXPS is given in ESI Table S1.† Four samples were molecular liquids (water, ethanol = EtOH, acetonitrile = MeCN and propylene carbonate = PC), 11 samples were solutes in molecular liquids (water and MeCN), and eight samples were neat ILs. Coordinates for the molecules/ions for which DFT calculations were performed are given in ESI Section 2;† to capture the liquid phase, a polarisable continuum model was used (see Section 2.6 for more details). Separation of solute and solvent contributions to the valence electronic structure was achieved using valence RXPS. The factors that underpin valence RXPS intensity were probed. It is challenging to determine the factors for many of the solutes studied here due to the dominance of solvent contributions; hence, we mainly focused in this part on neat solvents, i.e. molecular liquids and ILs. By combining knowledge from: (i) EB matches from valence RXPS and NRXPS and (ii) comparisons of valence RXPS traces to pDOS from calculations, we demonstrate that the one-electron picture for valence XPS holds for closed shell molecules/ions in the liquid phase. The cyano-based anions are ideal for testing the one-electron picture, as these anions gave sharp and well-defined features in valence NRXPS. Lastly, insights into bonding were gained by making comparisons between experimental RXPS traces. The use of valence RXPS traces as fingerprints for both intramolecular and intermolecular bonding/interactions is investigated; this knowledge is important to understand interactions of ions/molecules with both other solutes and surfaces.
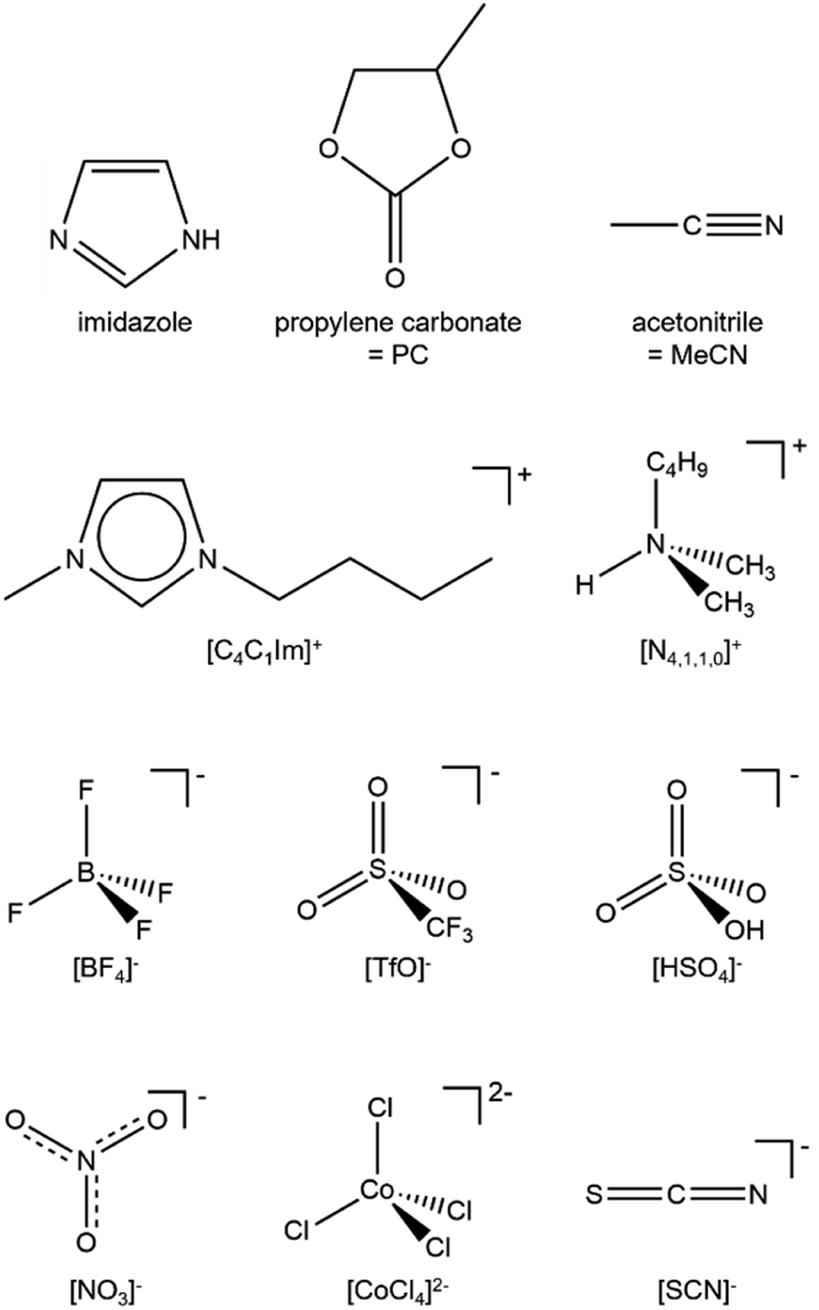 | ||
| Fig. 2 The key molecules, cations and anions studied. | ||
2. Experimental
2.1 Sample synthesis
Details of sample synthesis/preparation are given in ESI Section 1.†2.2 Lab XPS apparatus
Laboratory-based XPS for the IL [C4C1Im][BF4] was carried out using a Thermo K-alpha spectrometer utilising Al Kα radiation (hν = 1486.6 eV), as detailed in ref. 68. No impurities were observed after Ar+ ion sputtering (ESI Fig. S21†).2.3 Synchrotron XPS apparatus
The measurement for 15 solutions was performed on the U49/2-PGM 1 beamline with SOL3PES end-station81 at BESSY II (Germany). Solutes were weighed and mixed with a corresponding mass of solvent to achieve the desired concentration. Liquid-jet apparatus was used. XP and RXP spectra were acquired using a Scienta Omicron R4000 HIPP-2 hemispherical electron analyser.The measurements for two ILs ([C8C1Im]2[CoCl4] and [C8C1Im]2[Co(NTf2)4]) were performed on the B07-C (VerSoX) beamline at Diamond Light Source (UK).82 A thin film (less than 0.1 mL, essentially so that no drop could be observed) of the IL sample was placed on a tantalum sample holder. The “T-pot” analyser chamber was used, which allowed for rastering of the sample continually, perpendicular to the analyser entrance nozzle during X-ray irradiation. Rastering allowed for the maximum X-ray flux of B07, as beam damage did not occur during the raster experiments (unlike during static experiments, see ref. 69). XP and RXP spectra were acquired using a PHOIBOS 150 NAP hemispherical electron analyser supplied by SPECS. No sputtering was applied to the samples prior to measurement (no contaminants were observed by NRXPS).
The measurements for five ILs ([C8C1Im][C(CN)3], [C4C1Im][SCN], [P6,6,6,14][NO3], [N2,1,1,0][TfO] and [N4,1,1,0][HSO4]) were performed on the I311 beamline at MAX-lab (Sweden). The apparatus used at MAX-lab is explained in ref. 68.
The angle between the polarisation vector of the incoming X-rays and the analyser was different for the experimental set-ups used here (see ESI Table S1† for the angle each sample was run at). However, this angle is unimportant for valence RXPS, and will only affect the relative intensity of peaks in valence NRXPS.
2.4 Producing RXPS traces
Valence RXPS traces were produced using the subtraction procedure outlined in ref. 68. Briefly, traces of valence RXPS plus valence NRXPS had the valence NRXPS contributions subtracted. In some cases, this subtraction method gave negative features, due to either complications due to 2nd order light (e.g.Fig. 8c) or RXPS contributions being much lower intensity than the NRXPS contributions (e.g.Fig. 6d and 7e). The latter scenario causes a problem when a small percentage increase in the NRXPS contribution at the absorption edge (i.e. the hν at which the RXPS contribution occurs) relative to the hν at which only NRXPS contribution occurs that is used for subtraction, leading to negative peaks in the subtracted RXPS trace.2.5 Charge referencing methods for XP spectra
The valence NRXPS features were at constant EB with respect to hν for all experiments presented here, see Fig. 3a and ESI Fig. S2–S20.† Therefore, any changes in EB for resonant features relative to non-resonant features were not due to sample charging.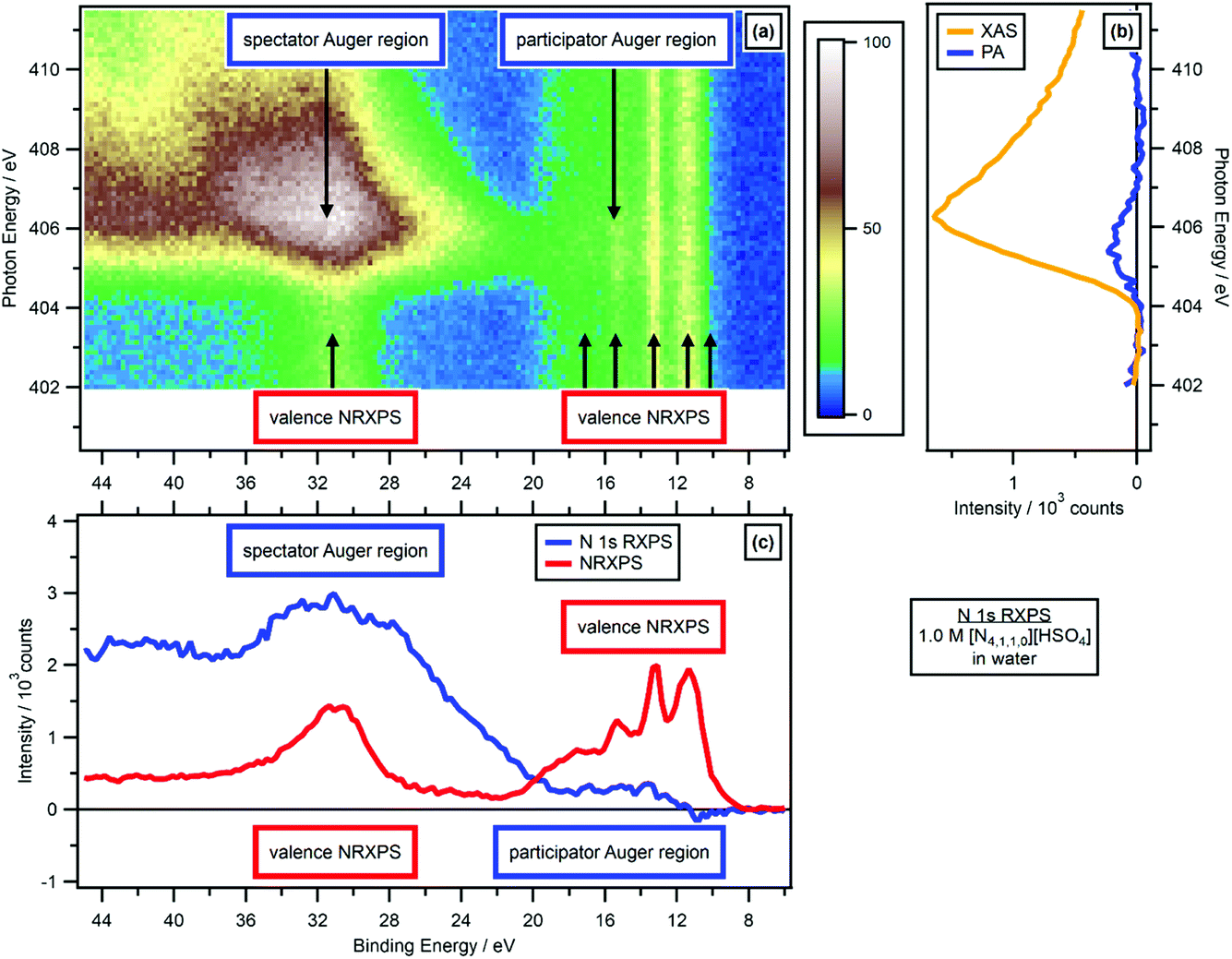 | ||
| Fig. 3 N 1s valence RXPS for 1.0 M [N4,1,1,0][HSO4] in water. (a) Heat map of photon energy against binding energy for the N 1s valence RXPS. (b) Partial electron yield N 1s NEXAFS spectra (spectator Auger region plus participator Auger region = XAS, and participator Auger region only). (c) Valence NRXPS below the N 1s absorption edge (hν = 403.0 eV) and N 1s valence RXPS (hν = 405.6 eV). The valence RXPS trace was produced using the procedure outlined in ref. 68. All electron spectra were charge referenced using procedures outlined in Section 2.5. | ||
For the 11 aqueous solutions, all RXPS spectra were charge referenced to EB(1b1) = 11.33 eV by fitting the valence NRXPS83 measured at hν just below the relevant absorption edge. No charge referencing was applied to RXPS for neat MeCN, neat EtOH and neat PC, given no literature EB values could be found for valence NRXPS. For 0.5 M [C4C1Im][OAc] in MeCN, the O 1s RXPS was charge referenced to the O 1s RXPS spectrum for 1.3 M [C4C1Im][OAc] in water to allow for visual comparisons. All IL spectra were effectively charge referenced to EB(Calkyl 1s) = 289.58 eV.84 Further small EB shifts were then applied to allow visual comparisons to the RXPS measured in molecular solvents.
2.6 Calculation methods
DFT calculations were carried out at the B3LYP-D3(BJ)/6-311+G(d,p) level; further details can be found in ESI Section 2.† Structures were optimised as individual, isolated molecules/ions. A generalised solvation model has been employed (SMD), and the parameters employed are reported in ESI Table S2.† Coordinates for the molecules/ions studied are given in ESI Section 10.†3. Results
3.1 Separating solute and solvent contributions to OVSs
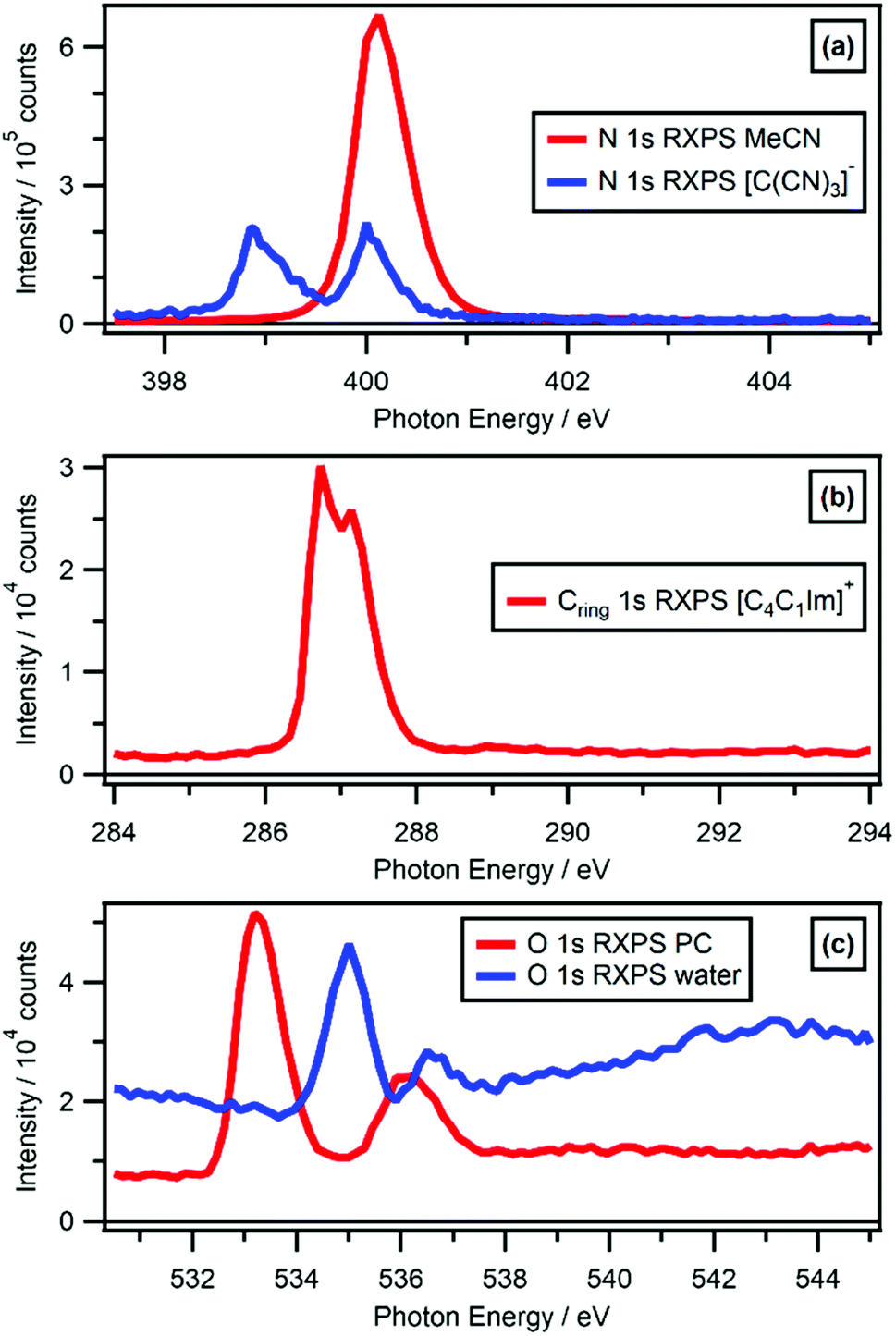
O 1s RXPS for solutes is a particular challenge, given oxygen features in many common solvents, especially those with the properties required for successful liquid jet XPS experiments, e.g. solvents with large relative permittivity. O 1s RXPS for [OAc]− in water worked very well, the O 1s → π* X-ray absorption energy for [OAc]− (hν ∼ 533 eV) was significantly lower than the O 1s → σ* X-ray absorption energies for water (hν > 534 eV). Therefore, the PA contributions for [OAc]− could be separated from the PA contributions for water. RXPS maps and traces for a number of key solvents (water, ethanol, propylene carbonate, acetonitrile) are given in ESI Section 4.† This data is a useful resource, especially for determining whether solute contributions can be distinguished from solvent contributions using RXPS.
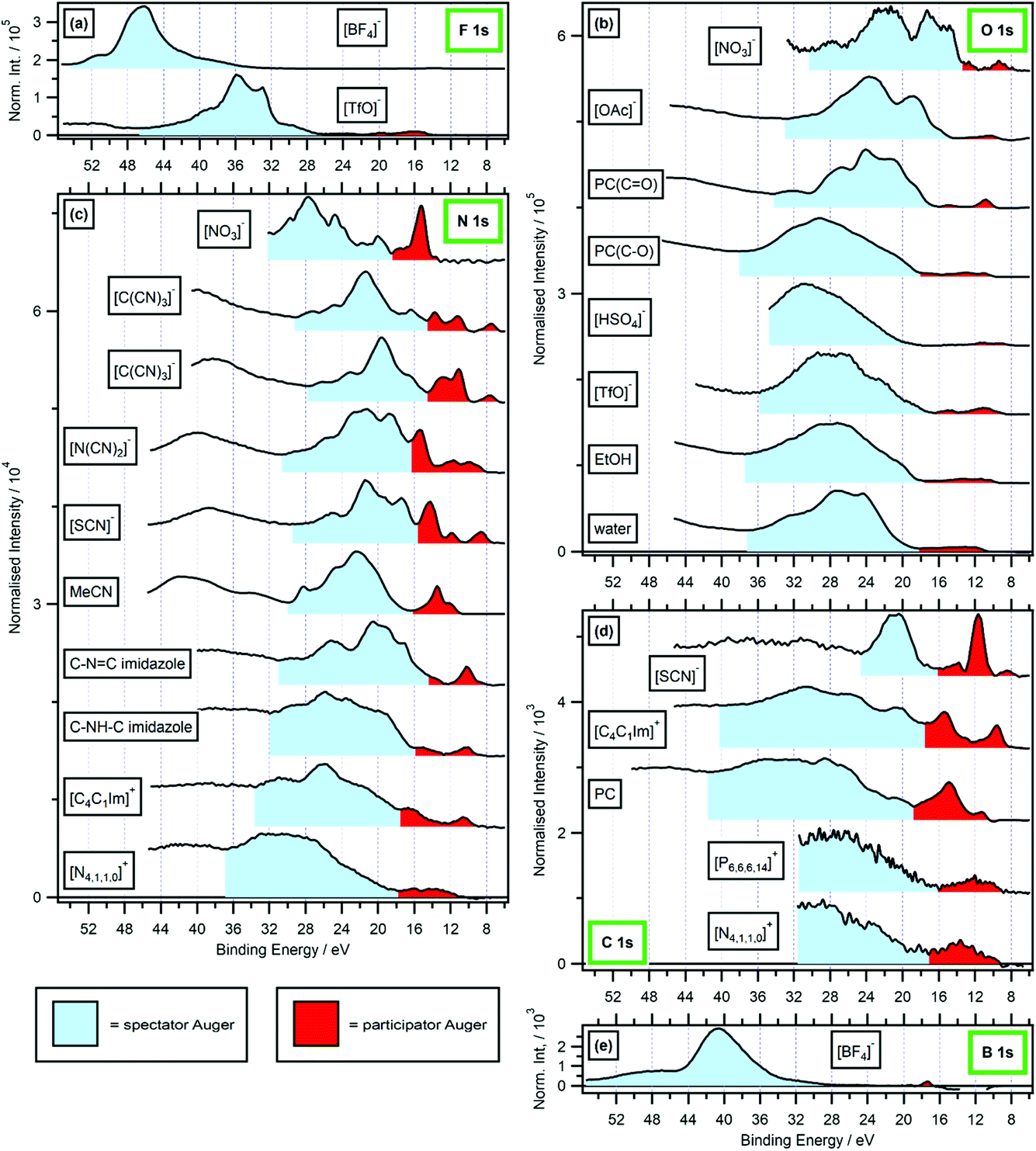 | ||
| Fig. 4 Valence RXPS at different absorption edges. Shaded regions are shown for estimated spectator Auger (SA, in light blue) and participator Auger (PA, in red) transition contributions; backgrounds have not been included, so the SA region in particular will give an overestimate of the true SA area. (a) F 1s valence RXPS for 0.5 M [C4C1Im][TfO] in water (hν = 690.6 eV) and 0.5 M [C4C1Im][BF4] in water (hν = 696.4 eV). (b) O 1s valence RXPS for [P6,6,6,14][NO3] (hν = 531.9 eV), 1.3 M [C4C1Im][OAc] in water (hν = 532.7 eV), propylene carbonate (PC, hν = 533.3 eV for CO absorption and hν = 536.2 eV for C–O absorption), [N4,1,1,0][HSO4] (hν = 536.8 eV), [N2,2,1,0][TfO] (hν = 535.3 eV), ethanol (EtOH, (hν = 535.4 eV)) and water (hν = 535.0 eV). (c) N 1s valence RXPS for [P6,6,6,14][NO3] (hν = 405.2 eV), [C8C1Im][C(CN)3] (hν = 399.0 eV and hν = 400.0 eV), 0.5 M [C4C1Im][N(CN)2] in water (hν = 400.1 eV), 0.5 M [C4C1Im][SCN] in water (hν = 399.8 eV), acetonitrile (MeCN, (hν = 400.1 eV)), 1.8 M imidazole in water (hν = 400.2 eV for C–NC absorption and hν = 401.9 eV for C–NH–C absorption), 0.5 M [C4C1Im]I in water (hν = 402.1 eV) and 3.3 M [N4,1,1,0][HSO4] in water (hν = 405.6 eV). (d) C 1s valence RXPS for 0.5 M K[SCN] in water (hν = 287.5 eV), 0.5 M [C4C1Im]I in water (hν = 287.0 eV), propylene carbonate (PC, hν = 290.5 eV), [P6,6,6,14][NO3] (hν = 287.6 eV) and [N4,1,1,0][HSO4] (hν = 288.5 eV). (e) B 1s valence RXPS for 0.5 M [C4C1Im][BF4] in water (hν = 201.4 eV). The valence NRXPS contributions were subtracted using the procedure outlined in ref. 68. All valence XP spectra were charge referenced using procedures outlined in Section 2.5. The upper EB limit for the participator Auger region was chosen based on a combination of the relative intensity compared with other features from participator/spectator transitions and variation in peak EB with respect to hν. The upper EB limit for the spectator Auger region was chosen based on the Auger dip (where measured, otherwise the upper limit of the measurement); the lower EB limit for the spectator Auger region was the same as the upper limit for the participator Auger region. | ||
For the anions [BF4]− and [TfO]− the F 1s RAES/RXPS spectra gave very low intensity features due to PA transitions at 24 eV > EB > 0 eV (Fig. 4a), giving very small PA:SA intensity ratios. For the anion [SCN]−, three cations and PC, the C 1s RAES/RXPS spectra gave much larger PA:SA intensity ratios than observed for F 1s (Fig. 4d and a). The PA:SA intensity ratios for O 1s RAES/RXPS spectra (Fig. 4b) and N 1s RAES/RXPS spectra (Fig. 4c) were, on average across all samples, between the two extremes of F 1s RAES/RXPS and C 1s RAES/RXPS. The correlation between the element electronegativity and PA:SA intensity ratio holds for B 1s RAES/RXPS for the solids BN and B2O3, where large PA:SA intensity ratios were observed.43–45 However, for B 1s RAES/RXPS for [BF4]− the PA:SA intensity ratio was very small indeed (Fig. 4e). It is expected that [BF4]− is a special case, as the difference in electronegativity is so large between B and F.85 Boron had little localised electron density/OVSs, leading to the observed weak PA signal; this observation is backed up by the boron pDoS for [BF4]− (Fig. 7e), demonstrating that the bonding in the individual ion/molecule needs to be taken into account when considering the PA:SA intensity ratio and electronegativity. For most samples, the PA:SA intensity ratios follow the order from largest to smallest: B 1s > C 1s > N 1s > O 1s > F 1s. This trend matches the element electronegativity,85 as noted for four gas phase small organic molecules.31 Overall, electronegativity gives a good guide to the PA:SA intensity ratio, at least for the period 2 elements from B to F.
When comparisons are made for different samples focusing on the same element edge (i.e. carbon versus carbon, nitrogen versus nitrogen and oxygen versus oxygen), the PA:SA intensity ratio was larger for samples with π* valence states than samples with σ* valence states (Fig. 4b–d). However, the PA:SA intensity ratio for [N4,1,1,0]+ N 1s → σ* RAES/RXPS (Fig. 4c) was significantly larger than the PA:SA intensity ratio for PC(CO) O 1s → π* RAES/RXPS (Fig. 4b). This finding demonstrates that the effect on the PA:SA intensity ratio of the bonding type was not nearly so large as the effect of the electronegativity.
The overall RXPS enhancement is captured with the RXPS:NRXPS intensity ratio for any EB region (Fig. 5, 6b and ESI Table S4†). For the data in Fig. 5 and 6b, the NRXPS intensity can be determined from the off-resonant signal below the absorption edge; the RXPS intensity can be determined from the on-resonant signal. By far the largest RXPS:NRXPS intensity ratios were observed for N 1s RXPS for MeCN (Fig. 5a and ESI Fig. S6†), e.g. for the HOVS of MeCN the RXPS:NRXPS intensity ratio was ∼100 (ESI Table S4†). Cring 1s RXPS for [C4C1Im]+ (i.e. the three carbon atoms in the imidazolium ring) and N 1s RXPS for [C(CN)3]− both gave RXPS:NRXPS intensity ratios of ∼20 (Fig. 5a, b and ESI Table S4†). All other RXPS:NRXPS intensity ratios were below 10 (ESI Table S4†). In particular, the RXPS:NRXPS intensity ratios were in the range of 1 to 2 for functional groups with little or no π* bonding and only σ* bonding, e.g. O 1s RXPS for water gave a RXPS:NRXPS intensity ratio of ∼2 (Fig. 5c and ESI Table S4†).
 | ||
| Fig. 5 Partial electron yield NEXAFS spectra. (a) N 1s edge for acetonitrile (MeCN, 12.6 eV > EB > 11.2 eV) and 0.5 M [C4C1Im][SCN] in water (9.4 eV > EB > 7.4 eV). (b) C 1s edge for 0.5 M [C4C1Im]I in water (10.0 eV > EB > 9.2 eV). (c) O 1s edge for propylene carbonate (PC, 11.4 eV > EB > 10.0 eV) and water (12.4 eV > EB > 10.8 eV). | ||
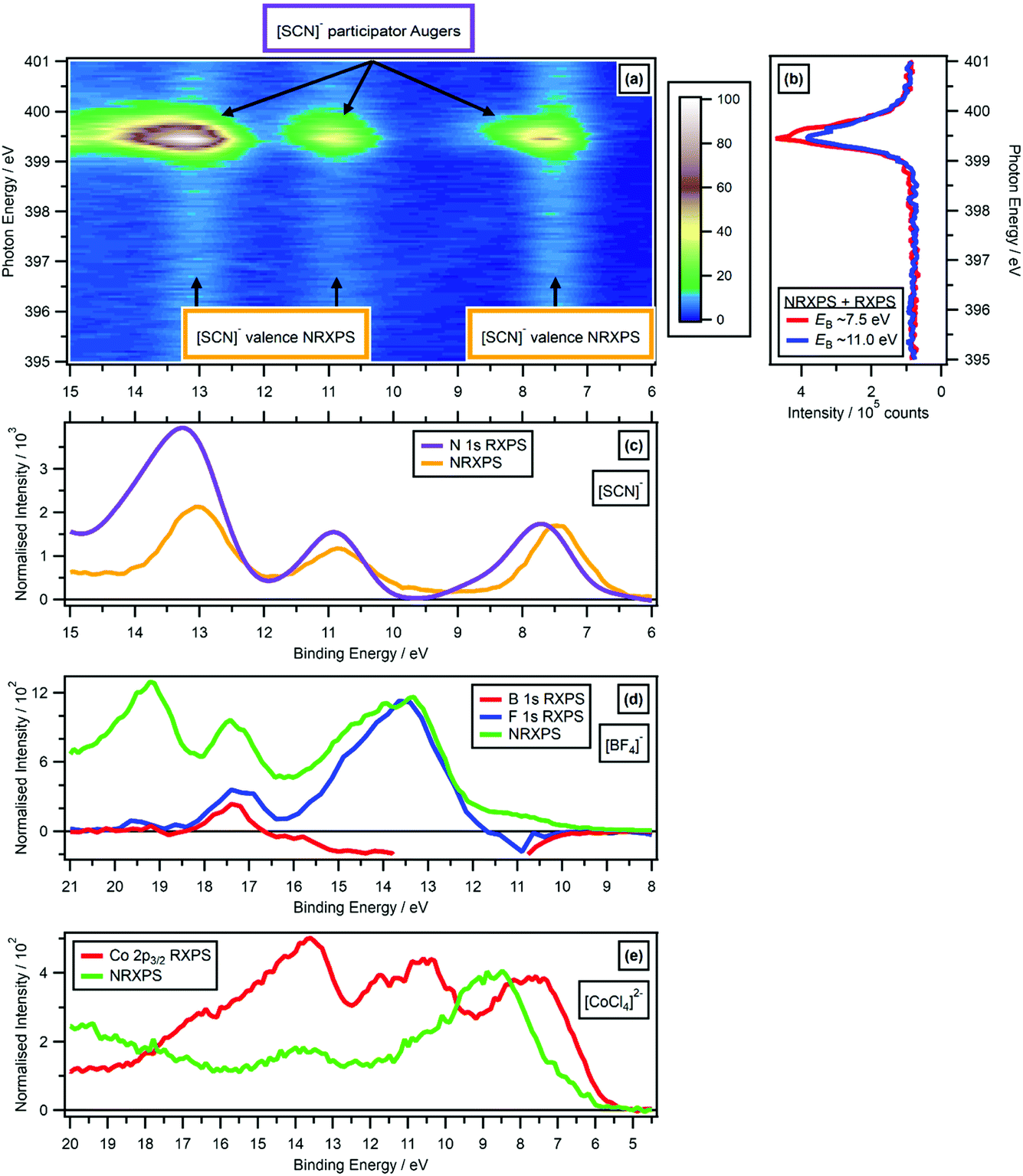 | ||
| Fig. 6 Comparing the valence RXPS and valence NRXPS for the anion N 1s → π* absorption for [C4C1Im][SCN]: (a) N 1s valence RXPS heat map for [C4C1Im][SCN]; (b) partial electron yield N 1s NEXAFS spectra for two regions (EB ∼ 7.5 eV and EB ∼ 11.0 eV). (c) N 1s valence RXPS for [C4C1Im][SCN] (hν = 399.5 eV), compared to the valence NRXPS for [C4C1Im][SCN] (hν = 1486.6 eV). The [C4C1Im]+ contribution to the valence NRXPS was negligible at hν = 1486.6 eV.68,69 (d) B 1s valence RXPS (hν = 201.4 eV) and F 1s valence RXPS (hν = 696.4 eV) for 0.5 M [C4C1Im][BF4] in water, compared to the valence NRXPS for [C4C1Im][BF4] (hν = 1486.6 eV). (e) Co 2p3/2 valence RXPS for [C8C1Im]2[CoCl4] (hν = 774.9 eV), compared to the valence NRXPS for [C8C1Im]2[CoCl4] (hν = 773.0 eV, intensity multiplied by 4). The valence NRXPS contributions to valence RXPS were subtracted using the procedure outlined in ref. 68. All electron spectra were charge referenced using procedures outlined in Section 2.5. | ||
Overall, the RXPS:NRXPS intensity ratios did not match to the PA:SA intensity ratios. For example, for MeCN N 1s RXPS the RXPS:NRXPS intensity ratio was the largest value by far, but for MeCN N 1s RXPS the PA:SA intensity ratio was less than a number of other samples, e.g. [SCN]− C 1s RXPS. Therefore, it appears that the RXPS:NRXPS intensity ratio was determined by a combination of the NEXAFS absorption cross-section and the competition between PA and SA transitions. However, it is difficult to draw wide-ranging conclusions given the dataset of RXPS:NRXPS intensity ratios available; especially problematic is the sparse data focused on functional groups with only σ* bonding (and only those at the F 1s or O 1s edges, where the PA:SA intensity ratios are very small).
3.2 Demonstrating the one-electron picture holds for valence RXPS
For [C(CN)3]−, two distinct N 1s(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) → π* absorptions were observed at hν = 399.0 eV and hν = 400.0 eV, both of which originate from the same core state, as the three nitrogen atoms in [C(CN)3]− are equivalent (see ref. 86 for more details). The two N 1s RXPS traces for [C(CN)3]− were very similar (Fig. 4c); furthermore, the SA features were broadly similar in terms of feature shape (Fig. 4c). Unsurprisingly, the OVSs that contributed to the two N 1s RXPS traces were essentially the same. For the two N 1s RXPS contributions at different hν for [C(CN)3]−, the feature at lowest EB from the HOVS was different by EB ∼ 0.15 eV, most likely due to subtle differences in final state vibrational contributions for the two PA transitions. Furthermore, for [C(CN)3]− mismatches were observed between the NRXPS and N 1s RXPS features for the HOVS; EB ∼ 0.2 eV for hν = 399.0 eV and EB ∼ 0.35 eV for hν = 400.0 eV (ESI Fig. S8e†). Again, these differences must be due to final state vibrational contributions in the PA transitions not present in the valence NRXPS.
N) → π* absorptions were observed at hν = 399.0 eV and hν = 400.0 eV, both of which originate from the same core state, as the three nitrogen atoms in [C(CN)3]− are equivalent (see ref. 86 for more details). The two N 1s RXPS traces for [C(CN)3]− were very similar (Fig. 4c); furthermore, the SA features were broadly similar in terms of feature shape (Fig. 4c). Unsurprisingly, the OVSs that contributed to the two N 1s RXPS traces were essentially the same. For the two N 1s RXPS contributions at different hν for [C(CN)3]−, the feature at lowest EB from the HOVS was different by EB ∼ 0.15 eV, most likely due to subtle differences in final state vibrational contributions for the two PA transitions. Furthermore, for [C(CN)3]− mismatches were observed between the NRXPS and N 1s RXPS features for the HOVS; EB ∼ 0.2 eV for hν = 399.0 eV and EB ∼ 0.35 eV for hν = 400.0 eV (ESI Fig. S8e†). Again, these differences must be due to final state vibrational contributions in the PA transitions not present in the valence NRXPS.
For 0.5 M [C4C1Im][BF4] in water, signals from valence NRXPS for the solute were dwarfed by signal from the solvent (ESI Fig. S11†). Therefore, comparisons are made between the valence NRXPS for neat IL [C4C1Im][BF4], which were dominated by anion contributions (in this case, F 2p contributions) due to photoionisation cross-section effects.69,87 These comparisons show a very good visual match at 18 eV > EB > 11 eV between the valence NRXPS for neat IL [C4C1Im][BF4] and both B 1s RXPS and F 1s RXPS for [BF4]− (Fig. 6d).
For the IL [C8C1Im]2[CoCl4], valence NRXPS and Co 2p3/2 RXPS showed significant differences in both EB for the main features and the spectral shapes (Fig. 6e). However, there were far more features observed in Co 2p3/2 RXPS than in valence NRXPS, particularly at 18 eV > EB > 12 eV. The kinetic energy EK was ∼764 eV for the Co L3M4,5L4,5 Auger transition from NRXPS,69 which gives an apparent EB ∼ 11 eV at hν = 775 eV, meaning that features due to SA transitions would be expected in the same EB region that the resonant features were detected for [CoCl4]2−. However, all of the resonant features remained at approximately constant EB when hν was increased, strongly suggesting that PA transitions dominated over SA transitions. It is possible that for the RXPS features at 12 eV > EB > 6 eV the one-electron picture held, but determining that is beyond the scope of this article. However, for the features at 18 eV > EB > 12 eV the one-electron picture certainly did not hold.
These comparisons for closed shell ions all demonstrate that the one-electron picture broadly held for valence RXPS. The lack of matching in terms of EB for the NRXPS and RXPS features, strongly indicates that the one-electron picture failed for open shell complexes such as [CoCl4]2−.
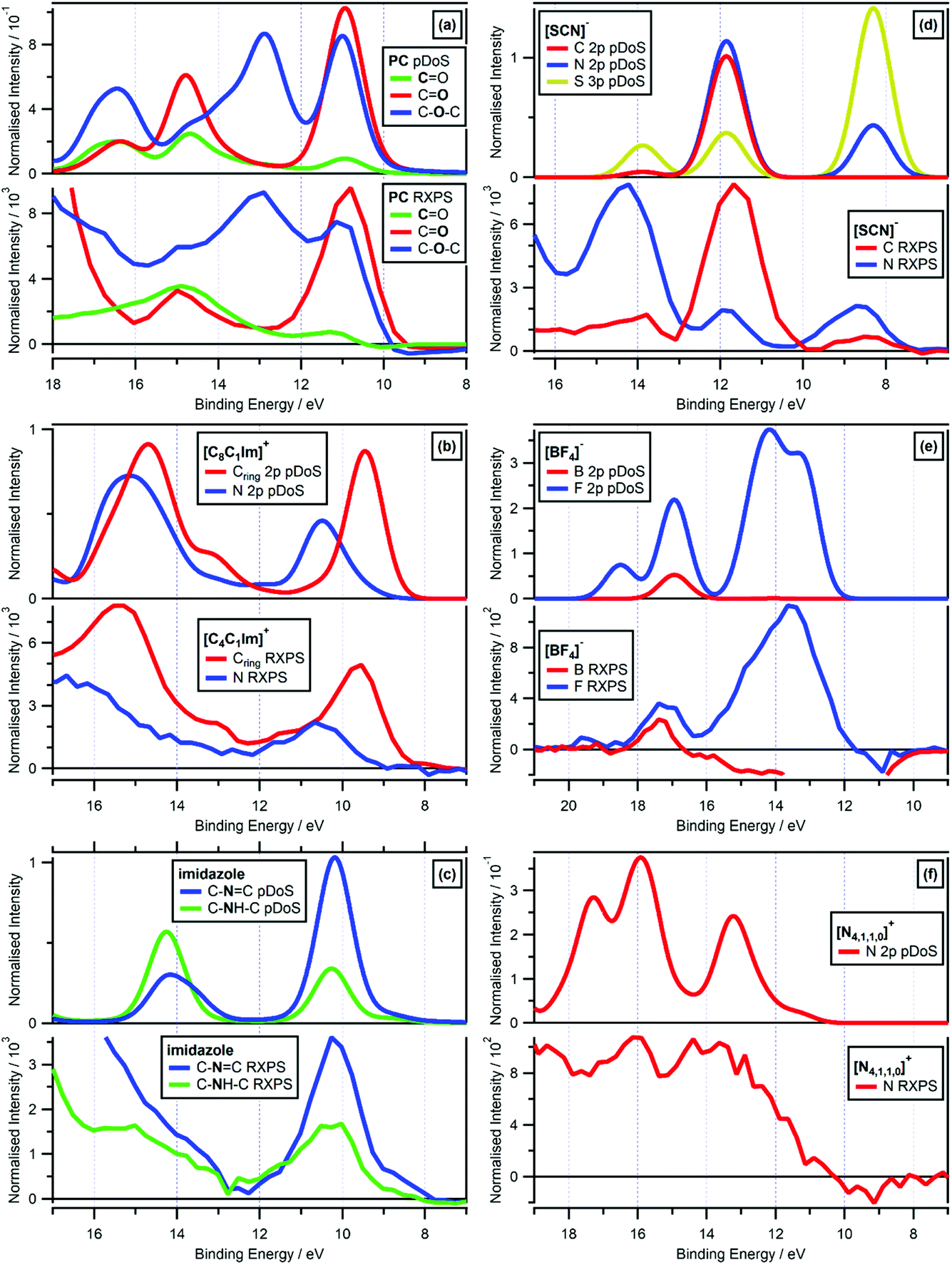 | ||
| Fig. 7 Valence RXPS at different absorption edges compared to molecule/lone ion SMD pDoS calculations. (a) O 1s valence RXPS traces for propylene carbonate (PC) at both the CO and C–O–C absorption edges (hν = 533.3 eV and hν = 536.2 eV respectively), and C 1s valence RXPS trace for propylene carbonate (PC) at the CO absorption edge (hν = 290.5 eV), compared to CO O pDoS, C–O–C O pDoS and CO C pDoS calculated for PC in an SMD. (b) N 1s valence RXPS (hν = 402.1 eV) and C 1s valence RXPS (hν = 287.0 eV) traces for 0.5 M [C4C1Im]I in water, compared to Cring 2p (i.e. the three carbon atoms in the imidazolium ring) and N 2p pDoS calculated for lone [C8C1Im]+ in an SMD. (c) N 1s valence RXPS traces for 1.8 M imidazole in water at both the C–NC and C–NH–C absorption edges (hν = 400.2 eV and hν = 401.9 eV respectively), compared to C–NC and C–NH–C N pDoS calculated for imidazole in an SMD. (d) N 1s valence RXPS trace for 0.5 M [C4C1Im][SCN] in water (hν = 399.8 eV) and C 1s valence RXPS trace for 0.5 M K[SCN] in water (hν = 287.5 eV), compared to C 2p, N 2p and S 3p pDoS calculated for lone [SCN]− in an SMD. (e) B 1s valence RXPS (hν = 201.4 eV) and F 1s valence RXPS (hν = 696.4 eV) traces for 0.5 M [C4C1Im][BF4] in water, compared to B 2p and F 2p pDoS calculated for lone [BF4]− in an SMD. (f) N 1s valence RXPS trace for 3.3 M [N4,1,1,0][HSO4] in water (hν = 405.6 eV), compared to N 2p pDoS calculated for lone [N4,1,1,0]+ in an SMD. The valence NRXPS contributions were subtracted using the procedure outlined in ref. 68. All electron spectra were charge referenced using procedures outlined in Section 2.5. | ||
The most impressive matches of RXPS traces and pDoS was for samples for which more than one RXPS trace was recorded, i.e. PC, [C4C1Im]+, imidazole, [SCN]−, [BF4]− and [NO3]− (Fig. 7 and ESI Section 9†). The visual match for PC was remarkable, as a comparison we showed that the pDoS matched the very small (but reliable, given they are measured in the same O 1s experiment with no charging effects observed) shift in EB for the lowest EB features for CO and C–O–C (Fig. 7a). Furthermore, the match of the EB peak separation for the features at lowest EB for carbon and nitrogen for [C4C1Im]+ was also very noteworthy; from both experimental RXPS and pDoS the EB peak separation was ∼1.0 eV (Fig. 7b), further emphasising the excellent match of the cheap, simple calculations and experimental RXPS. Lastly, for [BF4]− F 1s RXPS and F 2p pDoS gave excellent visual matches for the two main features at EB ∼ 17 eV and EB ∼ 13 eV (Fig. 7e) and [BF4]− B 1s RXPS and B 2p pDoS gave an excellent visual match at EB ∼ 17 eV (Fig. 7e).
For three different organic cations ([N4,1,1,0]+, [N2,2,1,0]+ and [P6,6,6,14]+) and both water and ethanol, which all contain only σ-bonding, there were reasonable matches of valence RXPS and pDoS calculations (see ESI Section 9†), but far less satisfactory than the other systems explained previously here. For all five of these ions/molecules, the RXPS traces showed a broad feature with no clear peaks, unlike all of the ion/molecules with π bonding (and [BF4]− and [TfO]− too) which showed clear, readily identifiable peaks due to PA transitions. Furthermore, for all five of these ions/molecules the relevant pDoS showed clear, readily identifiable peaks; hence, the matches were reasonable but clearly poorer than other systems studied here. For example, N 1s RXPS for [N4,1,1,0]+ gave a broader feature than the N 2p pDoS (Fig. 7f), agreeing with observations for a perovskite with an organic, protic ammonium cation.60 The small RXPS:NRXPS intensity ratios are probably not the explanation, otherwise all ions/molecules that contain only σ-bonding would show the same reasonable matches. All five of the ions/molecules that gave reasonable matches all had hydrogen atoms covalently bonded to the atoms studied using RXPS, e.g. one N–H bond for [N4,1,1,0]+. Therefore, final state vibrational contributions were likely a significant contribution to the broad experimental RXPS traces. Furthermore, it is possible that a range of ground state liquid phase environments not captured by the single calculated conformer (i.e. conformational flexibility and/or hydrogen bonding) also contributed for some ions/molecules. To untangle this problem further, RXPS calculations including a core-hole may be required, which are very challenging for condensed systems, particularly for solutes; these types of calculations have only been achieved for a small number of systems, with a focus on gas phase molecules with few atoms (e.g. formic acid,31 HNCO,88 CH4,89 and pyrimidine89).
[SCN]− N 1s RXPS and N 2p pDoS gave excellent visual matches for the three main features at lowest EB (Fig. 7d). Furthermore, [SCN]− C 1s RXPS and C 2p pDoS also gave excellent visual matches for the three main features at lowest EB (Fig. 7d). One small difference is the feature at EB ∼ 8 eV, which has an experimental C 1s RXPS contribution but not matching C 2p pDoS contribution, also found by other calculations.90,91 This mismatch was most likely due to subtle differences of experiments and calculations, i.e. the HOMO and HOMO−1 for [SCN]− have no C 2p contribution but significant N 2p and S 3p contributions, and it is these N 2p and S 3p contributions that most likely gave rise to the feature in the experimental C 1s RXPS data for [SCN]−. Essentially, how the density is assigned in pDoS calculations and the selection rules for RXPS do not quite match, which is not a surprising observation; furthermore, given the lack of success establishing selection rules for valence RXPS31,52,55 and the difficulty performing RXPS calculations31,88,89 solving this mismatch will not be straightforward.
Overall, these excellent matches of experimental valence RXPS and pDoS demonstrate that the one-electron picture of valence RXPS holds for these closed shell molecules/ions. Moreover, these excellent matches support the use of RXPS to identify atomic contributions to OVSs.
3.3 Insights into bonding
O) matched well in terms of EB to the O 1s NRXPS traces for CO and C–O–C; all three RXPS traces gave features at EB ∼ 11 eV and 15 eV (Fig. 7a). All these excellent matches strongly demonstrate that the one-electron picture held for these systems.
For both [C4C1Im]+ and imidazole (which both have rings in their structure), the same element gave two core state → π* NEXAFS absorptions, C 1s for [C4C1Im]+ and N 1s for imidazole (ESI Fig. S14b and S8b†). For both [C4C1Im]+ and imidazole, the two absorptions were caused by two different core states,92–95 and involved absorption to the same π* previously unoccupied valence state (see ESI Section 8† for more details). For both [C4C1Im]+ and imidazole, the RXPS traces recorded for the two core state → π* NEXAFS absorptions gave excellent matches of features/peaks and were essentially identical (ESI Fig. S14d† and 7c respectively), demonstrating that the same OVSs contributed to both RXPS traces. It can be concluded that the same OVSs had similarly good overlap with different core-holes in the ring structures, pointing towards delocalisation of the valence states over the ring structures.
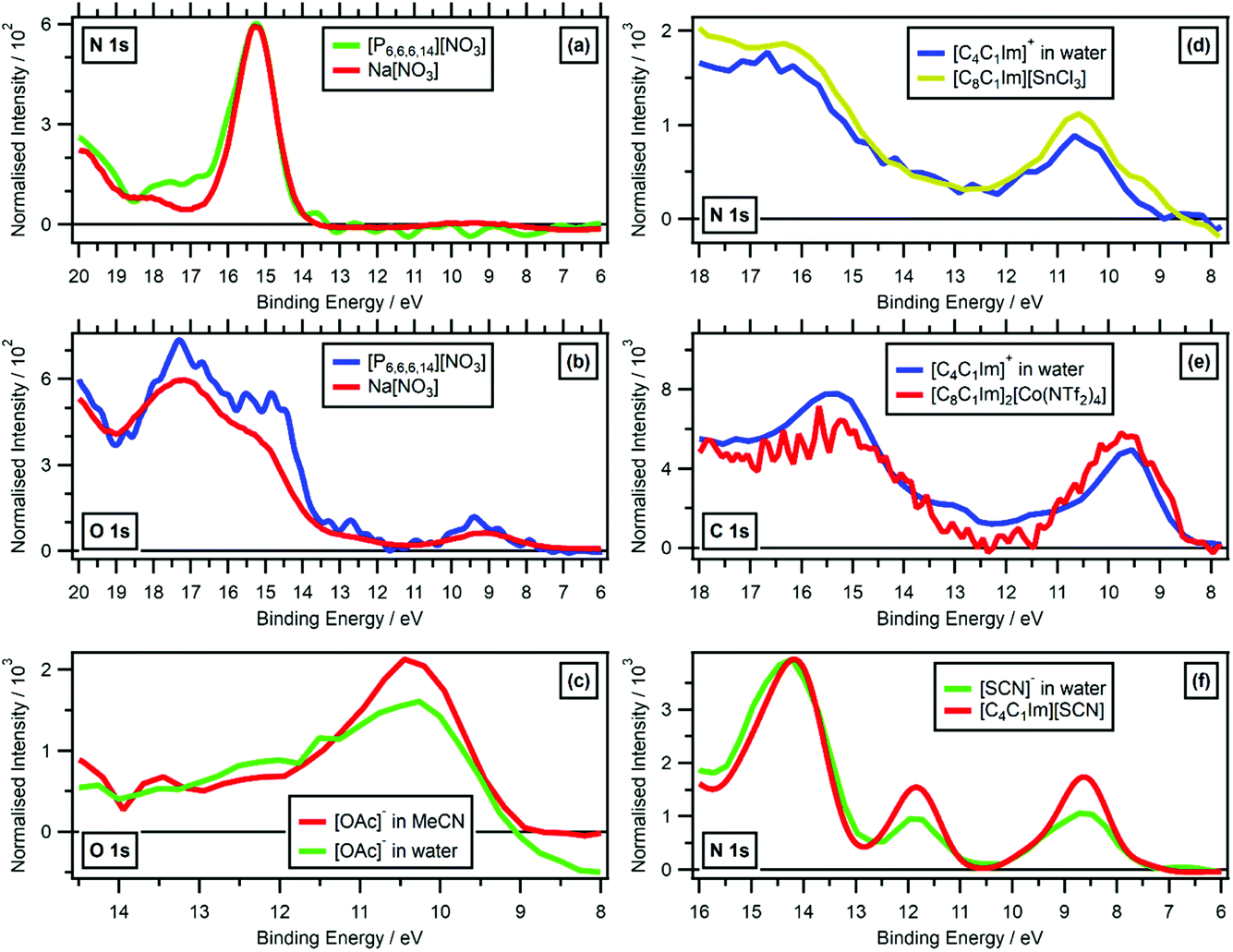 | ||
| Fig. 8 Valence RXPS at different absorption edges. (a) N 1s valence RXPS traces for solid Na[NO3] (hν = 405.0 eV, data from ref. 41) and [P6,6,6,14][NO3] (hν = 405.0 eV). (b) O 1s valence RXPS traces for solid Na[NO3] (hν = 532.0 eV, data from ref. 41) and [P6,6,6,14][NO3] (hν = 405.2 eV). (c) O 1s valence RXPS traces for 1.3 M [C4C1Im][OAc] in water (hν = 532.7 eV) and 0.5 M [C4C1Im][OAc] in MeCN (hν = 532.7 eV). (d) N 1s valence RXPS traces for 0.5 M [C4C1Im]I in water (hν = 402.1 eV) and [C8C1Im][SnCl3] (hν = 401.8 eV, data taken from ref. 69). (e) C 1s valence RXPS traces for 0.5 M [C4C1Im]I in water (hν = 287.0 eV) and [C8C1Im]2[Co(NTf2)4] (hν = 285.5 eV, data taken from ref. 69). (f) N 1s valence RXPS traces for 0.5 M [C4C1Im][SCN] in water (hν = 399.8 eV) and [C4C1Im][SCN] (hν = 399.5 eV). The valence NRXPS contributions were subtracted using the procedure outlined in ref. 68. All electron spectra were charge referenced using procedures outlined in Section 2.5. | ||
While varying solvation environments appear to have little effect on RXPS, small changes in the local covalent environment have been found to lead to significant differences in RXPS for a small number of organic molecules in the gas phase.31 The four ions/molecules studied here that contain at least one cyano functional group ([SCN]−, [N(CN)2]−, [C(CN)3]− and MeCN), the N 1s RXPS spectra were very different (Fig. 4c), matching the literature findings. Other examples where the same/similar functional group in two different samples gave very different RXPS spectra include: N 1s RXPS for [CnC1Im]+versus imidazole (Fig. 4c), O 1s RXPS for ethanol versus water (Fig. 4b), O 1s RXPS for PC(CO) versus [OAc]− (Fig. 4b) and O 1s RXPS for [HSO4]−versus [TfO]− (Fig. 4b). These findings match literature examples of small gas phase organic molecules.31
Broadly, it has been shown that changing the covalent, intermolecular bonding of an ion/molecule had a significant impact on the RXPS, but changing the intramolecular solvation environment had far less of an impact on the RXPS. In ref. 31, the extreme sensitivity of RXPS to changes in intermolecular covalent bonding was seen as a downside, as the technique could not be used for structural analysis of functional groups. However, the RXPS spectra published here are the beginnings of a dataset of fingerprint RXPS spectra of commonly studied ions and molecules.
4. Conclusions and future work
RXPS can be used to identify atomic contributions to OVSs. This approach is particularly valuable for complicated systems, e.g. solutes at low concentration. Closed shell ions/molecules with π bonding are particularly suited to RXPS studies, both in terms of giving high intensity RXPS features, but also giving well-defined RXPS features too.Very importantly, all evidence shows that the one-electron picture holds for valence RXPS for closed-shell ions/molecules. Valence RXPS data matches well in terms of EB to valence NRXPS, with the small differences that sometimes appear due to final state vibrational contributions. This finding demonstrates that the one-electron picture of RXPS holds for closed shell ions/molecules. However, for [CoCl4]2− the valence RXPS and valence NRXPS were very different, demonstrating that the one-electron picture of RXPS does not hold for open shell ions/molecules for all PA transitions, although the one-electron picture may hold for certain PA transitions. RXPS matched very well with pDoS for a wide range of samples and element edges. Therefore, RXPS data provides an excellent benchmark for validation of ground state calculations of OVSs. This finding reinforces that the one-electron picture of RXPS holds for closed shell ions/molecules. Furthermore, insight can be gained into bonding by comparisons of RXPS and pDoS.
Our experimental set-ups are well-suited to measuring small signals, e.g. PA transitions for fluorine, due to the constantly refreshed sample environment, by liquid jet or rastering respectively. Significantly, a measurable PA signal was observed for every molecule/ion studied here. For example, for F-containing anions, a F 1s PA signal was observed, whereas no F 1s PA signal was observed for solid CaF2.36,38 As far as we are aware, this represents the first measurement of an F 1s PA transition. Furthermore, the F 1s PA signal for both [BF4]− and [TfO]− matched very well to calculated pDoS data.
Two guidelines for ions/molecules to give large RXPS signal are: (i) the element studied having low electronegativity, (ii) π bonding involving the element studied. For example, F 1s valence RXPS for [TfO]− gave a small RXPS signal, even though the SO3 functional group contains π bonding, because the CF3 functional group contains only σ bonding. Therefore, O 1s valence RXPS of water as a solute will be challenging given the small and broad RXPS signal; conversely, water is a good solvent for studying O 1s valence RXPS of solutes dissolved in water. Overall, to obtain high quality data for RXPS for solutes with the same element as the solvent, the ideal is solutes that give large RXPS signal and solvents that give small RXPS.
An area of RXPS studies that has potential is studying functional groups of ions/molecules that contain only σ bonding. RXPS measurements of e.g. alkylamines and alkanes to determine their PA:SA intensity ratios would be instructive, to determine the relative importance of low electronegativity elements versus the bonding type. Furthermore RXPS for functional groups which only have σ bonding, e.g. tetraalkylammonium, alkanes, ethers, to determine the relative importance of covalent bonds to hydrogen for RXPS.
RXPS traces were used to demonstrate the strong impact of covalent bonding on RXPS, but the weak impact of non-covalent intermolecular interactions on RXPS. The dataset of fingerprint RXPS spectra of commonly studied ions and molecules presented here will prove very useful for understanding interactions of these ions and molecules with surfaces. Many simple molecules have been studied as adsorbates on surfaces for many years,96 along with larger adsorbates,62 but most of the complex ions presented here in particular are yet to be studied as adsorbates using RXPS.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KRJL acknowledges support from a Royal Society University Research Fellowship (URF\R\150353). JMS acknowledges support from a Royal Society University Research Fellowship Enhancement Award (RGF\EA\180089). EG acknowledges support from a Royal Society Research Grant for Research Fellows (RGF\R1\180053). AL acknowledges part funding for a studentship from Johnson Matthey plc. DH, GW, WQ, and RS thank the Deutsche Forschungsgemeinschaft (German Research Foundation, DFG) for support through an Emmy-Noether grant (SE 2253/3-1). MAX-lab are thanked for awarding beamtime on I311. Measurements were carried out at the SOL3PES instrument (beamline U49-2_PGM-1) at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin für Materialien und Energie. We acknowledge Diamond Light Source for time on Beamline B07 (VERSOX) under Proposals SI24304 and SI25929. The research leading to this result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. Bernd Winter is acknowledged for inspirational support and advice. Matthijs Van Spronsen is acknowledged for beamtime support on Proposal SI25929. The authors are grateful to Jacek Osiecki and Karsten Handrup (MAX-lab, Sweden) for assistance with experiments.References
- I. Fleming, Molecular Orbitals and Organic Chemical Reactions, Wiley, Chichester, 2010 Search PubMed.
- K. Fukui, Angew. Chem., Int. Ed. Engl., 1982, 21, 801–809 CrossRef.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
- R. G. Pearson, J. Org. Chem., 1989, 54, 1423–1430 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- H. D. Lim, B. Lee, Y. Zheng, J. Hong, J. Kim, H. Gwon, Y. Ko, M. Lee, K. Cho and K. Kang, Nat. Energy, 2016, 1, 16066 CrossRef CAS.
- X. D. Ren, P. Y. Gao, L. F. Zou, S. H. Jiao, X. Cao, X. H. Zhang, H. Jia, M. H. Engelhard, B. E. Matthews, H. P. Wu, H. Lee, C. J. Niu, C. M. Wang, B. W. Arey, J. Xiao, J. Liu, J. G. Zhang and W. Xu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28603–28613 CrossRef CAS PubMed.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS PubMed.
- E. Vedejs and S. E. Denmark, Lewis Base Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2016 Search PubMed.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, DOI:10.18434/T4D303, accessed 6 December 2021.
- F. A. Bulat, J. S. Murray and P. Politzer, Comput. Theor. Chem., 2021, 1199, 113192 CrossRef CAS.
- W. Liu, Y. D. Jiang, K. H. Dostert, C. P. O’Brien, W. Riedel, A. Savara, S. Schauermann and A. Tkatchenko, Sci. Adv., 2017, 3, e1700939 CrossRef PubMed.
- J. T. Li, S. Yang, J. C. Ren, G. R. Su, S. Li, C. J. Butch, Z. G. Ding and W. Liu, J. Phys. Chem. Lett., 2019, 10, 6755–6761 CrossRef CAS PubMed.
- T. D. Spivey and A. Holewinski, J. Am. Chem. Soc., 2021, 143, 11897–11902 CrossRef CAS PubMed.
- X. D. Ren, L. F. Zou, S. H. Jiao, D. H. Mei, M. H. Engelhard, Q. Y. Li, H. Y. Lee, C. J. Niu, B. D. Adams, C. M. Wang, J. Liu, J. G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 896–902 CrossRef CAS.
- R. A. Miranda-Quintana and J. Smiatek, Electrochim. Acta, 2021, 384, 138418 CrossRef CAS.
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- E. R. Fadel, F. Faglioni, G. Samsonidze, N. Molinari, B. V. Merinov, W. A. Goddard, J. C. Grossman, J. P. Mailoa and B. Kozinsky, Nat. Commun., 2019, 10, 3360 CrossRef PubMed.
- V. Pande and V. Viswanathan, J. Phys. Chem. Lett., 2019, 10, 7031–7036 CrossRef CAS PubMed.
- B. Winter and M. Faubel, Chem. Rev., 2006, 106, 1176–1211 CrossRef CAS PubMed.
- B. Winter, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601, 139–150 CrossRef CAS.
- T. Fransson, Y. Harada, N. Kosugi, N. A. Besley, B. Winter, J. J. Rehr, L. G. M. Pettersson and A. Nilsson, Chem. Rev., 2016, 116, 7551–7569 CrossRef CAS PubMed.
- R. Seidel, B. Winter and S. E. Bradforth, Annu. Rev. Phys. Chem., 2016, 67, 283–305 CrossRef CAS PubMed.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H. P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158–5190 CrossRef CAS PubMed.
- M. N. Pohl, E. Muchová, R. Seidel, H. Ali, S. Sršeň, I. Wilkinson, B. Winter and P. Slavíček, Chem. Sci., 2019, 10, 848–865 RSC.
- B. Jagoda-Cwiklik, P. Slavíček, D. Nolting, B. Winter and P. Jungwirth, J. Phys. Chem. B, 2008, 112, 7355–7358 CrossRef CAS PubMed.
- M. N. Piancastelli, R. F. Fink, R. Feifel, M. Bassler, S. L. Sorensen, C. Miron, H. Wang, I. Hjelte, O. Björneholm, A. Ausmees, S. Svensson, P. Salek, F. K. Gel’mukhanov and H. Agren, J. Phys. B: At., Mol. Opt. Phys., 2000, 33, 1819–1826 CrossRef CAS.
- I. Hjelte, M. N. Piancastelli, R. F. Fink, O. Björneholm, M. Bassler, R. Feifel, A. Giertz, H. Wang, K. Wiesner, A. Ausmees, C. Miron, S. L. Sorensen and S. Svensson, Chem. Phys. Lett., 2001, 334, 151–158 CrossRef CAS.
- E. E. Rennie, U. Hergenhahn, O. Kugeler, A. Rüdel, S. Marburger and A. M. Bradshaw, J. Chem. Phys., 2002, 117, 6524–6532 CrossRef CAS.
- U. Hergenhahn, A. Rüdel, K. Maier, A. M. Bradshaw, R. F. Fink and A. T. Wen, Chem. Phys., 2003, 289, 57–67 CrossRef CAS.
- P. Bolognesi, P. O’Keeffe, Y. Ovcharenko, L. Avaldi and V. Carravetta, J. Chem. Phys., 2012, 136, 154308 CrossRef CAS PubMed.
- A. Kikas, R. Ruus, A. Saar, E. Nommiste, T. Kaambre and S. Sundin, J. Electron Spectrosc. Relat. Phenom., 1999, 101, 745–749 CrossRef.
- H. Peisert, I. Biswas, L. Zhang, B. E. Schuster, M. B. Casu, A. Haug, D. Batchelor, M. Knupfer and T. Chasse, J. Chem. Phys., 2009, 130, 194705 CrossRef CAS PubMed.
- R. H. Temperton, N. W. Rosemann, M. Y. Guo, N. Johansson, L. A. Fredin, O. Prakash, K. Warnmark, K. Handrup, J. Uhlig, J. Schnadt and P. Persson, J. Phys. Chem. A, 2020, 124, 1603–1609 CrossRef CAS PubMed.
- T. Tiedje, K. M. Colbow, D. Rogers and W. Eberhardt, Phys. Rev. Lett., 1990, 65, 1243–1246 CrossRef CAS PubMed.
- E. Kukk, S. Aksela, H. Aksela, E. Nommiste, A. Kikas, A. Ausmees and M. Elango, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 9079–9085 CrossRef CAS PubMed.
- H. Aksela, E. Kukk, S. Aksela, A. Kikas, E. Nommiste, A. Ausmees and M. Elango, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 3116–3123 CrossRef CAS PubMed.
- M. Elango, A. Kikas, A. Maiste, R. Ruus and A. Saar, Phys. B, 1995, 208, 47–48 CrossRef.
- A. S. Vinogradov, A. B. Preobrajenski, S. L. Molodtsov, S. A. Krasnikov, R. Szargan, A. Knop-Gericke and M. Havecker, Chem. Phys., 1999, 249, 249–258 CrossRef CAS.
- A. B. Preobrajenski, A. S. Vinogradov, S. L. Molodtsov, S. K. Krasnikov, T. Chasse, R. Szargan and C. Laubschat, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 205116 CrossRef.
- A. B. Preobrajenski, A. S. Vinogradov, S. L. Molodtsov, S. A. Krasnikov, R. Szargan and C. Laubschat, Chem. Phys. Lett., 2003, 368, 125–131 CrossRef CAS.
- A. B. Preobrajenski, A. S. Vinogradov, E. Kleimenov, A. Knop-Gericke, S. A. Krasnikov, R. Szargan and N. Mårtensson, Phys. Scr., 2005, T115, 1071–1073 CrossRef CAS.
- A. B. Preobrajenski, A. S. Vinogradov and N. Mårtensson, J. Electron Spectrosc. Relat. Phenom., 2005, 148, 59–64 CrossRef CAS.
- H. Shimada, N. Matsubayashi, M. Imamura, T. Sato, Y. Kakudate, S. Fujiwara and A. Nishijima, J. Electron Spectrosc. Relat. Phenom., 1996, 79, 211–214 CrossRef CAS.
- R. Dudde, M. L. M. Rocco, E. E. Koch, S. Bernstorff and W. Eberhardt, J. Chem. Phys., 1989, 91, 20–28 CrossRef CAS.
- D. Menzel, G. Rocker, H. P. Steinrück, D. Coulman, P. A. Heimann, W. Huber, P. Zebisch and D. R. Lloyd, J. Chem. Phys., 1992, 96, 1724–1734 CrossRef CAS.
- T. Porwol, G. Domotor, H. J. Freund, R. Dudde, C. M. Liegener and W. Vonniessen, Phys. Scr., 1992, T41, 197–207 CrossRef CAS.
- M. Mauerer, P. Zebisch, M. Weinelt and H. P. Steinrück, J. Chem. Phys., 1993, 99, 3343–3352 CrossRef.
- W. Wurth and D. Menzel, J. Electron Spectrosc. Relat. Phenom., 1993, 62, 23–31 CrossRef CAS.
- S. L. Sorensen, S. J. Osborne, A. Ausmees, A. Kikas, N. Correia, S. Svensson, A. N. deBrito, P. Persson and S. Lunell, J. Chem. Phys., 1996, 105, 10719–10724 CrossRef CAS.
- J. J. Gallet, F. Bournel, S. Kubsky, G. Dufour, F. Rochet and F. Sirotti, J. Electron Spectrosc. Relat. Phenom., 2002, 122, 285–295 CrossRef CAS.
- D. V. Vyalikh, V. V. Maslyuk, A. Bluher, A. Kade, K. Kummer, Y. S. Dedkov, T. Bredow, I. Mertig, M. Mertig and S. L. Molodtsov, Phys. Rev. Lett., 2009, 102, 098101 CrossRef CAS PubMed.
- J. Kikuma and B. P. Tonner, J. Electron Spectrosc. Relat. Phenom., 1996, 82, 41–52 CrossRef CAS.
- J. J. Gallet, F. Jolly, F. Rochet, F. Bournel, G. Dufour, P. A. Avila, F. Sirotti and P. Torelli, J. Electron Spectrosc. Relat. Phenom., 2002, 122, 11–25 CrossRef CAS.
- R. Friedlein, S. L. Sorensen, A. Baev, F. Gel’mukhanov, J. Birgerson, A. Crispin, M. P. de Jong, W. Osikowicz, C. Murphy, H. Agren and W. R. Salaneck, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 125204 CrossRef.
- H. S. Kato, M. Furukawa, M. Kawai, M. Taniguchi, T. Kawai, T. Hatsui and N. Kosugi, Phys. Rev. Lett., 2004, 93, 086403 CrossRef CAS PubMed.
- L. Wang, K. Q. Zhang, X. Y. Gao, X. Y. Liu and A. T. S. Wee, Appl. Phys. Lett., 2006, 89, 013902 CrossRef.
- M. Gliboff, D. Sulas, D. Nordlund, D. W. deQuilettes, P. D. Nguyen, G. T. Seidler, X. S. Li and D. S. Ginger, J. Phys. Chem. C, 2014, 118, 5570–5578 CrossRef CAS.
- G. J. Man, C. M. Sterling, C. Kamal, K. A. Simonov, S. Svanstrom, J. Acharya, F. O. L. Johansson, E. Giangrisostomi, R. Ovsyannikov, T. Huthwelker, S. M. Butorin, P. K. Nayak, M. Odelius and H. Rensmo, Phys. Rev. B, 2021, 104, L041302 CrossRef CAS.
- M. Kot, K. Wojciechowski, H. Snaith and D. Schmeisser, Chem.–Eur. J., 2018, 24, 3539–3544 CrossRef CAS PubMed.
- J. Schnadt, P. A. Brühwiler, L. Patthey, J. N. O’Shea, S. Södergren, M. Odelius, R. Ahuja, O. Karis, M. Bässler, P. Persson, H. Siegbahn, S. Lunell and N. Mårtensson, Nature, 2002, 418, 620–623 CrossRef CAS PubMed.
- J. J. Gallet, F. Bournel, S. Carniato, G. Dufour, F. Rochet, S. Rangan and F. Sirotti, Surf. Sci., 2007, 601, 552–561 CrossRef CAS.
- F. Petraki, H. Peisert, I. Biswas, U. Aygul, F. Latteyer, A. Vollmer and T. Chasse, J. Phys. Chem. Lett., 2010, 1, 3380–3384 CrossRef CAS.
- C. Sauer, M. Wiessner, A. Scholl and F. Reinert, New J. Phys., 2015, 17, 043016 CrossRef.
- C. Sauer, M. Wiessner, A. Scholl and F. Reinert, Phys. Rev. B, 2016, 93, 045101 CrossRef.
- S. Thürmer, I. Unger, P. Slavíček and B. Winter, J. Phys. Chem. C, 2013, 117, 22268–22275 CrossRef.
- R. M. Fogarty, R. G. Palgrave, R. A. Bourne, K. Handrup, I. J. Villar-Garcia, D. J. Payne, P. A. Hunt and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2019, 21, 18893–18910 RSC.
- J. M. Seymour, E. Gousseva, A. I. Large, C. J. Clarke, P. Licence, R. M. Fogarty, D. A. Duncan, P. Ferrer, F. Venturini, R. A. Bennett, R. G. Palgrave and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2021, 23, 20957–20973 RSC.
- S. Thürmer, R. Seidel, W. Eberhardt, S. E. Bradforth and B. Winter, J. Am. Chem. Soc., 2011, 133, 12528–12535 CrossRef PubMed.
- R. Seidel, K. Atak, S. Thürmer, E. F. Aziz and B. Winter, J. Phys. Chem. B, 2015, 119, 10607–10615 CrossRef CAS PubMed.
- S. S. N. Lalithambika, K. Atak, R. Seidel, A. Neubauer, T. Brandenburg, J. Xiao, B. Winter and E. F. Aziz, Sci. Rep., 2017, 7, 40811 CrossRef CAS PubMed.
- S. S. N. Lalithambika, R. Golnak, B. Winter and K. Atak, Inorg. Chem., 2019, 58, 4731–4740 CrossRef PubMed.
- K. D. Mudryk, R. Seidel, B. Winter and I. Wilkinson, Phys. Chem. Chem. Phys., 2020, 22, 20311–20330 RSC.
- R. H. Temperton, W. Quevedo, R. Seidel, J. Uhlig, J. Schnadt and P. Persson, Phys. Rev. Res., 2021, 3, 033030 CrossRef CAS.
- H. Ali, R. Seidel, M. N. Pohl and B. Winter, Chem. Sci., 2018, 9, 4511–4523 RSC.
- H. Ali, R. Seidel, A. Bergmann and B. Winter, J. Mater. Chem. A, 2019, 7, 6665–6675 RSC.
- A. Y. Klyushin, M. M. Brzhezinskaya, A. V. Generalov, R. Puttner and A. S. Vinogradov, Fullerenes, Nanotubes, Carbon Nanostruct., 2010, 18, 600–604 CrossRef CAS.
- U. Gelius and K. Siegbahn, Faraday Discuss., 1972, 54, 257–268 RSC.
- U. Gelius, J. Electron Spectrosc. Relat. Phenom., 1974, 5, 985–1057 CrossRef CAS.
- R. Seidel, M. N. Pohl, H. Ali, B. Winter and E. F. Aziz, Rev. Sci. Instrum., 2017, 88, 073107 CrossRef PubMed.
- G. Held, F. Venturini, D. C. Grinter, P. Ferrer, R. Arrigo, L. Deacon, W. Q. Garzon, K. Roy, A. Large, C. Stephens, A. Watts, P. Larkin, M. Hand, H. C. Wang, L. Pratt, J. J. Mudd, T. Richardson, S. Patel, M. Hillman and S. Scott, J. Synchrotron Radiat., 2020, 27, 1153–1166 CrossRef CAS PubMed.
- S. Thürmer, S. Malerz, F. Trinter, U. Hergenhahn, C. Lee, D. M. Neumark, G. Meijer, B. Winter and I. Wilkinson, Chem. Sci., 2021, 12, 10558–10582 RSC.
- G. Greczynski and L. Hultman, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- L. C. Allen, J. Am. Chem. Soc., 1989, 111, 9003–9014 CrossRef CAS.
- R. M. Fogarty, R. P. Matthews, M. T. Clough, C. R. Ashworth, A. Brandt-Talbot, P. J. Corbett, R. G. Palgrave, R. A. Bourne, T. W. Chamberlain, T. Vander Hoogerstraete, P. B. J. Thompson, P. A. Hunt, N. A. Besley and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2017, 19, 31156–31167 RSC.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- F. Holzmeier, T. J. A. Wolf, C. Gienger, I. Wagner, J. Bozek, S. Nandi, C. Nicolas, I. Fischer, M. Guhr and R. F. Fink, J. Chem. Phys., 2018, 149, 034308 CrossRef CAS PubMed.
- G. Grell and S. I. Bokarev, J. Chem. Phys., 2020, 152, 074108 CrossRef CAS PubMed.
- S. E. Bradforth, E. H. Kim, D. W. Arnold and D. M. Neumark, J. Chem. Phys., 1993, 98, 800–810 CrossRef CAS.
- S. Rangan, J. Viereck and R. A. Bartynski, J. Phys. Chem. B, 2020, 124, 7909–7917 CrossRef CAS PubMed.
- C. Ehlert, M. Holzweber, A. Lippitz, W. E. S. Unger and P. Saalfrank, Phys. Chem. Chem. Phys., 2016, 18, 8654–8661 RSC.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. L. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797–2808 RSC.
- M. J. Thomason, C. R. Seabourne, B. M. Sattelle, G. A. Hembury, J. S. Stevens, A. J. Scott, E. F. Aziz and S. L. M. Schroeder, Faraday Discuss., 2015, 179, 269–289 RSC.
- D. Nolting, N. Ottosson, M. Faubel, I. V. Hertel and B. Winter, J. Am. Chem. Soc., 2008, 130, 8150–8151 CrossRef CAS PubMed.
- O. Bjorneholm, A. Sandell, A. Nilsson, N. Martensson and J. N. Andersen, Phys. Scr., 1992, T41, 217–225 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fd00117e |
| This journal is © The Royal Society of Chemistry 2022 |