 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Corrosion inhibition in acidic environments: key interfacial insights with photoelectron spectroscopy
Kiran
Kousar
a,
Michael
Dowhyj
ab,
Monika S.
Walczak
a,
Thomas
Ljungdahl
c,
Alexander
Wetzel
c,
Hans
Oskarsson
c,
Alex S.
Walton
 bd,
Paolo
Restuccia
e,
Nicholas M.
Harrison
e and
Robert
Lindsay
*ab
bd,
Paolo
Restuccia
e,
Nicholas M.
Harrison
e and
Robert
Lindsay
*ab
aCorrosion and Protection Centre, Department of Materials, The University of Manchester, Sackville Street, Manchester, M13 9PL, UK. E-mail: robert.lindsay@manchester.ac.uk
bPhoton Science Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cNouryon, Hamnvägen 2, SE-444 85 Stenungsund, Sweden
dDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PY, UK
eDepartment of Chemistry and Institute for Molecular Science and Engineering, Imperial College London, White City Campus, 80 Wood Lane, London W12 0BZ, UK
First published on 6th January 2022
Abstract
In many engineering scenarios, surface-active organic species are added to acidic solutions to inhibit the corrosion of metallic components. Given suitable selection, such corrosion inhibitors are highly effective, preventing significant degradation even in highly aggressive environments. Nevertheless, there are still considerable gaps in fundamental knowledge of corrosion inhibitor functionality, severely restricting rational development. Here, we demonstrate the capability of X-ray photoelectron spectroscopy (XPS), supported by ab initio modelling, for revealing key details of inhibited substrates. Attention is focussed on the corrosion inhibition of carbon steel through the addition of an exemplar imidazoline-based corrosion inhibitor (OMID) to aqueous solutions of both HCl and H2SO4. Most notably, it is demonstrated that interfacial chemistry varies with the identity of the acid. High resolution Fe 2p, O 1s, N 1s, and Cl 2p XPS spectra, acquired from well-inhibited carbon steel in 1 M HCl, show that there are two different singly protonated OMID species bound directly to the metallic carbon steel substrate. In sharp contrast, in 0.01 M H2SO4, OMID adsorbs onto an ultra-thin surface film, composed primarily of a ferric sulfate (Fe2(SO4)3)-like phase. Such insight is essential to efforts to develop a mechanistic description of corrosion inhibitor functionality, as well as knowledge-based identification of next generation corrosion inhibitors.
Introduction
Electrochemical corrosion of metallic engineering components continues to be a bête-noire of our modern world, resulting in significant costs, both economic and beyond.1 Given that this phenomenon is almost always thermodynamically inevitable, slowing material degradation down through controlling reaction kinetics is the best that can be achieved. To pursue this goal, corrosion engineers typically adopt one or more of three basic approaches, i.e., material selection, coating application, or operando chemical treatment. In the latter category, one option is the addition of corrosion inhibitor species, that reduce corrosion by inducing a change in the surface termination of the metallic substrate.2 This approach is well established, and highly effective in many real-world applications. There are, however, significant gaps in the fundamental understanding of corrosion inhibitor functionality, severely restricting rational development; selection of corrosion inhibitors at present is largely a trial-and-error exercise. In this contribution, it will be demonstrated that X-ray photoelectron spectroscopy (XPS), supported by ab initio modelling, can be exploited to gain fresh insight into inhibited interfaces in acidic environments, offering the possibility of more knowledge-based development of next generation corrosion inhibitors.In aqueous acidic solution, organic surface-active species are invariably employed as corrosion inhibitors.3,4 As illustrated in Fig. 1(a), it is typically assumed that these species function through binding to the metallic substrate, forming either a monolayer or bilayer.4 It should be emphasised, however, that this cartoon is rather speculative, with relatively little direct experimental evidence acquired to date, e.g., the nature of the surface of the metallic substrate is generally unknown. XPS is one technique that can be applied to begin to remedy this situation, as it can deliver chemically sensitive information about such interfaces. Here, XPS is employed to characterise inhibited interfaces formed on carbon steel in acidic solutions in the presence of an exemplar imidazoline-based surfactant, which will henceforth be referred to as OMID; a ball and stick model of OMID is depicted in Fig. 1(b).
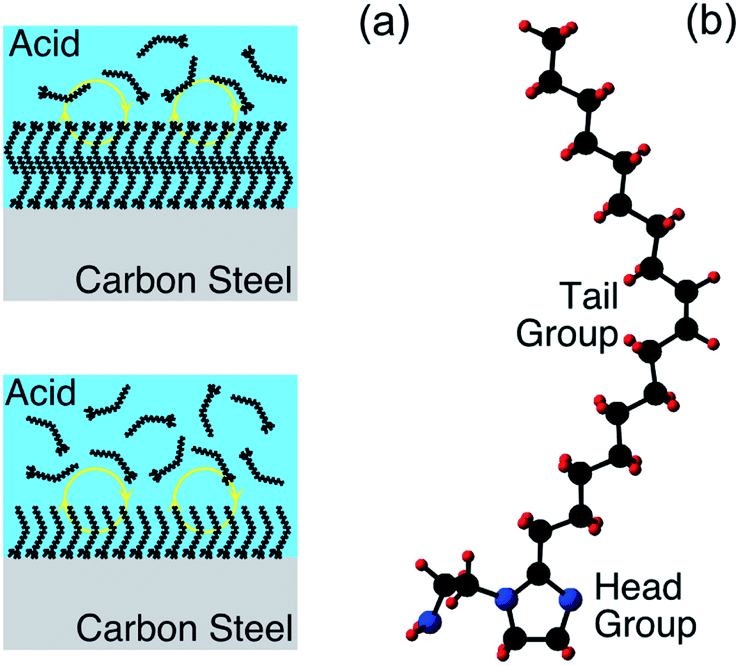 | ||
| Fig. 1 (a) Cartoons of organic corrosion inhibitor species forming a monolayer (bottom) or bilayer (top) on a metallic substrate (carbon steel) in acidic solution. The yellow circles with arrows indicate that the adsorbed layers are dynamic, i.e., they dissipate in the absence of corrosion inhibitor in the bulk solution. (b) Ball and stick model of OMID, where the black, blue, and red spheres represent carbon, nitrogen, and hydrogen atoms, respectively. The head group, which is expected to be involved in substrate bonding, comprises a heterocyclic ring (C3N2H4) and a pendant side group (C2H4NH2). The tail group is an unbranched aliphatic chain (C17H33). | ||
Previous studies have demonstrated that OMID is a very efficient corrosion inhibitor for carbon steel in acidic solution.5–8 Moreover, we have already applied XPS to characterise the interaction of OMID with carbon steel in 1 M HCl, concluding that the well-inhibited interface comprises OMID bound to film-free substrate, i.e., there is no surface oxide/salt phase.9 Furthering this work, in the following sections we present a study exploring the impact of varying the composition of the acidic solution (see Table 1 for a list of the solutions) on both the corrosion inhibition performance of OMID at the critical micelle concentration (CMC),† and the nature of the interface derived from XPS data; we focus on measurements at the CMC, as it is expected that OMID should perform optimally at this concentration.4 Density functional theory (DFT) calculations have been undertaken to aid data analysis/interpretation.
| Acid solutions |
|---|
| 1 M HCl |
| 0.01 M HCl |
| 1 M H2SO4 |
| 0.01 M H2SO4 |
Experimental
A single batch of carbon steel samples (10 mm × 10 mm × 1.5 mm), purchased from RCSL Corrosion Monitoring Ltd, were used as substrates. For preparation of the acidic solutions listed in Table 1, analytical grade reagents from Fischer Scientific were employed, namely 10.2 M HCl and 18.4 M H2SO4. OMID (97.7% purity) was synthesised in our laboratories; see ref. 8 for details of the synthetic route. The CMC of OMID in each acidic solution was determined through pendant drop surface tensiometry, using a Krüss GmbH Drop Shape Analyzer, DSA100.To assess the performance of OMID as a corrosion inhibitor for carbon steel in each acidic solution, corrosion rates were determined after 4 h of immersion at T ∼ 298 K, in both the absence and presence of OMID (at CMC); 4 h was adopted as the standard immersion period, as it was found that all samples had achieved a steady state by this point. A combination of electrochemical techniques, i.e., linear polarisation resistance (LPR) and potentiodynamic polarisation (PDP),10 were used to provide a quantitative estimate of corrosion rate. For these measurements, a standard three-electrode setup was employed in a 1 L glass cell containing the solution of interest; LPR (PDP) data were collected with a computer-controlled potentiostat (Ivium Compactstat), using a scan rate of 10 mV min−1 (120 mV min−1) over the range −10 mV to +10 mV (−250 mV to +250 mV) relative to open circuit potential (OCP). Before insertion of a carbon steel sample (working electrode) into the glass cell, it was mounted in epoxy resin, ground with a series of SiC papers (240 grit, 400 grit, 600 grit, and 800 grit), washed with ethanol and de-ionized water, and dried.
XPS spectra were recorded using a Kratos Axis Ultra spectrometer, equipped with an Al Kα X-ray source (hν = 1486.6 eV), a 165 mm hemispherical energy analyser, and an electron flood gun; samples were exposed to low energy electrons (≤3 eV) during data acquisition to minimize charge accumulation. Spectra were recorded at normal emission (θE = 0°), using pass energies of 80 eV and 20 eV for overview spectra and higher resolution single core level spectra, respectively. The metallic Fe 2p3/2 peak at a binding energy (BE) of 706.7 eV was used for calibration (±0.1 eV).11
Carbon steel samples for XPS analysis were ground with a series of SiC papers (400 grit, 600 grit, 800 grit, 1200 grit, 2400 grit, 4000 grit), and then polished with diamond paste (1 μm) to a mirror finish. Subsequently, each sample was rinsed with acetone and de-H2O, and then dried. To avoid air exposure, a nitrogen filled glove box directly attached to the XPS instrument, as shown in Fig. 2(a), was used for sample immersion. Following solution exposure for 4 h, the sample was retrieved, blown dry with nitrogen to minimise physical surface deposition of non-volatile solution constituents, and inserted directly into the XPS load-lock chamber. Implementation of this sample handling procedure, which is reported in detail in ref. 13, is essential for mitigating post-immersion substrate oxidation, which can lead to misinterpretation of inhibited interface chemistry. Fig. 2(b) and (c) demonstrate the advantage of this approach through comparison of Fe 2p and O 1s core level XPS spectra from highly inhibited carbon steel samples formed in 1 M HCl in the presence of the corrosion inhibitor 2-mercaptobenzimidazole (MBI).12,13 Blue (red) spectra were recorded from samples that had been removed from solution into a fully (partially) N2-purged glove box. The partially N2-purged data display features consistent with interface oxidation, i.e., signals assigned to O2−/OH− and Fe2+/3+, which are absent from the fully N2-purged data, indicating that post-immersion oxidation can have a significant bearing on XPS core level profiles.
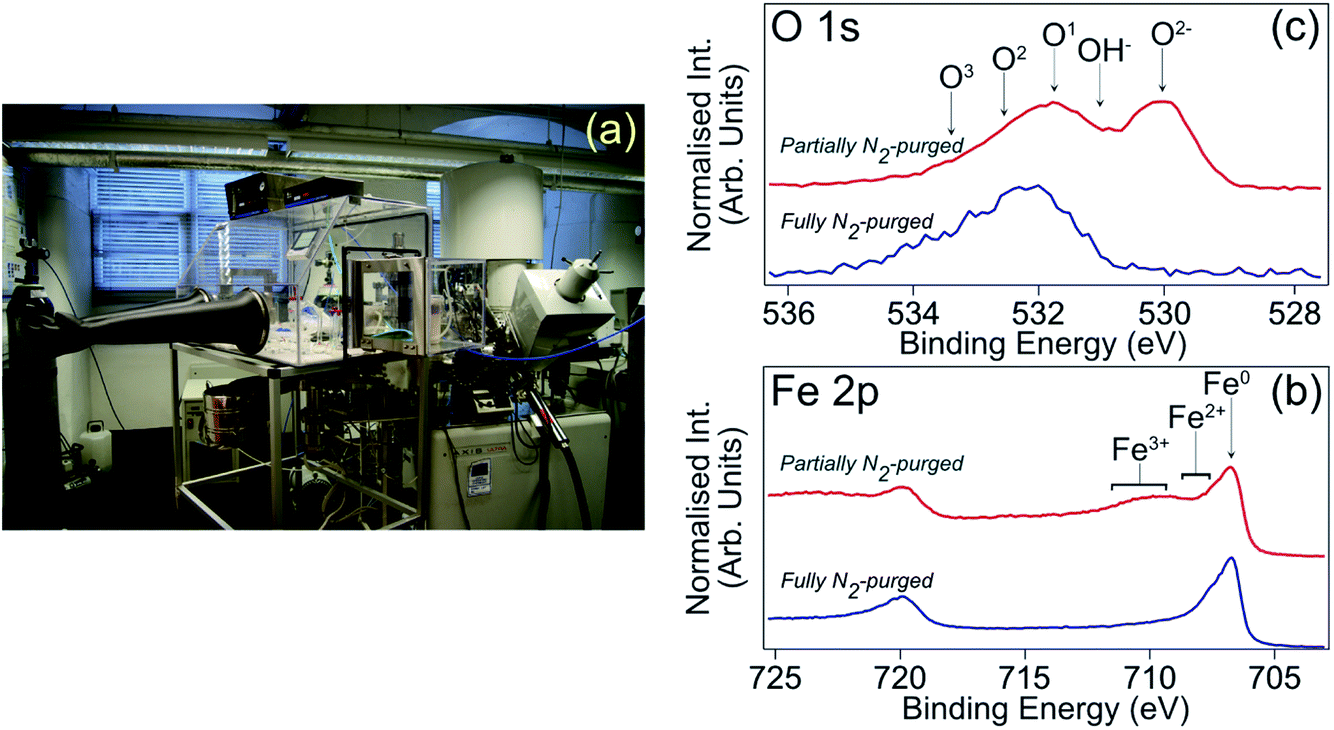 | ||
| Fig. 2 (a) Photograph of N2-filled glove box attached to load lock of XPS spectrometer to allow transfer of samples without post-immersion air exposure. This setup was employed for all XPS data presented in this study. (b) Fe 2p XPS spectra (hν = 1486.6 eV, θE = 0°) acquired from carbon steel samples that had been immersed in 1 M HCl inhibited with MBI.12,13 The blue (red) spectrum is from a sample transferred through a fully (partially) N2-purged glove box.12,13 (c) O 1s XPS spectra acquired under analogous conditions; O1, O2 and O3 arise from adsorbed OH (O1) and an adventitious carbon layer (O1–O3). | ||
Concerning ab initio DFT modelling, calculations have been undertaken on both isolated OMID head group (HG-OMID) molecules and a 5-layer slab of (110) oriented Fe decorated with chlorine atoms, using the CRYSTAL 17 program14,15 that expands molecular and crystal orbitals as a linear combination of atom centered Gaussian functions. For molecules, electronic exchange and correlation were approximated using the hybrid HSE06 functional, which describes the strong on-site electronic interactions of electrons localised in molecular orbitals well. Quadruple valence basis sets with polarisation function were used for the C, N, and H atoms, i.e., 6-31111++G** for C and N, and 31111++G** for H. Following energy minimisation through geometry optimisation, projected electronic density of states (PDOS) and proton affinities (PAs) were computed. The 2D periodic slab calculations of the surface were performed as previously outlined in ref. 9, with electron exchange and correlation approximated with the PBE exchange–correlation functional that describes the delocalised states of the metal adequately. All electron basis sets with polarisation were used for the Fe and Cl atoms, i.e., 8-64111d41G quadruple valence for Fe and 86-311G triple valence with the addition of a diffuse d-symmetry polarisation function for Cl. Again, following geometry optimisation, the PDOS was computed.
Results and discussion
Table 2 shows the CMC value determined for OMID in each acidic solution, along with the corrosion rate (CR) derived from LPR/PDP data after 4 h of substrate (carbon steel) immersion in both the absence ([OMID] = 0 mM) and presence of OMID ([OMID] = CMC); a standard methodology was employed to determine CRs in mm per year.10 Furthermore, the corrosion inhibitor efficiency (η%) displayed by OMID in each solution is listed, which was determined from the following equation:where U and I are the uninhibited and inhibited corrosion rates, respectively.2 This figure of merit for corrosion inhibitor performance is commonly used for selection purposes in engineering applications, with species displaying η% ≥ 95% typically being identified as high performing.16
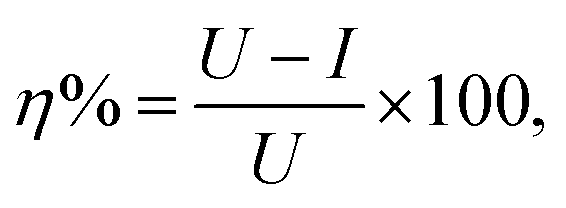
| Solution | OMID CMC (mM) | CR, 0 mM [OMID] (mm per year) | CR, CMC [OMID] (mm per year) | η% (%) |
|---|---|---|---|---|
| 1 M HCl | 0.18 ± 0.03 | 3.75 ± 0.07 | 0.11 ± 0.01 | 97.1 ± 0.3 |
| 0.01 M HCl | 5.90 ± 1.50 | 1.96 ± 0.12 | 0.28 ± 0.03 | 85.7 ± 1.8 |
| 1 M H2SO4 | 0.22 ± 0.05 | 52.71 ± 0.80 | 1.09 ± 0.07 | 97.9 ± 0.1 |
| 0.01 M H2SO4 | 0.70 ± 0.12 | 4.43 ± 0.70 | 0.20 ± 0.01 | 95.5 ± 0.8 |
Surveying the η% values in Table 2, it is evident that OMID is a high performing corrosion inhibitor (i.e., η% ≥ 95%) for carbon steel in 3 out of the 4 acidic solutions considered. Moreover, the absolute inhibited corrosion rate is ≤0.2 mm per year for two of these solutions, which we specify as an additional condition for a high performing corrosion inhibitor in this study.
Focusing on the two well-inhibited solutions, namely 1 M HCl with OMID at CMC and 0.01 M H2SO4 with OMID at CMC, which will henceforth be referred to as 1MHCl/OMID and 0.01MH2SO4/OMID, respectively, Fig. 3 shows overview XPS spectra acquired from carbon steel samples subject to 4 h of immersion in each of these solutions; a spectrum acquired from a polished carbon steel substrate is also displayed. Concerning the latter spectrum, features originating from photoemission out of Fe 2p, O 1s, and C 1s core levels are clearly visible. The Fe signal originates from the carbon steel substrate, O from both a surface oxidic film and adsorbed species, and C is a result of adventitious surface contamination.9,12
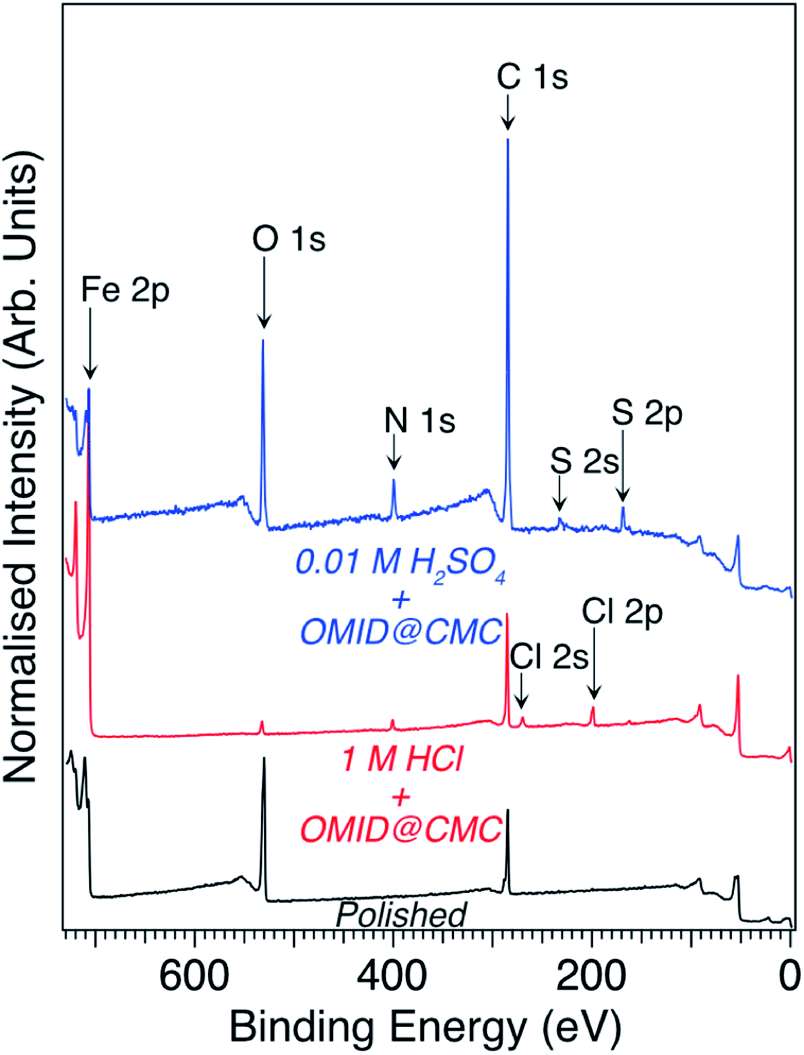 | ||
| Fig. 3 Overview XPS spectra (hν = 1486.6 eV, θE = 0°) acquired from carbon steel subsequent to immersion for 4 h in either 1MHCl/OMID (red line) or 0.01MH2SO4/OMID (blue line). The bottom-most spectrum (black line) was acquired from a polished carbon steel substrate. | ||
Immersion in either acidic solution induces modification of the overview spectral profile. One common change is the appearance of a N 1s feature, which is consistent with the adsorption of OMID, as it contains three nitrogen atoms (see Fig. 1). Fig. 4(a) shows corresponding higher resolution N 1s core level XPS spectra; the data shown in red were acquired from carbon steel following immersion in 1MHCl/OMID, and those shown in blue following immersion in 0.01MH2SO4/OMID. Evidently, OMID adsorption is not identical in the two cases, as the spectra differ significantly.
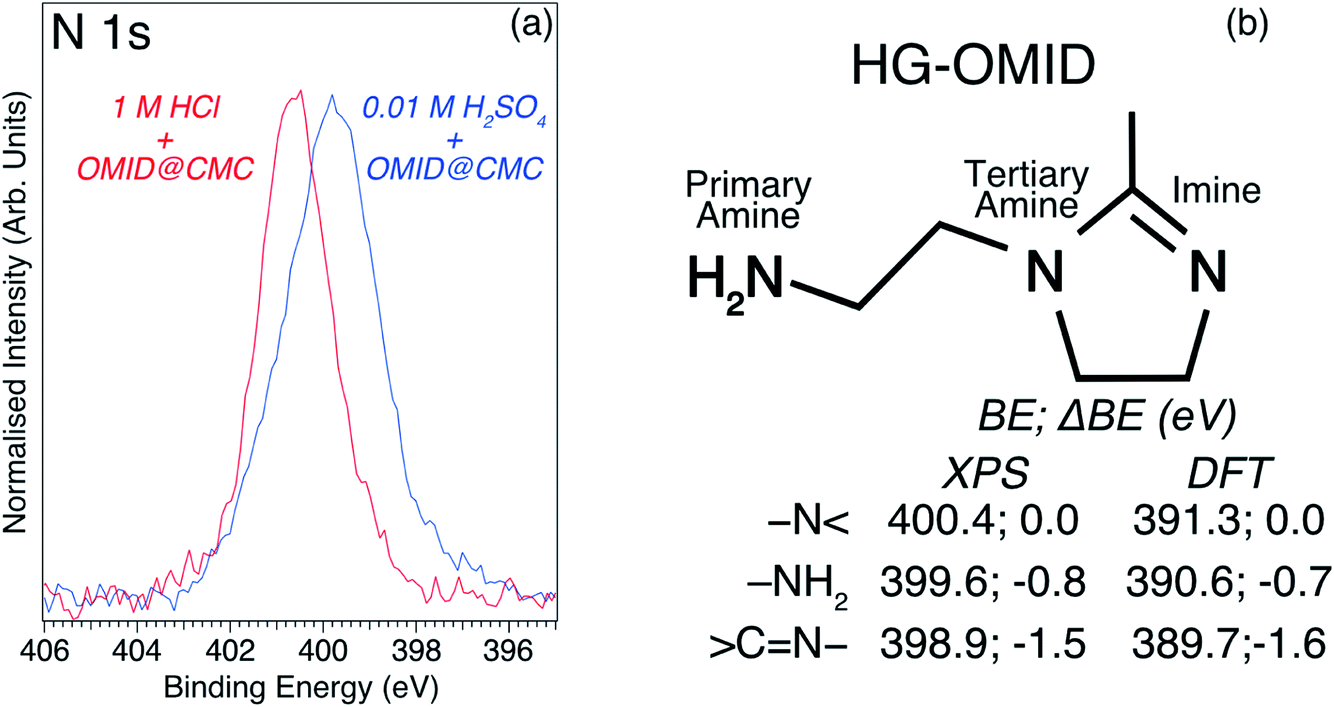 | ||
Fig. 4 (a) N 1s core level XPS spectra (hν = 1486.6 eV, θE = 0°) acquired from carbon steel following immersion for 4 h in either 1MHCl/OMID (red line) or 0.01MH2SO4/OMID (blue line). (b) Skeletal model of OMID head group (HG-OMID), and BEs, plus ΔBEs relative to –N![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) , for each nitrogen atom, derived from either XPS or DFT calculations. , for each nitrogen atom, derived from either XPS or DFT calculations. | ||
To aid interpretation of such N 1s XPS data, we presented in ref. 8 a N 1s core level spectrum acquired from a multilayer of the molecule depicted in Fig. 4(b), which is the OMID head group (HG-OMID). It was demonstrated that the core level signal was composed of three components (BEs = 398.9 eV, 399.6 eV, and 400.4 eV) of equal intensity, consistent with the three chemically distinct N atoms in OMID, i.e., imine (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), primary amine (–NH2), and tertiary amine (–N). In accord with earlier work,17–19 the following BE assignments were proposed: CN– to BE = 398.9 eV, –NH2 to BE = 399.6 eV, and –N to BE = 400.4 eV.
N–), primary amine (–NH2), and tertiary amine (–N). In accord with earlier work,17–19 the following BE assignments were proposed: CN– to BE = 398.9 eV, –NH2 to BE = 399.6 eV, and –N to BE = 400.4 eV.
In order to corroborate these suggested N 1s XPS BE attributions, an ab initio DFT calculation has been undertaken of isolated HG-OMID. Following geometric optimisation, the binding energy of each N 1s core level was extracted from computed PDOS. The values obtained are compared to the experimental XPS BEs in Fig. 4(b). As expected, given that the calculation is performed for the ground state of the molecule and single particle eigenvalues used to approximate the excitation energy, the absolute values differ. There is excellent agreement, however, in terms of relative energies, which are expressed in terms of binding energy shifts (ΔBE) relative to –N (ΔBE = 0 eV). This result suggests that the N 1s XPS assignments are reasonable.
Using the ΔBE data summarised in Fig. 4(b) as a starting point, Fig. 5 (lower spectrum) shows the best fit to the N 1s core level spectrum acquired from a carbon steel sample post immersion in 1MHCl/OMID; a combination of Gaussian–Lorentzian (GL(p))‡ line shape functions and a Shirley-type background function were employed to model the photoelectron peaks and secondary electron background, respectively. 5 GL(30) functions, each having a full width at half maximum (FWHM) of ∼1.2 eV, were required to achieve a satisfactory fit to the data. As discussed previously8,9 and illustrated in Fig. 5, this fit can be reconciled with the adsorption of OMID in two different singly protonated (OMIDH+) forms, where either the primary amine (–NH2 → –NH3+) or imine (CN– → CNH+–) is protonated. We note that OMID is expected to be doubly protonated, –NH3+ plus CNH+–, in bulk solution in both 1 M HCl and 0.01 M H2SO4;8 the tertiary amine group (–N) remains unprotonated. Hence, adsorption results in a reduction in the degree of protonation, i.e., OMID2H+ → OMIDH+.
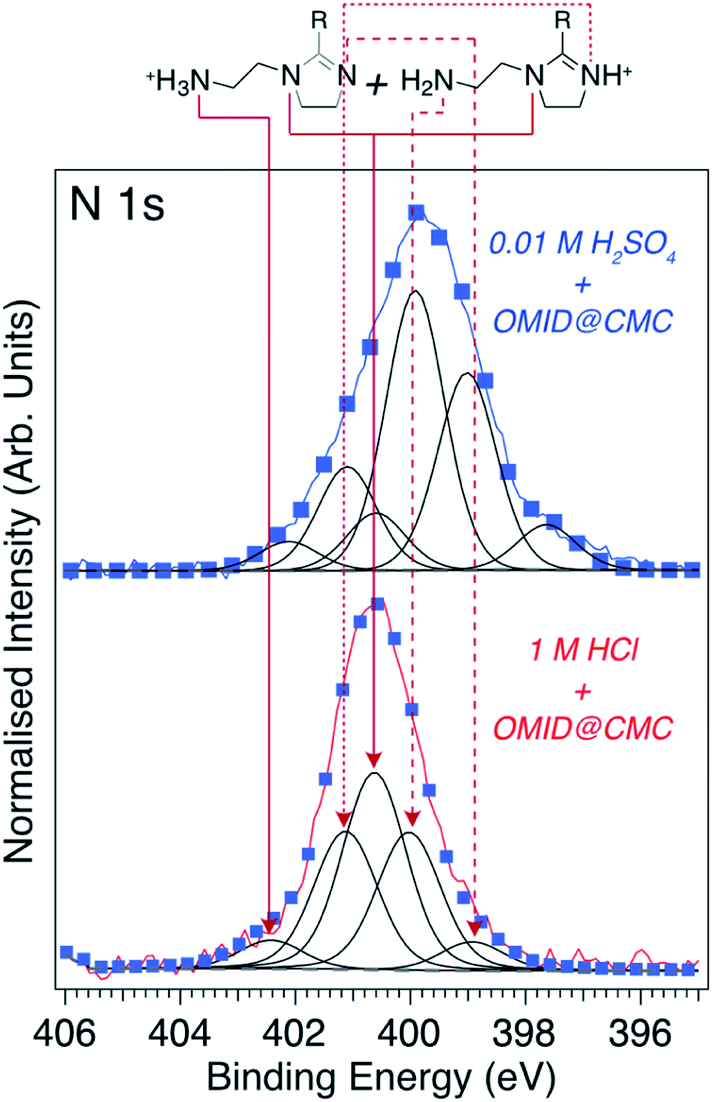 | ||
| Fig. 5 N 1s core level XPS spectra (hν = 1486.6 eV, θE = 0°) acquired from carbon steel following immersion for 4 h in either 1MHCl/OMID (lower spectrum) or 0.01MH2SO4/OMID (upper spectrum). Blue markers show best fits to the experimental spectra (red/blue lines), using GL(30) (black lines) and Shirley-type (broken grey lines) functions. Peak assignments, consistent with OMID being adsorbed on carbon steel in 1MHCl/OMID in the two OMIDH+ forms shown at the top of this figure, are also indicated. R = C17H33. | ||
From the relative intensity of the –NH2 and CNH+– components in Fig. 5 (lower spectrum), it can be concluded that the majority of OMID adsorbed on carbon steel in 1MHCl/OMID possesses a protonated imine group (CNH+–). Interestingly, this result agrees with the PA deduced from DFT total energy differences for protonation of each N in HG-OMID (HG-OMID → HG-OMIDH+). For optimised HG-OMID geometries it was found that (CN–) displayed the greatest PA, i.e., PA(CN) ∼ 713 kJ mol−1 > PA(–NH2) ∼ 613 kJ mol−1 > PA(–N) ∼ 582 kJ mol−1, and so CNH+– is expected to be the dominant protonated group for OMIDH+. We point out that the ab initio modelling is performed on isolated species and so takes no account of either solvent effects or OMID–substrate/OMID–solution interactions, but is nevertheless consistent with interpretation of the XPS data.
Turning to the N 1s XPS spectrum recorded from carbon steel subsequent to immersion in 0.01MH2SO4/OMID, we have attempted to reproduce the experimental data with the same 5 GL(30) functions used for 1MHCl/OMID. To achieve the fit shown in Fig. 5 (upper spectrum), the BE of each peak was allowed to shift by up to ∼0.3 eV relative to those determined for 1MHCl/OMID, and an additional GL(30) component was required (BE = 397.6 eV). Assuming these peaks correspond to the assignments above, the most intense component is a result of photoemission from the –NH2 group, which is not consistent with adsorption of the two OMIDH+ species illustrated in Fig. 5. If only these two species were adsorbed, then the peak attributed to –N (BE = 400.7 eV) would always be the most intense, as it is common to both OMIDH+ species.
Further effort to interpret the N 1s spectrum for 0.01MH2SO4/OMID has involved both consideration of other adsorbed species, e.g., OMID2H+ and OMID, as well as reassignment of components to other N atoms. To date, no physically reasonable solution has been found, indicating that the adsorption of OMID onto carbon steel in 0.01MH2SO4/OMID is significantly more complex than in 1MHCl/OMID.
The information derived from the N 1s spectra in Fig. 5 is summarised in Fig. 6. As illustrated, these data do not reveal anything about the nature of the substrate terminations. To begin to gain insight into this aspect of the interfaces, we return to the overview spectra in Fig. 3. Comparing the spectrum of the polished carbon steel to that of the sample immersed in 1MHCl/OMID, there is a clear reduction in O 1s signal, suggesting depletion of the surface oxidic layer. Moreover, features assigned to Cl XPS core levels (2s and 2p) appear, which almost certainly arise from the interaction of the substrate with Cl−(aq) anions. Concerning the sample exposed to 0.01MH2SO4/OMID, an intense O 1s feature is apparent, as well as S 2s and S 2p peaks, which suggest the presence of substrate bound SO42− anions.
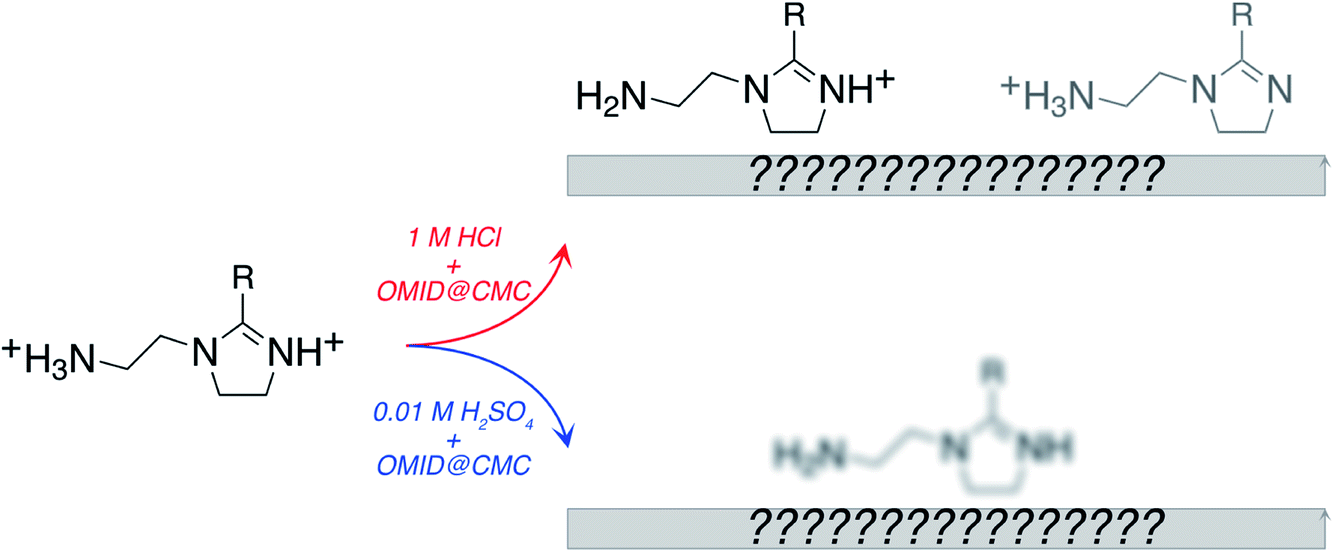 | ||
| Fig. 6 Cartoon summarising information obtained from N 1s XPS spectra acquired from carbon steel following immersion for 4 h in either 1MHCl/OMID or 0.01MH2SO4/OMID. For 1MHCl/OMID, two OMIDH+ species are concluded to be adsorbed, with the dominant one containing CNH+–. For 0.01MH2SO4/OMID, the exact nature of the adsorbed OMID remains ill-defined. R = C17H33. | ||
To facilitate more detailed understanding of the substrate terminations, higher resolution Fe 2p, O 1s, Cl 2p and S 2p core level XPS spectra, and corresponding best fits, acquired from carbon steel following immersion in either 1MHCl/OMID or 0.01MH2SO4/OMID are shown in Fig. 7; a Cl 2p (S 2p) spectrum is not shown for 0.01MH2SO4/OMID (1MHCl/OMID) as there was no signal above background. For the two common core levels (Fe 2p and O 1s), there are clear differences between the two acids, suggesting that not only does the adsorbed state(s) of OMID vary with solution chemistry, but so too does the substrate termination.
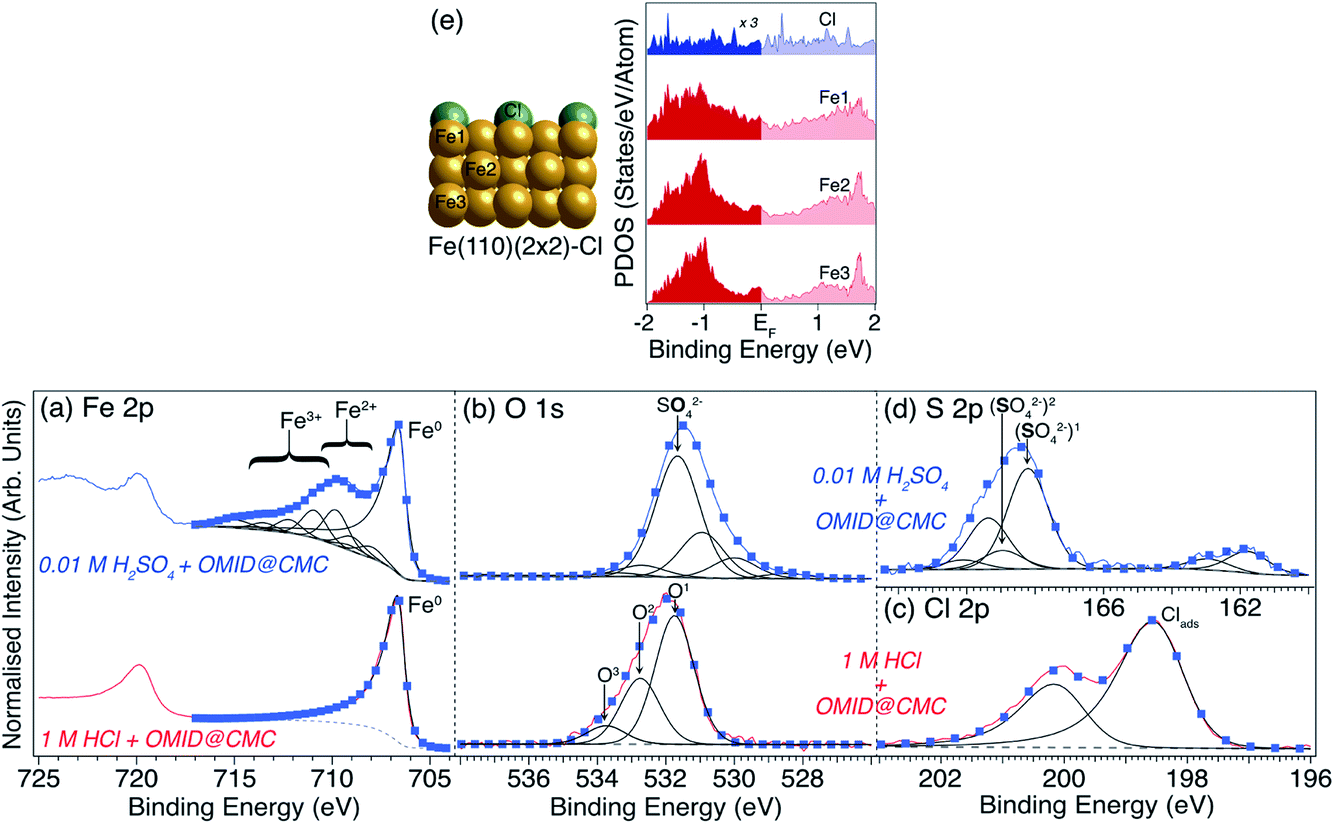 | ||
| Fig. 7 (a) Fe 2p, (b) O 1s, (c) Cl 2p and (d) S 2p core level XPS spectra acquired from carbon steel following immersion for 4 h in either 1MHCl/OMID (lower row) or 0.01MH2SO4/OMID (upper row). Spectra have been normalised such that minima are 0, and maxima are 1. Blue markers show best fits to the experimental spectra (solid red/blue lines), using GL (black lines), LF (black lines), and Shirley-type (broken grey lines) functions. Peak assignments are also indicated. For the Cl 2p and S 2p spin–orbit split doublets (2p1/2 and 2p3/2), the BE differences and intensity ratios are consistent with expected values.11 (e) On the left, side view of the structure of ordered (2 × 2) overlayer of Cl atoms on Fe(110) that was optimised in DFT calculations to produce PDOS for Cl, Fe1, Fe2, and Fe3. The corresponding PDOS are plotted on the right; both Fe and Cl display finite PDOS around EF. | ||
Concentrating on the 1MHCl/OMID data in Fig. 7, the Fe 2p spectrum is consistent with an oxide/salt-free metallic termination, as only features associated with Fe0 are visible; a single Gaussian-convoluted Lorentzian based asymmetric (LF) function20 has been used to fit the Fe 2p3/2 feature. 3 GL(30) components, labelled O1, O2, and O3, have been employed to fit the O 1s signal, which we have previously ascribed to adsorbed OH (O1) and adventitious RCxOy (O1, O2, and O3) species.9 The Cl 2p spectrum exhibits a single spin–orbit doublet, with the Cl 2p3/2 component located at BE ∼ 198.6 eV, which has been interpreted as resulting from surface adsorbed Cl atoms (Clads).9,12
Reflecting on our fitting of the Cl 2p spectrum, a notable detail is that two asymmetric LF line shape functions have been used, rather than GL functions. Such line shapes are more typically associated with the XPS core levels of metallic materials, e.g., Fe 2p of carbon steel, as per above. In metals, the origin of this asymmetry depends upon there being a finite density of states at the Fermi level (EF). Upon photoexcitation of a core level, this electronic structure gives rise to a weighted continuum of final states through electron–hole pair generation, and so a spectral profile with a tail to higher BE.21 As argued in ref. 12, analogous line shapes are expected for Clads, as there is appreciable hybridisation between substrate Fe0 and Clads resulting in a finite PDOS located on Clads in the vicinity of the EF. The existence of these PDOS is demonstrated in Fig. 7(e), which shows the results of DFT calculations performed for a (2 × 2) ordered overlayer of atomic chlorine on Fe(110).
Moving to the Fe 2p spectrum (Fig. 7(a)) of carbon steel following immersion in 0.01MH2SO4/OMID, the profile is substantially different to that of the sample exposed to 1MHCl/OMID. As annotated, there are features that can be assigned to the presence of both Fe2+ and Fe3+, with the latter being the major cationic species; the presented Fe2p3/2 fit comprises a LF line shape function for Fe0, along with a series of GL functions to describe the Fe2+/3+ multiplet states and satellites.22,23 Evidently, in sharp contrast to the inhibited interface formed in 1MHCl/OMID, the substrate termination is not oxide/salt-free metallic carbon steel following exposure to 0.01MH2SO4/OMID.
Comparing the O 1s spectra (Fig. 7(b)) of carbon steel immersed in either 1MHCl/OMID or 0.01MH2SO4/OMID, they are far from identical. For the latter, constraining each GL(30) function to have the same FWHM (i.e. ∼1.5 eV), six components were required to achieve a satisfactory fit to the experimental data. Unique assignment of each of these peaks has proven to be unfeasible at this juncture, but we propose that the most intense signal, labelled SO42− (BE ∼ 531.7 eV), originates from substrate bound SO42− anions.24,25 In addition, the peaks located at BEs of ∼530.0 eV and ∼531.0 eV are likely to be associated with surface iron oxides and hydroxides, respectively.9,12
As regards the existence of surface SO42−, the S 2p spectrum in Fig. 7(d) supports this deduction. More explicitly, the signal observed in the range 166 eV ≲ BE ≲ 171 eV is well known to be due to photoemission from highly oxidised sulfur species.24,25 Two S 2p doublets were used to fit this region, with S 2p3/2 BEs of ∼168.2 eV (labelled (SO42−)1) and ∼169.0 eV (labelled (SO42−)2), which are concluded to be both due to surface bound SO42−.24,25 The reason for two SO42− related doublets is uncertain but may be due to significant heterogeneity at the carbon steel surface. It should be noted that reduction of S to a lower formal oxidation state is a less likely explanation, as the BE difference would almost certainly be larger.25 There is also a lower intensity doublet in Fig. 7(d) located at much lower BE (S 2p3/2 BE ∼ 161.8 eV), which is attributed to surface sulfides, potentially arising from solution contaminants or possibly a SO42− reduction reaction.
Merging our analysis of the Fe 2p, O 1s, and S 2p core level XPS spectra acquired from carbon steel following immersion in 0.01MH2SO4/OMID, we posit the substrate termination depicted in Fig. 8. OMID is adsorbed onto an ultra-thin oxide/salt film, composed primarily of a ferric sulfate (Fe2(SO4)3)-like phase, which is formed atop the carbon steel. Also shown in Fig. 8 is the inhibited interface concluded to form on carbon steel in 1MHCl/OMID. In this case, OMIDH+ adsorbs onto an oxide/salt-free carbon steel surface decorated with chlorine atoms (Clads).
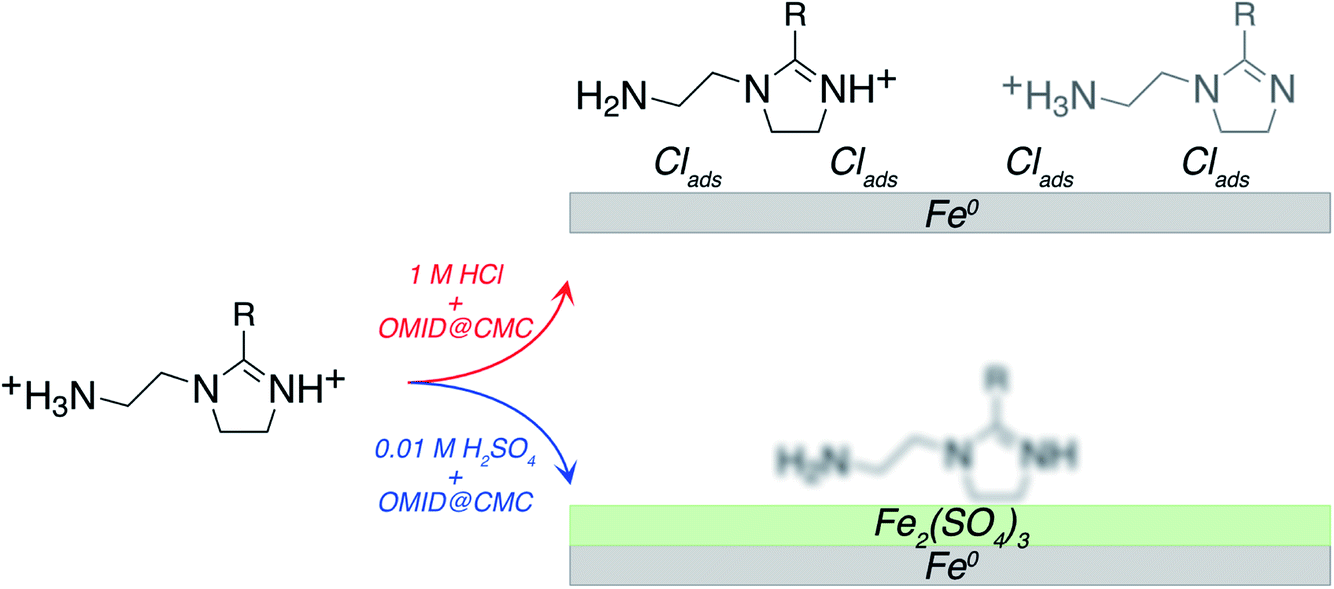 | ||
| Fig. 8 Cartoon summarising interpretation of N 1s, Fe 2p, O 1s, Cl 2p, and S 2p XPS spectra acquired from carbon steel following immersion for 4 h in either 1MHCl/OMID or 0.01MH2SO4/OMID. For 1MHCl/OMID, two OMIDH+ species are concluded to be adsorbed onto an oxide/salt-free surface, decorated with chlorine atoms. For 0.01MH2SO4/OMID, the exact nature of the adsorbed OMID remains ill-defined, but it is adsorbed onto an ultra-thin oxide/salt film, composed primarily of a ferric sulfate (Fe2(SO4)3)-like phase. R = C17H33. | ||
Regarding the above results, to our knowledge, these are the first data to explicitly demonstrate that the identity of the aqueous acid can determine the chemistry of a well-inhibited interface. Hence, speculation about the origin of this variation is of value. With regards to the surface termination being a ferric sulfate like layer in 0.01MH2SO4/OMID, one potentially salient point is that carbon steel is protected from corrosion by a layer of ferrous sulfate in highly concentrated H2SO4 (≳ 9 M);26 no analogous protective iron chloride film forms on carbon steel in concentrated HCl. On this basis, in 0.01MH2SO4/OMID, perhaps sorption of OMID limits dissolution of the ferric sulfate through formation of a protective hybrid surface film, thus inhibiting corrosion of the carbon steel.
Concerning 1MHCl/OMID, one topic of interest is the role of the adsorbed chlorine atoms, i.e., ‘Is Clads essential to corrosion inhibition or simply a spectator adsorbate?’. Referring to Table 2, the η% of OMID at CMC in 0.01 M HCl is ∼86%, which is a decrease of about 11% compared to 1 M HCl. This trend suggests that Clads does play a role in the adsorption/performance of OMID as a corrosion inhibitor in HCl. To test this hypothesis, we have determined η% for carbon steel in a solution consisting of OMID at CMC, 0.01 M HCl and 1 M NaCl, so that the total Cl− concentration is 1.01 M, which is similar to that in 1 M HCl. It was found that η% = 94.6 ± 1.8%, suggesting that Clads is required to ensure that OMID exhibits excellent corrosion inhibition in hydrochloric acid. We note that contrary to this variation in OMID performance with HCl concentration, Table 2 indicates that there is little loss in η% as the concentration of H2SO4 is reduced from 1 M to 0.01 M. This distinction is probably a consequence of a rather different inhibition mechanism, as discussed above.
Finally, we would like to make some comments on the approach adopted for both acquisition and analysis/interpretation of the presented XPS data. For the former, the importance of using the N2-filled glove box for sample transfer into the XPS facility, to avoid post-immersion oxidation, cannot be over emphasised. Similar measures are apparently not taken in many other related studies, including some conducted quite recently,27–29 which almost certainly leads to flawed interface characterisation. It should be borne in mind, however, that even using the glove box, the spectra have been acquired ex situ, and interface modification remains a concern, e.g., adventitious carbon, which is almost inevitable, may modify corrosion inhibitor adsorption geometry, if not the chemistry. Furthermore, the process of removing a sample from solution may strip off weakly bound corrosion inhibitors, e.g., those forming the outer part of an adsorbed bilayer (Fig. 1(a)).
As to data analysis/interpretation, our goal is to achieve a physically/chemically reasonable description of the inhibited interfaces through fitting of the spectral profiles with model line shape functions. We attempt to assign each peak to a specific state, although this can prove intractable on occasion, e.g., the N 1s peak acquired in 0.01MH2SO4/OMID (Fig. 5). Given the complexity of the systems studied, in our opinion, this lack of clarity, does not invalidate the effort, but indicates that other techniques are likely required to complete characterisation, e.g., X-ray absorption spectroscopy. In addition, atomic scale modelling may also facilitate more complete interface elucidation. Here, we have already demonstrated that ab initio DFT calculations can be used to support the assignment of core level peaks for HG-OMID, as well as predict the most favourable protonated states (HG-OMID → HG-OMIDH+). Furthermore, DFT calculations have allowed us to justify the use of an asymmetric line shape function for fitting Cl 2p features assigned to Clads (Fig. 7(c)). As highlighted in another recent study,30 we suggest that proper selection of model line shape functions will become increasingly important moving forward, as system complexity increases, e.g., in near ambient pressure XPS studies, multiple components are commonly required to fit an XPS core level spectrum.
Conclusions
In this study, it has been demonstrated that, through application of rigorous methodology, XPS can be employed to effectively characterise interfaces formed through surface interaction of organic corrosion inhibitors with metallic substrates in acidic solutions. Most notably, it has been determined that for corrosion inhibition of carbon steel by OMID, the nature of the well-inhibited interface is dependent on the acid. In 1 M HCl, it is concluded that OMID, assisted by the presence of Clads, adsorbs as two different singly protonated species onto an oxide/salt-free carbon steel surface. In sharp contrast, in 0.01 M H2SO4, OMID adsorbs onto an ultra-thin surface film, composed primarily of a ferric sulfate (Fe2(SO4)3)-like phase.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding and technical support from AkzoNobel and Nouryon through a collaboration with The University of Manchester is acknowledged. KK and MD are appreciative of funding from the EPSRC (EP/L01680X/1) through the Materials for Demanding Environments (M4DE) Centre for Doctoral Training (CDT). Furthermore, we are grateful for additional funding and technical support from bp through the bp International Centre for Advanced Materials (bp-ICAM) and EPSRC through the prosperity partnership (EP/G036850/1).References
- G. Koch, J. Varney, N. Thompson, O. Moghissi, M. Gould and J. Payer, NACE 2016 Impact Study, International Measures of Prevention Application, and Economics of Corrosion Technologies Study, 2016 Search PubMed.
- R. Lindsay and S. B. Lyon, in Shreir’s Corrosion, vol. 4, Management and Control of Corrosion, ed. R. A. Cottis, M. J. Graham, R. Lindsay, S. B. Lyon, J. A. Richardson, J. D. Scantlebury and F. H. Stott, Elsevier, Amsterdam, 2010, pp. 2891–2899 Search PubMed.
- M. Finšgar and J. Jackson, Corros. Sci., 2014, 86, 17–41 CrossRef.
- Y. Zhu, M. L. Free, R. Woollam and W. Durnie, Prog. Mater. Sci., 2017, 90, 159–223 CrossRef CAS.
- O. Olivares-Xometl, N. V. Likhanova, M. A. Domínguez-Aguilar, J. M. Hallen, L. S. Zamudio and E. Arce, Appl. Surf. Sci., 2006, 252, 2139–2152 CrossRef CAS.
- O. Olivares-Xometl, N. V. Likhanova, R. Martínez-Palou and M. A. Domínguez-Aguilar, Mater. Corros., 2009, 60, 14–21 CrossRef.
- M. Yadav, D. Behera and U. Sharma, Arabian J. Chem., 2016, 9, S1487–S1495 CrossRef CAS.
- K. Kousar, T. Ljungdahl, A. Wetzel, M. Dowhyj, H. Oskarsson, A. S. Walton, M. S. Walczak and R. Lindsay, J. Surfactants Deterg., 2020, 23, 225–234 CrossRef CAS.
- K. Kousar, M. S. Walczak, T. Ljungdahl, A. Wetzel, H. Oskarsson, P. Restuccia, E. A. Ahmad, N. M. Harrison and R. Lindsay, Corros. Sci., 2021, 180, 109195 CrossRef CAS.
- R. A. Cottis, in Shreir’s Corrosion, vol. 2, Corrosion in Liquids, Corrosion Evaluation, ed. R. A. Cottis, M. J. Graham, R. Lindsay, S. B. Lyon, J. A. Richardson, J. D. Scantlebury and F. H. Stott, Elsevier, Amsterdam, 2010, pp. 1341–1373 Search PubMed.
- ThermoFisher Scientific, https://www.thermofisher.com/uk/en/home/materials-science/learning-center/periodic-table.html, accessed November 20, 2021.
- P. Morales-Gil, M. S. Walczak, R. A. Cottis, J. M. Romero and R. Lindsay, Corros. Sci., 2014, 85, 109–114 CrossRef CAS.
- M. S. Walczak, P. Morales-Gil, T. Belashehr, K. Kousar, P. Arellanes Lozada and R. Lindsay, J. Visualized Exp., 2017, 55163 Search PubMed.
- R. Dovesi, V. Saunders, C. Roetti, R. Orlando, C. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. Harrison, I. Bush, P. D’Arco, M. Llunell, M. Causà, Y. Noël, L. Maschio, A. Erba, M. Rerat and S. Casassa, CRYSTAL17 User’s Manual, University of Torino, 2017 Search PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1360 Search PubMed.
- M. S. Walczak, P. Morales-Gil and R. Lindsay, Corros. Sci., 2019, 155, 182–185 CrossRef CAS.
- J. S. Stevens, S. J. Byard and S. L. M. Schroeder, Cryst. Growth Des., 2010, 10, 1435–1442 CrossRef CAS.
- J. S. Stevens, A. C. de Luca, M. Pelendritis, G. Terenghi, S. Downes and S. L. M. Schroeder, Surf. Interface Anal., 2013, 45, 1238–1246 CrossRef CAS.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2014, 16, 1150–1160 RSC.
- N. Fairley, CasaXPS Manual 2.3.15 Spectroscopy 1.3, Casa Software Ltd, 2009 Search PubMed.
- G. Wertheim and P. Citrin, in Topics in Applied Physics, Photoemission in Solids I, General Principles, ed. M. Cardona and L. Ley, Springer-Verlag, Berlin, Heidelberg, New York, 1978, pp. 197–236 Search PubMed.
- P. Gupta and S. K. Sen, Phys. Rev. B: Solid State, 1975, 12, 15–19 CrossRef.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Appl. Surf. Sci., 2000, 165, 288–302 CrossRef CAS.
- M. Wahlqvist and A. Shchukarev, J. Electron Spectrosc. Relat. Phenom., 2007, 156–158, 310–314 CrossRef CAS.
- Z. Panossian, N. L. de Almeida, R. M. F. de Sousa, G. de S. Pimenta and L. B. S. Marques, Corros. Sci., 2012, 58, 1–11 CrossRef CAS.
- J. Haque, V. Srivastava, M. A. Quraishi, D. Singh Chauhan, H. Lgaz and I.-M. Chung, Corros. Sci., 2020, 172, 108665 CrossRef CAS.
- A. Espinoza-Vázquez, F. J. Rodríguez-Gómez, G. E. Negrón-Silva, R. González-Olvera, D. Ángeles-Beltrán, M. Palomar-Pardavé, A. Miralrio and M. Castro, Corros. Sci., 2020, 174, 108853 CrossRef.
- L. Zhu, J. Fan, H. Huang, L. Guo, M. Zhu, X. Zheng and I. B. Obot, J. Mol. Struct., 2021, 1243, 130852 CrossRef CAS.
- C. R. O’Connor, M. A. van Spronsen, M. Karatok, J. Boscoboinik, C. M. Friend and M. M. Montemore, J. Phys. Chem. C, 2021, 125, 10685–10692 CrossRef.
Footnotes |
| † The critical micelle concentration (CMC) is the concentration of surfactant above which micelles (aggregates of surfactant molecules) start to form. |
| ‡ p, which lies in the range 0 ≤ p ≤ 100, describes the degree of Gaussian–Lorentzian mixing, where GL(100) (GL(0)) is a pure Lorentzian (Gaussian) line shape. |
| This journal is © The Royal Society of Chemistry 2022 |