
Reactive halogen radicals in saline water promote photochemically-assisted formation of manganese oxide nanosheets†
Zhenwei
Gao
 ,
Charlie
Skurie
and
Young-Shin
Jun
*
,
Charlie
Skurie
and
Young-Shin
Jun
*
Department of Energy, Environmental and Chemical Engineering, Washington University in St. Louis, One Brookings Drive, Campus Box 1180, St. Louis, Missouri 63130, USA. E-mail: ysjun@seas.wustl.edu; Fax: +(314) 696 1223; Tel: +(314) 935 4539 Web: https://encl.engineering.wustl.edu/
First published on 30th July 2022
Abstract
Halide ions are naturally abundant in oceans and estuaries. Large amounts of highly saline efflux are also made and discharged to surface water from desalination processes and from unconventional oil and gas recovery. These highly concentrated halides can generate reactive halogen radicals. However, the redox reactions of halogen radicals with heavy metals or transition metals have received little attention. Here, we report undiscovered fast oxidation of manganese ions (Mn2+) by reactive halogen radicals. Hydroxyl radicals (˙OH) are produced by nitrate photolysis. While ˙OH radicals play a limited role in the direct oxidation of Mn2+, ˙OH can react with halide ions to generate reactive halogen radicals to oxidize Mn2+. In addition, more Mn2+ was oxidized by bromide (Br) radicals generated from 1 mM Br− than by chloride (Cl) radicals generated from 500 mM Cl−. In the presence of Br radicals, the abiotic oxidation rate of Mn2+ to Mn(IV)O2 nanosheets is greatly promoted, showing a 62% increase in Mn2+(aq) oxidation within 6 h of reaction. This study advances our understanding of natural Mn2+ oxidation processes and highlights unexpected impacts of reactive halogen radicals on redox activities with heavy metals and corresponding nanoscale solid mineral formation in brine. This work suggests a new, environmentally-friendly, and facile pathway for synthesizing MnO2 nanosheets.
Environmental significanceReactive halogen radicals form from abundant halides in saline surface water and in the highly saline water of energy–water recovery systems. Here, we demonstrate an undiscovered manganese oxidation pathway by reactive halogen radicals. This is the first report of reactive halogen radicals oxidizing transition metal ions and forming oxidized solids under environmentally relevant pH. Bromide radicals in particular greatly increase the rate of abiotic manganese oxidation. These findings advance our understanding of abiotic Mn2+ oxidation and Mn oxide nanosheet formation mechanisms, especially in oceans, estuaries, and brine water systems, which will play a significant role in global biogeochemical and elemental redox cycling. Given the ever-increasing amounts of halide-containing efflux from desalination water treatment and subsurface energy recovery systems, halides and reactive halogen species are becoming even more important in the formation of redox active minerals via mechanisms similar to Mn oxidation. This study emphasizes the unforeseen impact of reactive halogen radicals and predicts their reactions with heavy metal ions other than manganese in environmental systems. |
1. Introduction
Interdependent energy–water engineered systems inherently produce large volumes of briny water. To meet rapidly growing clean water demands, desalination processes are increasingly used, consistently generating highly concentrated brine efflux (1.5–2 times as salty as seawater) that is directly released into adjacent lakes, rivers, and near shore seawater.1,2 In unconventional oil and gas recovery through hydraulic fracturing, large amounts of concentrated flowback brine (0.9–4.17 M Cl−, and 9–20 mM Br−) can accumulate in the surface impoundment before further treatment.3 As examples, Marcellus and Fayetteville hydraulic fracturing flowback fluids contain about 160 g L−1 (4.5 M) of Cl− and 1.8 g L−1 (22.5 mM) of Br−.4 The contaminated surface water from the effluent disposal sites in Pennsylvania contain about 60 g L−1 (1.7 M) of Cl− and 40 mg L−1 (0.5 mM) of Br−.4 These highly concentrated halides can be oxidized by the hydroxyl radicals (˙OH) and triplet state dissolved organic matter to form abundant reactive halogen radicals (e.g., X˙, X2˙−, XY˙−, where X, Y = Cl, Br, or I).5,38 In seawater, which usually contains about 0.54 M Cl− and 0.8 mM Br−, the concentrations of their corresponding halogen radicals, e.g., Br2˙− and ClBr˙−, can be 3–4 orders of magnitude higher than that of ˙OH.5,6Studies thus far have focused on the effects of reactive halogen radicals on the degradation and transformation of organic pollutants, such as microcystins in estuarine water,7 and various pharmaceuticals and personal care products (PPCPs) in water treatment processes.8 In particular, compared to fresh water, seawater increased the photodegradation kinetics of domoic acid (a cis–trans diene) because the generated reactive halogen radicals selectively targeted dienes.6 In addition, reactive halogen radicals can participate in natural oxidative reactions of mercury (Hg) in the atmosphere. A study conducted in Alaska reported that atmospheric elemental Hg was rapidly converted to reactive gaseous oxidized Hg species by reactive halogen radicals.9 However, the roles of reactive halogen radicals in inorganic aqueous systems, such as redox reactions with heavy metal and transition metal ions, remain largely unknown.
Manganese (Mn) oxidation by reactive halogen radicals under the environmental relevant conditions and the roles of reactive halogen radicals in the formation of Mn oxide minerals have never been reported. Mn oxide solids are excellent scavengers of redox-active species and heavy metals in nature, and they play significant roles in geochemical element redox cycling, carbon mineralization, and biological metabolisms.10–12 In nature, Mn oxide solids are known to be formed by oxidation of Mn2+(aq). In such processes, hitherto, O2˙− has been considered the most effective radical accomplishing Mn oxidation in both biotic and abiotic systems.13–17 However, Mn oxidation by reactive halogen radicals in nature has not been considered fully. Laurence et al. (1973) suggested that Br2˙− and Cl2˙−, generated by short duration (<0.1 second) flash photolysis of trihalide ions, oxidized Mn2+(aq) at an extremely low pH (∼1), and Mn3+(aq) was proposed to form rather than Mn(IV) oxide solids.18 Nevertheless, Mn3+(aq) is not stable at circumneutral pH in the absence of a ligand,19 so it is unclear whether these reactions can occur under environmentally relevant pH conditions. Further, it is unknown whether reactive halogen radicals can facilitate the oxidation of Mn2+(aq) and form nanoscale Mn(IV) oxide solids as the final products for a longer light illumination time rather than within one second.
Here, we show previously unknown effects of reactive halogen radicals on the oxidation of Mn2+(aq) and the fast kinetics of nanoscale Mn(IV) oxide solids formation. Reactive oxygen species (ROS), including O2˙− and ˙OH, were produced during the photolysis of solution containing nitrate.20 When halides were present in the solution, reactive halogen radicals can be generated by reactions between the halides and ˙OH, increasing the oxidation rate of Mn2+ and also increasing the rate and extent of Mn oxide nanosheets formation, compared with the rates of phenomena in the absence of halides. More Mn2+ was oxidized by Br radicals (Br˙ and Br2˙−) generated from 1 mM NaBr than by Cl radicals (Cl˙ and Cl2˙−) generated from 500 mM NaCl. Ultimately, δ-MnO2 nanosheets were formed by reactive halogen species. The crystallinity of the formed Mn oxide nanosheets was affected by the ionic strength. This newly discovered reaction mechanism between Mn2+(aq) and reactive halogen radicals under environmental relevant conditions can advance our understanding of nanoscale Mn oxide solids formation in the environment, and can help to predict its redox activities, such as scavenging and oxidation of heavy metals (As, Pb, and Cr) and geochemical elements (S, N, and P).
2. Materials and methods
2.1 Chemicals and materials
All chemicals used in this study were at least American Chemical Society grade. Manganese chloride (MnCl2, 97%, anhydrous) was purchased from Beantown Chemical Co. (NH, USA). Crystal sodium nitrate (NaNO3, ≥99.0%) was obtained from Avantor Performance Materials, Inc. (PA, USA). Leucoberbelin blue I (LBB, 65%), potassium permanganate (KMnO4, >99%), sodium sulfate (Na2SO4, ≥99%), sodium bromide (NaBr, ≥99.99%), superoxide dismutase bovine (SOD, >90%), and 2-bromoacetophenone (BrAP, >98%) were purchased from Sigma-Aldrich. Sodium hydroxide (NaOH, >97%) and sodium chloride (NaCl, >99%) were bought from VWR International LLC. Deionized (DI) water (resistivity ≥18.2 MΩ cm, Barnstead Ultrapure water systems) was used to prepare the solutions for all experiments. 0.2 μm polytetrafluoroethylene (PTFE) membrane was bought from VWR International, LLC.2.2 Photo-oxidation experiments
Photochemical experiments for Mn2+ oxidation were carried out in a 150 mL quartz reactor under a 450 W Xe-arc lamp (Newport 6279NS) equipped with a water optical filter to remove infrared light. The lamp provided stable light exposure with an irradiance intensity of ∼3.7 kW m−2. The light spectrum of the xenon arc lamp is shown in Fig. S1.† Although the high irradiance intensity helped accelerate the reaction, the mechanism presented in this work is still representative of that under sunlight conditions. The effects of light intensity on the photolysis of nitrate and subsequent Mn2+ oxidation merit systematic future studies. Solutions of MnCl2, NaNO3, NaBr, NaCl, and SOD were exposed to the light for 6 hours.0.1 mM MnCl2 was used as the Mn source. The concentrations of dissolved Mn in natural waters can range from 10 μg L−1 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg L−1 (0.18 μM–0.18 mM).21–23 Mandernack et al. examined Mn oxide mineral formation under environmentally relevant Mn concentrations of 10 μM–10 mM.24 The Mn2+ concentrations used in this study might be higher than those in natural surface waters, however, these results can still be useful in understanding diverse geochemical situations. In addition, 1 mM NaNO3 was used in this study. According to a 2011 World Health Organization report on nitrate and nitrite in drinking water, in 1986, nitrate concentrations higher than 44 mg L−1 (0.71 mM) were detected in many surface water supplies.25 Nitrate concentration can reach high levels in industrial areas or from contamination by human or animal wastes. For example, nitrate concentration can easily come up to several hundred mg L−1 (several mM) due to agricultural activities.25 Moreover, the ammonium in Marcellus and Fayetteville hydraulic fracturing flowback fluids can reach 420 mg L−1 (30 mM),4 and it can be further converted to nitrate.
000 μg L−1 (0.18 μM–0.18 mM).21–23 Mandernack et al. examined Mn oxide mineral formation under environmentally relevant Mn concentrations of 10 μM–10 mM.24 The Mn2+ concentrations used in this study might be higher than those in natural surface waters, however, these results can still be useful in understanding diverse geochemical situations. In addition, 1 mM NaNO3 was used in this study. According to a 2011 World Health Organization report on nitrate and nitrite in drinking water, in 1986, nitrate concentrations higher than 44 mg L−1 (0.71 mM) were detected in many surface water supplies.25 Nitrate concentration can reach high levels in industrial areas or from contamination by human or animal wastes. For example, nitrate concentration can easily come up to several hundred mg L−1 (several mM) due to agricultural activities.25 Moreover, the ammonium in Marcellus and Fayetteville hydraulic fracturing flowback fluids can reach 420 mg L−1 (30 mM),4 and it can be further converted to nitrate.
To study the effects of different reactive halogen radicals on Mn oxidation, 1 mM NaBr and 500 mM NaCl were added either separately or together into a solution. These halide concentrations represent a highly saline aqueous environment, including seawater and other water bodies with similar concentrations. Adding 500 mM NaCl into 1 mM NaBr greatly increased the ionic strength (IS) of the solution. Thus, for better comparison, the same IS was maintained between different experimental conditions. Although previous studies have substituted sodium perchlorate (NaClO4) for NaCl to maintain the same IS and Na+ concentration in a solution,6 NaClO4 was not used in this study because it could oxidize Mn2+ during photochemical reactions by forming O2˙−, even without nitrate (Fig. S2†). Instead, Na2SO4 was used to control the IS. To scavenge O2˙−, 0.5 μM SOD was utilized. We did not consider another halide ion, such as iodine, in this study because of its low concentrations in seawater (450 to 500 nM)26 and in contaminated surface waters from the brine treatment disposal sites for hydraulic fracturing (e.g., in Pennsylvania hydraulic fracturing site, [I−] ≤ 7.9 μM, [Cl−] = 1.7 M, [Br−] = 0.5 mM).4
Based on the water quality criteria set by the U.S. Environmental Protection Agency (EPA), freshwater pH ranges from 6.5 to 9.27,28 To mimic environmentally relevant conditions, in this study, the initial pH value was adjusted to 9.0 ± 0.1 using 10 mM NaOH and the pH decreased within 6 hours as the Mn oxide solids formed. To understand the pH of the reaction systems, the pH values were continuously monitored. No pH buffer was utilized to maintain the pH, because Mn3+(aq) forms strong complexation with pH buffers, such as pyrophosphate, EDTA, and citrate.29,30 The presence of pyrophosphate also affected the crystalline structure, morphology, and oxidation kinetics of MnO2.29 In addition, carbonate ions were not used to control the pH because they can react with ˙OH and reactive halogen radicals to form carbonate radicals,31,32 which would affect Mn2+(aq) oxidation processes. After 6 h of reaction, the solution pH dropped to the range of 5–7, as shown in Fig. 2B and S8.† As proved in our previous study, the pH drop was due mainly to the formation of Mn oxide solids rather than CO2 dissolution.17 As shown in Fig. 2A(5), during the photolysis of 0.1 mM MnCl2, 1 mM NaNO3, and 0.5 μM SOD, O2˙− was scavenged by SOD, but ˙OH and dissolved CO2 from the air were still present in the solution and could form carbonate radicals. However, no Mn oxidation was observed. Thus, we expect that the concentration of CO2 from dissolution in air in 6 h was not sufficient to play an important role in generating carbonate radicals to induce Mn oxidation. Furthermore, this CO2 dissolution process occurred under all experimental conditions and was offset when the results were compared. So the effect of carbonate radicals on Mn oxidation was not considered in this study.
2.3 Quantification of formed nanoscale Mn oxide solids
Concentrations of MnO2 were quantified by using 0.004% (w/v) LBB as reported before.17,33,34 LBB reduced the high oxidation states of Mn(IV) and Mn(III) in Mn oxide solids to aqueous Mn2+, and then displayed a blue color (Fig. S3B†). The intensity of oxidized LBB at 625 nm was proportional to the concentration of Mn oxide solids. For LBB analyses, different concentrations of KMnO4 solutions were used to obtain a linear calibration curve. In these measurements, LBB analyses cannot, however, distinguish whether the detected oxidized Mn came from the solid phase of Mn oxides or aqueous oxidized Mn. Thus, 0.2 μm PTFE membrane was used to filter out the formed Mn oxide solids and leave aqueous oxidized Mn in the solution. In this way, the existence of aqueous oxidized Mn can be quantified by measuring the solutions after membrane filtration.2.4 Phase characterization of Mn oxide solids
Photochemically-assisted synthesized solid MnO2 products were collected by centrifugation at 10000 rpm for 30 min. To remove impurities from the liquid supernatant, the solid products were sequentially washed, resuspended in DI water, and centrifuged five times at 5000 rpm for 5 min. The resulting solid products were dried at room temperature for further characterization.
To identify the mineral phase of Mn oxides samples, we used high-resolution X-ray diffraction (HRXRD, Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å)). Because of its low saturation index (SI) of −1.20 and solubility product constant (ksp) of 1.6 × 10−13,35 Mn(OH)2 (s) was not expected to form under above experimental conditions. MINEQL+ Version 4.6 was used to calculate the saturation indices for typical Mn(III)/Mn(IV) oxides based on pH ranging from pH 9 to 6. Common Mn oxide minerals, for example, birnessite (δ-MnO2), pyrolusite (β-MnO2), hausmannite (Mn3O4), and manganite (MnOOH) have SI values of 9.48–5.89, 11.54–7.95, 15.26–4.16, and 6.96–3.29, respectively, making them possible precipitates.
Mn oxidation states in the Mn oxides samples were identified by X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe II, UlvacPHI with monochromatic Al Kα radiation (1486.6 eV)). The C 1s peak (284.8 eV) was employed as the reference peak. To determine the ratio of Mn(II), Mn(III) and Mn(IV), the Mn 2p3/2 spin orbit was fitted with Mn(II) (640.8 eV), Mn(III) (641.8 eV) and Mn(IV) (642.2 eV) by the Gauss–Lorentz fitting method. Sources for the fitting reference values of Mn(II), Mn(III), and Mn(IV) are listed in Table S1.† An environmental scanning electron microscope (ESEM, ThermoFisher Quattro S) with an energy dispersive X-ray spectrometer (EDX) was used to characterize the surface morphology and material composition. Mn oxides samples were placed on a silicon wafer and visualized at a 10 mm working distance with a 10 kV accelerating voltage.
To characterize the crystalline phases and morphology of Mn oxide solids, high resolution transmission electron microscopy (HRTEM, JEOL-2100F) was used. The Mn oxide solids were well dispersed by sonication for about 15 min. Then a droplet of the suspension was placed on an ultrathin lacey carbon film coated-Cu grid (LC400-Cu-UL, Electron Microscopy Science, PA) for imaging. Lattice fringes and selective area electron diffraction images were obtained to identify the phases of Mn oxide solids. To measure the height (thickness) of MnO2 nanosheets, tapping mode atomic force microscopy (AFM) using a Nanoscope V multimode scanning probe microscope (SPM, Veeco Inc., NY) in the air was used. To measure the hydrodynamic diameter and zeta potential of Mn oxide nanosheets in solutions, a Zetasizer Nano (Malvern Nano ZS) was used.
2.5 Alternative method to generate Br radicals
Photolysis of BrAP was used in an alternative method to generate Br radicals using a solution of 0.1 mM MnCl2, 1 mM BrAP, and 0.5 μM SOD under photolysis. Mn oxide concentration and pH were measured every minute. At least duplicate tests were conducted for each condition.3. Results and discussion
3.1 ROS responsible for Mn2+ oxidation
Fig. 1A shows the reaction pathways that generate nitrogen oxide radicals (NO˙ or NO2˙), oxide radical anion (O˙−), O2˙−, and ˙OH during nitrate photolysis.20 Among these products, O2˙− and ˙OH are the most common and most strongly reactive oxidants. We recently proved that the production of ROS by nitrate photolysis can lead to fast oxidation of Mn2+ to nanoscale Mn oxide solids by O2˙−, even without organics or microorganisms.17 In the current study, we found that a solution containing 0.1 mM MnCl2 and 1 mM NaNO3 at initial pH 9 changed the solution color from transparent to yellowish (Fig. S3A†). Using LBB analysis, about 13.4 ± 1.0 μM of Mn oxide solids (based on Mn(IV) oxidation state) were formed within 6 h reaction, as shown in Fig. 2A(2). With 0.5 μM of SOD, an O2˙− scavenger, no Mn oxidation was observed (Fig. 2A(5)), confirming that O2˙− radicals are majorly responsible for Mn oxidation, which is similar to biotic Mn oxidation by extracellular O2˙− radicals.13–16 We have systematically proved ˙OH radicals have little contribution to Mn oxide formation in our previous study.17,33 An addition of tert-butyl alcohol, a scavenger of ˙OH did not suppress Mn oxide formation and indeed increased the oxidation, supporting that Mn2+ is majorly oxidized by O2˙− rather than ˙OH.17,33 While ˙OH radicals are powerful oxidants, they have a much shorter diffusion length and half-life (nanoseconds) compared with O2˙− (seconds).36,37 These limitations may influence Mn2+ oxidation by ˙OH. When the pH became lower, slower Mn oxidation was observed. Hence, Mn oxidation was preferred at higher pH.38 Moreover, the decay of O2˙− can be faster at lower pH values, inhibiting Mn oxidation.39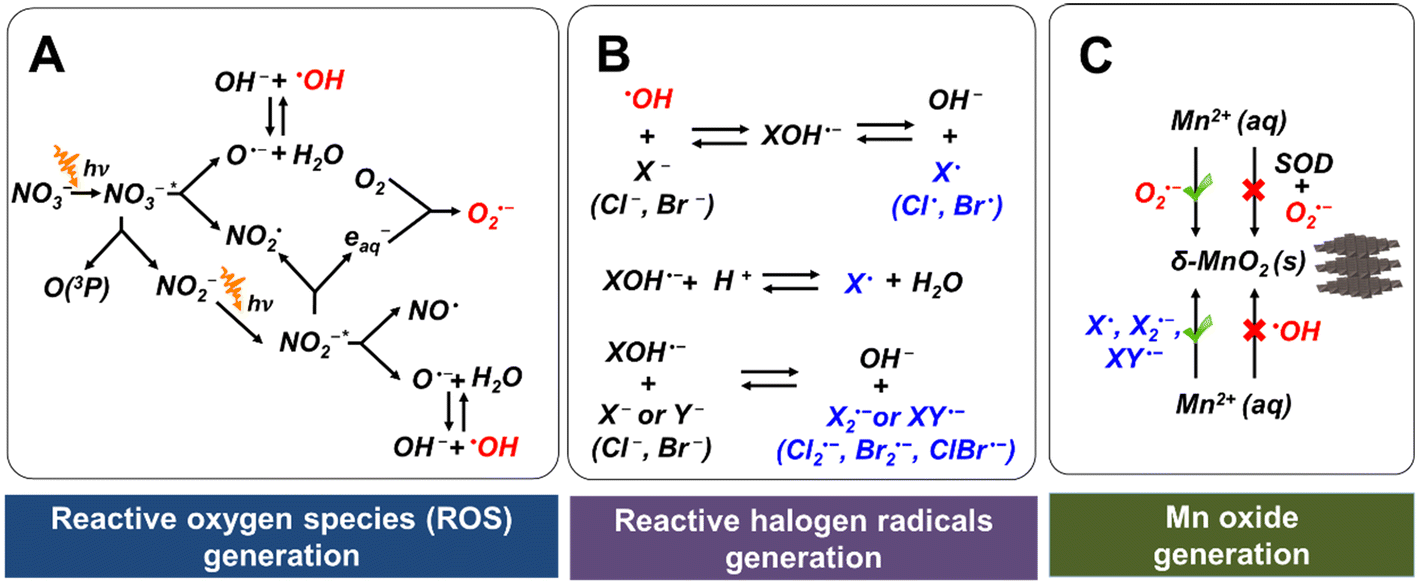 | ||
| Fig. 1 Schematics of the generation pathways of ROS and reactive halogen radicals, and Mn2+ oxidation pathways. (A) Chain reactions in the generation of O2˙− and ˙OH during nitrate photolysis. (B) Chain reactions in the generation of reactive halogen radicals. (C) Mn2+(aq) oxidation by O2˙− and reactive halogen radicals. | ||
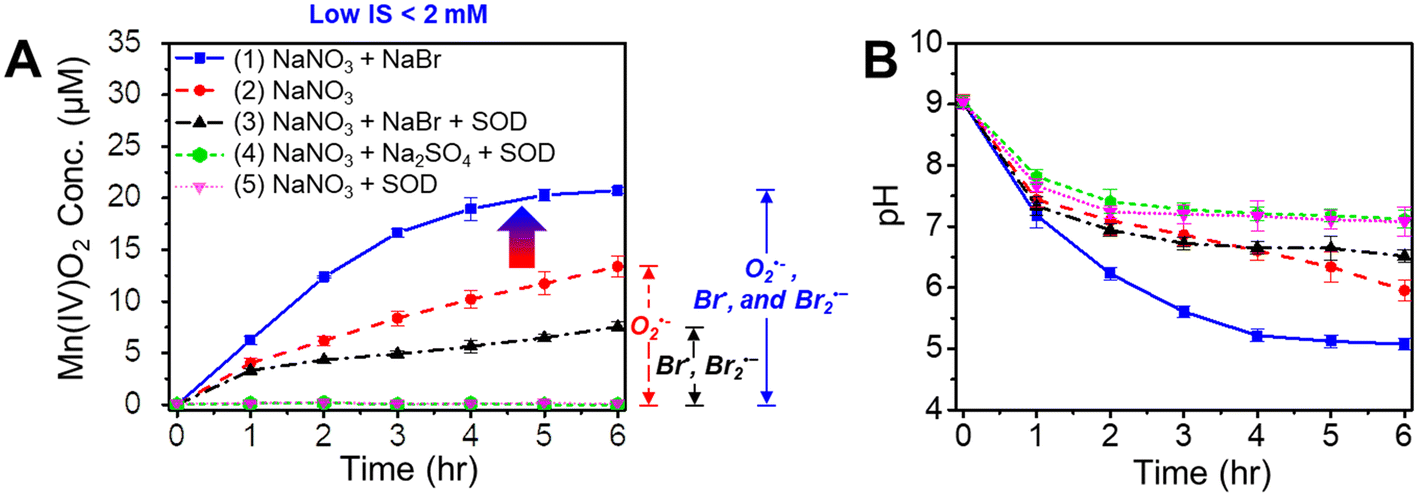 | ||
| Fig. 2 Oxidation of Mn2+(aq) to Mn oxide solids by O2˙− or/and Br radicals. (A) Oxidized Mn concentrations and (B) pH values during 6 h of photochemical reactions of solutions containing (1) 0.1 mM MnCl2, 1 mM NaNO3, and 1 mM NaBr; (2) 0.1 mM MnCl2 and 1 mM NaNO3; (3) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 0.5 μM SOD; (4) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM Na2SO4, and 0.5 μM SOD; and (5) 0.1 mM MnCl2, 1 mM NaNO3, and 0.5 μM SOD. Error bars represent standard errors of the data obtained from duplicate experiments. | ||
3.2 Faster Mn2+ oxidation in the presence of bromide radicals
Interestingly, in the presence of an additional 1 mM NaBr during nitrate photolysis, 62% more Mn2+(aq) was oxidized within 6 h of reaction than that without NaBr (Fig. 2A(1)). Also, the pH of a solution of 0.1 mM MnCl2, 1 mM NaNO3, and 1 mM NaBr decreased to ∼5.0 by 6 h reaction (Fig. 2B), faster than that without NaBr (decreased to ∼pH 6.0), due to the facilitated generation of Mn oxide solids. Fig. 2A(3) shows that Mn oxide solids formed even in a solution of 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 0.5 μM SOD, indicating that new oxidant species, rather than O2˙−, were oxidizing Mn2+(aq) to Mn oxide solids. We hypothesized that the additional Mn oxidation could be attributed to the Br radicals formed by the reactions between ˙OH and Br−. To test this hypothesis, Na2SO4 was substituted for NaBr. Mn oxidation was not detected in a solution of 0.1 mM MnCl2, 1 mM NaNO3, 1 mM Na2SO4, and 0.5 μM SOD (Fig. 2A(4)). Additionally, no Mn oxidation was observed in a solution of 0.1 mM MnCl2 and 1 mM NaBr during photolysis (Fig. S4A†), indicating that NaBr cannot directly oxidize Mn2+ during photolysis. Thus, we concluded that the additional Mn oxide solids are formed by Br radicals generated from the series of reactions between NaBr and ˙OH (Fig. 1B). In the presence of Br radicals, 62% more Mn oxidation was observed than that without Br−, suggesting that abiotic Mn oxidation by reactive radicals makes a significant contribution in nature. Halogen radicals can also transform to nonradical halogen oxidants (e.g., hypohalous acid, HOX), as shown in eqn (1) and (2) below,5 which can then oxidize Mn2+ to Mn oxide solids.40| X˙ + ˙OH → HOX | (1) |
| X2˙− + ˙OH → HOX + X− | (2) |
In addition to Mn oxide solid phase formation, we further explored whether reactive halogen radicals can oxidize Mn2+ to stable aqueous species, i.e., Mn(III)–halide complexation at circumneutral pH. Aqueous samples were taken every hour from a solution containing 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 0.5 μM SOD. The samples were filtered through 0.2 μm polytetrafluoroethylene (PTFE) filters three times to remove all Mn oxide solids in the solution before measuring the Mn(III)–halide concentration. This filtration is necessary because the LBB method only detects the oxidized Mn concentration but cannot distinguish whether it is in the solid or aqueous phase. In the filtered solution (Fig. S4B†), no oxidized aqueous Mn species was detected via the LBB method, which confirmed the absence of aqueous Mn(III) complex in the solution. The initial pH of the solution also affected the Mn oxidation rate (Fig. S5†). Compared with an initial pH of 9, a slower Mn oxidation rate was observed at an initial pH of 6.
3.3 Validation of Mn oxidation by Br radicals
To further validate the finding that Mn2+ can be oxidized by reactive halogen radicals, BrAP was used to directly produce Br˙ by photolysis (Fig. 3),42 rather than generating it from the reaction between halides and ˙OH formed by nitrate photolysis. Because the generated Br˙ can rapidly react with OH− to form ˙OH, and further produce O2˙−via chain reactions (Fig. 3A),6 the potentially formed O2˙− was scavenged by adding SOD. Fig. 3B shows the results of 4 min of photolysis of a solution containing 0.1 mM MnCl2, 1 mM BrAP, and 0.5 μM SOD. About 3 μM Mn oxide solids (based on Mn(IV) oxidation state) were produced, and the solution pH dropped from 5.5 to 4.4, further proving that Mn2+ was indeed oxidized by reactive halogen radicals. Taken together, the above results clearly demonstrated that reactive halogen radicals can contribute significantly to the formation of Mn oxide solids from aqueous Mn2+.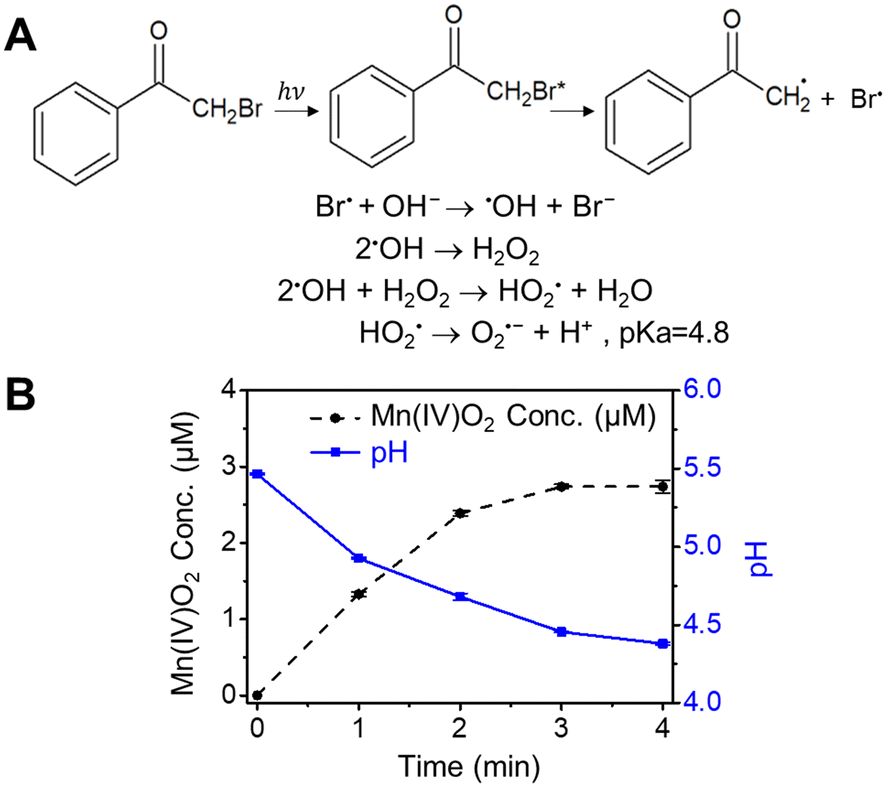 | ||
| Fig. 3 Mn oxidation by Br radicals generated from photolysis of bromoacetophenone (BrAP). (A) Photolysis of BrAP can produce Br˙, which can react with OH− to form ˙OH. O2˙− is potentially formed in the sequential reactions. (B) Oxidized Mn concentration and pH value during photolysis of solution containing 0.1 mM MnCl2, 1 mM BrAP, and 0.5 μM SOD. Error bars represent standard errors of the data obtained from duplicate experiments. | ||
3.4 The different contributions of Cl and Br radicals to Mn2+ oxidation
While ˙OH had little contribution to oxidizing Mn2+(aq) to Mn oxide solids, ˙OH can react with halide ions (Cl− or Br−) to form reactive halogen radicals that oxidize Mn2+(aq). As shown in Fig. 1B, halide ions can be oxidized to monatomic radicals (Cl˙ or Br˙) by ˙OH.43–45 In addition, diatomic radicals (Cl2˙−, Br2˙−, or ClBr˙−) can be generated.43–45 To understand the effects of different halide ions contribution and different ion concentrations on Mn oxidation, we tested Mn oxidation using a solution containing 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, or/and 500 mM NaCl. These NaBr and NaCl concentrations were chosen as a representative condition to study a highly saline aqueous environment. The IS of the solution was greatly changed in the solution containing 0.1 mM MnCl2 and 1 mM NaNO3, and 500 mM of NaCl (IS ∼500 mM), compared with that of a solution containing 0.1 mM MnCl2 and 1 mM NaNO3, and 1 mM NaBr (IS ∼2 mM). As a result, Na2SO4 was added to maintain the same IS between different experimental conditions for better comparison. A high IS of 500 mM strongly hindered the Mn oxidation process. Only ∼7 μM Mn2+ was oxidized by O2˙− after 6 h reaction during the photolysis of a solution containing 0.1 mM MnCl2, 1 mM NaNO3, and 166.7 mM Na2SO4 (IS ∼500 mM) (Fig. 4A(4)), which was only a half the Mn2+ oxidation amount in the solution without Na2SO4 at a lower IS (IS ∼1 mM) (Fig. 2A(2)). Because the higher IS reduced the activity coefficient and the saturation index of precursors with respect to forming Mn oxide solids, it provided a lower thermodynamic driving force for Mn oxide nucleation and could result in a slower formation kinetics.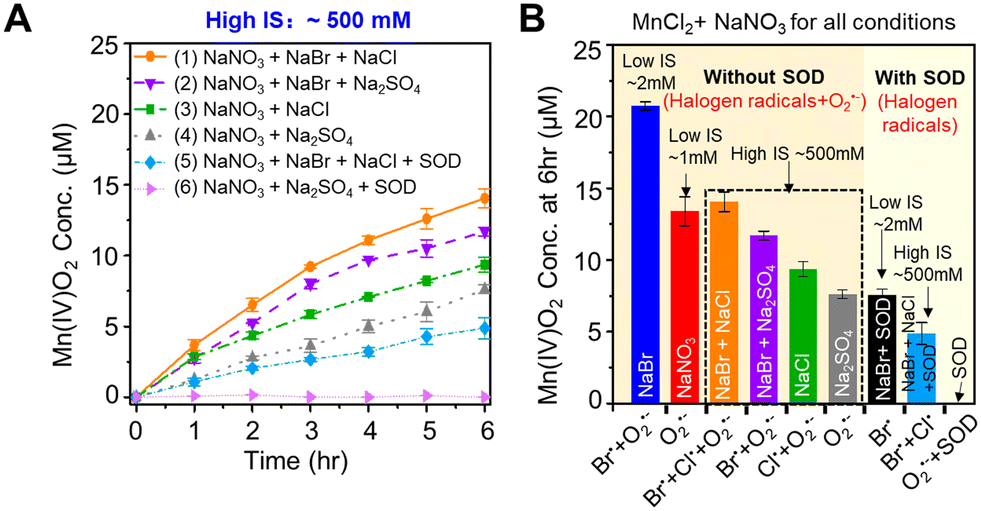 | ||
| Fig. 4 Comparison of the kinetics of Mn2+(aq) oxidation by O2˙− or/and reactive halogen radicals (Br or/and Cl radicals). (A) Mn oxide generation over 6 h of photolysis reaction of 0.1 mM MnCl2, 1 mM NaNO3 with different halide compositions at the same IS (∼500 mM) without/with SOD. (1) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 500 mM NaCl; (2) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 166.7 mM Na2SO4; (3) 0.1 mM MnCl2, 1 mM NaNO3, and 500 mM NaCl; (4) 0.1 mM MnCl2, 1 mM NaNO3, and 166.7 mM Na2SO4; (5) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, 500 mM NaCl, and 0.5 μM SOD; and (6) 0.1 mM MnCl2, 1 mM NaNO3, 166.7 mM Na2SO4, and 0.5 μM SOD. (B) Summary of Mn2+ oxidation amounts of all conditions after 6 h reaction. In the horizontal axis title, Br˙ stands for Br radicals (Br˙ and Br2˙−), Cl˙ stands for Cl radicals (Cl˙ and Cl2˙−), and Br˙ + Cl˙ stands for all Cl and Br radicals (Cl˙, Br˙, Cl2˙−, Br2˙−, and ClBr˙−). | ||
The contributions of Cl− and Br− to Mn oxidation differed due to their different concentrations in surface water and their efficacies in reactivity. Although NaBr had a 500-fold lower concentration than NaCl, at the same IS, more Mn2+ was oxidized by Br radicals generated from NaBr than by Cl radicals generated from NaCl, as seen in Fig. 4A(2 and 3). Most of the BrOH˙−, the intermediate radical generated from the reaction between ˙OH and Br−, as shown in Fig. 1B, can further transform to other reactive halogen radicals, such as Br˙ and Br2˙−,5,44,46 oxidizing Mn2+. On the other hand, for Cl radicals, more than 99.98% of ClOH˙− reverted to ˙OH and Cl− instead of forming Cl˙ and Cl2˙−.5,47–49 Thus, Cl radicals caused less Mn2+ oxidation.
When 1 mM NaBr and 500 mM NaCl were dissolved together (Fig. 4A(1)), more Mn oxide formation was observed than that from adding 1 mM NaBr or 500 mM NaCl separately (Fig. 4A(2) or (3)) at the same IS. The higher oxidation of Mn2+ could result from the sum of Br and Cl radicals compared with single halide radicals. In addition, the promoted Mn oxide formation could result from the reaction product in the co-presence of Br− and Cl−. When Br− and Cl− co-existed, ClBr˙− can be formed at a higher rate constant than the formation rates of Br˙ and Br2˙− from a Br− only system.5 Thus, we postulated that the coexistence of Br− and Cl− in our experimental system would generate ClBr˙−, which can also oxidize Mn2+ to Mn oxide solids faster.
3.5 Characterization of MnO2 solids formed by O2˙− or/and Br radicals
HRXRD was used to obtain the crystalline structure of Mn oxide formed by Br radicals, O2˙−, and Br radicals together with O2˙−, with the results shown in Fig. 5A. The 2θ XRD peaks at about 12.3°, 24.7°, and 36.7° indicated that birnessite, δ-MnO2 (ICDD PDF-4 #00-043-1456), was formed in all three above conditions. δ-MnO2 produced by Br radicals was quite amorphous and less crystalline than that formed by O2˙−. δ-MnO2 synthesized in the co-presence of Br radicals and O2˙− presented the highest crystallinity. HRTEM images (Fig. 5B) show a thin, nanosheet morphology, with the edge rolling up due to high surface tension. In Fig. 5C, crystalline lattices with d-spacings of 0.32 nm and 0.25 nm indicated the (002) and (110)/(200) planes of δ-MnO2, respectively. The (001) interlayer plane was observed with a 0.51 nm d-spacing, which was smaller than that calculated from the XRD measurement (∼0.7 nm), probably owing to the loss of interlayer water in the high vacuum environment of TEM.50 The selected area electron diffraction (SAED) patterns in the Fig. 5B inset were also consistent with the d-spacings of the (001), (002), and (110)/(200) planes, which further confirmed the formation of δ-MnO2. The AFM images shown in Fig. S6† were used to measure the height (thickness) of the MnO2 nanosheets. The lateral dimension of the MnO2 solids was about 1.5 μm and the thickness was 4–5 nm, values that confirmed its nanosheet morphology. Because a single layer of MnO2 is ∼0.7 nm thick in ambient pressure, a 4–5 nm thickness corresponds to 6–7 layers. The hydrodynamic diameters and zeta potential value of the Mn oxide solids were measured by electrophoretic mobility measurements to provide additional physicochemical information. The hydrodynamic diameter of MnO2 solids formed via 6 h photolysis of a solution containing 0.1 mM MnCl2, 1 mM NaNO3, and 1 mM NaBr was 2540 ± 166 nm, which reflects the average lateral dimension sizes of MnO2 nanosheets. The formed MnO2 nanosheets had a negative zeta potential value of −18.9 ± 0.4 mV at pH 5.1. The ESEM image shows a rough layered morphology, and EDX results suggest a slight percentage (0.2%) of Na+ incorporation into the structure (Fig. S7†). The XPS image in Fig. 5D suggested that formed Mn oxide solids mainly contained Mn(III) and Mn(IV), with a very small fraction of Mn(II). In general, Mn oxidation states did not show much practical difference between Mn oxide solids formed with and without NaBr. Slightly higher proportions of Mn(II) and Mn(III) were detected in δ-MnO2 produced in the presence of NaBr, resulting in a slightly lower Mn average oxidation state (3.67) in Fig. 5F than that in the absence of NaBr (3.73) in Fig. 5E. Please note that the Mn oxide solids generation results shown in Fig. 2A were calculated by the LBB method, which assumed that the Mn oxide solids consisted of only Mn(IV). Because the average oxidation state of Mn was lower than 4 in the XPS results, the actual concentrations of Mn oxide solids could be higher than the values in Fig. 2A.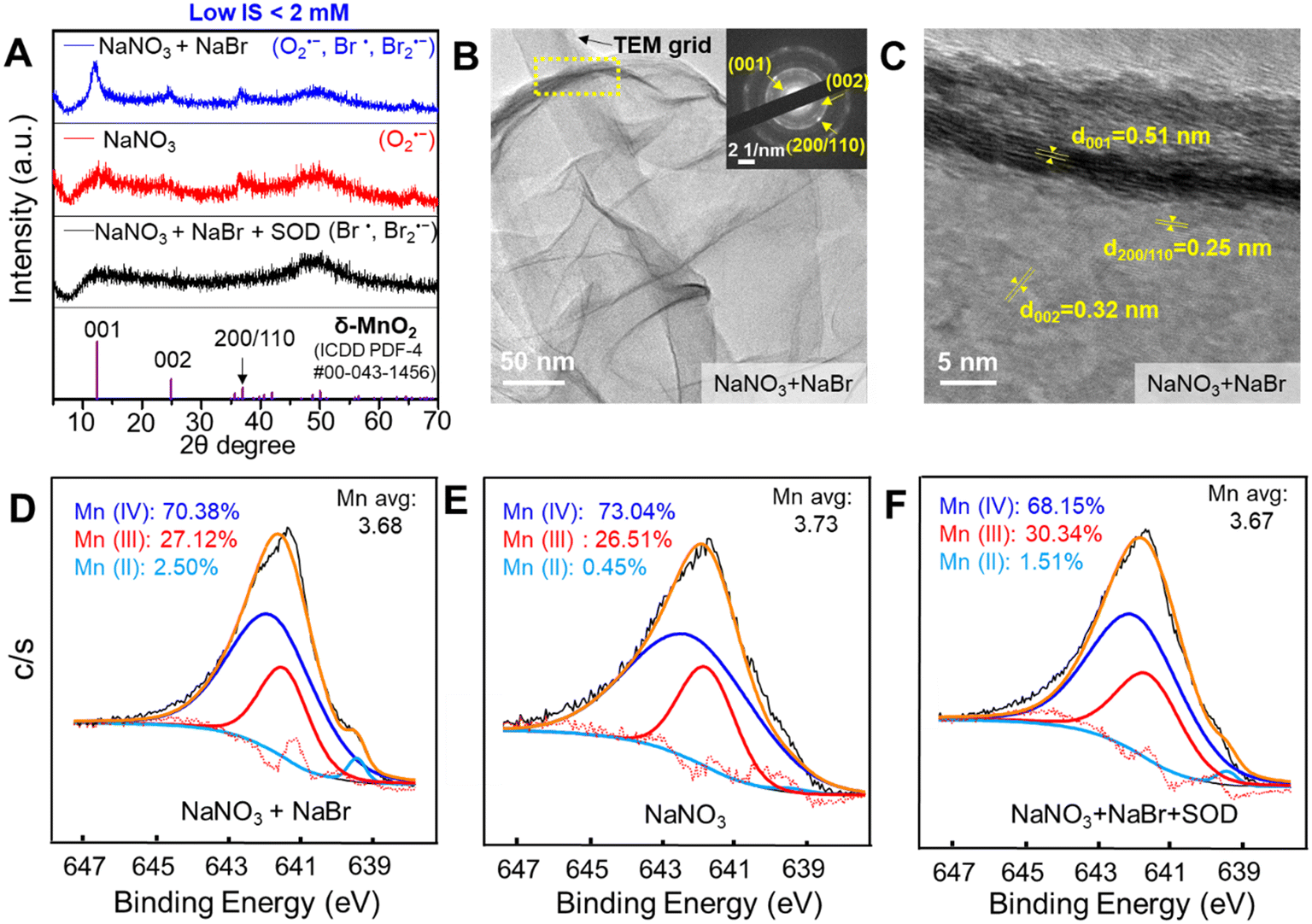 | ||
| Fig. 5 MnO2 nanosheets formed by Br radicals. (A) δ-MnO2 formation measured by HRXRD under the conditions of 0.1 mM MnCl2, 1 mM NaNO3, and 1 mM NaBr (blue); 0.1 mM MnCl2 and 1 mM NaNO3 (red); and 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 0.5 μM SOD (black) after 6 h of reaction. (B and C) HRTEM images of the synthesized Mn oxide nanosheets under the condition of 0.1 mM MnCl2, 1 mM NaNO3, and 1 mM NaBr. The SAED patterns in (B) confirmed the formation of δ-MnO2. (D–F) Average Mn oxidation states of the Mn 2p3/2 spectrum, calculated via Gaussian–Lorentzian fitting. Mn oxide solids were generated under the conditions listed in (A). At least duplicate tests were conducted for each condition. At least five different spots in the TEM grids were measured. | ||
3.6 Effects of O2˙− or/and reactive halogen radicals on Mn2+ oxidation and subsequent Mn oxide nanosheet formation
To investigate the singular contributions of reactive halogen radicals (Cl and Br radicals) to Mn oxidation without O2˙−, SOD was added to scavenge O2˙− in the solution. As shown in Fig. 4A(6), without any halides, no Mn oxidation was observed during the photolysis of a solution containing 0.1 mM MnCl2, 1 mM NaNO3, 166.7 mM Na2SO4, and 0.5 μM SOD, because neither reactive halogen radicals nor O2˙− were present to oxidize Mn2+. Substituting 1 mM NaBr and 500 mM NaCl for the 166.7 mM Na2SO4 caused the generation of reactive halogen radicals and Mn oxide solids formation (Fig. 4A(5)). Fig. 4B displays the Mn2+ oxidation amounts after 6 h reaction under different conditions at low or high IS: O2˙−, Br and/or Cl radicals, and O2˙− with Br and/or Cl radicals. At the same IS (the dashed box in Fig. 4B), the contribution of O2˙−, Br radicals, and Cl radicals to Mn oxide solids generation can be fairly compared. More Mn oxide nanosheets were formed during nitrate photolysis in the presence of additional halide ions. As seen in Fig. S8,† for all experiments at about 500 mM IS, the solution pH changed with time, but all showed the same pH trend, decreasing from an initial pH 9.0 to pH 7.0–6.0 after 6 h of photolysis. A slightly faster pH drop was observed with a larger amount of Mn oxide nanosheet formation.In the coexistence of Cl and Br radicals, the HRXRD results in Fig. 6A(1) and (3) confirmed that Mn oxide had a higher crystallinity than that produced by Br radicals, as shown in Fig. 6A(2) and (4), respectively. This finding suggested that higher IS promoted the crystallization of Mn oxide, although slower kinetics for Mn2+ formation by Cl radicals together with Br radicals was observed than for Br radicals alone (navy blue vs. orange and black vs. blue in Fig. 4B). Based on XPS analysis, the average Mn oxidation states of Mn oxide produced by O2˙− and/or Br radicals was 3.68–3.73 (Fig. 5D–F) under low IS conditions (i.e., less than 2 mM), while they decreased to 3.57–3.63 (Fig. 6B) under high IS conditions (∼500 mM) because of larger Mn(III) and smaller Mn(IV) portions. The insignificantly lower Mn oxidation states may result from charge compensation for more Na+ incorporation into the structure of Mn oxide nanosheets (3.4% of Na+, Fig. 6D) at high IS compared with that at low IS (0.2% of Na+, Fig. S7†). The ESEM image in Fig. 6C showed that Mn oxide samples presented rumpled and wrinkled surface morphology. Zhang et al. (2018) reported that intercalated Na+ tended to shift and coordinate with two unsaturated O from the MnO6 layer via strong electrostatic interactions rather than staying in between two MnO6 layers to balance the interactions, resulting in unbalanced supporting interstitial distances between MnO6 layers and causing a rough surface.51
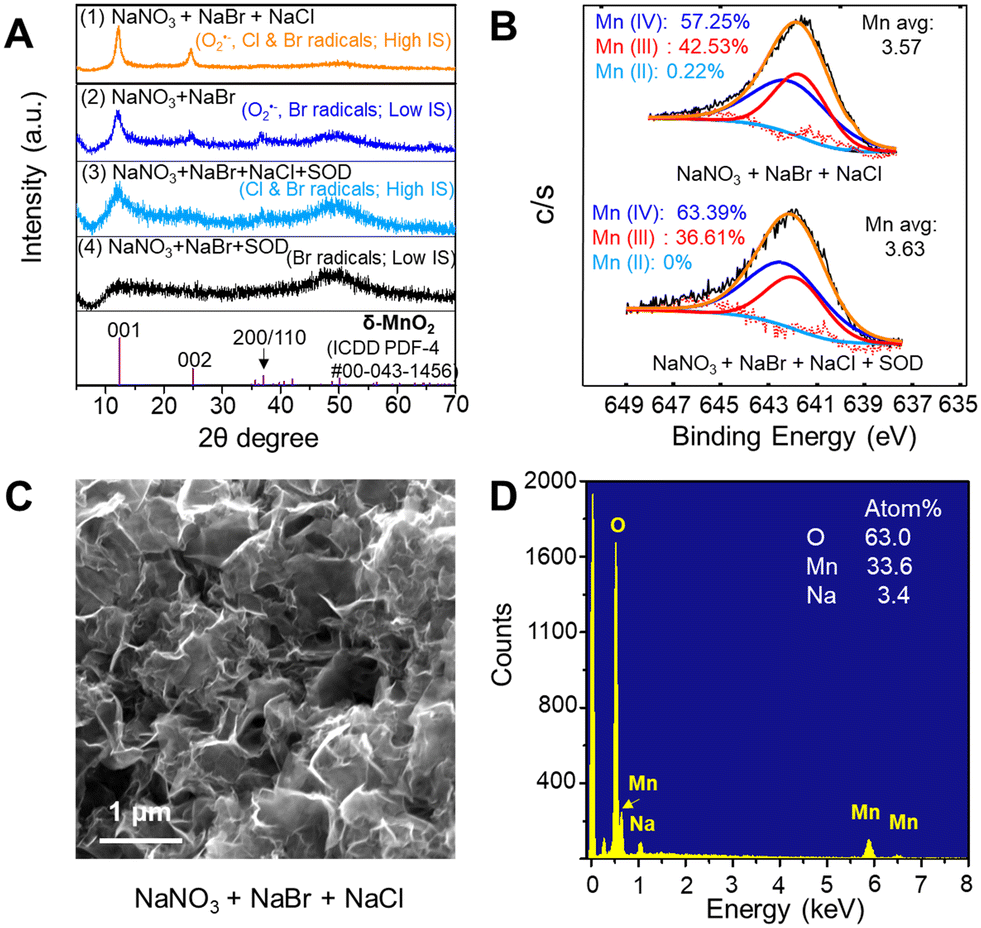 | ||
| Fig. 6 Comparison of O2˙− or/and reactive halogen radicals on the characteristics of newly formed Mn oxide solids. (A) Comparison of HRXRD spectra of Mn oxide solids formed under the conditions of (1) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 500 mM NaCl; (2) 0.1 mM MnCl2, 1 mM NaNO3, and 1 mM NaBr; (3) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, 500 mM NaCl, and 0.5 μM SOD; and (4) 0.1 mM MnCl2, 1 mM NaNO3, 1 mM NaBr, and 0.5 μM SOD. (B) Average Mn oxidation state of Mn 2p3/2 spectrum calculated via Gaussian–Lorentzian fitting. Mn oxide solids were formed under the conditions of (1) and (3). (C and D) Images showing surface morphology and elemental composition of Mn oxide formed under the condition of (1). At least duplicate tests were conducted for each condition. At least five different spots were measured for SEM imaging. | ||
4. Conclusions and environmental implications
Here we show the hitherto unexplored oxidation of Mn2+ by reactive halogen radicals. Compared to the abiotic oxidation rate of Mn2+ without Br radicals, in the presence of Br radicals, the abiotic oxidation rate of Mn2+ to Mn oxide nanosheets was greatly facilitated, exhibiting a 62% increase within 6 h. In the natural environment, NaBr had a much lower concentration than NaCl, however, at the same experimental IS, more Mn2+ was oxidized by Br radicals generated from 1 mM NaBr than by Cl radicals generated from 500 mM NaCl. δ-MnO2 nanosheets were formed by reactive halogen species, and increasing the IS caused more highly crystalline Mn oxides to form. Although the pH, the concentrations of Mn2+, nitrate, and halides, and the intensity of light illumination obviously vary in different water bodies, this study provides a good starting point for understanding how halogen radicals are generated and affect Mn2+ oxidation processes. In addition, this work highlights the importance of underappreciated reactive halogen radicals in inorganic redox reactions in aqueous environmental systems. In nature, reactive halogen radicals can be produced via oxidation of halides by ˙OH, SO4˙−, or triplet state dissolved organic matter (3DOM*).5 The natural contribution of reactive halogen radicals to Mn2+ oxidation and MnO2 formation merit future systematic experiments. The redox reaction mechanism between reactive halogen radicals and Mn2+(aq) can be applicable to other redox-sensitive transition metal ions, such as Fe2+, As3+, and Cr3+. Furthermore, the results are also critically valuable to understanding aerosol reactions involving halides in the atmosphere. Halide ions are preferentially present at air–water interfaces rather than in bulk solution,52 where surface halide ions can actively react with ˙OH to generate halogen radicals.53 In the atmosphere, this behavior can amplify the effects of reactive halogen species on the oxidation of redox active metals, forming nanoscale mineral aerosols that can act as cloud condensation nuclei and affect the albedo. Moreover, compared with bulk MnO2 particles, nanoscale MnO2 shows an improved catalytic performance for electrochemical oxygen reduction.54 This study suggests a novel, environmentally-friendly, and facile pathway for the synthesis of MnO2 nanosheets.Author contributions
Z. G., C. S., and Y.-S. J. conducted the experiments. Z. G. and Y.-S. J. designed the experimental setup and procedures and wrote the manuscript. All authors discussed the results and collectively revised the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by the U.S. National Science Foundation's Environmental Chemical Sciences program (CHE-1905077). The authors would like to acknowledge the Institute of Materials Science & Engineering (IMSE) of Washington University in St. Louis for the use of XPS, ESEM, and TEM, Professor Kimberly Parker for providing suggestions, and Professor James C. Ballard for carefully reviewing the manuscript.References
- N. Voutchkov, Overview of seawater concentrate disposal alternatives, Desalination, 2011, 273, 205–219 CrossRef CAS
.
- C. Zhang, Y. Shi, L. Shi, H. Li, R. Li, S. Hong, S. Zhuo, T. Zhang and P. Wang, Designing a next generation solar crystallizer for real seawater brine treatment with zero liquid discharge, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed
- K. B. Gregory, R. D. Vidic and D. A. Dzombak, Water management challenges associated with the production of shale gas by hydraulic fracturing, Elements, 2011, 7, 181–186 CrossRef
- J. S. Harkness, G. S. Dwyer, N. R. Warner, K. M. Parker, W. A. Mitch and A. Vengosh, Iodide, bromide, and ammonium in hydraulic fracturing and oil and gas wastewaters: environmental implications, Environ. Sci. Technol., 2015, 49, 1955–1963 CrossRef CAS PubMed
- K. Zhang and K. M. Parker, Halogen radical oxidants in natural and engineered aquatic systems, Environ. Sci. Technol., 2018, 52, 9579–9594 CrossRef CAS PubMed
- K. M. Parker and W. A. Mitch, Halogen radicals contribute to photooxidation in coastal and estuarine waters, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5868–5873 CrossRef CAS PubMed
- K. M. Parker, E. S. Reichwaldt, A. Ghadouani and W. A. Mitch, Halogen radicals promote the photodegradation of microcystins in estuarine systems, Environ. Sci. Technol., 2016, 50, 8505–8513 CrossRef CAS PubMed
- S. Cheng, X. Zhang, X. Yang, C. Shang, W. Song, J. Fang and Y. Pan, The multiple role of bromide ion in PPCPs degradation under UV/chlorine treatment, Environ. Sci. Technol., 2018, 52, 1806–1816 CrossRef CAS PubMed
- C. R. Stephens, P. B. Shepson, A. Steffen, J. W. Bottenheim, J. Liao, L. G. Huey, E. Apel, A. Weinheimer, S. R. Hall and C. Cantrell, The relative importance of chlorine and bromine radicals in the oxidation of atmospheric mercury at Barrow, Alaska, J. Geophys. Res.: Atmos., 2012, 117, D14 CrossRef
- C. R. Myers and K. H. Nealson, Bacterial manganese reduction and growth with manganese oxide as the sole electron acceptor, Science, 1988, 240, 1319–1321 CrossRef CAS PubMed
- B. M. Tebo, J. R. Bargar, B. G. Clement, G. J. Dick, K. J. Murray, D. Parker, R. Verity and S. M. Webb, Biogenic manganese oxides: properties and mechanisms of formation, Annu. Rev. Earth Planet. Sci., 2004, 32, 287–328 CrossRef CAS
- C. N. Butterfield, A. V. Soldatova, S.-W. Lee, T. G. Spiro and B. M. Tebo, Mn(II, III) oxidation and MnO2 mineralization by an expressed bacterial multicopper oxidase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11731–11735 CrossRef CAS PubMed
- D. Learman, B. Voelker, A. Vazquez-Rodriguez and C. Hansel, Formation of manganese oxides by bacterially generated superoxide, Nat. Geosci., 2011, 4, 95–98 CrossRef CAS
- C. M. Hansel, C. A. Zeiner, C. M. Santelli and S. M. Webb, Mn(II) oxidation by an ascomycete fungus is linked to superoxide production during asexual reproduction, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12621–12625 CrossRef CAS PubMed
- D. Learman, S. Wankel, S. Webb, N. Martinez, A. Madden and C. Hansel, Coupled biotic–abiotic Mn(II) oxidation pathway mediates the formation and structural evolution of biogenic Mn oxides, Geochim. Cosmochim. Acta, 2011, 75, 6048–6063 CrossRef CAS
- J. M. Diaz, C. M. Hansel, B. M. Voelker, C. M. Mendes, P. F. Andeer and T. Zhang, Widespread production of extracellular superoxide by heterotrophic bacteria, Science, 2013, 1237331 Search PubMed
- H. Jung, T. S. Chadha, D. Kim, P. Biswas and Y.-S. Jun, Photochemically assisted fast abiotic oxidation of manganese
and formation of δ-MnO2 nanosheets in nitrate solution, Chem. Commun., 2017, 53, 4445–4448 RSC
- G. S. Laurence and A. T. Thornton, Kinetics of oxidation of transition-metal ions by halogen radical anions. Part III. The oxidation of manganese(II) by dibromide and dichloride ions generated by flash photolysis, J. Chem. Soc., Dalton Trans., 1973, 1637–1644 RSC
- G. Davies, Some aspects of the chemistry of manganese(III) in aqueous solution, Coord. Chem. Rev., 1969, 4, 199–224 CrossRef CAS
- J. Mack and J. R. Bolton, Photochemistry of nitrite and nitrate in aqueous solution: a review, J. Photochem. Photobiol., A, 1999, 128, 1–13 CrossRef CAS
- WHO, Concise international chemical assessment document 12, Manganese and its compounds, World Health Organization, 1999 Search PubMed.
- WHO, Concise international chemical assessment document 63, Manganese and its Compounds: Environmental Aspects, World Health Organization, 2004 Search PubMed.
- M. Williams, G. D. Todd, N. Roney, J. Crawford, C. Coles, P. R. McClure, J. D. Garey, K. Zaccaria and M. Citra, Toxicological profile for manganese, 2013 Search PubMed.
- K. W. Mandernack, J. Post and B. M. Tebo, Manganese mineral formation by bacterial spores of the marine Bacillus, strain SG-1: Evidence for the direct oxidation of Mn(II) to Mn(IV), Geochim. Cosmochim. Acta, 1995, 59, 4393–4408 CrossRef CAS
- WHO, Nitrate and nitrite in drinking-water, World Health Organization, 2011 Search PubMed.
- R. Chance, A. R. Baker, L. Carpenter and T. D. Jickells, The distribution of iodide at the sea surface, Environ. Sci.: Processes Impacts, 2014, 16, 1841–1859 RSC
- EPA, Quality criteria for water, 1986 Search PubMed.
-
EPA, pH, https://www.epa.gov/caddis-vol2/caddis-volume-2-sources-stressors-responses-ph, (accessed January 18, 2021)
- H. Jung, T. S. Chadha, Y. Min, P. Biswas and Y.-S. Jun, Photochemically-assisted synthesis of birnessite nanosheets and their structural alteration in the presence of pyrophosphate, ACS Sustainable Chem. Eng., 2017, 5, 10624–10632 CrossRef CAS
- J. K. Klewicki and J. J. Morgan, Kinetic behavior of Mn(III) complexes of pyrophosphate, EDTA, and citrate, Environ. Sci. Technol., 1998, 32, 2916–2922 CrossRef CAS
- R. E. Huie, C. L. Clifton and P. Neta, Electron transfer reaction rates and equilibria of the carbonate and sulfate radical anions, Radiat. Phys. Chem., 1991, 38, 477–481 CrossRef CAS
- S. Yan, Y. Liu, L. Lian, R. Li, J. Ma, H. Zhou and W. Song, Photochemical formation of carbonate radical and its reaction with dissolved organic matters, Water Res., 2019, 161, 288–296 CrossRef CAS PubMed
- Z. Gao, D. Zhang and Y.-S. Jun, Does tert-butyl alcohol really terminate the oxidative activity of •OH in inorganic redox chemistry?, Environ. Sci. Technol., 2021, 55, 10442–10450 CrossRef CAS PubMed
- M. R. Jones, G. W. Luther III, A. Mucci and B. M. Tebo, Concentrations of reactive Mn(III)-L and MnO2 in estuarine and marine waters determined using spectrophotometry and the leuco base, leucoberbelin blue, Talanta, 2019, 200, 91–99 CrossRef CAS PubMed
- R. K. Fox, D. F. Swinehart and A. Garrett, The equilibria of manganese hydroxide, Mn(OH)2, in solutions of hydrochloric acid and sodium hydroxide, J. Am. Chem. Soc., 1941, 63, 1779–1782 CrossRef CAS
- M. Levy, P. P. Chowdhury and P. Nagpal, Quantum dot therapeutics: a new class of radical therapies, J. Biol. Eng., 2019, 13, 48 CrossRef PubMed
- K. Krumova and G. Cosa, Overview of reactive oxygen species, 2016 Search PubMed.
- Z. Gao, J. Liu, C. Skurie, Y. Zhu and Y.-S. Jun, Photochemical reactions of dissolved organic matter and bromide ions facilitate abiotic formation of manganese oxide solids, Water Res., 2022, 222, 118831 CrossRef CAS PubMed
- J. Ma, H. Zhou, S. Yan and W. Song, Kinetics studies and mechanistic considerations on the reactions of
superoxide radical ions with dissolved organic matter, Water Res., 2019, 149, 56–64 CrossRef CAS PubMed
- S. Allard, L. Fouche, J. Dick, A. Heitz and U. von Gunten, Oxidation of manganese(II) during chlorination: role of bromide, Environ. Sci. Technol., 2013, 47, 8716–8723 CrossRef CAS PubMed
- E. M. Rodríguez and U. von Gunten, Generation of hydroxyl radical during chlorination of hydroxyphenols and natural organic matter extracts, Water Res., 2020, 177, 115691 CrossRef PubMed
- W. McGimpsey and J. Scaiano, Photochemistry of α-chloro-and α-bromoacetophenone. Determination of extinction coefficients for halogen–benzene complexes, Can. J. Chem., 1988, 66, 1474–1478 CrossRef CAS
- P. F. De Violet, Polyhalide radical anions as intermediates in chemistry, Rev. Chem. Intermed., 1981, 4, 121–169 CrossRef
- D. Zehavi and J. Rabani, Oxidation of aqueous bromide ions by hydroxyl radicals. Pulse radiolytic investigation, J. Phys. Chem., 1972, 76, 312–319 CrossRef CAS
- X.-Y. Yu and J. R. Barker, Hydrogen peroxide photolysis in acidic aqueous solutions containing chloride ions. I. Chemical mechanism, J. Phys. Chem. A, 2003, 107, 1313–1324 CrossRef CAS
- G. Merenyi and J. Lind, Reaction mechanism of hydrogen abstraction by the bromine atom in water, J. Am. Chem. Soc., 1994, 116, 7872–7876 CrossRef CAS
- G. Jayson, B. Parsons and A. J. Swallow, Some simple, highly reactive, inorganic chlorine derivatives in aqueous solution. Their formation using pulses of radiation and their role in the mechanism of the Fricke dosimeter, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 1597–1607 RSC
- A. Grigor'ev, I. Makarov and A. Pikaev, Formation of Cl2− in the bulk of solution during radiolysis of concentrated aqueous solutions of chlorides, Khimiya Vysokikh Ehnergij, 1987, 21, 123–126 Search PubMed
- U. K. Kläning and T. Wolff, Laser flash photolysis of HClO, ClO− , HBrO, and BrO− in aqueous solution. reactions of Cl- and Br-atoms, Ber. Bunsenges. Phys. Chem., 1985, 89, 243–245 CrossRef
- J. Zhu, Q. Li, W. Bi, L. Bai, X. Zhang, J. Zhou and Y. Xie, Ultra-rapid microwave-assisted synthesis of layered ultrathin birnessite K0.17MnO2 nanosheets for efficient energy storage, J. Mater. Chem. A, 2013, 1, 8154–8159 RSC
- T. Zhang, L. Liu, W. Tan, S. L. Suib, G. Qiu and F. Liu, Photochemical formation and transformation of birnessite: Effects of cations on micromorphology and crystal structure, Environ. Sci. Technol., 2018, 52, 6864–6871 CrossRef CAS PubMed
- P. Jungwirth and D. J. Tobias, Ions at the air/water interface, J. Phys. Chem. B, 2002, 106, 6361–6373 CrossRef CAS
- E. Knipping, M. Lakin, K. Foster, P. Jungwirth, D. Tobias, R. Gerber, D. Dabdub and B. Finlayson-Pitts, Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols, Science, 2000, 288, 301–306 CrossRef CAS PubMed
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, MnO2-based nanostructures as catalysts for electrochemical oxygen reduction in alkaline media, Chem. Mater., 2010, 22, 898–905 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2en00410k |
| This journal is © The Royal Society of Chemistry 2022 |