
Nanoconfinement matters in humidified CO2 interaction with metal silicates†
Siavash
Zare
 a,
K. M. Salah
Uddin
b,
Andreas
Funk
bc,
Quin R. S.
Miller
d and
Mohammad Javad
Abdolhosseini Qomi
*a
a,
K. M. Salah
Uddin
b,
Andreas
Funk
bc,
Quin R. S.
Miller
d and
Mohammad Javad
Abdolhosseini Qomi
*a
aDepartment of Civil and Environmental Engineering, Henry Samueli School of Engineering, University of California, Irvine, E4130 Engineering Gateway, Irvine, CA 92697-2175, USA. E-mail: mjaq@uci.edu
bDepartment of Civil Engineering, University of Kassel, Germany, Mönchebergstraße 19, 34125 Kassel, Germany
cChemistry Department, University of Siegen, Adolf-Reichwein-Straße 2, 57068 Siegen, Germany
dPhysical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA, USA
First published on 22nd August 2022
Abstract
With enigmatic observations of enhanced reactivity of wet CO2-rich fluids with metal silicates, the mechanistic understanding of molecular processes governing carbonation proves critical in designing secure geological carbon sequestration and economical carbonated concrete technologies. Here, we use the first principle and classical molecular simulations to probe the impact of nanoconfinement on physicochemical processes at the rock–water–CO2 interface. We choose nanoporous calcium–silicate–hydrate (C–S–H) and forsterite (Mg2SiO4) as model metal silicate surfaces that are of significance in cement chemistry and geochemistry communities, respectively. We show that while a nanometer-thick interfacial water film persists at unsaturated conditions consistent with in situ infrared spectroscopy, the phase behavior of the water–CO2 mixture changes from its bulk counterpart depending on the surface chemistry and nanoconfinement. We also observe enhanced solubility at the interface of water and CO2 phases, which could amplify the CO2 speciation rate. Through free energy calculations, we show that CO2 could be found in a metastable state near the C–S–H surface, which can potentially react with surface water and hydroxyl groups to form carbonic acid and bicarbonate. These findings support the explicit consideration of nanoconfinement effects in reactive and non-reactive pore-scale processes.
Environmental significanceNanoconfined environments in the structure of natural and synthetic metal silicates alter the bulk phase behavior of wet CO2-rich fluid mixtures and could impact the carbonation reaction rate in nano-porous geological materials and construction waste. This study shows that regardless of CO2 pressure, pore size, and surface chemistry, a nanometer-thick water layer persists on the surface of metal silicates under unsaturated conditions. Moreover, it is shown that CO2 molecules could reach the surface of calcium–silicate–hydrate and react with the surface water and hydroxyl groups to produce carbonic acid and bicarbonate with a relatively low energy barrier. The presence of adsorbed water film and surface carbon speciation in nanoconfined environments signify the benefits of carbon sequestration with humidified CO2-rich fluids. |
Introduction
Carbonation of Mg- and Ca-rich silicates promises scalable solutions to mitigate the calamitous impacts of anthropogenic carbon emissions. These solutions can be realized in geological settings through in situ carbonation of mafic and ultramafic lithologies,1–3 or above ground through well-controlled ex situ carbon mineralization facilities to produce value-added concrete products.4 The potential in situ CO2 storage capacity of continental flood basalts,5 oceanic igneous plateaus,6,7 and basalt ridges8,9 is greater by orders of magnitude than the estimated CO2 emissions from burning all fossil fuel resources on Earth.3 Ultramafic tailings,10,11 alkaline industrial residues,12 construction and demolition waste13,14 and naturally occurring minerals such as olivine,15 can also collectively offset up to 30 Gt of emissions a year via ex situ processes.16Whether in situ or ex situ, the permanent CO2 conversion to carbonate minerals is advantageous over storage through residual trapping in nonreactive sedimentary formations.17,18 However, the carbon mineralization extent and associated costs should be optimized to engender maximal CO2 uptake through a rapid and close-to-complete carbonation process. Therefore, it is critical to predict the carbonation processes, including rates and mechanisms, as they directly affect storage security, efficiency, and cost. Despite this urgency, our understanding of carbon mineralization kinetics and underlying molecular pathways remain limited, especially with humidified CO2-rich fluids.
A major challenge in determining the carbonation rate is the complexity arising from confinement effects on fluid–rock and fluid–fluid interactions in the pore space. Compared to the bulk phase, confined fluids express substantially different physicochemical attributes and reaction rates.19–21 For instance, recent experiments highlight the distinct thermodynamic properties of the fluids in shales,22–24 model nanoporous glasses,25 and mesoporous silicon.26 When confined in nanoporosity, fluid–solid interaction can also significantly impact the fluid phase behavior, sorption, capillary condensation, wettability, and imbibition.27–30 Based upon experiments performed on mesoporous silicon,26 a follow-up theoretical study27 showed that imbibition occurs when the relative humidity is above a critical value, well below the vapor saturation pressure. The fluid mixture phase behavior in the porous rock is particularly significant as it remains unclear to what extent the classical bulk aqueous-mediated dissolution–precipitation pathways can be applied to water-poor systems. Understanding wettability is also critical given its influences on sealing rock effectiveness31,32 and storage efficiency in a subsurface carbon storage scenario, including in mafic geologic formations.33,34
In this work, we study nanoconfined water–CO2 mixtures on two model metal silicates with wide-ranging environmental and technological significance: 1) forsterite (Mg2SiO4), the magnesium endmember of olivine, relevant to carbonation studies of mafic (e.g., basalt) and ultramafic (e.g., peridotite) lithologies, and 2) calcium–silicate–hydrate (C–S–H), the binding phase in the cement paste that is responsible for concrete's strength, fracture, and durability properties.35–37 Carbonation of concrete infrastructure is estimated to be the sink for about 2.5% of the anthropogenic carbon emissions, despite an unsettlingly high carbon footprint linked to cement production.38–40 Carbonation of C–S–H is also of prime importance for the well-bore integrity41–43 and valorization of construction and demolition wastes that places tangible pathways for net zero or even negative carbon footprint concrete technologies within reach.44 The motivation to conduct a comparative study between C–S–H and forsterite is two-fold and goes far beyond their technological significance. First, while forsterite is a natural mineral dominated by macroscopic fracture networks, C–S–H provides a nanoporous model system45 that readily lends itself to studies of confined fluids.46 Second, the water residence time in the first hydration shell of Mg2+ cations is at least three orders of magnitude longer than that around Ca2+ ions.47 Such extended residence time can potentially hinder dehydration and regulate interfacial processes in rock–water–CO2 systems.48
Herein, we employ molecular simulations to determine the competitive sorption of CO2 and H2O in metal silicate nanopores and address two basic questions surrounding the interactions of humidified CO2 with forsterite and C–S–H within the thermodynamic range of technological interest. First, we seek to quantify the impact of nanoporosity and chemical composition on the thickness of interfacial adsorbed water films. While molecular simulations delineate the structure and energetics of single- and multi-component fluids on metal silicate surfaces,49–51 quantitative measurements of the adsorbed water film thickness and its dependence on thermodynamic state variables of CO2–H2O mixture (pressure, temperature), surface chemistry and pore size remain to be understood. These gaps are difficult to address experimentally and are key for parameterizing realistic MD simulations of these interfaces.49,52–54 Although continual progress is being made in determining water film thicknesses at mineral–H2O–CO2 interfaces,54–64 including for forsterite surfaces, the initial hydration state of the mineral surface to measurements is often unknown, and the influence of pressure–temperature-composition on water film thicknesses has not been systematically explored. Second, we investigate whether carbonic acid and bicarbonate can potentially form at the water-solid interface. To this end, we determine the energetic penalty for displacing CO2 through interfacial water layers toward the surface and model its subsequent reaction with hydroxyl groups and adsorbed water molecules.
Results and discussion
Formation of interfacial water films on metal silicate surfaces in contact with wet CO2-rich fluids
When the CO2–H2O mixture invades the pore structure of the host rock (Me2+ silicates), the CO2–H2O molar fraction changes according to the surface chemistry and the confinement geometry. Therefore, for water-bearing CO2 sequestration applications where carbonation reactions occur at the confined thin water film on the Me2+ silicates, it is critical to understand the molar fractions of water (adsorbed water film thickness) and CO2 that exist in the pore.To unravel the CO2–H2O mixture composition in the slit-pore, we perform Grand Canonical Monte Carlo (GCMC) simulations of competitive CO2–H2O adsorption. Unlike the classical molecular dynamic (MD) that keeps the number of atoms and molecules fixed during the simulation, GCMC allows the exchange of species between a pre-defined reservoir and the simulation box, making it the proper computational platform to model adsorption phenomena. We perform GCMC calculations with four types of wet CO2-rich fluids on the surface of defective Hamid tobermorite65 (Ca2.25[Si3O7.5(OH)1.5]·1H2O) (001) at 1.7 Ca/Si ratio as an analog of C–S–H (ref. 35) and hydroxylated (010) surface of forsterite. In GCMC simulations, one fluid corresponds to the normal carbonation at ambient conditions; two are relevant to water-bearing supercritical CO2 fluids at geological conditions,3 and one relates to an intermediate gaseous CO2. For thermodynamic conditions corresponding to the above-mentioned four fluid mixtures, see Table 1, ESI† Note S1 and Fig. S1–S3. The relative humidity across these conditions is roughly 90%.
| T (K) | P (bar) | μ H2O (kJ mol−1) | μ CO2 (kJ mol−1) | ρ CO2-rich (g cm−3) |
|
|
|---|---|---|---|---|---|---|
| Ambient condition | 300 | 1 | −46.00 | −40.00 | 0.0002 | 0.9756 |
| CO2(g) at 20 bar | 300 | 20 | −46.00 | −33.25 | 0.0409 | 0.9982 |
| scCO2 at 100 bar | 348.15 | 100 | −47.00 | −35.75 | 0.2125 | 0.9942 |
| scCO2 at 200 bar | 348.15 | 200 | −47.00 | −34.50 | 0.5965 | 0.9956 |

The slit pore in C–S–H is varied between 26 Å to 54 Å to explore its nanoporous structure. The forsterite simulations focus on a 44 Å slit pore, representing a cleaved surface and proving large enough to prevent imbibition. These olivine nanopores, including grain boundaries and fluid inclusions, are important dynamic environments that host dissolution–precipitation and mass transfer reactions that promote volume changes and reactive cracking.66–69 More generally, these reactive nanopores are important in mafic rocks, including basalts, as Luhmann et al.70 (ultra)small-angle neutron scattering measurements showed that CO2-rich brine increases the basalt's porosity, leading to pore sizes in the range of ∼1 nm to 1 μm in response to secondary reactions and shrinkage-induced volume changes.
As presented in Fig. 1a–h, interfacial water films persist on metal silicate surfaces even at supercritical CO2 conditions and shield surfaces from direct contact with the segregated CO2-rich phase. Similar observations are also reported for clays50 and calcite,71,72 which suggest nanometer-thick water films should prevail on hydrophilic mineral surfaces when in contact with humidified CO2-rich fluids. Interestingly, for the case of scCO2 at 100 bar and 348 K, imbibition occurs in the slit-pore of C–S–H with a size as large as 44 Å, see Fig. 1b. However, when we increase the CO2 pressure to 200 bar, CO2 manages to enter the same slit-pore size, as shown Fig. 1a. On the other hand, water is only present as layers on the C–S–H surfaces at ambient (1 bar) and 20 bar CO2(g) at 300 K (Fig. 1c and d). These two observations show the critical role of pressure and temperature in diffusion-to-imbibition transition at unsaturated water vapor pressure. Also, in contrast to C–S–H, we observe that CO2 permeates into the slit-pore of forsterite at the CO2 pressure of 100 bar and temperature of 348 K, as shown in Fig. 1f. This emphasizes the role of surface chemistry in the fluid phase behavior in confined spaces, and it is a signature of the more hydrophilic surface of C–S–H than the {010} surface of forsterite.
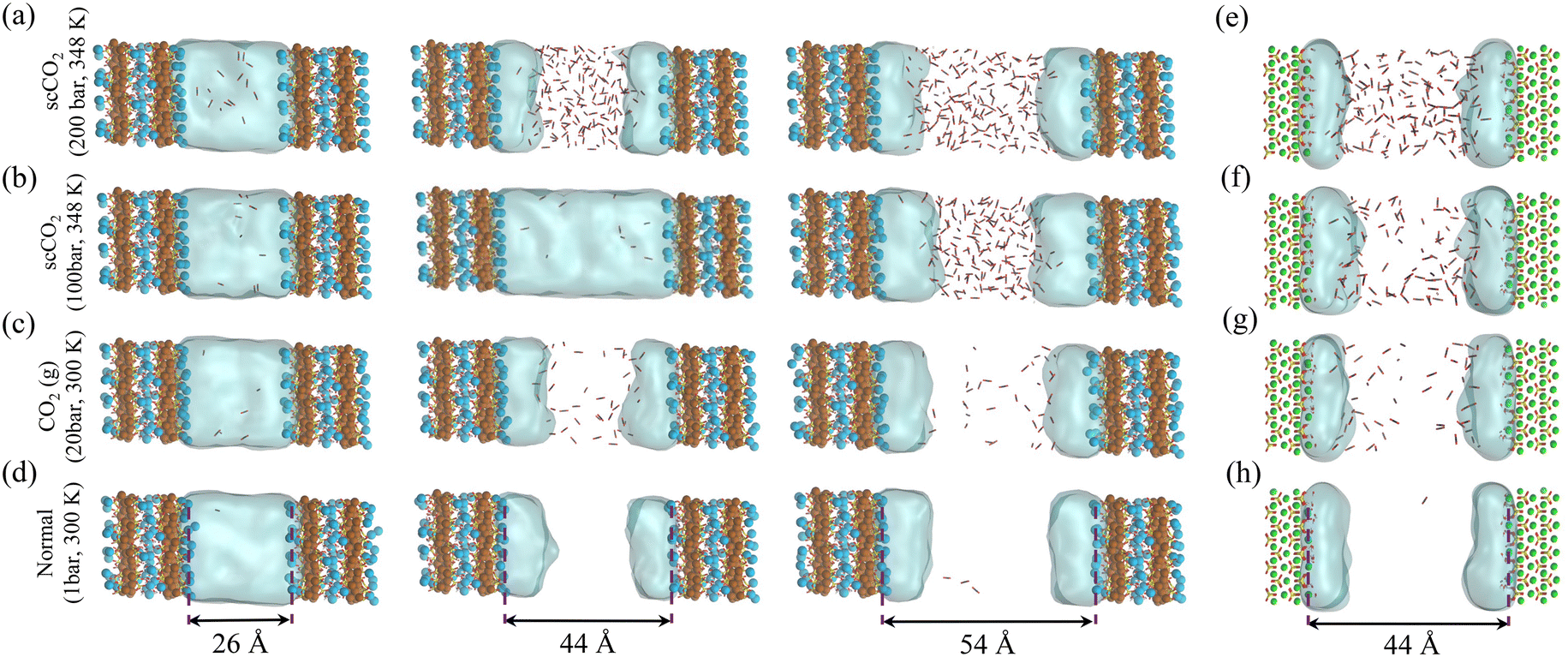 | ||
| Fig. 1 Competitive adsorption of H2O–CO2 mixtures in the slit C–S–H and forsterite porosity at normal, gaseous, and supercritical conditions. (a–d) Snapshots from the equilibrated two-phase GCMC adsorption simulations in C–S–H slit pores at different pore sizes and thermodynamic conditions. (e–h) Snapshots from equilibrated two-phase GCMC adsorption simulations in forsterite slit pores at different thermodynamic conditions. Blue and brown spheres represent intralayer and interlayer calcium in C–S–H, respectively. Green sphere represents magnesium. Silicate chains are depicted in yellow-red sticks. The continuous light blue cloud represents water and CO2 is shown by black and red sticks. | ||
In the temperature and pressure ranges considered here, CO2 could only penetrate the slit pore when the interlayer distance is more than 30 Å, while the interfacial water remains on the surface regardless of the distance, Fig. 2. Moreover, the total number of adsorbed water molecules converges to an asymptotic value at large pore sizes (>40 Å), as shown by the dashed line in Fig. 2. The number of adsorbed water molecules per volume on the surface of forsterite is, in general, lower than that on the surface of C–S–H, again showing the more hydrophilic surface of C–S–H than forsterite. Also, at the largest slit-pore distance (54 Å for C–S–H and 44 Å for forsterite), we observe that the number of adsorbed water molecules on the surface of forsterite/C–S–H at 200 bar and 348 K is lower compared to those at 100 bar and 348 K. Similar observations can be made for simulations at 300 K. Therefore, we can conclude that CO2 pressure can affect the amount of water adsorbed on the surface. It is also noteworthy that at interlayer distances of less than 20 Å, the density of capillary water is found to be higher than its bulk value (1 g cm−3), consistent with the simulations on the confined nature of water in C–S–H,73,74 see ESI† Fig. S4.
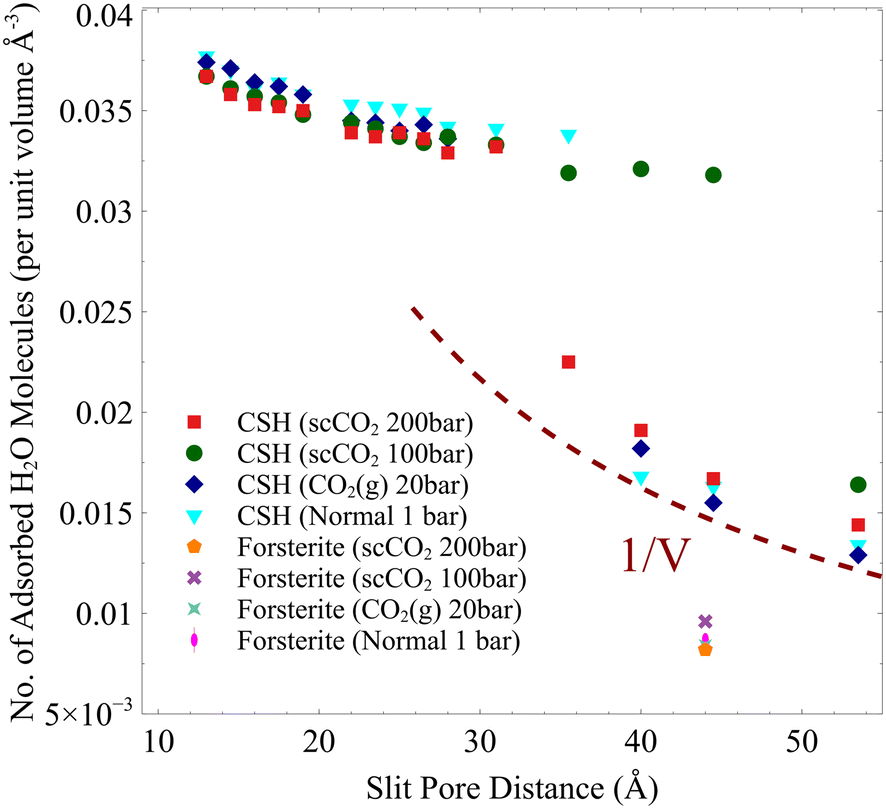 | ||
| Fig. 2 Number of adsorbed H2O molecules per unit volume based on the slit-pore distance. 1/V dashed line shows that the number of adsorbed water molecules is constant beyond a certain slit pore. | ||
To gain a more quantitative picture of the phase behavior, the resultant CO2 mole fractions are overlaid on the experimental and theoretical bulk mixture phase diagrams75,76 in Fig. 3. The CO2 molar fractions corresponding to H2O-rich and CO2-rich are respectively shown in Fig. 3a and b. Here, we show that the CO2 mole fraction in the slit-pore of C–S–H and forsterite with the size of less than 5 nm deviates from its bulk counterpart denoted by dashed lines in Fig. 3a and b. Also, although all GCMC simulations are initialized at CO2-rich conditions, the equilibrium nanoconfined fluid in pores smaller than ∼30 Å turns H2O-rich, as shown in Fig. 3a. The exact slit pore size at which CO2-rich fluid shifts to H2O-rich fluid depends on CO2 pressure, temperature, and surface chemistry, among others. The observed deviation of CO2 mole fraction from bulk values and the transition from CO2-rich to H2O-rich fluid in the confined space can be related to the hydrophilic nature of forsterite49 and C–S–H (ref. 46) surfaces and the effect of surface forces in nanoscale confinements on the fluid phase behavior.26,27 Beyond 30 Å, CO2 permeates within the slit pores, and the mixture composition shifts toward the bulk CO2-rich mixture (akin to the wet CO2-rich reservoirs), as shown in Fig. 3b. The observed confinement effect has implications, especially for multi-scale porous rocks such as C–S–H, which should be taken into account in continuum models and pore-scale models.77–79
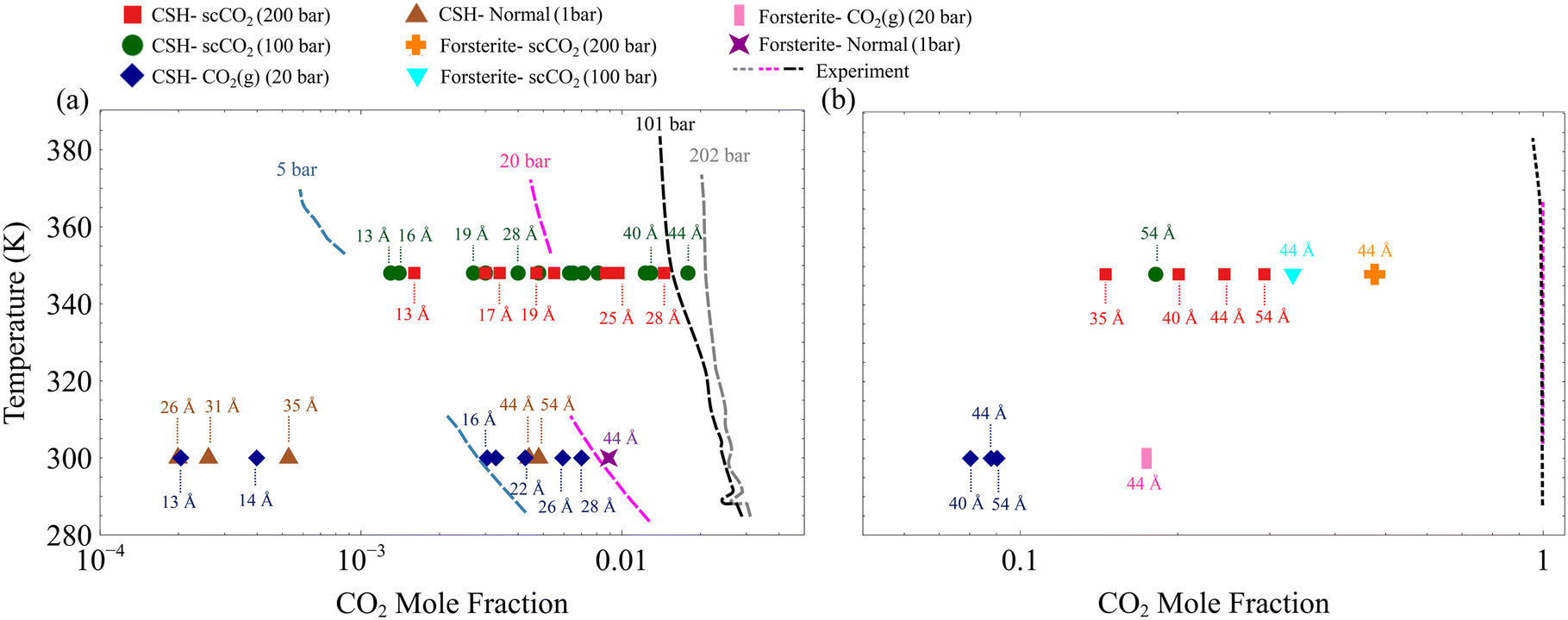 | ||
| Fig. 3 The H2O–CO2 phase coexistence diagram overlaid with the adsorbed mixture composition in the slit nano-porosities under various thermodynamic conditions computed via GCMC simulations. Dashed lines represent experimental and theoretical curves for the bulk mixture.75,76 (a) Phase diagrams for H2O–rich water–CO2 mixtures. (b) CO2–rich water–CO2 mixtures. The CO2 mole fractions in the slit-pore of C–S–H and forsterite derived from GCMC simulations are consistently lower than their bulk counterparts. | ||
Determining the thickness of the adsorbed water film on the surface of metal silicate minerals is critical as it is a first-order control for carbon mineralization rates and mechanisms but is difficult to probe experimentally. Table 2 is a compilation of water film thickness data from several experiments and our adsorption simulation results. As shown in this table, experiments and simulations show adsorbed water film with thicknesses in the nanometer range. According to our GCMC adsorption simulations, the average water film thickness on the forsterite surface is 0.6–0.8 nm, depending on the temperature and pressure of the simulation. They are in good agreement with experimental values, especially those reported in recent years, such as Exp_7, Exp_8, Exp_9, and Exp_12 in Table 2. Nevertheless, we observe slight variability in the average film thickness on the surface at different carbonation conditions. To address the variability of water film thickness based on CO2 pressure, a better picture could be achieved by calculating the distribution of species at variable carbonation conditions.
| Forsterite study | Experiment/simulation | Pressure (bar) | Temperature (K) | Relative humidity (%) | Reported water film thickness on forsterite (nm) |
|---|---|---|---|---|---|
| a Molar concentration of water determined using Beer's law in FTIR setup. b Measured by IR titration using H2O. c Measured by IR titration using D2O. d Calculated in this work via Grand Canonical Monte Carlo simulations. | |||||
| Loring et al. (2011)57 | Exp_1 | 182 | 323 | 136 | 2a |
| Loring et al. (2011)57 | Exp_2 | 182 | 323 | 95 | 1a |
| Loring et al. (2011)57 | Exp_3 | 182 | 323 | 81 | 0.1a |
| Thompson et al. (2014)56,93 | Exp_4 | 100 | 308 | 93 | 1.9a |
| Loring et al. (2015)58,92 | Exp_5 | 90 | 323 | 77 | 1.48b |
| Loring et al. (2019)59 | Exp_6 | 90 | 323 | 77 | 1.71b |
| Miller et al. (2019)59,61 | Exp_7 | 90 | 323 | 83 | 0.57b |
| Miller et al. (2019)59,61 | Exp_8 | 90 | 323 | 84 | 0.66b |
| Miller et al. (2019)59,61 | Exp_9 | 90 | 323 | 85 | 0.62b |
| Miller et al. (2019)59,61 | Exp_10 | 90 | 323 | 83 | 0.57c |
| Miller et al. (2019)59,61 | Exp_11 | 90 | 323 | 85 | 0.52c |
| Placencia-Gómez et al. (2020)54 | Exp_12 | 90 | 323 | 85 | 1b |
| Kerisit et al. (2021)62,86 | Exp_13 | 90 | 323 | 65 | 0.23b |
| This work | Sim_1 | 200 | 348 | 90 | 0.7d |
| This work | Sim_2 | 100 | 348 | 90 | 0.8d |
| This work | Sim_3 | 20 | 300 | 90 | 0.6d |
| This work | Sim_4 | 1 | 300 | 90 | 0.8d |
The water film thickness on the surface of C–S–H is higher than that on the hydroxylated surface of forsterite, as shown in Table 2. To clarify the latter point, we calculate the water adsorption energies for different numbers of water monolayers on both surfaces. The water adsorption energies for 1, 2, 3, and 4 monolayers of water on the {010} hydroxylated surface of forsterite and C–S–H are shown in Fig. 4. The water adsorption energies on the surface of C–S–H are shown to be consistently lower than that of the hydroxylated surface of forsterite. B3LYP calculations on gas phase clusters80 show that the hydration energies of Mg2+ cation are generally lower than that of Ca2+. However, AIMD simulations52 show that calcio-olivine (γ-Ca2SiO4), a crystalline calcium silicate structure, is more hydrophilic than forsterite when ½ monolayer to 2 monolayers of water is adsorbed on the surface. C–S–H is more disordered and has more defects than calcio-olivine, which could entail an even higher affinity to water.
 | ||
| Fig. 4 The calculated water adsorption energies on C–S–H and forsterite surfaces. Calorimetry measurements81 and previous classical MD simulation results49 on non-hydroxylated surface of forsterite are also presented for comparison. | ||
For comparison, the adsorption energies derived from calorimetry measurements81 and previous MD simulations on the non-hydroxylated {010} surface of forsterite49 are also included in Fig. 4. Our calculations overestimate the adsorption energies on the hydroxylated surface of forsterite by about 2–3 kcal mol−1 compared to the upper values of calorimetry experiments. This slight discrepancy could be potentially due to the forcefield artifacts or an indication that not all silicate groups dissociate water molecules adjacent to the surface. We should also emphasize that using the same forcefield, Kerisit et al.49 derive lower adsorption energy for associative adsorption of a single water monolayer on the non-hydroxylated surface of forsterite compared to experiments.
We also note that the water surface coverage for the first monolayer of hydroxylated forsterite and C–S–H is lower than that on the non-hydroxylated forsterite surface, Fig. 4. We can attribute this to the surface structure of these solids where dissociated water molecules are initially added to the surface resulting in the hydration of silicate groups and the presence of hydroxyl groups adjacent to metal cations. Therefore, the water surface coverage in hydroxylated structures with two monolayers is equivalent to one monolayer on the non-hydroxylated surface.
In addition, we calculate the adsorption energies of forsterite surface with the SPC/Fw water model,82 which is a modification of the SPC model with flexible bonds and angle, compared to the SPC/E water model that was originally incorporated in the modified ClayFF49 for the surface of forsterite. As pointed out in the methods section, our GCMC calculations are performed with the SPC water model, which has different oxygen and hydrogen charges than SPC/E, and, therefore, a different dipole moment. We use the SPC model in our GCMC calculations because it predicts water vapor pressure more accurately than SPC/E.83 Nevertheless, upon our calculation of water adsorption energies on the surface forsterite, SPC/Fw water model only slightly overestimates the adsorption energies compared to the SPC/E model, as shown in Fig. 4.
To resolve the spatial distribution of CO2 and H2O molecules and surface hydroxyl groups in the slit pore, we perform MD on fully equilibrated confined GCMC simulations. The details of these simulations are presented in the Methods section. The quantitative spatial distribution of water, CO2, and hydroxyl groups are presented in Fig. 5. The formation of nanometer-thick interfacial water layers is evident from distributions, acting as a barrier for CO2 molecules to contact the surface directly. Regardless of thermodynamic conditions, we observe four peaks in the water number density distributions on the surface of forsterite, see Fig. 5a–d. However, the furthest peak from the surface does not represent a fully formed water layer. This is consistent with a recent scCO2-forsterite experiment,61 which shows 3.5 water monolayers form on the forsterite surface at 85% relative humidity. On the other hand, we also observe water layers formed on the surface of C–S–H at all pore sizes, see Fig. 5e–h. However, the appearance of multiple peaks close to each other makes it hard to distinguish water monolayers, although the overall water film thickness on C–S–H is higher than that on forsterites. These irregularities of the water density peaks on the surface of C–S–H could partly result from the observation that surface calcium atoms are more labile and less attached to the surface than surface magnesium on forsterite. This enables the water molecules to penetrate the C–S–H gel and attach to the second calcium layer and silicate groups, as shown in Fig. 5e–h. Another reason for this irregularity on the C–S–H surface is a more disordered layering of calcium ions that attract water molecules and the defects and nanoscale cavities resulting from defective silicate chains. Therefore, hydrogen bonds form between water molecules, hydroxides, and hydrated silicates that are scattered at different elevations from the surface. The heterogeneity of hydrogen bonds in the disordered C–S–H structure has been studied through simulations and experiments.73,74,84,85
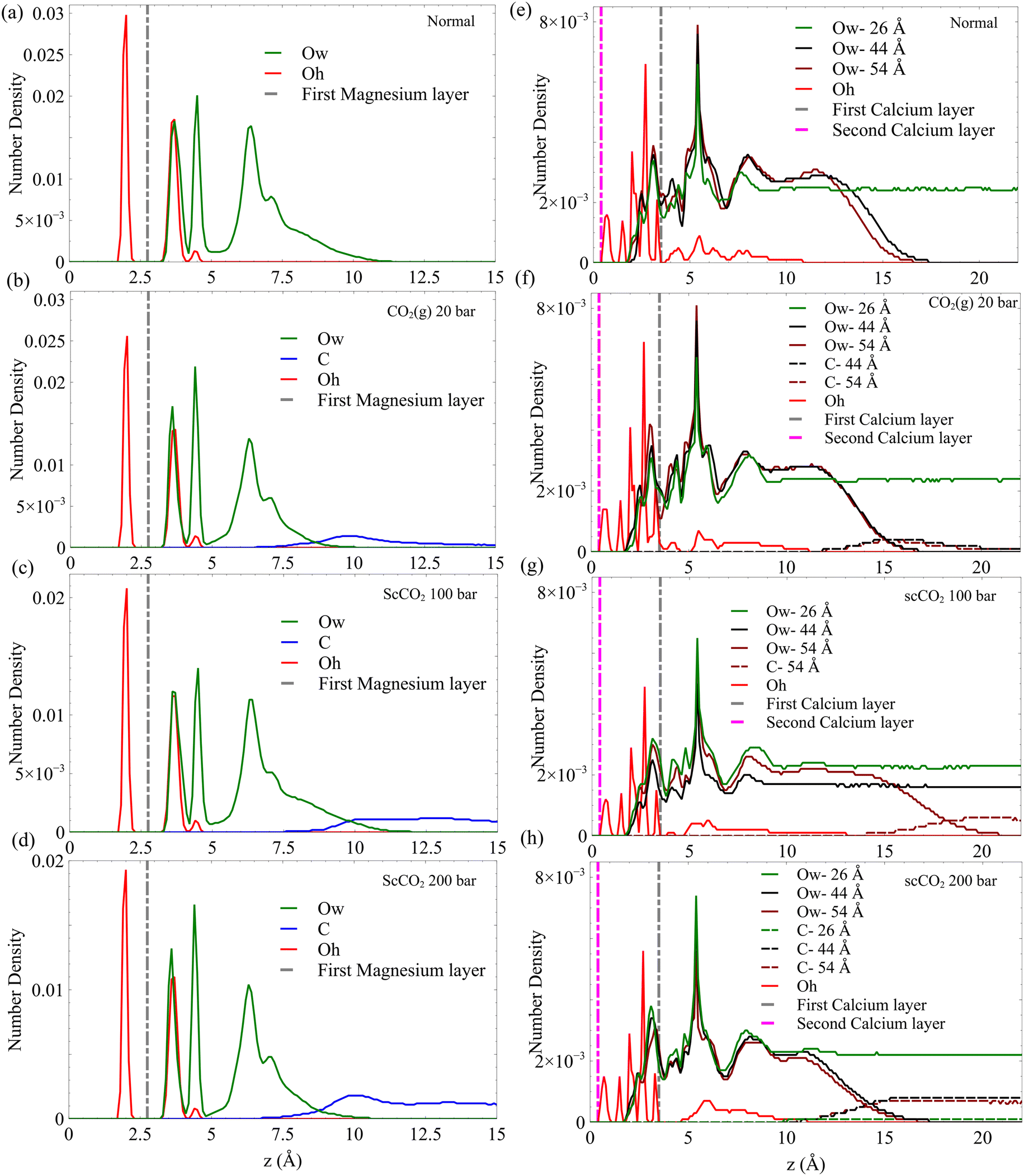 | ||
| Fig. 5 The average distribution of H2O, CO2, and OH-species in the slit-pore of forsterite and C–S–H. (a–d) Distribution of species in the slit-pore of forsterite at normal, CO2(g), scCO2 (100 bar), scCO2 (200 bar), respectively. (e–h) Distribution of species in the slit-pore of C–S–H at normal, sbCO2, scCO2 (100 bar), scCO2 (200 bar), respectively. The average location of the first magnesium layer on the surface of forsterite, as well as the location of the first and second calcium layers on C–S–H surface are shown by vertical dashed lines. Ow, C, and Oh represent water oxygen, carbon in carbon dioxide, and surface hydroxide oxygen, respectively. | ||
The formation of hydration layers has been previously reported for both forsterite49 and C–S–H (ref. 51) in unsaturated and saturated conditions, respectively. Additionally, recent Fast Force Mapping (FFM) experiments on the surface of boehmite86 illustrate the formation of the first water layer on the surface cavities (here, the silicate groups) and the second layer forming on top of hydroxide groups. This is consistent with our results on the surface of forsterite, as shown in Fig. 5a–d. For the surface of C–S–H, which is much more disordered, the appearance of multiple water density peaks around the surface demonstrates the same phenomena, as shown in Fig. 5e–h.
Another significant aspect of the distribution diagrams is that upon the entrance of the dense CO2 inside the pore, the last water layer contracts, and CO2 distribution peaks at the interface, as could be seen by comparing the distribution at ambient (1 bar) gaseous to supercritical CO2 (100, 200 bar) in Fig. 5. Although the number of water layers does not change, this could be a sign that CO2 pressure, if above a certain threshold, might alter the number of water layers. The appearance of interfacial peaks in CO2 distribution at all four conditions is a characteristic of immiscible mixtures verified through experiments and simulations.87–90 However, after juxtaposing water distributions with those of carbon dioxide, we confirm that although CO2 is not a good solvent for dipolar water, the appearance of overlapping shoulders between two species is indicative of enhanced mutual solubility at the interface. This is attributed to the interfacial capillary wave phenomenon and the Coulombic attraction between H2O and CO2 molecules.20 This could potentially amplify the rate of CO2 speciation at the interface. Another important feature of the resulted distributions is that the proximity of CO2 and hydroxides within the nanolayered hydration films, especially on the surface of C–S–H, may increase the bicarbonate and carbonate production rate. Although it is well established from previous simulations that the thin water film on hydrophilic material displaces CO2 away from the surface, this proximity of CO2 molecules and surface hydroxides motivates us to explore the energetics of CO2 physisorption on the surface.
Energetics of CO2 speciation on metal silicate surfaces
Our adsorption simulations are non-reactive. Thus, it is critical to resolve the mechanistic picture of CO2 speciation in adsorbed water nanofilms. CO2 may speciate to carbonic acid and bicarbonate within the nanofilm in the presence of dissolved cations.61 Moreover, it is probable that CO2 reacts with surface hydroxyl groups and surface cations. However, since CO2 molecules are displaced from the surface due to the presence of the adsorbed water film, it is necessary to quantify the free energy required to bring CO2 close to the surface. Since the residence time of water molecules around solvated Mg2+ ion is in the order of microseconds,47 the water coordination number around surface Mg2+ cations is important. Therefore, to sample the full phase space in our free energy calculation for the adsorption of CO2 on the forsterite surface, we consider the surface Mg water coordination number in addition to the perpendicular distance of CO2 from the surface. The coordination number is defined as: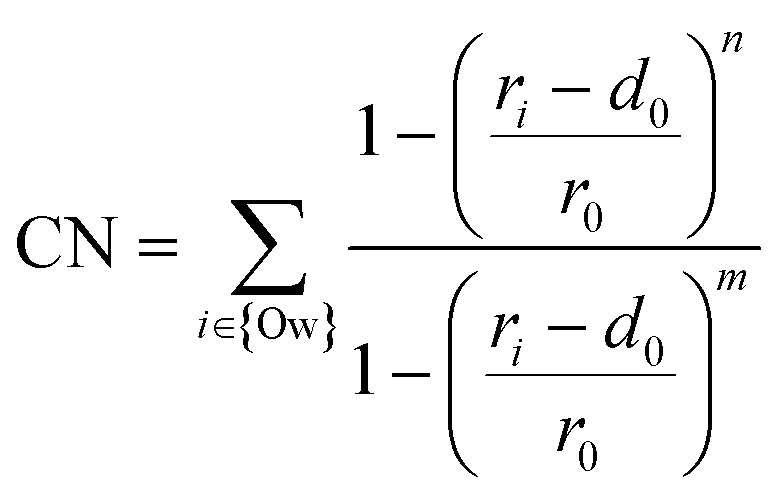 | (1) |
The resulting two-dimensional free energy landscape is shown in Fig. 6a. The minimum energy path (MEP) of the CO2 molecule as it goes from the solution to the vicinity of the surface is calculated via the Nudged Elastic Band technique (NEB) method.91 As shown in the figure, the coordination number of surface Mg changes from two to one as CO2 approaches the surface. Also, depending on the initial location of the CO2 molecule, three distinct MEPs are derived for the cases where CO2 is initially located at the 2nd, 3rd, and 4th layers of the adsorbed water, as shown in Fig. 6a. However, the energy that is required for the CO2 molecule to pass through the layers and reach the surface is not affected by the initial position of the CO2. This perhaps shows that regardless of the number of adsorbed water layers, which depends on the relative humidity and CO2 pressure, the energy of CO2 physisorption on the forsterite surface is unique. Also, the calculated free energy of CO2 adsorption is 2.5 kcal mol−1 higher than the adsorption free energy previously calculated by Kerisit et al.49 for the non-hydroxylated {010} surface of forsterite. However, the magnesium coordination number was not considered in the PMF calculation in that work. Therefore, this energy difference could either be the consequence of incorporating magnesium coordination number, the existence of a stronger hydrogen bond structure due to surface hydroxides, or the difference in computational methods used.
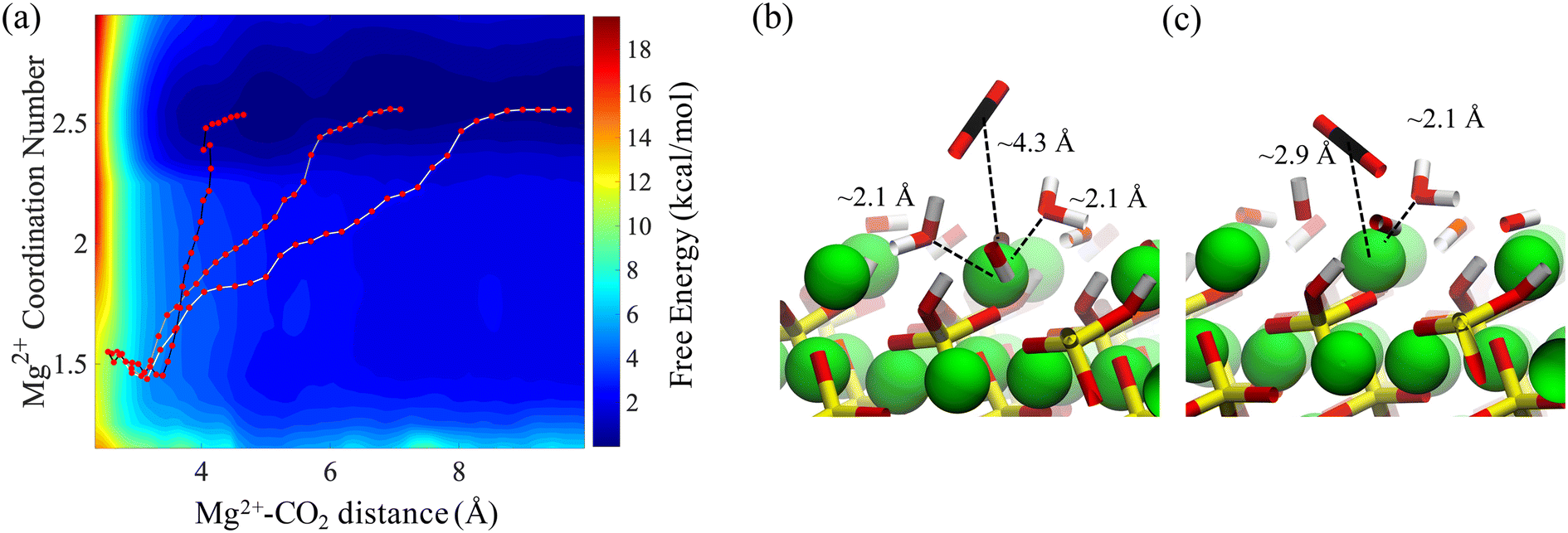 | ||
| Fig. 6 Potential-of-Mean-Force (PMF) for the adsorption of CO2 from the solution on the surface of forsterite. (a) 2D PMF for the adsorption CO2 on the surface. The three red-dotted paths represent minimum free energy paths corresponding to CO2 adsorption from the edge of the 2nd, 3rd and 4th adsorbed water layer. While these three paths are distinct, they show that Mg2+ dehydration is necessary for the adsorption of CO2 on the forsterite surfaces. Surface magnesium and the adsorbed CO2 molecule with (b) two and (c) one neighboring water molecule. Free energy calculations show the departure of one water molecule is thermodynamically necessary for CO2 adsorption. | ||
It is noteworthy that although we calculate the free energy of CO2 adsorption on the surface in contact with bulk water, it was previously shown that the structure of the water layers is not drastically different when the forsterite surface is in contact with a various number of water monolayers and supercritical CO2.49 These findings are consistent with in situ XRD measurements of H2O–CO2 sorption in hydrophilic montmorillonite,92,93 where CO2 intercalation was found limited when the hydration level goes beyond one water layer, regardless of isomorphous Me2+ exchange.
We also calculate the associative adsorption of CO2 at random surface sites of C–S–H, as shown in Fig. 7. We find that, like the forsterite case, CO2 is more stable in the solution than adsorbed on the surface. However, a metastable state is observed when the CO2 molecule and surface Ca2+ ions are separated by about 3.2 Å on the C–S–H surface. As shown in the inset, the oxygen of the CO2 molecule in the metastable state is oriented toward the surface calcium cation, reminiscent of a weakly chemisorbed state.94 Unlike the forsterite case where no minimum is observed, this unique characteristic of the C–S–H surface urges us to consider interfacial CO2 reactions.
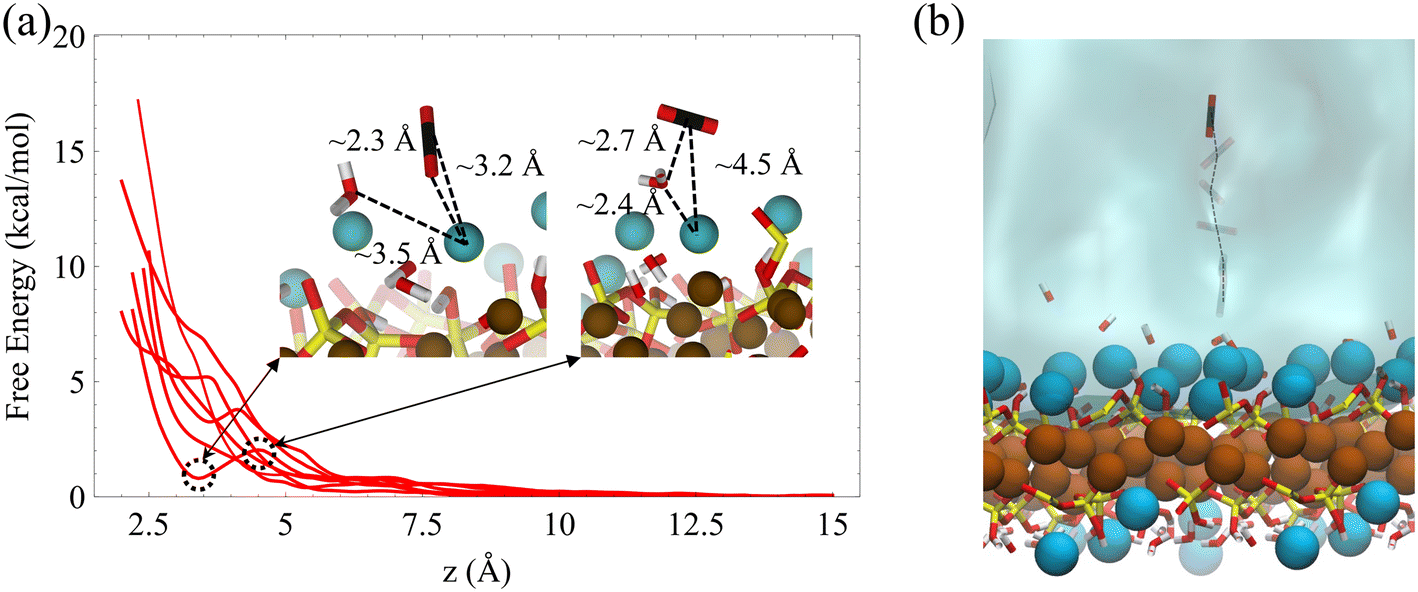 | ||
| Fig. 7 Non-reactive interaction of CO2 molecule with wet C–S–H. (a) Physisorption of a CO2 molecule on the surface of C–S–H. Multiple free energy curves are derived for random surface calcium atoms. The insets demonstrate a CO2 molecule on the surface at the metastable state and the transition state. (b) The umbrella sampling stages for the adsorption of CO2 on the surface. | ||
For the carbonation progression, the physisorbed CO2 must speciate to carbonic acid and bicarbonate by reacting with interfacial water molecules or surface-bound hydroxide. Here, we use Density Functional Theory (DFT) along with the Nudged Elastic Band (NEB) method to calculate the corresponding reaction paths, see the Methods section. Fig. 8a summarizes the three reaction paths. The adsorbed, transition, intermediate, and product states are schematically shown for each reaction path in Fig. 8b–e.
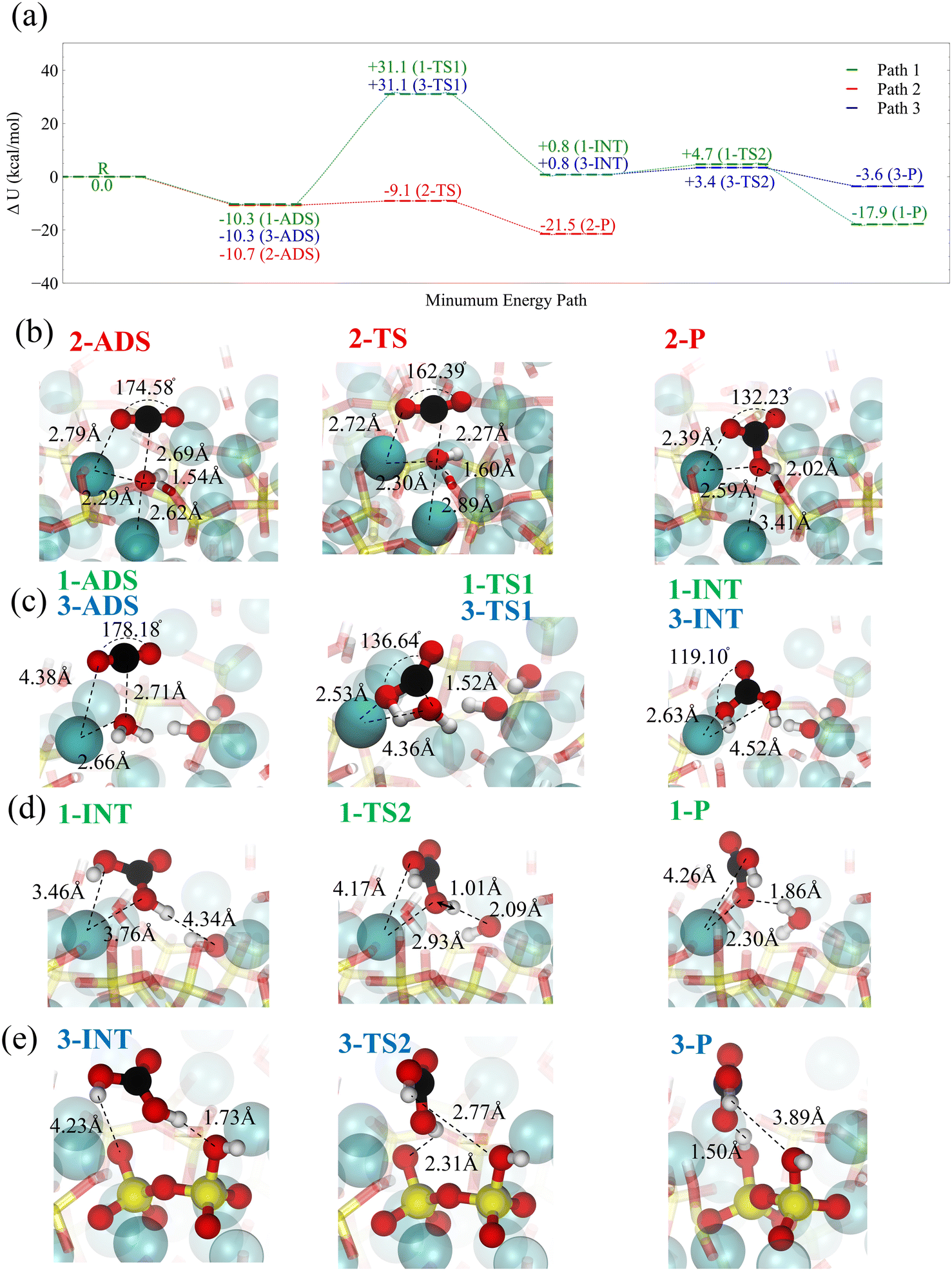 | ||
| Fig. 8 Reactive interaction of CO2 molecule with wet C–S–H. (a) DFT calculation of the reaction pathways between CO2 and C–S–H. (Path 1) The reaction between CO2 and bound water at the hydroxide site. (Path 2) The reaction between CO2 and surface-bound hydroxide. Strong physisorption is followed by a chemical reaction with a low barrier leading to the formation of bicarbonate. (Path 3) The reaction between CO2 and bound water at the dangling oxygen site. (b) Snapshots of the adsorbed state (ADS), transition state (TS) and product (P) for reaction paths 2. (c) Snapshots of the ADS, TS and intermediate (INT) state for the reaction of surface water and CO2 in path 1 and 3. d) Snapshots of the dissociation of carbonic acid to bicarbonate through reaction with surface hydroxide. e) Snapshots of the dissociation of carbonic acid to bicarbonate through reaction with dangling oxygen in silicates. | ||
The most significant reaction occurs at the hydroxide site (path 2), where strong physisorption is connected to a chemical reaction by a low barrier, leading to the formation of bicarbonate, see Fig. 8b. In a fully hydrated environment, it takes about 3–10 kcal mol−1 to bring CO2 to the hydroxide site on the surface of C–S–H, see Fig. 7a. The overall energy barrier associated with this path is lower than or in the same range of the free energy barrier for bicarbonate formation in solutions as calculated via ab initio MD.95 However, the abundance of hydroxide sites on the surface of C–S–H considerably impacts the overall rate of bicarbonate formation when compared to solution reactions. On the other hand, the reaction barrier of CO2 with water, according to paths 2 and 3, if seen from the adsorbate, is approximately 10 kcal mol−1 lower than the corresponding gas-phase reaction.96,97 The snapshots of this reaction are shown in Fig. 8c. Although our DFT simulations do not explicitly consider interfacial water molecules, these remarks indicate the catalytic role of the C–S–H surface. Furthermore, it should be noted that adding liquid water would further lower the barrier down to 19 kcal mol−1, according to coupled-cluster theory CCSD(T) calculations,96 without taking to account any surface catalytic effects. Once carbonic acid forms, it can dissociate, either transferring a proton to a neighboring unhydrated dangling silicate dimer oxygen on the C–S–H surface (path 3) or a neighboring hydroxide (path 1), see Fig. 8d and e. With a wetted surface, the first option for proton transfer is unlikely, and the proton transfer to hydroxide is far more exothermic. In both paths 1 and 3, the adsorbed CO2 reacts with surface water and forms carbonic acid in a concerted reaction.
It is noteworthy that ab initio molecular dynamics (AIMD) simulations in bulk aqueous water show that CO2 could react with water to form bicarbonate and a hydronium ion, followed by the formation of carbonic acid in a stepwise reaction.98 Similar routes for this reaction could be imagined to happen in the adsorbed water nanofilm on C–S–H and forsterite, leading to the formation of bicarbonate followed by the structural migration of excess proton to the surface hydroxide. Whether through reaction path 2 or in the adsorbed water nanofilm, the formation of bicarbonate is confirmed experimentally through in situ1H–13C Cross-Polarized NMR spectroscopy on forsterite nanoparticles.61 However, bicarbonates were not found on fused silica surfaces. As we demonstrated earlier in this work, it is energetically unlikely to bring CO2 to the surface of forsterite in the presence of surface water layers. Therefore, by ruling out the formation of carbonic acid through the direct reaction of CO2 with the surface hydroxide or surface water, there remain two possible reaction pathways: the reaction of CO2 with water 1) in the solvation shell of dissolved magnesium in the thin water film,61 or 2) at the interface of dense CO2 and water nanofilm. However, for the further progression of carbonation reaction to carbonate nucleation, whether on the C–S–H or forsterite, the carbonic acid needs to turn into bicarbonate and carbonate. The specific mechanisms for these deprotonation reactions are still not clear. We delve into the mechanistic picture of these reactions in a future paper.
Methods
For all DFT calculations, the program CP2K115 has been used with a combined Gaussian and plane waves ansatz. A double-ζ polarized basis set optimized for condensed phase116 was applied with a cutoff value of 500 Ry for the plane waves basis and GTH pseudopotentials.117–119 The Kohn–Sham equations were solved to an accuracy of 10−6 Eh with the revised PBE120,121 GGA functional and the D3 set of dispersion corrections.122 Extensive benchmark calculations show that revised PBE GGA functional with D3 dispersion correction is among the top performers in GGA functionals.123,124 The pre-optimized structures were fully optimized without any constraints, and the resulting structures were interpolated for NEB calculations. First, the NEB was optimized within the D-NEB125 framework until a maximum error of 5 mEh a0−1 in RMS gradients and 10−2 a0 in RMS displacement had been achieved. Next, the band optimization continued using IT-NEB126 for 10 steps before it switched to CI-NEB127 until the final convergence of 0.5 mEh a0−1 in RMS gradients and 10−3 a0 in RMS displacements had been achieved, which corresponds to an accuracy of at least 1 kJ mol−1. Minima were converged to at least 0.45 mEh a0−1 maximum gradient and 0.003 a0 maximum displacements. We note that the direct calculation of eigenvalues through the diagonalization of the Hessian matrix is imperative to confirm the transition state obtained vis the NEB method. In the case of NEB calculations in this work, the number of plane waves is very high in our DFT calculations to the level that the calculation of the Hessian matrix becomes impractical.
Competing interests
The authors declare no competing interests.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on work supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES), Chemical Sciences, Geosciences, and Biosciences Division through its Geosciences program at the University of California Irvine through an Early Career award to MJAQ (DE-SC0022301) and at Pacific Northwest National Laboratory (PNNL). Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect those of the funding agency. We also thank Dr. Russell Detwiler for fruitful discussions.References
- J. M. Matter and P. B. Kelemen, Nat. Geosci., 2009, 2, 837–841 CrossRef CAS.
- S. K. White, F. A. Spane, H. T. Schaef, Q. R. S. Miller, M. D. White, J. A. Horner and B. P. McGrail, Environ. Sci. Technol., 2020, 54, 14609–14616 CrossRef CAS PubMed.
- S. R. Gislason and E. H. Oelkers, Science, 2014, 344, 373–374 CrossRef CAS PubMed.
- M. F. Bertos, S. J. R. Simons, C. D. Hills and P. J. Carey, J. Hazard. Mater., 2004, 112, 193–205 CrossRef PubMed.
- B. P. McGrail, H. T. Schaef, A. M. Ho, Y.-J. Chien, J. J. Dooley and C. L. Davidson, J. Geophys. Res.: Solid Earth, 2006, 111(B12) DOI:10.1029/2005JB004169.
- D. S. Goldberg, T. Takahashi and A. L. Slagle, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9920–9925 CrossRef CAS PubMed.
- D. S. Goldberg, D. V. Kent and P. E. Olsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1327–1332 CrossRef CAS PubMed.
- C. Marieni, T. J. Henstock and D. A. H. Teagle, Geophys. Res. Lett., 2013, 40, 6219–6224 CrossRef CAS.
- S. Ó. Snæbjörnsdóttir, F. Wiese, T. Fridriksson, H. Ármansson, G. M. Einarsson and S. R. Gislason, Energy Procedia, 2014, 63, 4585–4600 CrossRef.
- I. M. Power, J. McCutcheon, A. L. Harrison, S. A. Wilson, G. M. Dipple, S. Kelly, C. Southam and G. Southam, Minerals, 2014, 4, 399–436 CrossRef.
- S. A. Wilson, A. L. Harrison, G. M. Dipple, I. M. Power, S. L. L. Barker, K. Ulrich Mayer, S. J. Fallon, M. Raudsepp and G. Southam, Int. J. Greenhouse Gas Control, 2014, 25, 121–140 CrossRef CAS.
- D. N. Huntzinger, J. S. Gierke, L. L. Sutter, S. K. Kawatra and T. C. Eisele, J. Hazard. Mater., 2009, 168, 31–37 CrossRef CAS PubMed.
- J. K. Stolaroff, G. V. Lowry and D. W. Keith, Energy Convers. Manage., 2005, 46, 687–699 CrossRef CAS.
- G. Rim, N. Roy, D. Zhao, S. Kawashima, P. Stallworth, S. G. Greenbaum and A.-H. A. Park, Faraday Discuss., 2021, 230, 187–212 RSC.
- W. K. O'Connor, D. C. Dahlin, G. E. Rush, C. L. Dahlin and W. K. Collins, Min. Metall. Explor., 2002, 19, 95–101 Search PubMed.
- National Academies of Sciences, Engineering, and Medicine, Negative Emissions Technologies and Reliable Sequestration: A Research Agenda, 2018 Search PubMed.
- I. C. Bourg, L. E. Beckingham and D. J. DePaolo, Environ. Sci. Technol., 2015, 49, 10265–10284 CrossRef CAS PubMed.
- J. Alcalde, S. Flude, M. Wilkinson, G. Johnson, K. Edlmann, C. E. Bond, V. Scott, S. M. V. Gilfillan, X. Ogaya and R. S. Haszeldine, Nat. Commun., 2018, 9, 2201 CrossRef PubMed.
- L. D. Gelb, K. E. Gubbins, R. Radhakrishnan and M. Sliwinska-Bartkowiak, Rep. Prog. Phys., 1999, 62, 1573–1659 CrossRef CAS.
- B. K. Peterson, J. P. R. B. Walton and K. E. Gubbins, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 1789–1800 RSC.
- E. E. Santiso, A. M. George, M. Sliwinska-bartkowiak, M. B. Nardelli and K. E. Gubbins, Adsorption, 2005, 1, 349–354 CrossRef.
- N. Chakraborty, Z. Karpyn, S. Liu, H. Yoon and T. Dewers, J. Nat. Gas Sci. Eng., 2020, 76, 103120 CrossRef CAS.
- A. Cihan, T. K. Tokunaga and J. T. Birkholzer, Langmuir, 2019, 35, 9611–9621 CrossRef CAS PubMed.
- T. K. Tokunaga, W. Shen, J. Wan, Y. Kim, A. Cihan, Y. Zhang and S. Finsterle, Water Resour. Res., 2017, 53, 9757–9770 CrossRef.
- A. Erfani, K. Szutkowski, C. P. Aichele and J. L. White, Langmuir, 2021, 37, 858–866 CrossRef CAS PubMed.
- O. Vincent, B. Marguet and A. D. Stroock, Langmuir, 2017, 33, 1655–1661 CrossRef CAS PubMed.
- A. Cihan, T. K. Tokunaga and J. T. Birkholzer, Water Resour. Res., 2021, 57, e2021WR029720 CrossRef.
- H. Li, E. Fratini, W.-S. Chiang, P. Baglioni, E. Mamontov and S.-H. Chen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 061505 CrossRef PubMed.
- R. Velasco, M. Pathak, P. Panja and M. Deo, in Unconventional Resources Technology Conference, Austin, Texas, 24–26 July 2017, Society of Exploration Geophysicists, American Association of Petroleum Geologists, Society of Petroleum Engineers, 2017, pp. 3248–3257 Search PubMed.
- K. Wu, Z. Chen, J. Li, X. Li, J. Xu and X. Dong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3358–3363 CrossRef CAS PubMed.
- P. Chiquet, D. Broseta and S. Thibeau, Geofluids, 2007, 7, 112–122 CrossRef CAS.
- E. A. Al-Khdheeawi, S. Vialle, A. Barifcani, M. Sarmadivaleh and S. Iglauer, Int. J. Greenhouse Gas Control, 2018, 68, 216–229 CrossRef CAS.
- S. Iglauer, C. H. Pentland and A. Busch, Water Resour. Res., 2015, 51, 729–774 CrossRef CAS.
- S. Iglauer and A. Al-Yaseri, Adv. Geo-Energy Res., 2021, 5, 347–350 CrossRef CAS.
- M. J. Abdolhosseini Qomi, K. J. Krakowiak, M. Bauchy, K. L. Stewart, R. Shahsavari, D. Jagannathan, D. B. Brommer, A. Baronnet, M. J. Buehler, S. Yip, F.-J. Ulm, K. J. Van Vliet and R. J.-M. Pellenq, Nat. Commun., 2014, 5, 4960 CrossRef CAS PubMed.
- M. Bauchy, B. Wang, M. Wang, Y. Yu, M. J. Abdolhosseini Qomi, M. M. Smedskjaer, C. Bichara, F.-J. Ulm and R. Pellenq, Acta Mater., 2016, 121, 234–239 CrossRef CAS.
- A. Morshedifard, S. Masoumi and M. J. Abdolhosseini Qomi, Nat. Commun., 2018, 9, 1785 CrossRef CAS PubMed.
- F. Xi, S. J. Davis, P. Ciais, D. Crawford-Brown, D. Guan, C. Pade, T. Shi, M. Syddall, J. Lv, L. Ji, L. Bing, J. Wang, W. Wei, K.-H. Yang, B. Lagerblad, I. Galan, C. Andrade, Y. Zhang and Z. Liu, Nat. Geosci., 2016, 9, 880–883 CrossRef CAS.
- T. A. Boden, R. J. Andres and G. Marland, Global, Regional, and National Fossil-Fuel CO2 Emissions (1751–2010) (V. 2013), Environmental System Science Data Infrastructure for a Virtual Ecosystem; Carbon Dioxide Information Analysis Center (CDIAC), Oak Ridge National Laboratory (ORNL), Oak Ridge, TN (United States), 2013 Search PubMed.
- L. Barcelo, J. Kline, G. Walenta and E. Gartner, Mater. Struct., 2014, 47, 1055–1065 CrossRef CAS.
- G. Rimmelé, V. Barlet-Gouédard, O. Porcherie, B. Goffé and F. Brunet, Cem. Concr. Res., 2008, 38, 1038–1048 CrossRef.
- V. Barlet-Gouédard, G. Rimmelé, O. Porcherie, N. Quisel and J. Desroches, Int. J. Greenhouse Gas Control, 2009, 3, 206–216 CrossRef.
- E. A. C. Panduro, M. Torsæter, K. Gawel, R. Bjørge, A. Gibaud, Y. Yang, S. Bruns, Y. Zheng, H. O. Sørensen and D. W. Breiby, Environ. Sci. Technol., 2017, 51, 9344–9351 CrossRef PubMed.
- P. Renforth, Nat. Commun., 2019, 10, 1401 CrossRef PubMed.
- P. J. M. Monteiro, G. Geng, D. Marchon, J. Li, P. Alapati, K. E. Kurtis and M. J. Abdolhosseini Qomi, Cem. Concr. Res., 2019, 124, 105806 CrossRef CAS.
- M. J. Abdolhosseini Qomi, M. Bauchy, F.-J. Ulm and R. J.-M. Pellenq, J. Chem. Phys., 2014, 140, 054515 CrossRef PubMed.
- H. Erras-Hanauer, T. Clark and R. van Eldik, Coord. Chem. Rev., 2003, 238–239, 233–253 CrossRef CAS.
- M. J. Abdolhosseini Qomi, Q. R. S. Miller, S. Zare, H. T. Schaef, J. P. Kaszuba and K. M. Rosso, Nat. Rev. Chem., 2022 DOI:10.1038/s41570-022-00418-1.
- S. Kerisit, J. H. Weare and A. R. Felmy, Geochim. Cosmochim. Acta, 2012, 84, 137–151 CrossRef CAS.
- A. Botan, B. Rotenberg, V. Marry, P. Turq and B. Noetinger, J. Phys. Chem. C, 2010, 114, 14962–14969 CrossRef CAS.
- S. Masoumi, S. Zare, H. Valipour and M. J. Abdolhosseini Qomi, J. Phys. Chem. C, 2019, 123, 4755–4766 CrossRef CAS.
- S. Kerisit, E. J. Bylaska and A. R. Felmy, Chem. Geol., 2013, 359, 81–89 CrossRef CAS.
- Q. R. S. Miller, J. P. Kaszuba, S. N. Kerisit, H. T. Schaef, M. E. Bowden, B. Peter McGrail and K. M. Rosso, Environ. Sci.: Nano, 2020, 7, 1068–1081 RSC.
- E. Placencia-Gómez, S. N. Kerisit, H. S. Mehta, O. Qafoku, C. J. Thompson, T. R. Graham, E. S. Ilton and J. S. Loring, Environ. Sci. Technol., 2020, 54, 6888–6899 CrossRef PubMed.
- C. J. Thompson, J. S. Loring, K. M. Rosso and Z. Wang, Int. J. Greenhouse Gas Control, 2013, 18, 246–255 CrossRef CAS.
- C. J. Thompson, P. F. Martin, J. Chen, P. Benezeth, H. T. Schaef, K. M. Rosso, A. R. Felmy and J. S. Loring, Rev. Sci. Instrum., 2014, 85, 044102 CrossRef PubMed.
- J. S. Loring, C. J. Thompson, Z. Wang, A. G. Joly, D. S. Sklarew, H. T. Schaef, E. S. Ilton, K. M. Rosso and A. R. Felmy, Environ. Sci. Technol., 2011, 45, 6204–6210 CrossRef CAS PubMed.
- J. S. Loring, J. Chen, P. Bénézeth, O. Qafoku, E. S. Ilton, N. M. Washton, C. J. Thompson, P. F. Martin, B. P. McGrail, K. M. Rosso, A. R. Felmy and H. T. Schaef, Langmuir, 2015, 31, 7533–7543 CrossRef CAS PubMed.
- J. S. Loring, Q. R. S. Miller, C. J. Thompson and H. T. Schaef, in Science of Carbon Storage in Deep Saline Formations, ed. P. Newell and A. G. Ilgen, Elsevier, 2019, pp. 63–88 Search PubMed.
- X. Zhang, A. S. Lea, A. M. Chaka, J. S. Loring, S. T. Mergelsberg, E. Nakouzi, O. Qafoku, J. J. De Yoreo, H. T. Schaef and K. M. Rosso, Nat. Mater., 2021, 1–7 Search PubMed.
- Q. R. S. Miller, D. A. Dixon, S. D. Burton, E. D. Walter, D. W. Hoyt, A. S. McNeill, J. D. Moon, K. S. Thanthiriwatte, E. S. Ilton, O. Qafoku, C. J. Thompson, H. T. Schaef, K. M. Rosso and J. S. Loring, J. Phys. Chem. C, 2019, 123, 12871–12885 CrossRef CAS.
- S. N. Kerisit, S. T. Mergelsberg, C. J. Thompson, S. K. White and J. S. Loring, Environ. Sci. Technol., 2021, 55, 12539–12548 CrossRef CAS PubMed.
- S. T. Mergelsberg, S. N. Kerisit, E. S. Ilton, O. Qafoku, C. J. Thompson and J. S. Loring, Chem. Commun., 2020, 56, 12154–12157 RSC.
- R. S. Smith, Z. Li, Z. Dohnálek and B. D. Kay, J. Phys. Chem. C, 2014, 118, 29091–29100 CrossRef CAS.
- S. A. Hamid, Z. Kristallogr., 1981, 154, 189–198 CAS.
- S. Peuble, M. Andreani, P. Gouze, M. Pollet-Villard, B. Reynard and B. Van de Moortele, Chem. Geol., 2018, 476, 150–160 CrossRef CAS.
- K. Marquardt and U. H. Faul, Phys. Chem. Miner., 2018, 45, 139–172 CrossRef CAS.
- T. Xing, W. Zhu, F. Fusseis and H. Lisabeth, Solid Earth, 2018, 9, 879–896 CrossRef.
- S. Erdmann, B. Scaillet, C. Martel and A. Cadoux, J. Petrol., 2014, 55, 2377–2402 CrossRef CAS.
- A. J. Luhmann, B. M. Tutolo, B. C. Bagley, D. F. R. Mildner, W. E. Seyfried Jr. and M. O. Saar, Water Resour. Res., 2017, 53, 1908–1927 CrossRef.
- A. Silvestri, E. Ataman, A. Budi, S. L. S. Stipp, J. D. Gale and P. Raiteri, Langmuir, 2019, 35, 16669–16678 CrossRef CAS PubMed.
- A. Silvestri, A. Budi, E. Ataman, M. H. M. Olsson, M. P. Andersson, S. L. S. Stipp, J. D. Gale and P. Raiteri, J. Phys. Chem. C, 2017, 121, 24025–24035 CrossRef CAS.
- M. J. Abdolhosseini Qomi, M. Bauchy, F.-J. Ulm and R. J.-M. Pellenq, J. Chem. Phys., 2014, 140, 054515 CrossRef PubMed.
- M. Youssef, R. J.-M. Pellenq and B. Yildiz, J. Am. Chem. Soc., 2011, 133, 2499–2510 CrossRef CAS PubMed.
- N. Spycher, K. Pruess and J. Ennis-King, Geochim. Cosmochim. Acta, 2003, 67, 3015–3031 CrossRef CAS.
- L. W. Diamond and N. N. Akinfiev, Fluid Phase Equilib., 2003, 208, 265–290 CrossRef CAS.
- Q. Li, C. I. Steefel and Y.-S. Jun, Environ. Sci. Technol., 2017, 51, 10861–10871 CrossRef CAS PubMed.
- G. W. Scherer and B. Huet, Int. J. Greenhouse Gas Control, 2009, 3, 731–735 CrossRef CAS.
- A. Raoof, H. M. Nick, T. K. T. Wolterbeek and C. J. Spiers, Int. J. Greenhouse Gas Control, 2012, 11, S67–S77 CrossRef CAS.
- M. Pavlov, P. E. M. Siegbahn and M. Sandström, J. Phys. Chem. A, 1998, 102, 219–228 CrossRef CAS.
- S. Chen and A. Navrotsky, Am. Mineral., 2010, 95, 112–117 CrossRef CAS.
- Y. Wu, H. L. Tepper and G. A. Voth, J. Chem. Phys., 2006, 124, 024503 CrossRef PubMed.
- J. Vorholz, V. I. Harismiadis, B. Rumpf, A. Z. Panagiotopoulos and G. Maurer, Fluid Phase Equilib., 2000, 170, 203–234 CrossRef CAS.
- D. Hou, H. Ma, Y. Zhu and Z. Li, Acta Mater., 2014, 67, 81–94 CrossRef CAS.
- B. Li, N. Li, H. J. H. Brouwers, Q. Yu and W. Chen, Constr. Build. Mater., 2020, 233, 117347 CrossRef CAS.
- E. Nakouzi, A. G. Stack, S. Kerisit, B. A. Legg, C. J. Mundy, G. K. Schenter, J. Chun and J. J. De Yoreo, J. Phys. Chem. C, 2021, 125, 1282–1291 CrossRef CAS.
- S. R. P. da Rocha, K. P. Johnston, R. E. Westacott and P. J. Rossky, J. Phys. Chem. B, 2001, 105, 12092–12104 CrossRef CAS.
- B. Kvamme, A. Graue, T. Buanes, T. Kuznetsova and G. Ersland, Int. J. Greenhouse Gas Control, 2007, 1, 236–246 CrossRef CAS.
- T. Kuznetsova and B. Kvamme, Phys. Chem. Chem. Phys., 2002, 4, 937–941 RSC.
- D. N. Espinoza and J. C. Santamarina, Water Resour. Res., 2011, 15, 707–719 Search PubMed.
- H. Jónsson, G. Mills and K. W. Jacobsen, in Classical and Quantum Dynamics in Condensed Phase Simulations, World Scientific, 1998, pp. 385–404 Search PubMed.
- H. T. Schaef, J. S. Loring, V.-A. Glezakou, Q. R. S. Miller, J. Chen, A. T. Owen, M.-S. Lee, E. S. Ilton, A. R. Felmy, B. P. McGrail and C. J. Thompson, Geochim. Cosmochim. Acta, 2015, 161, 248–257 CrossRef CAS.
- J. S. Loring, E. S. Ilton, J. Chen, C. J. Thompson, P. F. Martin, P. Bénézeth, K. M. Rosso, A. R. Felmy and H. T. Schaef, Langmuir, 2014, 30, 6120–6128 CrossRef CAS PubMed.
- U. Burghaus, Prog. Surf. Sci., 2014, 89, 161–217 CrossRef CAS.
- A. Stirling, J. Phys. Chem. B, 2011, 115, 14683–14687 CrossRef CAS PubMed.
- M. T. Nguyen, M. H. Matus, V. E. Jackson, V. T. Ngan, J. R. Rustad and D. A. Dixon, J. Phys. Chem. A, 2008, 112, 10386–10398 CrossRef CAS PubMed.
- S. Yamabe and N. Kawagishi, Theor. Chem. Acc., 2011, 130, 909–918 Search PubMed.
- A. Stirling and I. Pápai, J. Phys. Chem. B, 2010, 114, 16854–16859 CrossRef CAS PubMed.
- M. G. Martin, MCCCS Towhee: a tool for Monte Carlo molecular simulation, Mol. Simul., 2013, 39, 1212–1222 CrossRef CAS , (accessed 29 September 2018).
- G. C. A. M. Mooij, D. Frenkel and B. Smit, J. Phys.: Condens. Matter, 1992, 4, L255 CrossRef.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, in Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, Israel, April 13–16, 1981, ed. B. Pullman, Springer Netherlands, Dordrecht, 1981, pp. 331–342 Search PubMed.
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS.
- R. T. Cygan, J.-J. Liang and A. G. Kalinichev, J. Phys. Chem. B, 2004, 108, 1255–1266 CrossRef CAS.
- M. J. Abdolhosseini Qomi, K. J. Krakowiak, M. Bauchy, K. L. Stewart, R. Shahsavari, D. Jagannathan, D. B. Brommer, A. Baronnet, M. J. Buehler, S. Yip, F.-J. Ulm, K. J. V. Vliet and R. J.-M. Pellenq, Nat. Commun., 2014, 5, 4960 CrossRef CAS PubMed.
- H. Yan, C. Park, G. Ahn, S. Hong, D. T. Keane, C. Kenney-Benson, P. Chow, Y. Xiao and G. Shen, Geochim. Cosmochim. Acta, 2014, 145, 268–280 CrossRef CAS.
- H. E. King, M. Stimpfl, P. Deymier, M. J. Drake, C. R. A. Catlow, A. Putnis and N. H. de Leeuw, Earth Planet. Sci. Lett., 2010, 300, 11–18 CrossRef CAS.
- N. H. de Leeuw, S. C. Parker, C. R. A. Catlow and G. D. Price, Phys. Chem. Miner., 2000, 27, 332–341 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- R. T. Cygan, V. N. Romanov and E. M. Myshakin, J. Phys. Chem. C, 2012, 116, 13079–13091 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- R. Shahsavari, R. J.-M. Pellenq and F.-J. Ulm, Phys. Chem. Chem. Phys., 2011, 13, 1002–1011 RSC.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- A. Funk and H. F. R. Trettin, Ind. Eng. Chem. Res., 2013, 52, 2168–2173 CrossRef CAS.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- T. Gould, Phys. Chem. Chem. Phys., 2018, 20, 27735–27739 RSC.
- S. A. Trygubenko and D. J. Wales, J. Chem. Phys., 2004, 120, 2082–2094 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2en00148a |
| This journal is © The Royal Society of Chemistry 2022 |