
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing potential profiles in water electrolysers to minimise titanium use†
Hans
Becker
 a,
Edmund J. F.
Dickinson
a,
Xuekun
Lu
ab,
Ulf
Bexell
c,
Sebastian
Proch
c,
Claire
Moffatt
c,
Mikael
Stenström
c,
Graham
Smith
a and
Gareth
Hinds
*a
a,
Edmund J. F.
Dickinson
a,
Xuekun
Lu
ab,
Ulf
Bexell
c,
Sebastian
Proch
c,
Claire
Moffatt
c,
Mikael
Stenström
c,
Graham
Smith
a and
Gareth
Hinds
*a
aNational Physical Laboratory, Hampton Road, Teddington, TW11 0LW, UK. E-mail: gareth.hinds@npl.co.uk
bElectrochemical Innovation Lab, Department of Chemical Engineering, UCL, London, WC1E 7JE, UK
cSurface Research, Strategic Research, AB Sandvik Materials Technology, 81181 Sandviken, Sweden
First published on 10th May 2022
Abstract
The assumption of a highly corrosive potential at the anode bipolar plate (BPP) and porous transport layer (PTL) in a proton exchange membrane water electrolyser (PEMWE) stack often leads to selection of expensive materials such as platinum–coated titanium. Here, we develop a physicochemical model of electrochemical potential distribution in a PTL and validate it by in situ and ex situ electrochemical potential measurement. Model predictions suggest that, under typical PEMWE operating conditions, the corrosive zone associated with the anode polarisation extends only ≈200 μm into the PTL from the catalyst layer, obviating the need for highly corrosion-resistant materials through the bulk of the PTL and at the BPP. Guided by this observation, we present single cell PEMWE tests using anode current collectors fabricated from carbon-coated 316L stainless steel. The material is shown to be tolerant to potentials up to 1.2 V vs. RHE and when tested in situ for 30 days at 2 A cm−2 showed no evidence of degradation. These results strongly suggest that much of the titanium in PEMWEs may be substituted with cheaper, more abundant materials with no loss of electrolyser performance or lifetime, which would significantly reduce the cost of green hydrogen. The combined modelling and experimental approach developed here shows great promise for design optimisation of PEMWEs and other electrochemical energy conversion devices.
Broader contextComplete decarbonisation of the heating and transportation sectors is required in the next 25 years to limit global warming to below 2 °C, and preferably to 1.5 °C. Large scale energy storage is key to this goal and, while advances in Li-ion battery technology have enabled their use in passenger vehicles and short term electricity storage, alternative energy vectors such as hydrogen are more suited to heavy duty transport and seasonal energy storage for heating. Production of ‘green hydrogen’ by the electrolysis of water is a promising solution but is currently hampered by the high cost of the technology. Operating costs will continue to fall with decreasing renewable electricity prices so the main challenge is to reduce the capital cost of water electrolysers. This work points the way towards cost reduction of proton exchange membrane water electrolyser stacks by establishing the underpinning theory and experimental validation required to inform new design strategies for replacement of expensive component materials with cheaper alternatives. |
Introduction
Proton exchange membrane water electrolysers (PEMWEs) use renewable energy to produce green hydrogen, a key energy vector for advanced economies trying to achieve COP21 targets. The cost of green hydrogen is still high in comparison to both grey and blue hydrogen and achieving cost competitiveness in multiple sectors, expected at prices of ≈USD 2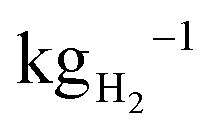 , remains a challenge.1 Much research is being performed to decrease the cost of hydrogen produced by PEMWE, often with a focus on reducing the cost of electrocatalysts2 or optimising cell design to increase performance.3 A range of reports indicate that, regardless of manufacturing scale, bipolar plates (BPPs) and porous transport layers (PTLs) are among the most expensive components in a PEMWE stack.4–6 Reducing the cost of these components is therefore key to meeting Hydrogen Europe's price target of €1000 per kg d−1 of H2 production by 2030.7
, remains a challenge.1 Much research is being performed to decrease the cost of hydrogen produced by PEMWE, often with a focus on reducing the cost of electrocatalysts2 or optimising cell design to increase performance.3 A range of reports indicate that, regardless of manufacturing scale, bipolar plates (BPPs) and porous transport layers (PTLs) are among the most expensive components in a PEMWE stack.4–6 Reducing the cost of these components is therefore key to meeting Hydrogen Europe's price target of €1000 per kg d−1 of H2 production by 2030.7
The BPP and PTL are critical components of the stack as they perform multiple functions, including distribution of electrical current and heat, mechanical support for the stack and catalyst coated membrane (CCM), and flow distribution to ensure effective mass transport to and from the catalytic sites. At the anode side of the electrolyser, the highly oxidising potential of the catalyst layer (CL) and the presence of molecular oxygen make corrosion resistance a required property for both BPPs and PTLs if they are to operate effectively for the >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h expected of a state-of-the-art PEMWE.8
000 h expected of a state-of-the-art PEMWE.8
Titanium is the conventional material of choice for the PTL and BPP since it readily forms a passivating oxide layer in contact with oxygen, which protects the bulk metal from corrosion. Unfortunately titanium is comparatively expensive to produce6 and machine.9 Titanium has also been identified by the European Union as a critical raw material, highlighting a potential risk to supply.10 Furthermore, titanium oxides have an unacceptably high contact resistance and have been reported to corrode and contaminate the CCM after operation for 1000 h under constant current or current cycling operation.11,12 Titanium is therefore typically coated with thin layers of more noble metals such as platinum,13 gold,14 and/or iridium,9,15 which reduce interfacial contact resistance (ICR) and mitigate titanium corrosion.13 Some alternative materials have been shown to degrade in the harsh environment of the anode and shorten PEMWE lifetime: for example, a carbon and 316L stainless steel-based PTL experienced significant corrosion after being operated for a relatively short period.16–18 If 316L is to be used as the PTL, an additional corrosion-resistant coating is needed.19–22
These limitations on the application of cheaper materials derive only from investigations in which the PTL is substituted in its entirety. The requirement for a highly corrosion-resistant material for the BPP is based on the commonly-held assumption that a uniformly aggressive corrosive environment applies throughout the anode electrolyte. This view is often reinforced by the results of ex situ testing at elevated potential, under which conditions many materials experience significant corrosive attack. Conversely, the authors’ recent report of in situ reference electrode measurements made at the PTL–current collector interface in a single cell (corresponding to the PTL–BPP interface in a stack) concluded that, due to the high resistivity of the PTL aqueous phase, the BPP remains at its open circuit potential (OCP), even when the cell voltage is >2 V.23 This implies a ‘decoupling’ of the BPP from the highly anodically polarised environment of the CL and raises the possibility that the BPP, and possibly some portion of the PTL more distant from the anode CL, can be safely substituted with cheaper materials.
In this work, we investigate the practicality of this suggestion by exploring the spatial profile and magnitude of the electrochemical potential (hereafter referred to simply as ‘potential’) through the thickness of the anode PTL, using theoretical modelling and in situ reference electrode measurements. Having predicted a much less aggressive environment at the BPP/PTL interface, we identify carbon-coated 316L stainless steel (C-316L) as a promising candidate material for replacing platinum–coated titanium (Pt–Ti), and evaluate it as the current collector material in a single cell PEMWE operated for 30 days. Due to the use of a single cell in the experimental part, the term current collector (CC), rather than BPP, will be used hereafter in this work when referring to the components behind the PTL (i.e. monopolar end plate and expanded titanium mesh) in the context of specific measurements reported. Systematic post-mortem and in situ measurements reveal no degradation of the C-316L samples during testing, encouraging further long-term, full-scale testing of this low-cost material.
Results and discussion
Potential profile across anode components
The potential of the anode CL in an operating PEMWE is necessarily in excess of 1.2 V vs. RHE in order to drive the oxygen evolution reaction. This is the reason that anode catalysts are limited to very oxidation-stable materials such as iridium oxide. However, it does not automatically follow that the potential of all components in the anode is also in excess of 1.2 V vs. RHE. While the CL, PTL and CC are all connected together with negligibly small electrical resistance, the ionic resistance between different parts of the anode may be high due to the low conductivity of the feed water. It is a combination of these resistances, with other parameters, that determines the extent to which the potential is dominated by local electrochemical reactions (such as corrosion processes) vs. those occurring in the CL (oxygen evolution). If the PTL aqueous phase were highly conductive, corrosion of the PTL material could proceed with the cathode CL (hydrogen evolution reaction) acting as counter electrode, because current density can easily be sunk from the PTL through the proton exchange membrane (PEM). Conversely, for a highly resistive PTL aqueous phase, the ionic resistance prevents the cathode CL from supporting corrosion of the PTL material – the available cathodic area is the PTL itself. The latter case is the situation of a corrosion potential, as would be encountered in an ex situ experiment where the material is unpolarised, and so the local potential would be expected to be close to its open circuit value. We describe the PTL potential in this case as being ‘decoupled’ from the anode CL.The water in a state-of-the art operating PEMWE is highly deionised, with a conductivity <1.0 μS cm−1 (typical specifications are ASTM Type II water or ISO 3696:1987 Grade 2), because the membrane and ionomers used as proton conductors in PEMWEs are vulnerable to foreign cation poisoning. The finite water resistivity means that the extent of local anodic polarisation of the PTL due to the applied cell voltage varies spatially, decreasing further away from the anode CL. This raises two related questions for the rational design of PTL geometry and selection of materials. First, can the potential at the CC be predicted theoretically as a function of the PTL properties? Second, for a given PTL, can the length scale be determined, theoretically and/or experimentally, over which the local potential transitions from the highly oxidative conditions of the anode CL to the more benign OCP of the CC?
To address the first question, a lumped theoretical physicochemical model was developed to describe the extent of decoupling between the anode CL and CC, using a “decoupling coefficient” suitable for situations when the PTL is made of the same material as the CC. The derivation and form of the decoupling coefficient are given in Supplementary Note 1 (ESI†). This model shows that the extent of decoupling is proportional to the electrolyte resistivity; it also depends on PTL geometry (electrochemically active surface area, porosity, tortuosity) as well as the kinetic properties of the electrochemical reaction that determines the OCP of the CC material in the electrolyte, described here simply with an exchange current density. When evaluated with suitable input parameters (Table S1, ESI†), the model prediction agrees with the previous observation of extensive decoupling under typical operating conditions.23
To evaluate the potential profile through the PTL, a simple physicochemical model was developed and parameterised using realistic, experimentally established values. Key assumptions are described briefly in the “Methods” section, with full mathematical detail and the chosen parameter values described fully in Supplementary Note 2 and Tables S2 and S3 (ESI†). Predicted potential profiles are shown in Fig. 1(a) parameterised for realistic PEMWE operating conditions of 2 A cm−2 at 60 °C. Various water conductivities are compared, ranging from that of Type I deionised water up to that of pH 1 H2SO4(aq) as an unrealistic upper limit. The results illustrate how the zone of appreciable anodic polarisation within the PTL is confined more closely to the CL as electrolyte resistivity increases. Experimental validation of this trend is presented in Fig. S1 and S2 (ESI†). For the more realistic case of deionised water, the partial thickness of the PTL that is polarised to 100 mV more positive than its OCP is only ≈70 μm for Type I water and only ≈300 μm for Type II water.
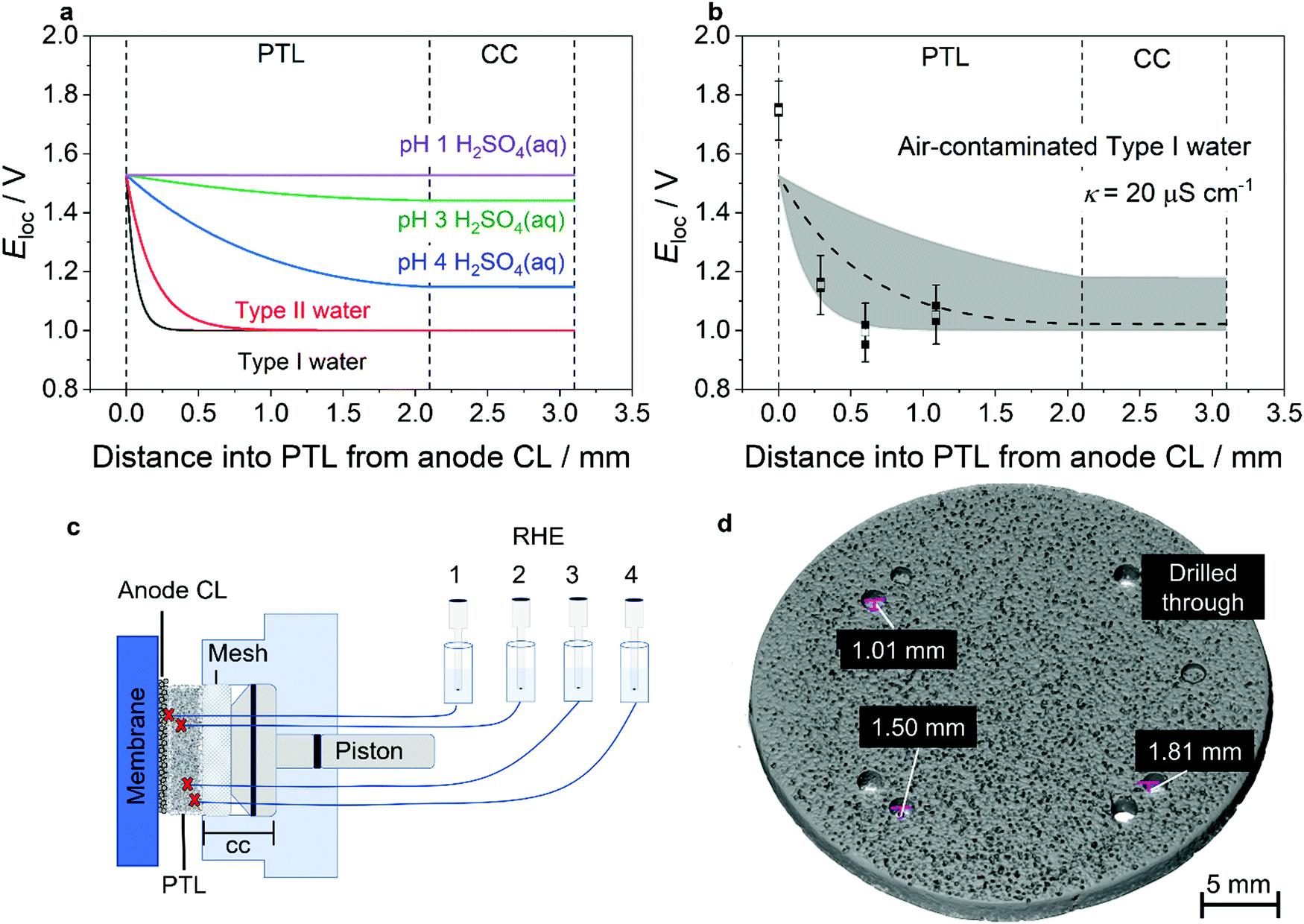 | ||
| Fig. 1 Potential profile through the thickness of an electrolyser anode. (a) Prediction from a physicochemical model of the potential profile through the thickness of an operating PEMWE anode (2 A cm−2 at 60 °C), showing that lowered ionic strength of the feed water results in the local potential tending to the ex situ OCP (set as 1 V vs. RHE) at shorter distances away from the CL. (b) Comparison of predicted (dashed line), measured (filled black squares), and average of measured (open squares) local potential as a function of distance into the PTL from the anode CL in air-contaminated Type I water. Each measurement was performed three times, with all data points (filled black squares) shown in the figure. The shaded area indicates the possible range of simulation results given uncertainty in input parameters. Error bars reflect the experimental measurement uncertainty, which is associated primarily with liquid junction potentials. Details of model parameterisation and uncertainty evaluation are given in Supplementary Note 2 and Tables S2 and S3 (ESI†). (c) Schematic showing the placement of in situ reference electrodes within the anode PTL of an operating single cell PEMWE and the measurement of Eloc as specified in Fig. S3 (ESI†), corresponding to the potential difference between the anode CC and any one RHE. (d) XCT of the anode PTL showing the holes used to locate the in situ reference electrodes at defined distances from the back of the PTL. | ||
Finally, the model was used to predict the extent of decoupling expected in a realistic worst-case scenario of a laboratory experimental cell containing air-contaminated Type I water. Here the water equilibrates with CO2 from the ambient air and therefore has a measured conductivity of 20 μS cm−1 at 60 °C (Fig. 1(b)), which is above the recommended acceptable water quality conductivity standard for PEMWEs of 1 μS cm−1 (ASTM Type II water).24 The model prediction is shown in Fig. 1(b) (dashed line) and demonstrates that the potential of the CC is determined almost solely by its OCP under these operating conditions. The parameterisation used here assumes that all components are Pt–Ti, which from experiment exhibited an OCP of 1.0 V vs. RHE in oxygen-saturated water at 60 °C (Fig. 2(a)). Note that this OCP is material-dependent so the potentials shown do not constitute predictions for all scenarios. Parameterisation of this model relied on a number of parameters for which experimentally determined information was limited, so a predicted range is shown to indicate the sensitivity of the model.
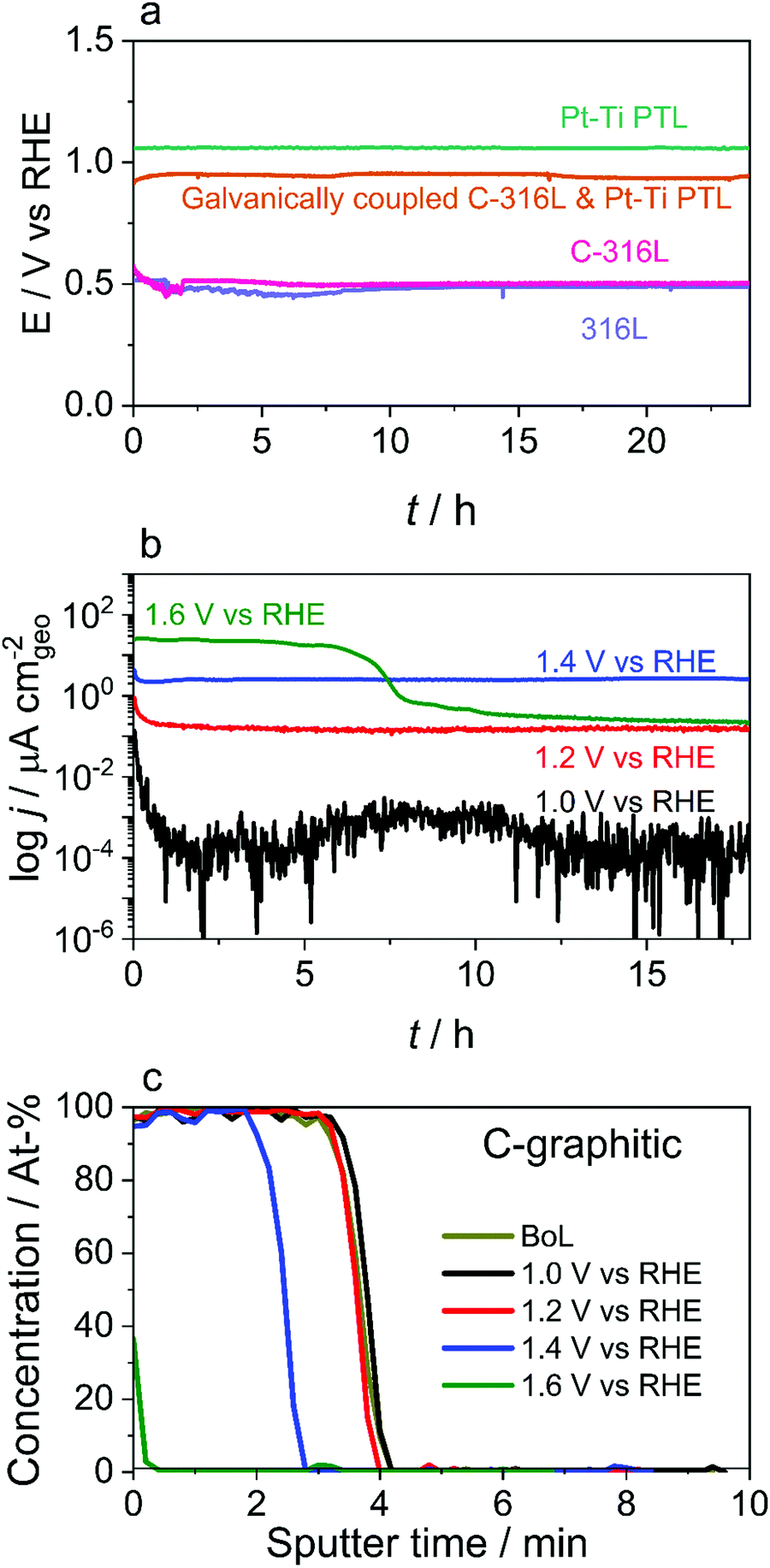 | ||
| Fig. 2 Evaluating alternative materials for suitability as anode components. (a) Ex situ test of the OCP of 316L, C-316L, Pt-coated Ti PTL, and galvanically coupled C-316L and Pt-coated Ti PTL in oxygen-saturated dilute aqueous H2SO4 (pH 4.5) at 60 °C. Precise values depend on surface history but general trends are observed: the thick (≈μm) coating in Pt-coated Ti makes its OCP close to that of Pt/O2, while the OCP of C-316L is close to that of 316L due to the much thinner and more porous carbon coating (≈nm). Faster kinetics of Pt pulls the OCP of the galvanically coupled C-316L and Pt–Ti PTL closer to the OCP of the Pt–Ti PTL. (b) Current density response of C-304L to a potential hold for 24 h in oxygen-saturated dilute aqueous H2SO4 (pH 3), 0.1 ppm HF, at 70 °C. Significant corrosion current density is only observed at ≥1.4 V vs. RHE. (c) AES sputter depth profiles of C-304L at beginning of life (BoL) and after the 18 h potential hold experiments. Measurable carbon coating removal is only observed at ≥1.4 V vs. RHE. | ||
Previous experimental evidence has shown decoupling of the CC from the CL, but has not resolved the potential profile closer to the CL, i.e. within the PTL. The model predictions were therefore experimentally validated using an operating single cell PEMWE fitted with four in situ reference electrodes at varying depths through the PTL. The in situ reference electrode was a reversible hydrogen electrode (RHE) immersed in 0.5 M H2SO4 connected to the water inside the operating cell via an insulated Nafion tube; the working principle of the in situ reference electrode has been established by prior work23,25 and is described further in the Methods section as well as in Supplementary Note 1 and Fig. S3 (ESI†). Each Nafion tube was inserted into a hole of a defined depth in the PTL and was therefore held at a known position relative to the CL as shown in Fig. 1(c). The respective distances of the tips of the reference electrodes from the CL, as measured using XCT (Fig. 1(d)), were: 0 μm (drilled through the PTL completely), 290 μm, 600 μm, and 1090 μm. The potential difference measured between the anode electrode/CC/PTL and the reference electrode is dominated by the potential of the material in the proximity of the reference electrode tip and in this manuscript is referred to as Eloc. Fig. 1(b) shows the predicted and measured Eloc as a function of PTL thickness in air-contaminated Type I water; corresponding comparisons of predicted and measured Eloc at different depths are shown for more conductive, less realistic solutions (0.05 mM H2SO4 and 0.5 mM H2SO4) in Fig. S1 and S2 (ESI†). Error bars are included to show systematic error on the measured values; this is estimated to be ± 100 mV, which we attribute principally to uncorrected liquid junction potentials associated with proton leakage from the acidified Nafion tip. Fig. S4 (ESI†) shows an ex situ experiment demonstrating that the magnitude of the liquid junction potential is of the order of 100 mV.
Good agreement is observed between the predicted and measured values, with the results in Fig. 1(b) together with Fig. S1 and S2 (ESI†) clearly showing that the potential drops from that observed at the CL (≈1.7 V vs. RHE) to ≈1.15 V vs. RHE within 300 μm in the case of the air-contaminated Type I water. The potential further decreases to a value typical of the OCP of Pt/O2 (≈1 V vs. RHE) at distances >600 μm from the anode CL. The measurements in Fig. S1 and S2 (ESI†) also validate the model predictions that the extent of the potential penetration increases with increasing electrolyte ionic conductivity. It is noted that the local potential measured at the PTL/CL interface is anomalously high, being close to the cell voltage. This is attributed to a combination of electrode shielding impacting the behaviour of the CL at the interface and pH effects on the liquid junction potential; it does not affect our confidence in the measured values within the PTL.
From the physicochemical model and in situ reference electrode experiments we conclude that, for a PEMWE stack operating at typical conditions of <1.0 μS cm−1 water conductivity, 2.0 A cm−2 current density, and 60 °C, the anode BPP and a large part of the PTL will experience a potential close to their OCP in oxygen-saturated water at 60 °C. This raises the commercially significant possibility of replacing a large proportion of the coated titanium materials used in PEMWEs with cheaper alternatives.
Ex situ evaluation of alternative BPP materials
The physicochemical model introduced above predicts that, under typical operating conditions, the CC material remains largely at its OCP, as does the PTL at sufficient distances from the CL. For a stable material in oxygen-saturated solution, the OCP is determined by the metal/metal oxide redox couple and/or the oxygen reduction reaction and is typically in the range 0.5–1.2 V vs. RHE, depending on the materials and conditions. Even these potentials are relatively oxidising, with most metals unstable with respect to oxidation. Carbon, however, is a possible candidate. While carbon is thermodynamically unstable at potentials above ≈0.2 V vs. RHE, it has been shown to be highly kinetically stable, with many highly graphitic carbons used in oxidising environments such as catalyst supports at the cathode of PEM fuel cells.26 Furthermore, as carbon does not form an oxide layer it maintains a low ICR at high potentials.27 Ready alternatives are carbon-coated stainless steels, which are ideally suited to volume manufacturing through stamping. These are cheaper than titanium and consequently are sold as BPPs in a range of fuel cell applications.27,28To evaluate the suitability of C-316L as a BPP material, a range of ex situ electrochemical experiments were performed. Samples were prepared using a high throughput physical vapor deposition technique that is scalable to high volume roll-to-roll production. Initially the OCP of C-316L was evaluated in oxygen-saturated dilute aqueous sulphuric acid (pH 4.5) simulating a ‘worst case’ electrolyser system (Fig. 2(a)). Here the OCP is ≈0.5 V vs. RHE, equivalent to that of uncoated 316L, suggesting that the carbon coating is sufficiently thin and porous that the OCP of the C-316L is governed largely by the underlying metal. In an operating cell, however, the CC is in electrical contact with the PTL. When this is simulated ex situ by galvanically coupling the C-316L and the Pt–Ti samples, the OCP is shifted to ≈0.90 V vs. RHE. An Evans diagram (Fig. S5, ESI†) shows that Pt exhibits much faster oxygen reduction kinetics compared to the oxidation kinetics of C-316L and this is responsible for pulling the mixed OCP more positive. It should be recognised that OCP can depend significantly on the mechanical and chemical exposure history of the surface as well as the material itself; here, we emphasise the observation that the carbon coating has a negligible effect, whereas galvanic coupling to a Pt-coated surface can displace the OCP to close to 1 V vs. RHE.
An accelerated stress test was performed to assess the stability of the carbon coating. Coated samples were polarised to potentials between 1.0 V and 1.6 V vs. RHE with the current monitored (Fig. 2(b)), and changes in the coating thickness were evaluated by Auger electron spectroscopy (AES) depth profiling after the tests (Fig. 2(c)). These experiments were carried out under conservative conditions, with an identical coating used on 304L stainless steel (C-304L), in a more corrosive electrolyte, specifically oxygen-saturated aqueous H2SO4 (pH 3) at 70 °C with 0.1 ppm HF added. HF was included to simulate plausible Nafion degradation products. 304L stainless steel is chosen for conservatism as it is recognised as a less corrosion-resistant alloy than the 316L stainless steel intended for use in the BPP; comparative dissolution properties of 304L and 316L stainless steels have been studied previously in electrolyser-relevant environments.29 A significant corrosion current and measurable loss in thickness are only detected at potentials ≥1.4 V vs. RHE, with almost complete removal of the carbon coating observed at 1.6 V vs. RHE (Fig. 2(c) and Fig. S6, ESI†). This suggests that carbon-coated stainless steels should be stable at the surface of the BPP, where Eloc would be between 0.9–1.0 V vs. vs. RHE, but not at the CL where the potential is >1.5 V vs. RHE; this is compatible with the previous reported degradation of a PTL made entirely of carbon.14 Contact resistance measurements (Table S4, ESI†) show a low ICR of 2.5 ± 0.2 mΩ cm2, meeting the 2020 DoE PEM fuel cell BPP ICR target of <10 mΩ cm2.30 The ICR value does not change for the sample held at 1.0 V vs. RHE, but increases slightly at higher potentials.
In situ evaluation of BPP materials
Based on the promising theory and results presented above, single cell PEMWE tests (Fig. 3) were performed with a C-316L anode CC to assess the short-term stability of a real cell with this material substitution. The cell voltage and Eloc of the C-316L anode CC are observed to remain decoupled throughout a 7 day test (Fig. 3(a)) with the Eloc of the C-316L anode CC maintaining a steady value of ≈1 V vs. RHE over the entire testing period. This is consistent with the simulation predictions. Similar behaviour is observed with a bare 316L anode CC (Fig. S7, ESI†). Irrespective of the CC material (C-316L, 316L, and Pt–Ti), a similar rate of cell voltage degradation (0.44–0.66 mV h−1) is observed in the 7 day tests (Fig. 3(a) and Fig. S7a, ESI†), indicating a degradation mechanism arising from suboptimal CCM design rather than choice of CC material. A further 30 day single cell test was conducted using C-316L as the anode CC material, this time without an in situ reference electrode. No significant degradation is identified, as indicated by the stable cell voltage (Fig. 3(b)) and polarisation curves at the beginning and end of the 30 day test (Fig. 3(c)), which show no significant increase in cell resistance.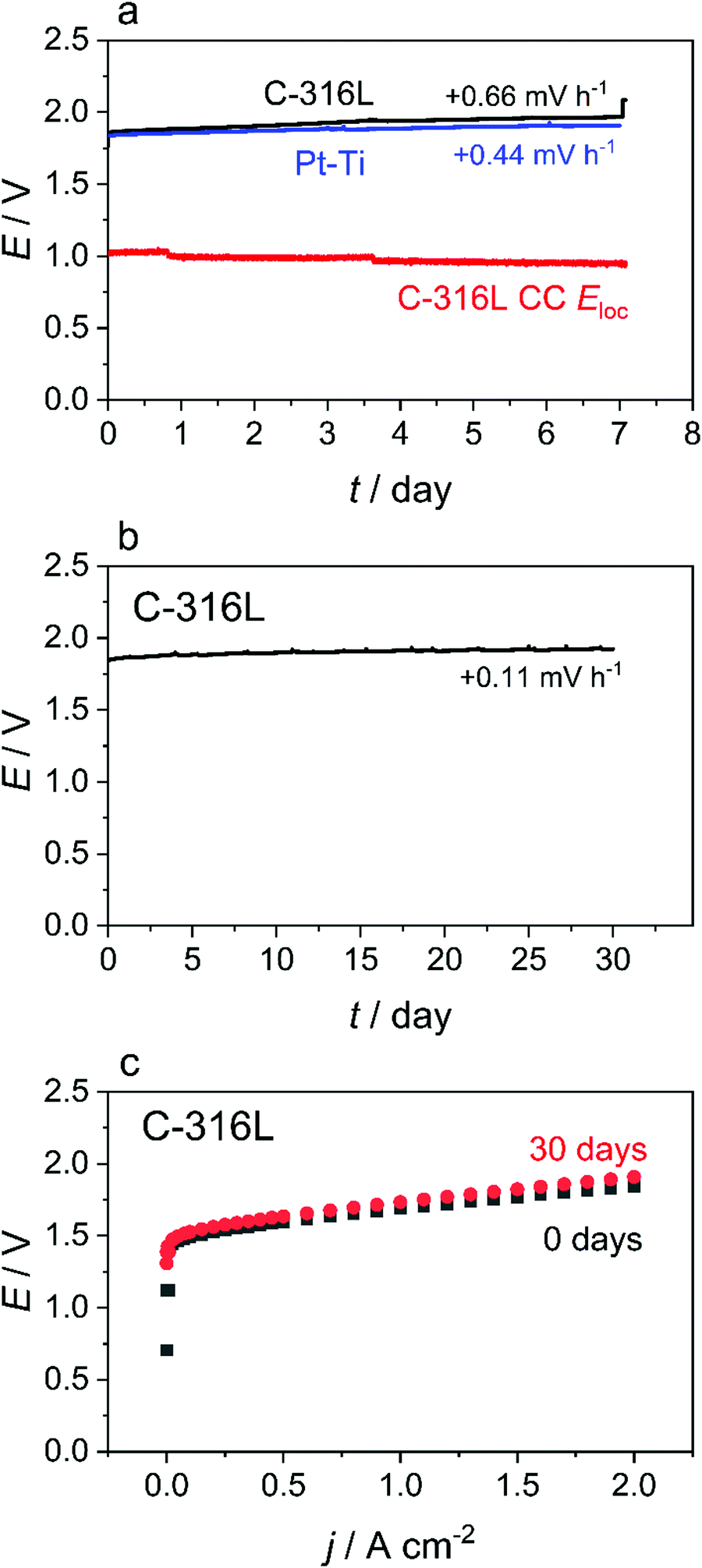 | ||
| Fig. 3 Evaluation of PEMWE cells made with a C-316L current collector (a) Evolution of cell voltage and Eloc in a single cell PEMWE with C-316L and Pt–Ti anode CCs operated at 2 A cm−2 for 7 days at 60 °C using Type I water allowed to equilibrate with air. The local potential at the CC is completely decoupled from the cell voltage and remains at ≈1.0 V vs. RHE throughout the test. (b) Evolution of cell voltage over 30 days for the same cell under identical conditions. The cell voltage maintains a steady value of ≈1.9 V, demonstrating no significant change in ICR at the CC/PTL interface over 30 days of operation. (c) Polarisation curves of the cell before and after the 30 day test, confirming no significant deterioration in cell performance. | ||
Post-mortem analysis was performed on the C-316L material. An element-by-element comparison of the AES depth profiles of all samples (as-received and after the 30 day test) reveals no oxidation of the C-316L substrate or loss of carbon thickness, though the testing did result in minor contamination by adventitious carbon (Fig. S8b and S9, ESI†). Post-mortem analysis of the CCM by acid digestion and inductively-coupled plasma – sector field mass spectrometry (ICP-SFMS) shows no build-up of typical elements present in 316L (Cr, Mn, Fe) even after 30 days of electrolyser operation (Table S5, ESI†). Furthermore, semi-quantitative ICP-MS analysis of the anode and cathode feed water before and after the 30 day test demonstrates that all elements remain at the ≈1 ppb level or lower (Table S5, ESI†), confirming the absence of meaningful corrosion of the 316L during the test. Interestingly, ruthenium contamination of the CC surface is observed for both C-316L (Fig. S8a, ESI†) and Pt–Ti (Fig. S8c, ESI†). This is due to the use of mixed iridium–ruthenium oxides in the anode catalyst of the CCMs used in this work; ruthenium is well known to be unstable under anode CL conditions resulting in dissolution.31–33 Redeposition of the ruthenium on the surface of the CC provides further indirect evidence of the more negative potential at this location.
While all of the experimental results reported here point strongly to the utility of C-316L as a stack BPP material, an experimental duration of 30 days is negligible compared to targeted PEMWE stack lifetimes of >100000 h.8 The theoretical treatment has also focused only on the potential under idealised steady-state operating conditions and has not considered the influence of transient conditions or the possibility of enhanced corrosion in the presence of trace chemical species such as H2O2, radical species, or HF, which may facilitate slow but significant corrosion processes. Experimental testing of the material in a larger stack configuration for many thousands of hours is therefore required; these experiments are ongoing.
Implications
Short-term tests on C-316L as a BPP material show excellent performance and promising indications of stability, supporting the possibility of substituting the Pt–Ti in PEMWE BPPs with a much cheaper material. This view is strengthened by the predictions of the model, which reinforce the conclusion that the BPP sits at its OCP even under conservative PEMWE operating conditions. The natural question arises: is material substitution also an option for the PTL, at least in part? At the PTL/CL interface, a highly corrosion-resistant material would still be required, but it may be possible to use a cheaper material outside the corrosive region.The ex situ results indicate that C-316L is stable below ≈ 1.2 V vs. RHE. Model predictions (Fig. S10, ESI†) for Type II water at 60 °C suggest that the local potential would only exceed this value within ≈200 μm of the CL. For traditional sintered PTLs with thickness in the range 1–2 mm, as in the single cell used in this work, the implication is that a cheaper material could be used for the bulk of the PTL, protected from the highly oxidising region by a thin microporous layer of corrosion-resistant material (such as Pt–Ti), as shown schematically in Fig. 4. The use of a microporous layer at the PTL/CL interface has been studied by various groups with the results broadly indicating that, notwithstanding material substitution advantages as considered in this paper, such a layer improves both the mass transport and electrode kinetics.34–39 Such a scheme may therefore have benefit both through enabling the use of cheaper materials and in improving performance.
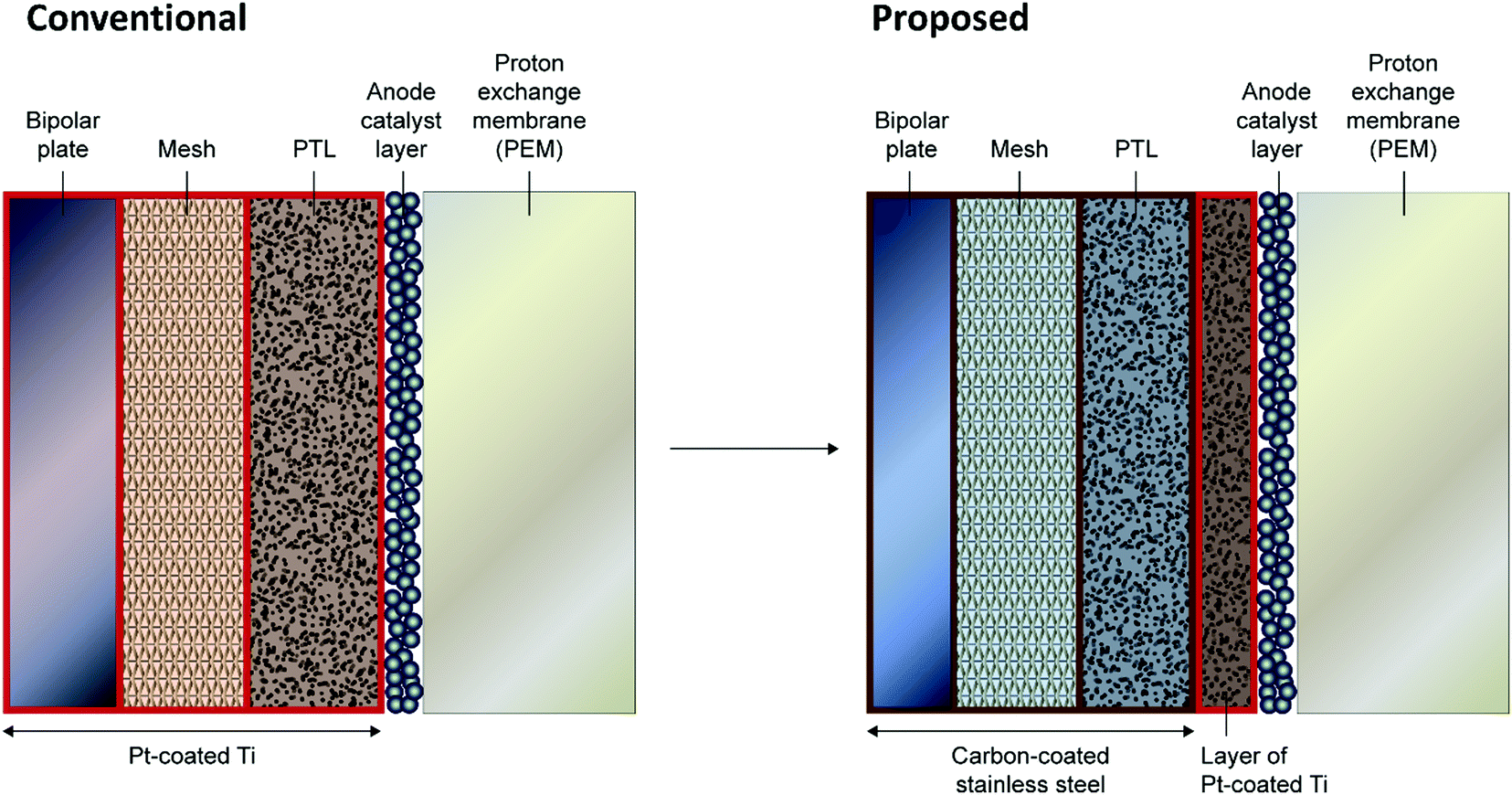 | ||
| Fig. 4 Illustration of potential material substitution in PEMWE stacks. Pt–coated Ti components are replaced with carbon-coated stainless steel in both the BPP and PTL, leaving only a thin microporous Pt–Ti layer at the anode PTL/CL interface. | ||
We recognise that component selection in state-of-the-art PEMWE stacks may diverge in design from the sintered PTL studied here. Stack design can nonetheless clearly benefit from awareness, at the design and material selection stage, of the spatial non-uniformity of aggressivity of electrochemical conditions through the PTL thickness, to an extent that depends on material properties, especially electrolyte conductivity. Application of both our theoretical methodology and in situ reference electrode techniques to these systems may result in improved understanding and optimised stack design, with contextual consideration for cost and performance requirements. While the case for material substitution within the PTL is more nuanced than for the BPP and will depend on existing stack design, material properties and operating conditions, this work has demonstrated a powerful new approach to design optimisation and cost reduction that can be applied over a wide range of PTL materials, thicknesses and geometries.
The analysis and model developed herein are also readily applicable to a wide range of other topical electrochemical energy conversion devices that have water or electrolyte circulating between an electrode and current collector. Such devices include redox flow batteries, CO2 reduction cells and anion exchange membrane electrolysers, where, for instance, there is still debate about the optimal nature and utility of the electrolyte.
Conclusions
A new predictive modelling approach to understanding potential profiles in PEMWE anode components has been developed and validated using in situ reference electrode measurements. The model predicts that under typical operating conditions the corrosive zone associated with the elevated potential of the anode electrode extends only ≈200 μm into the PTL due to the low ionic conductivity of the aqueous phase. Furthermore, single cell tests with carbon-coated stainless steel anode current collectors showed no evidence of corrosion or degradation after 30 days of operation at 2 A cm−2. The results demonstrate the potential for significant cost savings in PEMWE stack design by substitution of Pt–Ti with cheaper BPP and PTL materials. This combined modelling and experimental approach will also be applicable to design optimisation for other electrochemical energy conversion technologies, such as redox flow batteries, CO2 reduction cells and anion exchange membrane electrolysers.Physicochemical model
The physicochemical model predicts the spatial potential profile on the anode side of the cell in 1D (parallel to the current flow direction) subject to the following approximations:• Electrolyte current in the aqueous and ionomer phases obeys Ohm's law with a constant resistivity (different values for each phase).
• Electrical current in the electron-conducting PTL structure obeys Ohm's law with a constant resistivity.
• Mass transfer effects are neglected.
• The PEMWE is considered to be homogeneous in the plane of the electrodes.
• The oxygen evolution reaction at the anode has Tafel kinetics, fit to the experimentally measured polarisation curve.40
• The PTL corrosion process proceeds with linear kinetics as a function of polarisation, with a defined exchange current density. This approximation is chosen in the absence of a more detailed characterisation for the PTL surface electrochemistry; with respect to any nonlinear kinetic model, it is conservative with respect to potential penetration into the PTL (i.e. a linear model predicts a larger polarised region).
• The cathode is assumed to behave ideally (zero hydrogen evolution reaction overpotential).
• The cell is assumed to be isothermal (uniform temperature).
• All porous media are treated as locally homogenised, with their morphology expressed by a local porosity and tortuosity.
We choose a representative rate for the PTL passive corrosion kinetics of i0,PTL = 2 × 10−4 A m−2 based on reported values of exchange current density and activation energy for the Pt/PtO surface.41
The mathematical formulation of the current distribution model subject to the above approximations is given in Supplementary Note 2 (ESI†). All equations were solved in COMSOL Multiphysics 5.6 (COMSOL AB, Stockholm, Sweden).
Experimental
Materials
The catalyst coated membrane (CCM, FuelCellsEtc, USA) comprised a Nafion 115 membrane, 3 mg cm−2 IrRuOx anode, and 3 mg cm−2 Pt black cathode. The CCM was pre-treated by submerging in 1 M H2SO4 at 60 °C for 6 h, rinsing with Type I water three times, and finally immersing overnight in Type I water. The CCM was activated overnight at 1 A cm−2 before any measurement was performed.The metal sheet samples (82 μm thick) were bright-annealed 316L stainless steel and carbon-coated 316L stainless steel. The manufacturing process for the carbon-coated stainless steel has been described previously.27,28 In brief, the oxide layer of bright-annealed 316L was removed (plasma etching) and, subsequently, the stainless-steel strip was coated with a metallic adhesion layer and carbon in a coil-to-coil, high-throughput physical vapour deposition (PVD) coating line. The production process aimed to achieve a 35 nm carbon layer and a 95 nm adhesion layer, but some variation of these values is expected over large areas.
Pt-coated Ti was produced in a batch process as follows. Titanium sheets (Grade 1, 75 μm thick) were cleaned ultrasonically in 10–20 wt% NaOH + 5–10 wt% N,N-bis(carboxymethyl)-alanine trisodium salt aqueous solution (Trutest AK-13 Ultra OS, A Clean Partner International AB, Sweden) at 60 °C for 10 min. Afterwards, they were rinsed in warm tap water and finally in deionised water (10 MΩ cm). The samples were mounted in an E-beam vacuum chamber for batch coating. The chamber was pumped down to a pressure of 10−5 mbar and the Ti substrates were heated to 200 °C. During adhesion layer coating, the pressure was balanced to 8 × 10−5 mbar by Ar chamber back-fill. For the final Pt layer, Ar was turned off and the pressure stabilized at 2 × 10−5 mbar during coating. Nominal layer thicknesses were measured by a quartz crystal microbalance (QCM) as 70 nm for the adhesion layer and 40 nm for the Pt layer.
ASTM Type I water (Elga, Purelab Ultra, UK) was used in all experiments, with the exception of the PTL potential profile experiment. Solutions of 0.5 mM and 0.05 mM H2SO4 were prepared by diluting 0.5 M H2SO4 (Fluka, UK) with Type I water. The pH was measured using a Semi-Micro pH electrode connected to a pH meter with a temperature probe (Jenway 3520, Cole-Parmer, UK). The pH electrode was calibrated prior to measurement using NIST traceable buffer solutions (Sigma Aldrich, UK). Conductivity was measured using a conductivity meter (Jenway 4520 Conductivity meter, Cole Palmer, UK), which was calibrated prior to measurement using a 147 μS cm−1 NIST traceable conductivity solution (Sigma Aldrich, UK).
X-ray computed tomography (XCT)
The PTL disk was mounted onto the sample holder using adhesive putty and then transferred to the rotation stage of a lab-based X-ray micro-CT system (Zeiss Xradia Versa 620 microscope, Carl Zeiss, CA, USA). 2001 X-ray projections of the PTL disk were acquired over a full rotation of 360° at a voxel size of 16.9 μm. The exposure time of each projection was 1.5 s with a source voltage of 140 kV. The projections were then reconstructed using the built-in software Zeiss XMReconstructor, based on standard filtered back-projection algorithms. Commercial software Avizo V9.5 (Avizo, Thermo Fisher Scientific, Waltham, MA, USA) was used for the post-processing. An automatic thresholding algorithm was employed to segment the volume into pore and solid phase for porosity analysis. The tortuosity measurement was conducted in the open-source software TauFactor.42In situ reference electrode
The in situ reference electrode consisted of a reversible hydrogen electrode (RHE, Hydroflex, Gaskatel, Germany) immersed in 0.5 M H2SO4 and connected to the single cell with an acid-filled Nafion tubing. Further details have been reported in our previous work.23,25 In this work, holes were drilled in the anode piston to accommodate either 1 single reference electrode or 4 reference electrodes in a cross configuration. As the cell area is small, inhomogeneity due to lateral flow distribution is assumed to be negligible. For the 7 day in situ reference electrode measurement, the Nafion tube end tip was positioned at the interface of the metal sheet and mesh. Metal sheet samples were sandwiched between the piston and the gridded mesh. To measure the potential profile within the PTL, holes in the PTL were drilled to different depths and the Nafion tube end tips were positioned at the bottom of the respective holes. Measurements at each point within the PTL were conducted three times in each electrolyte (0.5 mM H2SO4, 0.05 mM H2SO4 and Type II water) to ensure repeatability.Measurement of Eloc with the reference electrode was made in a two-electrode arrangement by connecting the working electrode cable to the anode piston and the reference electrode cable to the RHE. Polarisation curves were recorded by firstly ramping the current density at defined values with a 5 min dwell time up to 2 A cm−2 and then lowering it at defined current density steps with 1 min dwell time and 30 s recording time.
Electrolyser testing
The electrolyser single cell consisted of an acrylic circular cell-housing with an active area of 8 cm2. The titanium cell components comprised a piston, mesh (1 mm thick), and sintered porous transport layer (PTL, 2.1 mm thick) coated with 100 nm of Pt (Teer Coatings, UK). All components were ultrasonicated sequentially in acetone (VWR Chemicals, UK), isopropanol (VWR Chemicals, UK), and Type I water for 15 min each before assembly. An extra step of ultrasonication in hexane (VWR Chemicals, UK) for 15 mins prior to the other organic solvents was added for the C-316L and 316L metal sheets to remove impurities arising from sample manufacture. In all tests, the metal sheet was sandwiched between the piston and the mesh. The hardware was tightened using 4 M6 bolts, nuts, and spring washers at a torque of 1 N m and then pneumatically compressed using 20 bar nitrogen from the cathode side. 80 ml min−1 of feed water was circulated in a closed loop to both the anode and cathode using a peristaltic pump (Watson Marlow, UK), with individually separated tanks for each electrode. The water was pre-heated to 60 °C using a water bath (Grant Optima TC120, Grant Instruments, UK).Scanning electron microscopy (SEM)
The as-received and tested materials were characterised using high resolution scanning electron microscopy (FEG-SEM, Zeiss Sigma VP or Zeiss Ultra 55) in combination with energy dispersive X-ray spectroscopy (EDS, Oxford Aztec software and X-max 50 mm2 SDD detector or Oxford Inca energy 450 system). The SEM and EDS analyses were performed using a primary electron energy of 5 keV.Auger electron spectroscopy (AES)
AES analyses were performed with an ULVAC-PHI 700 Xi scanning Auger nanoprobe with an acceleration voltage of 10 kV and a primary beam current of 10 nA. Auger depth profiles were performed with Ar+ ion sputtering at an accelerating voltage of 2 kV, giving a sputter rate of 27 nm min−1 at the sample position (as measured on a 100 nm thick Ta2O5 reference material at the same position) using 1 mm × 1 mm rastering of the ion beam. On uncoated samples a 2 mm × 2 mm raster was used, giving a sputter rate of 8.3 nm min−1. PHI MultiPak (Physical Electronics, Chanhassen, MN, USA) was used to evaluate the Auger depth profile data. Linear least squares fitting was used to extract different chemical states of the elements found.Trace metal analysis
Analysis of trace metals in the CCM was conducted by an accredited lab (ALS, Luleå, Sweden); the details have been previously described by Novalin et al.28 In addition, samples of both anode and cathode feed water before and after the 30 day test with a C-316L anode CC were analysed for trace metals using an inductively coupled plasma mass spectrometer (ICP-MS, Agilent 8900). The analysis was semi-quantitative, giving results as indicative mass fractions for a range of elements. The samples were matrix-matched in 1% HNO3 to a semi-quantitative standard, which was PA tuning solution (Agilent).Ex situ OCP measurement
Prior to ex situ measurement, the reverse of the metal sheet samples was ground with sandpaper and connected to an ethylene tetrafluoroethylene (ETFE)-insulated copper wire (RS Components, UK) using a silver epoxy resin (RS Components, UK). The silver resin connection was dried at room temperature overnight followed by final curing at 65 °C for 2 h. The connection and the sample reverse face were then covered with an epoxy (Araldite Rapid Epoxy Adhesive, RS Components, UK) and left to dry overnight followed by curing at 60 °C for 2 h.The OCP of the sample was measured against a RHE immersed in 0.5 M H2SO4 with Nafion tubing as the bridge to the electrolyte in a water-heated electrochemical cell. The electrolyte was dilute H2SO4 (pH 4.5) to mimic the electrolyser water condition.23 The electrolyte was oxygen-sparged and heated to 60 °C. The galvanically coupled potential of a Pt–Ti PTL contacting the C-316L metal sheet was measured by immersing both samples and combining the electrical connection as the working electrode. In order to obtain the Evans diagram, the potential of a Pt working electrode (∅ = 2 mm, CH Instruments, USA) was scanned from OCP to −0.5 V vs. OCP at 10 mV s−1 and that of a C-316L working electrode (encapsulated with Araldite leaving an active area ∅ = 13 mm) was scanned from OCP to +0.5 V vs. OCP at 10 mV s−1. The counter electrode was a Pt wire (20 cm, ∅ = 1 mm, Goodfellow, UK) and the reference electrode was the RHE/Nafion tubing configuration described above.
Ex situ corrosion testing
Ex situ corrosion testing of 316L, C-316L and C-304L coupons was conducted using an electrochemical setup accommodating eight working electrodes with a shared electrolyte and a Pt–Ti mesh counter electrode (Fuel Cell Store, Texas, USA), referred to as “MultiCell”. The working electrodes were coupons made from a (coated) metal strip and were sealed against the cell body (polytetrafluoroethylene, PTFE) with a Viton O-ring exposing a 3 × 3 cm2 geometric area to the electrolyte to avoid edge effects (uncoated portions due to cutting) for coated samples. Areas on the coupons not exposed to the electrolyte served as reference (not exposed to electrochemical conditions). A RHE (MiniHydrogen, Gaskatel, Germany) was placed in the middle of the exposed area of each working electrode at a distance of <1 mm. All eight RHEs were combined into one reference electrode signal at the potentiostat (ECi-211, Nordic Electrochemistry, Denmark), which helps to compensate for bubble formation and unstable reference potential. The cell was heated via a jacketed glass mantle and a thermostat (CC-205B High Precision Thermoregulation, Huber, Offenburg, Germany). The cell was initially heated to 70 °C over the course of 2.5 h and only when the correct temperature had been reached was the electrolyte added. Here, 217 μl of H2SO4 and 1.7 μl of HF (40% EMSURE® ISO for analysis, Merck Chemicals and Life Sciences, Solna, Sweden) were added to achieve pH 3 H2SO4 (49 ppm) and 0.1 ppm HF. This electrolyte is recommended by the US Department of Energy (DoE) to simulate the local BPP environment in PEM fuel cells.26 In another experiment to mimic the single cell PEMWE water conditions, 6.7 μl of H2SO4 (>98%, EMSURE® for analysis, Merck Chemicals and Life Sciences, Solna, Sweden) were added to achieve pH 4.5.All coupons investigated in the “MultiCell” were cleaned by subsequent sonication in toluene (≥98%, Technical grade, VWR Chemicals, Sweden), acetone (≥99.8%, AnalaR NORMAPUR® ACS, VWR Chemicals, Sweden), ethanol (absolute, ≥99.5%, TechniSolv®, VWR Chemicals, Sweden), and ultrapure water (18 MΩ cm, Millipore Elix® Essential 15 UV and Milli-Q® Integral 3 system, Merck Chemicals and Life Science AB, Solna, Sweden) for 10 min each and dried in an argon stream (Laboratory Argon 5.7, 99.9997%, Linde Gas AB, Solna, Sweden) immediately before the experiment. After the coupons had been sealed against the cell, 8 L of ultrapure water was added and oxygen sparging (≥99.5%, Linde Gas AB, Solna, Sweden, 1000 ml min−1, three inlets) commenced.
Author contributions
H. B., E. J. F. D., G. S., G. H. and S. P. conceived the initial ideas to perform the experiments. H. B. performed all electrolyser experiments and ex situ OCP experiments. C. M. performed carbon-stability and ex situ ICR measurements. E. J. F. D. developed the theory, and H. B. and E. J. F. D. undertook theoretical calculations. X. L. performed the XCT imaging. H. B, E. J. F. D., G. S., and G. H. analysed all the experimental data. M. S. manufactured the Pt–Ti, 316L and C-316L samples. U. B. and C. M. performed the post-mortem analysis of the Pt–Ti, 316L and C-316L samples. H. B., E. J. F. D., U. B., C. M., S. P., G. S., and G. H. wrote the manuscript, while all authors discussed the results and contributed during manuscript preparation.Data availability statement
Source data are provided with this paper.Conflicts of interest
U. B., S. P., C. M., and M. S. are employees of SMT. All other authors declare no competing interest in this work.Acknowledgements
This work was funded by the National Measurement System of the UK's Department of Business, Energy & Industrial Strategy and by Sandvik Materials Technology (SMT). The authors acknowledge funding acquisition from SMT by Carlos Bernuy-Lopez and Jörgen Westlinder.References
- Path to hydrogen competitiveness: a cost perspective. (Hydrogen Council, 2020); https://hydrogencouncil.com/wp-content/uploads/2017/06/Hydrogen-Council-Vision-Document.pdf.
- S. Anwar, F. Khan, Y. Zhang and A. Djire, Recent development in electrocatalysts for hydrogen production through water electrolysis., Int. J. Hydrogen Energy, 2021, 46, 32284–32317 CrossRef CAS.
- P. Shirvanian and F. Berkel, Novel components in Proton Exchange Membrane (PEM) Water Electrolyzers (PEMWE): Status, challenges and future needs. A mini review., Electrochem. Commun., 2020, 114, 106704 CrossRef CAS.
- Q. Feng, et al., A review of proton exchange membrane water electrolysis on degradation mechanisms and mitigation strategies., J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- K. Ayers, The potential of proton exchange membrane–based electrolysis technology., Curr. Opin. Electrochem., 2019, 18, 9–15 CrossRef CAS.
- A. Mayyas, et al., Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, National Renewable Energy Laboratory, 2019.
- Addendum to the Multi - Annual Work Plan 2014 - 2020. Fuel Cells and Hydrogen 2 Joint Undertaking (FCH 2 JU) (2018); https://www.fch.europa.eu/sites/default/files/MAWPfinalversion:endorsedGB15062018%28ID3712421%29.pdf. (Accessed: 26th March 2021).
- The Future of Hydrogen. (International Energy Agency, 2019); https://www.iea.org/reports/the-future-of-hydrogen.
- C. Liu, et al., Performance enhancement of PEM electrolyzers through iridium-coated titanium porous transport layers., Electrochem. Commun., 2018, 97, 96–99 CrossRef CAS.
- Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions. Critical Raw Materials Resilience: Charting a Path towards greater Security and Sustainability. COM(2020) 474 final (European Union: European Commission, 2020).
- C. Rakousky, et al., An analysis of degradation phenomena in polymer electrolyte membrane water electrolysis., J. Power Sources, 2016, 326, 120–128 CrossRef CAS.
- C. Rakousky, et al., Polymer electrolyte membrane water electrolysis: Restraining degradation in the presence of fluctuating power., J. Power Sources, 2017, 342, 38–47 CrossRef CAS.
- C. Rakousky, G. P. Keeley, K. Wippermann, M. Carmo and D. Stolten, The stability challenge on the pathway to high-current-density polymer electrolyte membrane water electrolyzers., Electrochim. Acta, 2018, 278, 324–331 CrossRef CAS.
- H.-Y. Jung, S.-Y. Huang, P. Ganesan and B. N. Popov, Performance of gold-coated titanium bipolar plates in unitized regenerative fuel cell operation., J. Power Sources, 2009, 194, 972–975 CrossRef CAS.
- Z. Kang, S. M. Alia, M. Carmo and G. Bender, In-situ and in-operando analysis of voltage losses using sense wires for proton exchange membrane water electrolyzers., J. Power Sources, 2021, 481, 229012 CrossRef CAS.
- J. L. Young, Z. Kang, F. Ganci, S. Madachy and G. Bender, PEM electrolyzer characterization with carbon-based hardware and material sets., Electrochem. Commun., 2021, 124, 106941 CrossRef CAS.
- J. Mo, et al., Study on corrosion migrations within catalyst-coated membranes of proton exchange membrane electrolyzer cells., Int. J. Hydrogen Energy, 2017, 42, 27343–27349 CrossRef CAS.
- J. Mo, et al., Electrochemical investigation of stainless steel corrosion in a proton exchange membrane electrolyzer cell., Int. J. Hydrogen Energy, 2015, 40, 12506–12511 CrossRef CAS.
- A. S. Gago, et al., Protective coatings on stainless steel bipolar plates for proton exchange membrane (PEM) electrolysers., J. Power Sources, 2016, 307, 815–825 CrossRef CAS.
- P. Lettenmeier, et al., Coated stainless steel bipolar plates for proton exchange membrane electrolyzers., J. Electrochem. Soc., 2016, 163, F3119–F3124 CrossRef CAS.
- P. Lettenmeier, et al., Low-cost and durable bipolar plates for proton exchange membrane electrolyzers., Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- S. Stiber, et al., High-performance, durable and low-cost proton exchange membrane electrolyser with stainless steel components., Energy Environ. Sci., 2022, 15, 109–122 RSC.
- H. Becker, L. Castanheira and G. Hinds, Local measurement of current collector potential in a polymer electrolyte membrane water electrolyser., J. Power Sources, 2020, 448, 227563 CrossRef CAS.
- NEL Hydrogen. M series containerized PEM electrolyser. (2020).
- E. Brightman, J. Dodwell, N. Van Dijk and G. Hinds, In situ characterisation of PEM water electrolysers using a novel reference electrode., Electrochem. Commun., 2015, 52, 1–4 CrossRef CAS.
- J. Zhao, Z. Tu and S. H. Chan, Carbon corrosion mechanism and mitigation strategies in a proton exchange membrane fuel cell (PEMFC): A review., J. Power Sources, 2021, 488, 229434 CrossRef CAS.
- S. Proch, et al., Coated stainless steel as bipolar plate material for anion exchange membrane fuel cells (AEMFCs)., Int. J. Hydrogen Energy, 2020, 45, 1313–1324 CrossRef CAS.
- T. Novalin, et al., Trace-metal contamination in proton exchange membrane fuel cells caused by laser-cutting stains on carbon-coated metallic bipolar plates, Int. J. Hydrogen Energy, 2021, 46, 13855–13864 CrossRef CAS.
- X. Li, et al., Transient stainless-steel dissolution and its consequences on ex situ bipolar plate testing procedures., Int. J. Hydrogen Energy, 2020, 45, 984–995 CrossRef CAS.
- DOE Technical Targets for Polymer Electrolyte Membrane Fuel Cell Components; energy.gov/eere/fuelcells/doe-technical-targets-polymer-electrolyte-membrane-fuel-cell-components#bipolarplate.
- N. Hodnik, et al., New insights into corrosion of ruthenium and ruthenium oxide nanoparticles in acidic media., J. Phys. Chem. C, 2015, 119, 10140–10147 CrossRef CAS.
- S. Cherevko, et al., Dissolution of noble metals during oxygen evolution in acidic media., ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- S. Siracusano, et al., New insights into the stability of a high performance nanostructured catalyst for sustainable water electrolysis., Nano Energy, 2017, 40, 618–632 CrossRef CAS.
- M. Zlobinski, T. Schuler, F. N. Büchi, T. J. Schmidt and P. Boillat, Elucidation of fluid streamlining in multi-layered porous transport layers for polymer electrolyte water electrolyzers by operando neutron radiography., J. Electrochem. Soc., 2021, 168, 014505 CrossRef CAS.
- S. Stiber, et al., Porous transport layers for proton exchange membrane electrolysis under extreme conditions of current density, temperature, and pressure, Adv. Energy Mater., 2021, 11, 2100630 CrossRef CAS.
- T. Schuler, et al., Hierarchically structured porous transport layers for polymer electrolyte water electrolysis., Adv. Energy Mater., 2020, 10, 1903216 CrossRef CAS.
- P. Lettenmeier, et al., Comprehensive investigation of novel pore-graded gas diffusion layers for high-performance and cost-effective proton exchange membrane electrolyzers., Energy Environ. Sci., 2017, 10, 2521–2533 RSC.
- Z. Kang, et al., Investigation of thin/well-tunable liquid/gas diffusion layers exhibiting superior multifunctional performance in low-temperature electrolytic water splitting., Energy Environ. Sci., 2017, 10, 166–175 RSC.
- Z. Kang, et al., Developing titanium micro/nano porous layers on planar thin/tunable LGDLs for high-efficiency hydrogen production., Int. J. Hydrogen Energy, 2018, 43, 14618–14628 CrossRef CAS.
- D. S. Falcão and A. M. F. R. Pinto, A review on PEM electrolyzer modelling: Guidelines for beginners., J. Cleaner Prod., 2020, 261, 121184 CrossRef.
- A. Damjanovic, L. R. Yeh and J. F. Wolf, Formation of thin oxide films at platinum anodes in alkaline solutions: III. pH dependence., J. Electrochem. Soc., 1982, 129, 55–61 CrossRef CAS.
- S. J. Cooper, A. Bertei, P. R. Shearing, J. A. Kilner and N. P. Brandon, TauFactor: An open-source application for calculating tortuosity factors from tomographic data., SoftwareX, 2016, 5, 203–210 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee00876a |
| This journal is © The Royal Society of Chemistry 2022 |