
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe Ir–OOOO–Ir transition state and the mechanism of the oxygen evolution reaction on IrO2(110)†
Tobias
Binninger
 * and
Marie-Liesse
Doublet
* and
Marie-Liesse
Doublet
ICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: tobias.binninger.science@gmx.de
First published on 4th May 2022
Abstract
Carefully assessing the energetics along the pathway of the oxygen evolution reaction (OER), our computational study reveals that the “classical” OER mechanism on the (110) surface of iridium dioxide (IrO2) must be reconsidered. We find that the OER follows a bi-nuclear mechanism with adjacent top surface oxygen atoms as fixed adsorption sites, whereas the iridium atoms underneath play an indirect role and maintain their saturated 6-fold oxygen coordination at all stages of the reaction. The oxygen molecule is formed, via an Ir–OOOO–Ir transition state, by association of the outer oxygen atoms of two adjacent Ir–OO surface entities, leaving two intact Ir–O entities at the surface behind. This is drastically different from the commonly considered mono-nuclear mechanism where the O2 molecule evolves by splitting of the Ir–O bond in an Ir–OO entity. We regard the rather weak reducibility of crystalline IrO2 as the reason for favoring the novel pathway, which allows the Ir–O bonds to remain stable and explains the outstanding stability of IrO2 under OER conditions. The establishment of surface oxygen atoms as fixed electrocatalytically active sites on a transition-metal oxide represents a paradigm shift for the understanding of water oxidation electrocatalysis, and it reconciles the theoretical understanding of the OER mechanism on iridium oxide with recently reported experimental results from operando X-ray spectroscopy. The novel mechanism provides an efficient OER pathway on a weakly reducible oxide, defining a new strategy towards the design of advanced OER catalysts with combined activity and stability.
Broader contextThe vision of a hydrogen economy is more and more turning into reality after becoming one of the top priorities of industrial policies around the globe. Water electrolysis will likely become the workhorse for the massive generation of clean hydrogen, but the limited energy efficiency of the process is a major obstacle for the industrial upscaling. Whereas the cathodic hydrogen evolution reaction is relatively fast and efficient, the anodic oxygen evolution reaction (OER) is slow and responsible for a significant share of the energy losses, for which reason the OER is in the focus of current electrocatalysis research. Iridium oxide (IrO2) is the most important OER catalyst in acidic conditions, providing both high activity and stability. Therefore, a thorough understanding of the OER mechanism on IrO2 is the starting point for the search for cheaper alternatives. Surprisingly, we discovered that the oxygen molecule evolves from the IrO2 surface in an entirely different way than conventionally assumed, involving an intriguing Ir–OOOO–Ir transition state with a fourfold chain of oxygen atoms. Our findings have a critical impact on our understanding of the OER and reveal a new strategy towards the design of stable and active next-generation OER electrocatalysts. |
Introduction
The oxygen evolution reaction (OER) is the kinetic bottleneck of aqueous electrolysis,1 and, as such, of utmost importance for the energy efficiency of the electrochemical production of sustainable hydrogen and chemical feedstock. Iridium oxide provides high electrocatalytic activity towards the OER combined with good stability under acidic OER conditions,2–5 for which reason it is the most important OER electrocatalyst applied in proton-exchange-membrane water electrolyzers. The scarcity and price of iridium, however, is a matter of concern6 and the development of OER electrocatalysts with reduced noble metal content is a mandate for present electrocatalysis research.7–9 The detailed investigation of the OER mechanism at an atomistic level is crucial for these efforts and led to the identification of “scaling relations” among the adsorption energies of OER intermediate species10,11 that provide an explanation for observed volcano-shaped trends in OER activities of various metal oxides and serve as a guide for the discovery of novel electrocatalyst materials.12Due to the importance of iridium oxide for OER electrocatalysis, the reaction mechanism on crystalline rutile IrO2 has attracted considerable interest both from the experimental5,13–18 and the computational/theoretical19–26 side, with a particular focus on the IrO2(110) surface, which has been identified as the thermodynamically stable orientation across a wide potential range27,28 and generally serves as a reference system for OER electrocatalysis. According to the “classical” OER mechanism proposed by Rossmeisl et al.,19 the O2 molecule is formed from two H2O molecules through a sequence of surface-adsorbed intermediate species involving four proton–electron transfer (PET) steps (RS: reaction step),
| Ir* + H2O → Ir*OH + H+ + e− | (RS1) |
| Ir*OH → Ir*O + H+ + e− | (RS2) |
| Ir*O + H2O → Ir*OOH + H+ + e− | (RS3) |
| Ir*OOH → Ir* + O2 + H+ + e− | (RS4) |
| Ir*O +Ir*O + H2O → Ir*OOH + Ir*OH | (RS5) |
| Ir*OOH + Ir*OH → Ir*OO +Ir*OH + H+ + e− | (RS6) |
| Ir*OO + Ir*OH → Ir*OO + Ir*O + H+ + e− | (RS7) |
| Ir*OO → Ir* + O2 | (RS8) |
Likewise, in essentially all OER mechanisms discussed to date, coordinately unsaturated transition-metal cations with a formally reduced oxidation state are left behind after the evolution of the oxygen molecule. This not only holds for classical “adsorbate-evolving mechanisms” as discussed above, but also for alternative “lattice oxygen mechanisms” that involve the formation of lattice oxygen vacancies in the surface layer of the metal oxide catalyst.1,29–35 As a second universal aspect, the first formation of an O–O bond at the metal oxide surface directly produces the OO motif that eventually evolves as an O2 molecule.
In contrast, our density-functional theory (DFT) computational results strongly suggest that the classical adsorbate-evolving mechanism of the OER on the IrO2(110) surface does not proceed through an O2 evolving step (RS4) or (RS8) with an unsaturated Ir* intermediate species. Instead, we demonstrate that the iridium surface atoms remain fully saturated with a 6-fold oxygen coordination at all stages of the OER pathway, and the O2 molecule evolves in an association step of the outer oxygen atoms of two adjacent Ir*OO entities via an Ir*OOOO*Ir transition state,
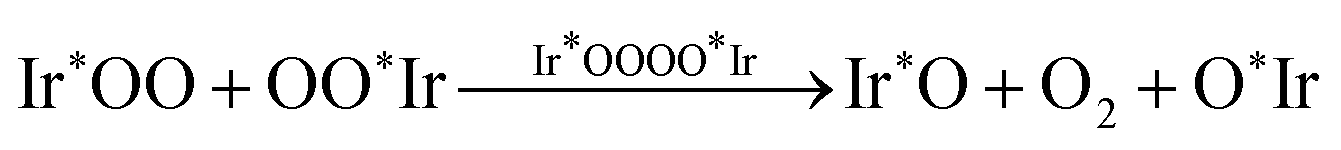 | (RS9) |
Computational methods
DFT calculations were performed, including spin-polarization, with the VASP package36 using the PAW method37 and the PBE38 GGA exchange–correlation functional. The plane-wave energy cutoff was 520 eV, Fermi-Dirac smearing with kT = 0.05 eV was set, and the D3(BJ) method39 was used to account for dispersion interaction. The PBE functional has been shown to provide an accurate description of the electronic properties of IrO2,40,41 with the density of states of bulk IrO2 obtained from our calculations in good agreement, see Fig. S1 (ESI†). Also the computed tetragonal lattice constants of the relaxed IrO2 rutile structure (comp.: a = 4.515 Å and c = 3.182 Å) agree within less than 1% with experimental values42 (exp.: a = 4.505 Å and c = 3.159 Å). Symmetric 5-layered IrO2(110) slabs were constructed from the relaxed bulk structure with the help of the pymatgen python package,43 comprising the 2 × 1 surface cell of the (110) orientation with 4 iridium atoms per layer. Slab calculations were performed with a Γ-centered (3 × 3 × 1) k-point mesh. Atomic positions of all systems were relaxed with a convergence threshold of 0.01 eV Å−1 for the forces, keeping the innermost IrO2 layer frozen. Periodic slab images were separated by a 20 Å-wide interspace region that was filled with implicit water described by the polarizable continuum model in the VASPsol implementation44 with a dielectric constant εr = 78.4 and using a critical density parameter nc = 0.0025 Å−3 to define the implicit solvent boundary. All structural images in the present work were produced using the VESTA software.45Water-derived adsorbates were placed symmetrically on both surfaces of the IrO2(110) slab (see Fig. S2 and S3, ESI†). The free energies of adsorbate structures were computed according to A = EDFT + Avib from the ground-state DFT energies EDFT by adding the vibrational free energies 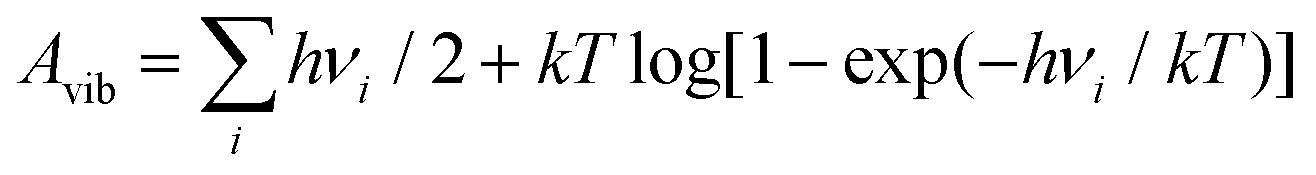 at T = 25 °C of the computed vibrational frequency spectrum νi in harmonic approximation. Here, Avib = Uvib − TSvib contains both the vibrational inner energy (incl. zero-point energy) and entropy contributions. The influence of electrochemical interface charging was simulated using the homogeneous background method46 in a recently developed version47 for the treatment of electrode surfaces in presence of adsorbate species. In short, the electron number N was varied with respect to N0 of the neutral cell, and the free energy was transformed to the grand potential Ω = A − EFermi(N − N0) for a potentiostatic setting, where the chemical potential is given by the Fermi energy. The electrode potential e
at T = 25 °C of the computed vibrational frequency spectrum νi in harmonic approximation. Here, Avib = Uvib − TSvib contains both the vibrational inner energy (incl. zero-point energy) and entropy contributions. The influence of electrochemical interface charging was simulated using the homogeneous background method46 in a recently developed version47 for the treatment of electrode surfaces in presence of adsorbate species. In short, the electron number N was varied with respect to N0 of the neutral cell, and the free energy was transformed to the grand potential Ω = A − EFermi(N − N0) for a potentiostatic setting, where the chemical potential is given by the Fermi energy. The electrode potential e![[scr E, script letter E]](https://www.rsc.org/images/entities/char_e140.gif) = (−e)ϕelyte − EFermi was determined as the “work function” of the electrode surface in the implicit water environment, where ϕelyte is the electrostatic potential level in the bulk implicit solvent. A reference value of 4.44 eV was used for the standard hydrogen electrode (SHE) potential.48 Electrochemical reaction free energies were determined using the computational hydrogen electrode method49 by referencing the chemical potential of proton-electron pairs at the potential of the reversible hydrogen electrode (RHE) to half the Gibbs free energy per molecule of hydrogen gas. Without considering effects of interface charging, all results were valid on the RHE scale at any pH value. However, this no longer holds when including interface charging, because the potential of zero charge of a given surface state does not shift with the RHE scale. We therefore report all results versus the normal hydrogen electrode (NHE), corresponding to the RHE at pH = 0. Gibbs free energies G = H − TS of liquid water, gaseous hydrogen and gaseous oxygen at T = 25 °C and p = 1 bar were computed from the ground-state DFT energies of relaxed H2O, H2, and O2 molecules, respectively, adding the vibrational zero-point energy, as well as the CODATA50 tabulated values of the corresponding entropic (−TS)-contribution, gas-phase enthalpy difference H (298.15 K) − H (0 K), and, for liquid water, heat of condensation HH2O,liq − HH2O,gas at 298.15 K.
= (−e)ϕelyte − EFermi was determined as the “work function” of the electrode surface in the implicit water environment, where ϕelyte is the electrostatic potential level in the bulk implicit solvent. A reference value of 4.44 eV was used for the standard hydrogen electrode (SHE) potential.48 Electrochemical reaction free energies were determined using the computational hydrogen electrode method49 by referencing the chemical potential of proton-electron pairs at the potential of the reversible hydrogen electrode (RHE) to half the Gibbs free energy per molecule of hydrogen gas. Without considering effects of interface charging, all results were valid on the RHE scale at any pH value. However, this no longer holds when including interface charging, because the potential of zero charge of a given surface state does not shift with the RHE scale. We therefore report all results versus the normal hydrogen electrode (NHE), corresponding to the RHE at pH = 0. Gibbs free energies G = H − TS of liquid water, gaseous hydrogen and gaseous oxygen at T = 25 °C and p = 1 bar were computed from the ground-state DFT energies of relaxed H2O, H2, and O2 molecules, respectively, adding the vibrational zero-point energy, as well as the CODATA50 tabulated values of the corresponding entropic (−TS)-contribution, gas-phase enthalpy difference H (298.15 K) − H (0 K), and, for liquid water, heat of condensation HH2O,liq − HH2O,gas at 298.15 K.
The energy of the oxygen molecule is known to be poorly described at the GGA-level,51 for which reason the free energy of gaseous O2 is commonly determined indirectly from the H2 and H2O free energies to yield the correct standard equilibrium potential of 1.229 VSHE for the OER. Using the DFT-derived free energy of O2, we obtain an equilibrium potential of 1.127 VSHE. The difference of 0.102 eV (per electron) could be compensated by an O2 energy correction of +0.408 eV. A similar energy shift has also been previously reported for the PBE functional.21,51 However, for the sake of consistency, we prefer to avoid ascribing the error in the computed equilibrium potential fully to the oxygen molecule. Moreover, we also compute energy barriers for the O2 formation steps, and it cannot be determined to which parts of the barrier the O2 energy correction would (entirely or partially) apply. We therefore treat all of O2, H2, and H2O at an equal footing and accept the energetic errors of the computational framework in the results presented in the figures of this article. We then discuss the robustness of our results against such errors by adding +0.408 eV to the energy of the O2 molecule. Although such correction was only established for the free double-bonded O2 molecule,51 we additionally consider a “worst-case” by adding the same correction per OO entity for all adsorbate states of the IrO2(110) surface involving such motifs. The results including the O2 and OO energy corrections are presented in corresponding figures in the ESI.† We already emphasize, however, that our results confirm previous findings21,26 on a superoxide *OO− character of adsorbed *OO species, which would not require such energy correction.26 The “worst-case” must therefore be considered a rather unrealistic scenario.
To validate the computational results in comparison with reported experimental results, cyclic voltammograms (CVs) were simulated in terms of the electrochemical interface capacitance as a function of the electrode potential. For this purpose, we first computed the grand partition function 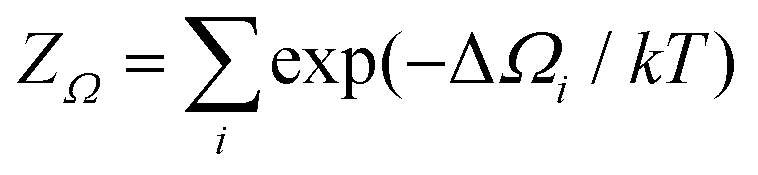 from the grand potential curves ΔΩi of the individual adsorbate configurations, see Fig. 1, from which we obtained the total grand potential Ω = −kT
from the grand potential curves ΔΩi of the individual adsorbate configurations, see Fig. 1, from which we obtained the total grand potential Ω = −kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(ZΩ), see the “concave hull” in Fig. 1. Charge and capacitance were then computed from the derivatives of the grand potential with respect to the electrode potential,47,52,53Q = −∂Ω/∂ and C = −∂2Ω/∂2.
log(ZΩ), see the “concave hull” in Fig. 1. Charge and capacitance were then computed from the derivatives of the grand potential with respect to the electrode potential,47,52,53Q = −∂Ω/∂ and C = −∂2Ω/∂2.
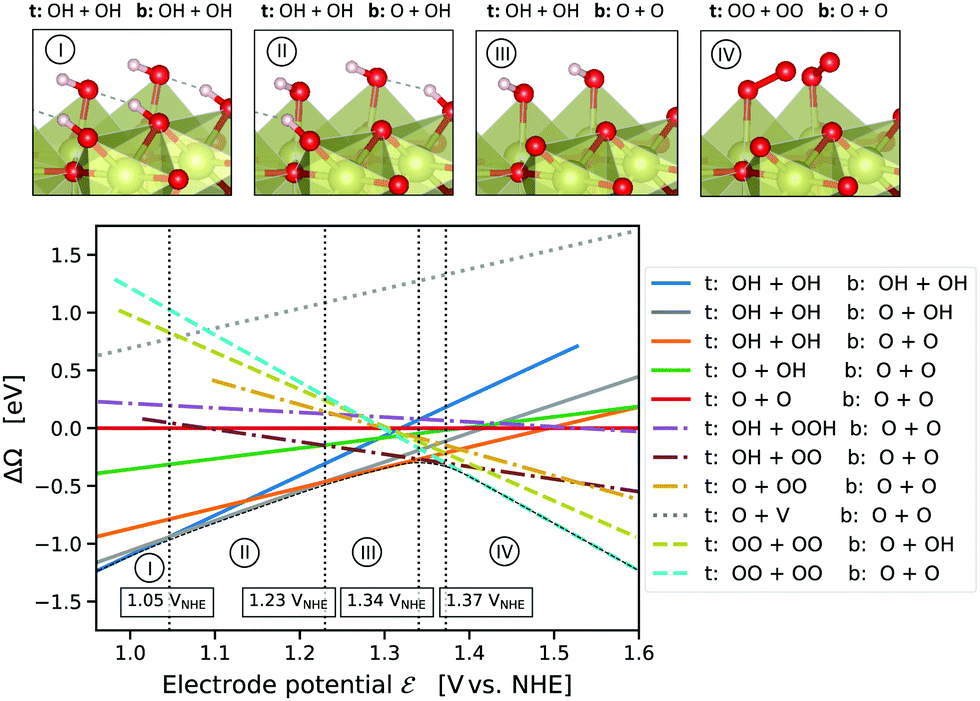 | ||
| Fig. 1 Computed grand-canonical stability diagram of adsorbate configurations on the IrO2(110) surface in aqueous environment at pH = 0 with the fully *O-covered surface as reference system. Vertical dotted lines indicate the equilibrium potentials for the transition between lowest-energy adsorbate states (shown above with oxygen: red; hydrogen: white; iridium: pale golden). The total grand potential of the system is the “concave hull”, shown as a thin black dashed curve. Naming scheme: species at the top (t) and bridge (b) oxygen sites (two of each per simulated surface cell); V: vacant top oxygen site. | ||
Activation energy barriers for the relevant reaction steps were obtained from the climbing-image nudged elastic band (CI-NEB) method.54 The barriers were computed at constant-charge conditions. The treatment of gas-phase entropy contributions in NEB calculations must be carefully assessed. Unlike in ref. 21, we consider it more consistent to not include O2 gas-phase entropy contributions at the transition-state of the oxygen evolution steps. Gas-phase entropy primarily results from the translational degrees of freedom of the “free” gas molecules in the final state. In contrast, the OO entity at the transition state of O2 evolution does not possess such translational freedom due to its linkage to a specific motif of surface sites from which it evolves. The gas-phase entropy thus only affects the entirely free product state of the evolved O2 molecule, which has no influence on the forward rate of the elementary reaction step. According to the basic rate law rf = cRkf of kinetic rate theory/transition-state theory,55 the forward rate rf of an elementary reaction step only depends on the reactant concentration cR and not the product concentration. The entropy of gas-phase O2 is dominated by the partial-pressure-dependent term −RlogpO2. Adding the respective −TS contribution to the free energy of the transition state would lower the barrier as a function of pO2, thus making the forward rate of the elementary O2 evolution step dependent on the product O2 concentration (partial pressure) and violating the basic rate law.
Results
Fig. 1 presents the computed grand-canonical stability diagram of the relevant aqueous adsorbate configurations on IrO2(110) at pH = 0. We note that the presented grand-canonical free energy (grand potential) ΔΩ is equivalent to the free energy of reaction for the formation of any given adsorbate configuration from the fully *O-covered reference state of the surface. Balance in oxgyen and hydrogen atom numbers is provided by water molecules, protons, and electrons in appropriate stoichiometries. The relative stability and equilibrium potentials between the mixed *O- and *OH-covered surface states are in excellent agreement with previously reported results21 (see Fig. S4, ESI†). Above 1.23 VNHE, the bridge oxygen sites are largely deprotonated and remain inactive, while the top sites are fully covered with *OH (labelled (III) in Fig. 1). Taking into account adsorbate states with *OOH and *OO entities, we find that the fully protonated top oxygen sites would transform into a completely *OO-covered state (labelled (IV) in Fig. 1) around 1.34–1.37 VNHE. These qualitative results are robust against the “worst-case” OO energy correction discussed above, which merely shifts the latter transition potential to around 1.5 VNHE as shown in Fig. S5 (ESI†). It is important to note that, as shown by the dotted curve in Fig. 1, the state with an oxygen vacancy (V) at the top site, corresponding to an unsaturated Ir* (see Fig. S2, ESI†), is thermodynamically out of reach. This is not surprising, because strong dissociative adsorption of water molecules to Ir* is known to occur at the stoichiometric IrO2(110) surface,21 for which we obtain a binding energy of 1.3 eV per H2O. However, this observation already indicates that, even if temporary, the formation of unsaturated Ir* in the course of the OER, as in steps (RS4) or (RS8), is rather unlikely.How then could the O2 molecule evolve from the surface without creating Ir*? As discussed above, the thermodynamically stable surface configuration in the OER-relevant potential range involves complete coverage of the top oxygen sites by *OO. As shown in the structural drawing (IV) of Fig. 1, the spatial extension of the *OO entities enables the outer oxygen atoms of two adjacent *OO to closely interact without requiring distortion of the underlying IrO2 lattice. We therefore propose reaction (RS9) as the actual O2-evolution step at the IrO2(110) surface, where the O2 molecule is formed by association of the outer oxygen atoms of neighboring *OO while preserving the full coordination of surface Ir cations. The complete catalytic cycle of the novel mechanism is schematically shown in Fig. 2, including two dissociative water adsorption steps, four oxidation steps by proton–electron removal, and the final step of formation and evolution of the oxygen molecule. Fig. 3 compares the novel pathway (blue) with the conventional one (golden) at an OER-relevant electrode potential of 1.53 VNHE in 3(a) and the equilibrium potential of 1.23 VNHE in 3(b). We note that different zero-energy reference states were chosen in (a) and (b), because the relative stability of these states changes between the corresponding potentials. Our results for the conventional pathway agree very well with previous reports,21,22,26 as discussed and compared in detail in the ESI† (see Tables S3–S5). As expected from the slight error in the computational equilibrium potential discussed above, the initial and final states in 3(b) are not aligned at the real equilibrium potential. Results including the commonly used O2 energy correction, with aligned initial and final states at 1.23 VNHE, are shown in Fig. S7 (ESI†) and do not affect the conclusions. The same holds when applying the “worst-case” OO energy correction, see Fig. S8 (ESI†). The grand-canonical free energies of all OER intermediate states shown in Fig. 3 are given in Tables S1 and S2 (ESI†). The novel mechanism shares the same steps as the conventional one until a top *OO entity is formed. According to the conventional mechanism, this *OO directly evolves as O2. In the novel mechanism, another adjacent *OO entity is first formed, and the outer oxygen atoms finally associate to evolve as O2. From the energetic alignment of the respective intermediate states, it becomes directly clear that the novel OER mechanism is favored over the conventional one at any potential. In fact, at 1.53 VNHE all steps of the novel mechanism are downhill, whereas the O2 evolution step of the conventional mechanism costs a significant uphill free energy of 0.46 eV.
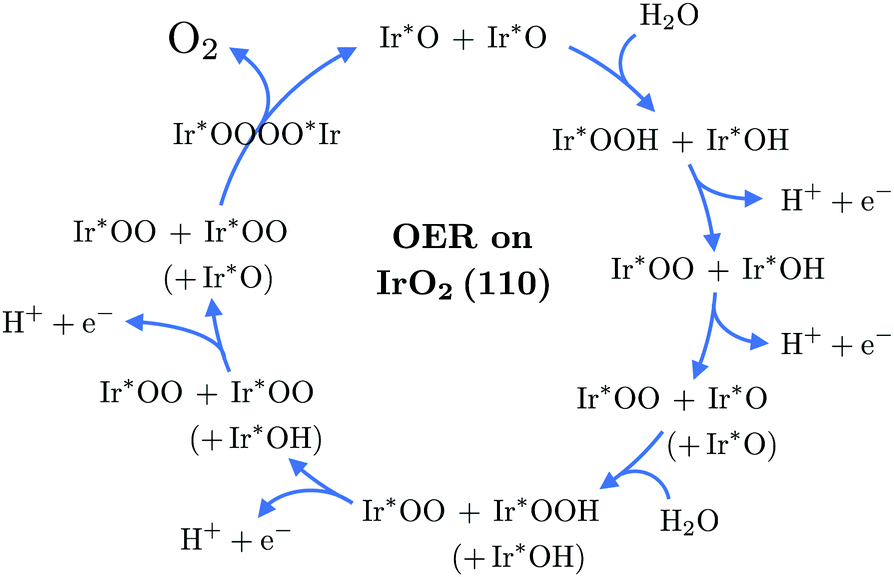 | ||
| Fig. 2 Schematic of the catalytic cycle of the OER on IrO2(110) according to the novel mechanism. For the dissociative adsorption of the second water molecule, a third surface oxygen entity is involved, indicated in brackets for the respective steps only. | ||
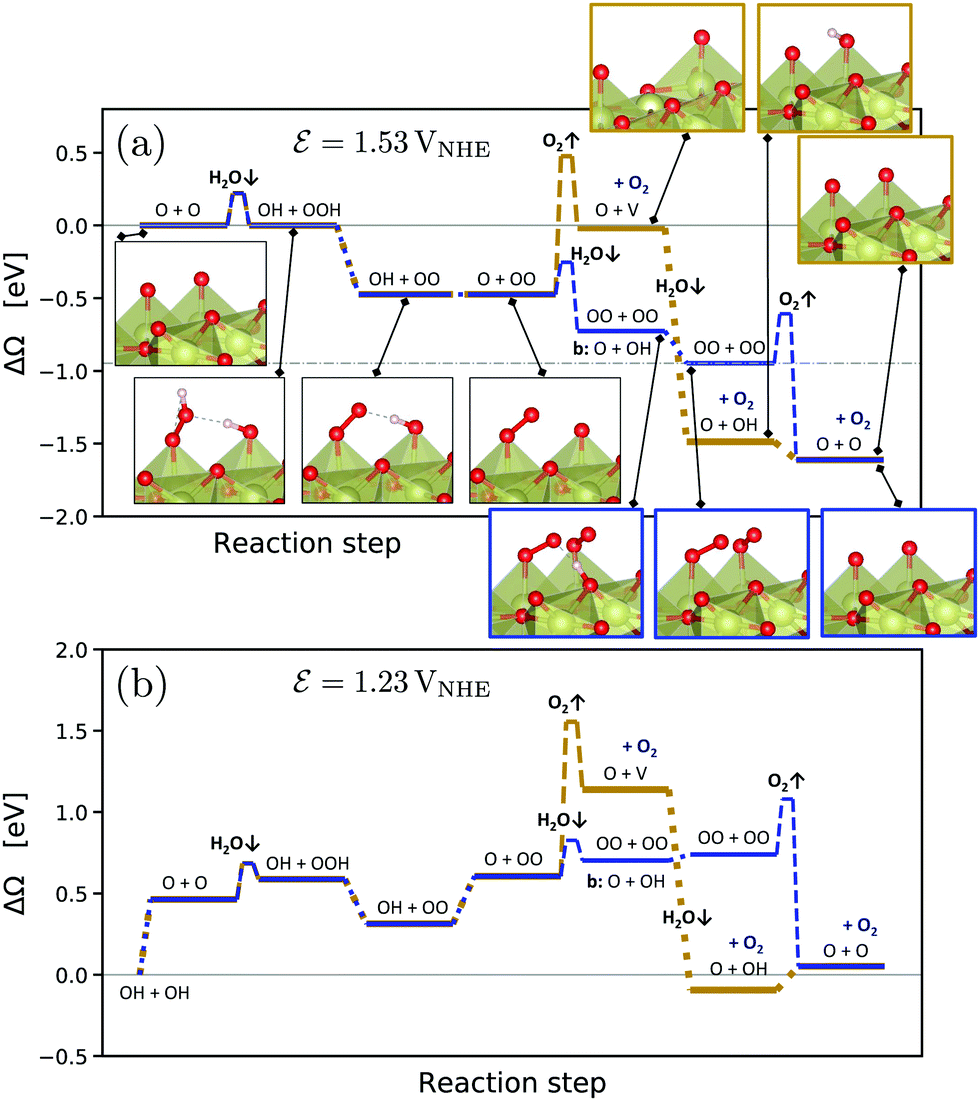 | ||
| Fig. 3 Comparison between the conventionally considered OER mechanism (golden color) and the novel mechanism (blue color) at pH = 0 and a potential of 1.53 VNHE (a) and 1.23 VNHE (b). Note that no correction of the O2 DFT energy was applied, see Computational Methods, for which reason the initial and final states at the correct equilibrium potential in (b) are slightly misaligned. Results including such corrections are presented in Fig. S7 and S8 (ESI†). The intermediate adsorbate configurations are shown and denoted by the occupation of the two top oxygen sites per surface cell. If not otherwise indicated, the bridge oxygen sites are occupied by O + O. Reaction barriers of dissociative water adsorption (H2O↓) and oxygen evolution (O2↑) steps were estimated from climbing-image NEB calculations for the neutral systems, see Fig. 4. The zero energy reference was chosen at the respective lowest-energy surface configuration involving only *O and *OH, see Fig. 1. The absolute lowest-energy configuration OO + OO at 1.53 VNHE is indicated by a dash-dotted line in (a). To facilitate conversion between different reference states, Table S6 (ESI†) presents their free energies versus the “clean” stoichiometric surface state as a common reference. | ||
To estimate the influence of the activation energy barriers, we used the climbing-image nudged elastic band (CI-NEB) method54 for dissociative water adsorption on the *O-covered surface (RS5), oxygen evolution by *OO–OO* association (RS9) (novel mechanism), and oxygen evolution by desorption (RS8) (conventional mechanism). We assume the barriers of simple PET steps in acidic conditions to be negligible. The corresponding energy barriers are shown in Fig. 4 together with images of the initial, final, and intermediate/transition states. The presented results were obtained for the neutral systems, but we also performed NEB calculations at a fixed negative surface charge as discussed below. For the conventional O2 evolution step by desorption, we obtained an activation energy of ΔEDFT = 0.95 eV, measured with respect to the initial state of a single adsorbed *OO, see Fig. 4(c). In contrast, the novel step of O2 evolution by *OO–OO* association only requires an activation energy of ΔEDFT = 0.34 eV, measured with respect to the ground-state of two adjacent *OO, see Fig. 4(b). Therefore, both thermodynamically and kinetically, the novel OER mechanism on IrO2(110) is favored over the conventional. As shown in Fig. S7 and S8 (ESI†), this conclusion also holds when adding the O2/OO energy penalties discussed above. For the single *OO adsorbate, we find an *O–O bond length of 1.28 Å, and essentially the same for the double *OO with 1.29 Å, which is indicative for a superoxide *OO− character in agreement with previous findings.21,26 Accordingly, no OO energy correction would be required for *OO adsorbates,26 making the scenario of Fig. S8 (ESI†) unrealistic.
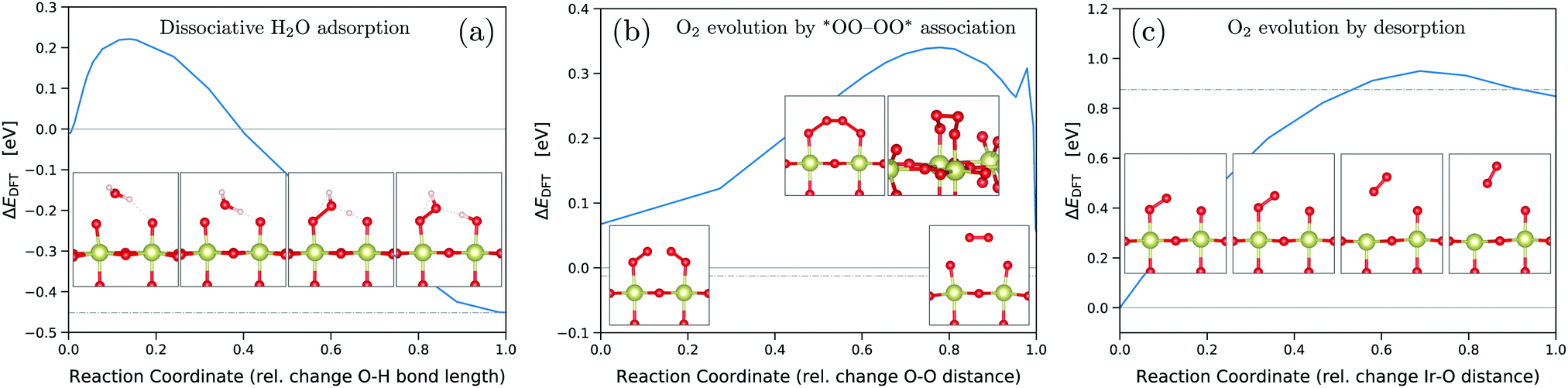 | ||
| Fig. 4 Activation energy barriers from climbing-image NEB54 calculations for dissociative water adsorption on the *O covered surface (a), see reaction (RS5), oxygen evolution by *OO–OO* association (b), see reaction (RS9), and oxygen evolution by desorption (c), see reaction (RS8). Initial and final state energies are indicated by horizontal solid and dash-dotted lines, respectively. NEB calculations were performed for uncharged systems. | ||
The transition state of the O2 evolution step is an intriguing Ir–OOOO–Ir entity with a chain of four oxygen atoms and three O–O bonds, see Fig. 4(b). The evolving O2 molecule results from creating a novel inner O–O bond while splitting the two outer O–O bonds, whereas the basal Ir–O bonds remain intact. The creation of the O–O bond of the evolving O2 molecule is shown in Fig. 5 at the Ir–OOOO–Ir state, where all three O–O bonds have an equal length of 1.44 Å, indicating peroxide-type bonding. Fig. 5(c) shows the σ-bonding charge density of the newly forming inner O–O bond, which becomes visible in the local density of states (LDOS) integrated over the energy range from −3.0 to −2.0 eV vs. EFermi. In the energy range from −2.0 to −1.0 eV vs. EFermi, we observe certain π-bonding characteristics, see Fig. 5(b), whereas the LDOS in the energy range directly below the Fermi level reveals a π*-antibonding character, see Fig. 5(a). We further note that the LDOS plots also show the crystal-field splitting of the iridium 5d orbitals. The shape of the LDOS around the iridium atom of the second layer in Fig. 5(a) (at the lower-half center) results from the superposition of the dxz and dyz orbitals that determine the density of states around the Fermi level, whereas the dx2−y2 orbital dominates in the energy range from −2.0 to −1.0 eV vs. EFermi, shown in Fig. 5(b). In the range from −3.0 to −2.0 eV vs. EFermi, all three orbitals dxz, dyz, and dx2−y2 have approximately equal contributions, resulting in the t2g-like LDOS visible in Fig. 5(c). The t2g-like iridium 5d orbitals are involved in π-type Ir–O interactions. These observations are in very good agreement with previously reported results for the crystal-field splitting and bonding in bulk IrO2.40
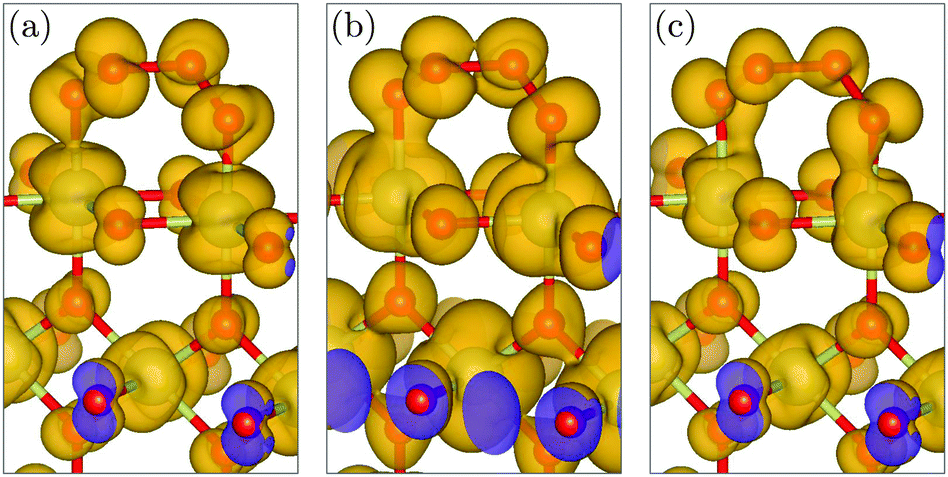 | ||
| Fig. 5 Creation of the final O–O bond of the evolving O2 molecule at the Ir–OOOO–Ir transition state: Isosurface plots of the local density of states (LDOS) integrated over different energy ranges (PARCHG file from VASP): (a) from −1.0 to 0.0 eV vs. EFermi, (b) from −2.0 to −1.0 eV vs. EFermi, and (c) from −3.0 to −2.0 eV vs. EFermi. | ||
The question remains for the rate-determining step (RDS) of the novel OER mechanism. The dissociative H2O adsorption on the *O-covered surface has been identified as RDS of the conventional mechanism.21 However, in this study, the O2-desorption barrier had been lowered by adding entropy contributions of the free O2 molecule to the transition state. As discussed in the Computational Methods section, we consider it more consistent with transition-state theory to relate the transition state to the initial state rather than the final state of the given elementary step. Without including the final-state entropy contribution, the O2-desorption barrier of ΔEDFT = 0.95 eV is significantly higher than the H2O-dissociation barrier, for which we obtained ΔEDFT = 0.22 eV, see Fig. 4(a), at the uncharged *O-covered surface (with a potential of zero charge of 2.59 VSHE). Although H2O dissociation according to reaction (RS5) could be regarded as a “chemical” step due to the absence of PET, Ping et al.21 found a marked potential dependence of the corresponding energy barrier. We therefore also performed a NEB calculation for the charged system with a constant excess surface charge of −0.6 e nm−2, corresponding to an electrode potential of 1.69 VSHE for the *O-covered surface in the initial state. Under these conditions, we obtained an H2O-dissociation barrier of ΔEDFT = 0.55 eV, significantly larger than for the neutral system, and in good agreement with the previously reported values at similar potentials.21 However, even when taking into account the influence of surface charging, the H2O-dissociation barrier remains significantly smaller than the energy barrier of the conventional O2-desorption step, so that the latter would be rate-limiting for the conventional OER mechanism. For the energy barrier of the novel O2-evolution step by *OO–OO* association, we found a negligible dependence on the charge state of the surface (ΔEDFT = 0.33 eV at −0.6 e nm−2vs. ΔEDFT = 0.34 eV for the uncharged system). From the comparison with the H2O-dissociation barrier, we conclude that H2O dissociation according to reaction (RS5) is the RDS of the novel OER mechanism on IrO2(110) in the OER-relevant potential range. Close to the OER equilibrium potential, however, the thermodynamic destabilization of the *OO-covered surface state compared to the *O-covered state can make the Ir–OOOO–Ir transition state of the O2-evolution step the highest point to be overcome along the free-energy profile of the OER pathway, see Fig. 3(b), and thus OER rate-determining. The RDS of the novel mechanism thus depends on the potential range considered.
Discussion
Given the extensive research in the field of OER electrocatalysis during the past decade, we acknowledge that the proposition of a major change in the “classical” mechanism on IrO2 might appear surprising. We emphasize that both our computational methodology and our results agree in all relevant aspects with the state of the art,21,22,26 as discussed and compared in detail in the ESI† (see Tables S3–S5). This holds, in particular, for the use of implicit solvent/electrolyte models21,26 and the comparison with reported results including explicit solvation effects.22 We therefore attribute the discovery of the novel mechanism primarily to it having been overlooked so far. The only relevant difference in our methodological approach concerns the treatment of the transition state for the conventional O2 desorption step. To the best of our knowledge, only Ping et al.21 also investigated the corresponding energy barrier. However, in their study the strongly negative entropic free energy contribution of the “free” gas-phase O2 molecule was added to the transition state of the desorption step, thus artificially lowering the energy barrier. As explained in the Computational methods, we consider this approach as inconsistent with the basic rate law of kinetic theory and transition-state theory. Without such artificial lowering of the O2 desorption barrier, we found that this barrier is significantly higher than the water dissociation barrier for both neutral and charged surface calculations. We therefore conclude that the O2 desorption step must be regarded as the RDS of the conventional mechanism on IrO2(110). Only the novel mechanism makes the formation of Ir* OOH by water dissociation become RDS by providing a fast bypass for the slow conventional O2 desorption step. At the same time, we emphasize that our general conclusion regarding the preference of the novel mechanism over the conventional one is independent from these subtle questions regarding RDS and energy barriers. Even when ignoring energy barriers and only considering the intermediate states shown in Fig. 3 (and Fig. S7 and S8, ESI†) the novel pathway is strongly favored over the conventional one.In the following, we first compare the cyclic voltammogram (CV) derived from our results with the experimental CV reported for the same system16 in order to confirm the general validity of our computational framework. Thereafter, we show that the novel OER pathway is strongly supported by the current experimental state of knowledge from in situ/operando X-ray spectroscopy. The computed CV of the IrO2(110) electrode at pH = 0 is plotted in Fig. 6(a) in terms of the capacitance C as a function of the electrode potential , see Computational Methods. The respective results including the OO energy correction are shown as dash-dotted curves. The computed CV is characterized by two broad pseudocapacitive peaks in the potential range 1.0–1.3 VNHE that correspond to the deprotonation of the two bridge oxygen sites of the simulated 2 × 1 surface cell, see Fig. 1. These features are in agreement with the broad pseudocapacitive region observed in the experimental CV of IrO2(110) in acidic electrolyte within the same potential range, as reported by Kuo et al.16 Also the integrated charge, shown in Fig. 6(b), agrees well with the experimental results. The plateau in the charge plot around 1.3–1.4 VNHE (before the final steep increase) corresponds to a charge of 1.6–1.7 electrons per 2 × 1 surface cell, which is in excellent agreement with a reported value of around 1.7 (0.85 electrons per surface top-site Ir).16 We conclude that the pseudocapacitive pre-OER region in the CV of IrO2(110) corresponds to the deprotonation of bridge oxygen sites, in agreement with the spectroscopic observation of the formation of “OI−” species within the same potential range reported by Saveleva et al.,18 which the authors assigned to deprotonated bridge oxygen species. However, based on our analysis, this process has no direct relation to the OER, because the bridge oxygen are “spectator” species during the OER cycle. The OER-relevant top oxygen sites are passivated due to protonation in the pre-OER region.
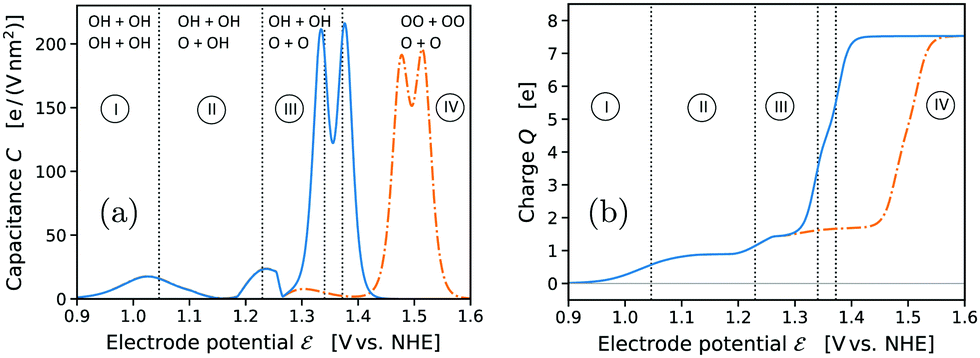 | ||
| Fig. 6 Computed cyclic voltammogram (a) of an IrO2(110) electrode at pH = 0, and integrated charge (b) per 2 × 1 surface cell with the fully *OH-covered surface (t: OH + OH b: OH + OH) as zero-charge reference state. The results including the “worst-case” OO energy penalty are shown as dash-dotted curves. Vertical dotted lines and numbered potential ranges have the same meaning as in Fig. 1. The most stable adsorbate configurations in the respective potential ranges are indicated with top and bridge occupations. | ||
The formation of the OER-active state of the top oxygen species occurs in the pronounced peak of the simulated CV between 1.3–1.4 VNHE, which, including the OO energy correction, shifts to the range 1.45–1.55 VNHE, in excellent agreement with the experimental OER onset on IrO2(110) in acidic electrolyte.16 Thermodynamically, the top oxygen state would transform from protonated *OH straight to *OO, see Fig. 1. However, the OER drives the surface out of equilibrium in this potential range, and the surface state becomes dependent on the kinetics and RDS of the OER mechanism. We argued that the O2-desorption step would be rate-limiting for the conventionally considered mechanism, with a significantly larger activation energy than the H2O-dissociation step. In such a case, the *OOH and *OO intermediates would equilibrate with all “upstream” species of the OER pathway, in particular the *OH and *O surface states, and the top surface sites would become fully covered with *OO, see Fig. 1. The same would hold for the novel mechanism if the O2-evolution step (RS9) was rate-limiting. However, we saw from the NEB calculations that at OER-relevant potentials the formation of *OOH in the H2O-dissociation step (RS5) is more likely the RDS of the novel mechanism, so that the surface state equilibrates primarily among the upstream species of *OH and *O. This would result in a dominant *O-coverage in the OER potential range above 1.5 VNHE, see Fig. S4 (ESI†), in agreement with the spectroscopic findings by Pedersen et al.56 The *OOH and *OO entities lie “downstream” of the RDS towards the OER product side, resulting in a low, kinetically controlled surface coverage, for which reason the spectroscopic detection of such species57 is challenging.
Different studies have highlighted the central role of electrophilic surface oxygen atoms with a certain radical character18,21,26,58,59 for the OER mechanism on iridium oxides. Based on the spectroscopic observation of “OI−” species, Pfeifer et al.59 and Saveleva et al.18 have suggested an oxygen-anion redox cycle, in contrast to the conventional notion of a transition-metal-cation redox cycle. However, none of the proposed OER mechanisms to date is consistent with such a perspective, because, even if temporarily in an “OI−” state, the active surface oxygen species follow a unidirectional oxidation sequence until they become part of the evolving O2 molecule. Furthermore, the existence of coordinately unsaturated Ir* species after the O2-evolution step is implied in any of these mechanisms, so that the actual redox cycle is provided by iridium cations rather than surface oxygen anions. The novel OER mechanism closes this consistency gap by establishing a fully closed anion redox cycle of top surface oxygen species on IrO2(110) that represent the fixed adsorption sites along the OER pathway. The Ir cations underneath the top oxygen sites preserve their full oxygen coordination at all stages and thus only indirectly participate in the OER mechanism. As a consequence, the saturated Ir species merely undergo screened, indirect changes of their oxidation state. In particular, the novel mechanism does not involve partially reduced “IrIII+” species (corresponding to Ir*) at any stage. These findings explain the spectroscopically observed constancy of the iridium oxidation state and the absence of iridium cation species in a reduced oxidation state during OER on IrO2.18,56 The novel mechanism thus completes the paradigm shift away from a metal-cation redox cycle to an oxygen-anion redox cycle as the key element of OER electrocatalysis on crystalline iridium oxide, with possible consequences also for other metal oxide catalysts. The suppression of the conventional O2-desorption step indicates a rather weak reducibility of crystalline IrO2. In other words, it is a certain resistance of IrO2 against reduction that favors the novel OER mechanism over the conventional one. This conclusion is supported by the fact that besides rutile-structure IrO2, no other binary oxides of Ir are known, in particular no reduced binary oxide like, e.g., Ir2O3.60 The novel mechanism provides an efficient pathway for the OER to proceed at the oxygen-covered surface of a weakly reducible oxide. We consider this insight to be of fundamental importance for strategies towards the discovery of advanced metal-oxide-based OER catalysts.
As a unique feature, the novel mechanism involves both the formation and the cleavage of O–O bonds. In fact, it is the splitting of the O–O bond in the Ir*OO entities during the O2-evolution step (RS9) which closes the redox cycle of the oxygen species in the remaining Ir*O entities. The electronic character of the stable top oxygen species must therefore not only enable a facile formation of an O–O bond by water nucleophilic attack in step (RS5), but also a facile splitting of this bond in the subsequent O2-evolution step (RS9). This aspect is highly relevant for an analysis of scaling relations and volcano-plots10,11,19,20,61 that typically employ the adsorption energies of *OH, *O, and *OOH intermediates on coordinately unsaturated transition metal atoms as descriptors. According to the novel mechanism, however, the Ir–O bond of the top Ir*O entities never gets broken. Instead, it is the adsorption energies of H, OH, and O on the fixed top oxygen atoms that govern the energetics along the OER pathway, while the strength of the Ir–O bond only plays an indirect role. Moreover, since the novel OER mechanism does not require the breakage of an Ir–O bond, it decouples OER kinetics from metal oxide stability, which is determined by the strength of the Ir–O bond. This explains why high OER activity can be combined with catalyst stability for the case of crystalline IrO2.5 It is interesting to note that mixed-metal-oxide catalysts, such as La2LiIrO6/La2IrO6, which contain iridium cations in a higher formal oxidation state up to IrVI+, involve an iridium-cation redox cycle with the formation of oxygen vacancies during OER, resulting in structural decomposition of the catalyst.58 In contrast, the stable oxygen-anion redox cycle on the weakly reducible IrO2 enables the unique combination of OER activity and structural stability. The combination of an only weak reducibility of the bulk oxide with an electrophilic oxygen surface termination thus appears a promising principle for the design and discovery of advanced OER catalysts providing both superior activity and stability.
Finally, our findings could also have consequences for the discovery of novel electrocatalyst materials for the oxygen reduction reaction (ORR). We established the deprotonated *O-covered IrO2(110) surface as the stable active state of the electrocatalyst during OER. By reversing the direction of the novel OER mechanism, it appears possible, at least in principle, to design electrocatalysts for the reverse ORR that operate in the oxidized surface state. This could enable higher operation potentials than conventional ORR catalysts, which require reduced transition-metal sites to be available.
Conclusions
In summary, we found that the “classical” OER mechanism on IrO2(110) proceeds differently than presently assumed. Instead of the conventionally considered O2-desorption step, the O2 molecule evolves in an association step from two neighboring Ir*OO entities via an intriguing Ir*OOOO*Ir transition state. At practical OER potentials, the dissociative water adsorption at the *O-covered surface is the rate-determining step, but close to the equilibrium potential, the availability of Ir*OO surface motifs for the O2-evolution step becomes the overall thermodynamic bottleneck along the OER pathway. The fixed top oxygen atoms at the IrO2(110) surface are the active sites of the catalytic cycle, and the surface iridium atoms remain fully coordinated at all stages. The novel mechanism thus corresponds to a fully closed oxygen-anion redox cycle, representing a paradigm shift in OER electrocatalysis and consistently explaining recently reported results from in situ/operando X-ray spectroscopy experiments. Furthermore, our results provide an explanation for the surprising combination of activity and stability for crystalline IrO2, because the OER occurs without requiring the breaking of Ir–O bonds. Optimizing the strength of the metal–oxygen bond for stability therefore does not necessarily jeopardize the OER activity, which is highly relevant for the design of advanced OER catalysts combining performance in both activity and stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. B. acknowledges financial support in the form of a research fellowship grant funded by the SNSF (Swiss National Science Foundation).Notes and references
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- C. C.-L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, K.-C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y.-T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS PubMed.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- T. Weber, V. Vonk, D. Escalera-López, G. Abbondanza, A. Larsson, V. Koller, M. J. Abb, Z. Hegedüs, T. Bäcker, U. Lienert, G. S. Harlow, A. Stierle, S. Cherevko, E. Lundgren and H. Over, ACS Catal., 2021, 11, 12651–12660 CrossRef CAS.
- C. Minke, M. Suermann, B. Bensmann and R. Hanke-Rauschenbach, Int. J. Hydrogen Energy, 2021, 46, 23581–23590 CrossRef CAS.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J.-J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- C. W. Song, J. Lim, H. B. Bae and S.-Y. Chung, Energy Environ. Sci., 2020, 13, 4178–4188 RSC.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- M. Busch, N. B. Halck, U. I. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- K. S. Exner, ACS Appl. Energy Mater., 2019, 2, 7991–8001 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169–7172 CrossRef CAS PubMed.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 1636–1641 CrossRef CAS PubMed.
- D.-Y. Kuo, J. K. Kawasaki, J. N. Nelson, J. Kloppenburg, G. Hautier, K. M. Shen, D. G. Schlom and J. Suntivich, J. Am. Chem. Soc., 2017, 139, 3473–3479 CrossRef CAS PubMed.
- O. Kasian, J.-P. Grote, S. Geiger, S. Cherevko and K. J.-J. Mayrhofer, Angew. Chem., Int. Ed., 2018, 57, 2488–2491 CrossRef CAS PubMed.
- V. A. Saveleva, L. Wang, D. Teschner, T. Jones, A. S. Gago, K. A. Friedrich, S. Zafeiratos, R. Schlögl and E. R. Savinova, J. Phys. Chem. Lett., 2018, 9, 3154–3160 CrossRef CAS PubMed.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- Z. Xu, J. Rossmeisl and J. R. Kitchin, J. Phys. Chem. C, 2015, 119, 4827–4833 CrossRef CAS.
- Y. Ping, R. J. Nielsen and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 149–155 CrossRef CAS PubMed.
- J. A. Gauthier, C. F. Dickens, L. D. Chen, A. D. Doyle and J. K. Nørskov, J. Phys. Chem. C, 2017, 121, 11455–11463 CrossRef CAS.
- L. G.-V. Briquet, M. Sarwar, J. Mugo, G. Jones and F. Calle-Vallejo, ChemCatChem, 2017, 9, 1261–1268 CrossRef CAS.
- K. S. Exner and H. Over, ACS Catal., 2019, 9, 6755–6765 CrossRef CAS.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- D. González, J. Heras-Domingo, M. Sodupe, L. Rodríguez-Santiago and X. Solans-Monfort, J. Catal., 2021, 396, 192–201 CrossRef.
- Y. Ping, W. A. Goddard and G. A. Galli, J. Am. Chem. Soc., 2015, 137, 5264–5267 CrossRef CAS PubMed.
- F. Nattino and N. Marzari, Phys. Chem. Chem. Phys., 2020, 22, 10807–10818 RSC.
- C. Spöri, J. T.-H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T.-M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- O. Kasian, S. Geiger, T. Li, J.-P. Grote, K. Schweinar, S. Zhang, C. Scheu, D. Raabe, S. Cherevko, B. Gault and K. J.-J. Mayrhofer, Energy Environ. Sci., 2019, 12, 3548–3555 RSC.
- K. Schweinar, B. Gault, I. Mouton and O. Kasian, J. Phys. Chem. Lett., 2020, 11, 5008–5014 CrossRef CAS PubMed.
- A. Zagalskaya, I. Evazzade and V. Alexandrov, ACS Energy Lett., 2021, 6, 1124–1133 CrossRef CAS.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- S. Czioska, A. Boubnov, D. Escalera-López, J. Geppert, A. Zagalskaya, P. Röse, E. Saraçi, V. Alexandrov, U. Krewer, S. Cherevko and J.-D. Grunwaldt, ACS Catal., 2021, 11, 10043–10057 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. M. Kahk, C. G. Poll, F. E. Oropeza, J. M. Ablett, D. Céolin, J.-P. Rueff, S. Agrestini, Y. Utsumi, K. D. Tsuei, Y. F. Liao, F. Borgatti, G. Panaccione, A. Regoutz, R. G. Egdell, B. J. Morgan, D. O. Scanlon and D. J. Payne, Phys. Rev. Lett., 2014, 112, 117601 CrossRef CAS PubMed.
- Y. Ping, G. Galli and W. A. Goddard, J. Phys. Chem. C, 2015, 119, 11570–11577 CrossRef CAS.
- A. A. Bolzan, C. Fong, B. J. Kennedy and C. J. Howard, Acta Crystallograph. Sect. B, 1997, 53, 373–380 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J.-S. Filhol and M. Neurock, Angew. Chem., Int. Ed., 2006, 45, 402–406 CrossRef CAS PubMed.
- A. Hagopian, M.-L. Doublet, J.-S. Filhol and T. Binninger, J. Chem. Theory Comput., 2022, 18, 1883–1893 CrossRef PubMed.
- IUPAC, in Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”), ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 1997 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. D. Cox, D. D. Wagman and V. A. Medvedev, CODATA Key Values for Thermodynamics, Hemisphere Publishing Corp., New York, 1989 Search PubMed.
- E. Sargeant, F. Illas, P. Rodríguez and F. Calle-Vallejo, J. Electroanal. Chem., 2021, 896, 115178 CrossRef CAS.
- W. Schmickler and E. Santos, Interfacial Electrochemistry, Springer, Berlin, Heidelberg, 2010 Search PubMed.
- N. G. Hörmann, O. Andreussi and N. Marzari, J. Chem. Phys., 2019, 150, 041730 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- A. F. Pedersen, M. Escudero-Escribano, B. Sebok, A. Bodin, E. Paoli, R. Frydendal, D. Friebel, I. E.-L. Stephens, J. Rossmeisl, I. Chorkendorff and A. Nilsson, J. Phys. Chem. B, 2018, 122, 878–887 CrossRef PubMed.
- N. Sivasankar, W. W. Weare and H. Frei, J. Am. Chem. Soc., 2011, 133, 12976–12979 CrossRef CAS PubMed.
- A. Grimaud, A. Demortière, M. Saubanère, W. Dachraoui, M. Duchamp, M.-L. Doublet and J.-M. Tarascon, Nat. Energy, 2016, 2, 16189 CrossRef.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143–2149 RSC.
- M.-S. Miao and R. Seshadri, J. Phys.: Condens. Matter, 2012, 24, 215503 CrossRef PubMed.
- M. Busch, Curr. Opin. Electrochem., 2018, 9, 278–284 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computed DOS of bulk IrO2; pictures of investigated adsorbate configurations at IrO2(110); computed grand-canonical stability diagrams; computed OER pathways including O2/OO energy penalties; computed grand-canonical free energies of OER intermediate states; comparison of free energy steps for the conventional OER mechanism with previous literature. See DOI: https://doi.org/10.1039/d2ee00158f |
| This journal is © The Royal Society of Chemistry 2022 |