
A niobium oxide with a shear structure and planar defects for high-power lithium ion batteries†
Tongtong
Li‡
ab,
Gyutae
Nam‡
a,
Kuanting
Liu
c,
Jeng-Han
Wang
 c,
Bote
Zhao
*ad,
Yong
Ding
a,
Luke
Soule
a,
Maxim
Avdeev
ef,
Zheyu
Luo
a,
Weilin
Zhang
a,
Tao
Yuan
a,
Panpan
Jing
a,
Min Gyu
Kim
*g,
Yanyan
Song
*b and
Meilin
Liu
*a
c,
Bote
Zhao
*ad,
Yong
Ding
a,
Luke
Soule
a,
Maxim
Avdeev
ef,
Zheyu
Luo
a,
Weilin
Zhang
a,
Tao
Yuan
a,
Panpan
Jing
a,
Min Gyu
Kim
*g,
Yanyan
Song
*b and
Meilin
Liu
*a
aSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30332-0245, USA. E-mail: meilin.liu@mse.gatech.edu
bCollege of Sciences, Northeastern University, Shenyang, 110004, China. E-mail: yysong@mail.neu.edu.cn
cDepartment of Chemistry, National Taiwan Normal University, 88, Sec. 4 Ting-Zhou Road, Taipei 11677, Taiwan, Republic of China
dSchool of Environment and Energy, South China University of Technology, Guangzhou 510006, China. E-mail: botezhao@scut.edu.cn
eAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organization, Sydney, Australia
fSchool of Chemistry, The University of Sydney, Sydney 2006, Australia
gBeamline Research Division, Pohang Accelerator Laboratory (PAL), Pohang University of Science and Technology (POSTECH), Pohang, Republic of Korea. E-mail: mgkim@postech.ac.kr
First published on 16th November 2021
Abstract
The development of anode materials with high-rate capability is critical to high-power lithium batteries. T-Nb2O5 has been widely reported to exhibit pseudocapacitive behavior and fast lithium storage capability. However, the other polymorphs of Nb2O5 prepared at higher temperatures have the potential to achieve even higher specific capacity and tap density than T-Nb2O5, offering higher volumetric power and energy density. Here, micrometer-sized H-Nb2O5 with rich Wadsley planar defects (denoted as d-H-Nb2O5) is designed for fast lithium storage. The performance of H-Nb2O5 with local rearrangements of [NbO6] octahedra blocks surpasses that of T-Nb2O5 in terms of specific capacity, rate capability, and stability. A wide range variation in the valence of niobium ions upon lithiation was observed for defective H-Nb2O5via operando X-ray absorption spectroscopy. Operando extended X-ray absorption fine structure and ex situ Raman spectroscopy analyses reveal a large and reversible distortion of the structure in the two-phase region. Computation and ex situ X-ray diffraction analysis reveal that the shear structure expands along major lithium diffusion pathways and contracts in the direction perpendicular to the shear plane. Planar defects relieve strain through perpendicular arrangements of blocks, minimizing volume change and enhancing structural stability. In addition, strong Li adsorption on planar defects enlarges intercalation capacity. Different from nanostructure engineering, our strategy to modify the planar defects in the bulk phase can effectively improve the intrinsic properties. The findings in this work offer new insights into the design of fast Li-ion storage materials in micrometer sizes through defect engineering, and the strategy is applicable to the material discovery for other energy-related applications.
Broader contextFaster charge lithium-ion batteries with higher capacity are desired for consumer electronics and electrified transportation. In particular, global warming due to CO2 emission drives the desire for wide-spread adoption of electric vehicles. Slow charge rate and limited range of Li-ion battery vehicles are bottlenecks to replace gasoline vehicles. Developing electrode material that enables fast and safe lithium-ion storage could address these issues. Niobium oxides have shown promise as anode materials with larger capacity and higher rate capability than commercialized lithium titanium oxide. The design of a high-performance niobium-based oxide electrode using a straightforward strategy remains a big challenge. Here a unique structure of H-Nb2O5 is found that could achieve fast lithium storage by engineering its Wadsley planar defect. d-H-Nb2O5 with particle size in micrometers achieves significantly enhanced electrochemical performance compared to pure H-Nb2O5 and T-Nb2O5. d-H-Nb2O5 exhibits excellent rate capability, delivering over 40% of its capacity in 20 seconds without obvious capacity decay for 4000 cycles. This work not only reports a highly promising anode for fast charge and long-life lithium-ion batteries but also proposes a new strategy to enhance battery performance through the use of defect engineering. |
Introduction
The increasing demand of portable devices and electric vehicles is driving the development of new-generation lithium-ion batteries (LIBs) with faster charge capability and longer lifespan.1,2 In particular, electric vehicles must be fully charged within a few minutes to make the process comparable to refueling conventional gasoline vehicles. Substantial effort has been devoted to the development of new materials with high-rate electrochemical energy storage capability during the last decade.3–7 Li4Ti5O12 (LTO) has been regarded as one of the most promising high-rate anodes for LIBs. However, several drawbacks of LTO (e.g., relatively low capacity and gassing problem) limit the broad application of LTO on the commercial market.8 Niobium-based materials, including niobium pentoxide,3,9–13 niobium tungsten oxide4,14,15 and titanium niobium oxide,16–18 have been reported to exhibit fast lithium storage capability as anodes. These niobium-based materials are electrochemically active in a similar operating voltage range to LTO but with a higher specific capacity.4,19Niobium-based materials can be classified into two structural categories: the bronze structure represented by the T-phase (orthorhombic) Nb2O5 and Wadsley–Roth crystallographic shear structure represented by H-phase (monoclinic) Nb2O5.14,20–22 In T-Nb2O5, the NbO6 or NbO7 polyhedra are edge- or corner-shared within the (001) plane and exclusively corner-shared along the [001] direction (Fig. S1, ESI†). The remaining ∼5% niobium atoms are randomly distributed and coordinated to 9 oxygen atoms in interstitial sites between the (001) planes to balance charge.23 H-Nb2O5 crystallizes in shear structures with two kinds of blocks, 3 × 4 and 3 × 5 blocks in the (010) plane and infinite in the third dimension (b direction).3,24 These blocks are built of corner-sharing octahedra and connect to the adjacent block by edge-sharing. The remaining voids in the structure are filled by corner-sharing tetrahedra. In the early period, Kumagai et al. investigated the effect of polymorphs of Nb2O5 on cell behavior.25 Furthermore, they studied the electrochemical lithium intercalation process into the crystal lattice of T-Nb2O5, the one that has the best reversibility among Nb2O5 family proven by their previous work at that time.26–28 For the class of Wadsley–Roth phases such as H-Nb2O5, their performance in secondary lithium cells and structure change upon lithium insertion were studied by R. J. Cava in the same period.22 Recently T-Nb2O5 has been reported in the literature to exhibit fast lithium-ion storage through a facile two-dimensional (2D) lithium-ion diffusion pathway along the (001) interplane3,19,29–33 while the lithium intercalation behavior of H-Nb2O5 is yet to be studied systematically.22 Compared to T-Nb2O5, the higher sintering temperature of H-Nb2O5 results in larger particles. Accordingly, it is harder to synthesize nanostructured H-Nb2O5, leading to fewer studies on the structure and properties of H-Nb2O5. Recent investigations of ternary shear phases in the Nb2O5–TiO2 and Nb2O5–WO3 systems give hints that H-Nb2O5 might display greater rate capability. These ternary crystallographic shear compounds have close structural relationships to H-Nb2O5 and despite having more complex compositions,34 they share the common structural feature of Wadsley planar defects by the intergrowth of shear phases. H-Nb2O5 has the potential to deliver higher rate capability and greater tap density offering higher volumetric power and energy density than T-phase Nb2O5 by engineering the morphology and orientation of particles and crystal defects.
In this work, we designed and prepared a series of H-Nb2O5 phases with and without Wadsley planar defects by adjusting the calcination temperatures. The effects of these defects on their electrochemical properties were investigated systematically. We demonstrate that the defective d-H-Nb2O5 prepared at the optimal temperature (i.e. 950 °C) exhibits a much higher intercalation capacity than well-crystallized H-Nb2O5. The electrochemical properties of micrometer-sized d-H-Nb2O5 (such as specific capacity, rate capability, and durability) are better than those of T-Nb2O5 with much smaller particle sizes, which is validated by a wider variation in the valence of niobium ions and a greater reversible structural change upon lithiation of d-H-Nb2O5 in operando X-ray absorption spectroscopy (XAS). Operando extended X-ray absorption fine structure (EXAFS), ex situ XRD, and Raman spectroscopy reveal that strong lithium adsorption in this shear structure induces distortion to accommodate more lithium at high Li concentration; the cell volume shows anisotropic change in the two-phase region, with the contraction in a–c plane constrained by the shear plane and the expansion in the b direction along the lithium diffusion pathway. DFT calculations show that strong adsorption energy of Li ions (Eads(Li+)) on defects results in a higher intercalation capacity of d-H-Nb2O5 among other Nb2O5. The open structure of d-H-Nb2O5 created by the planar defects alleviates the build-up of strain via b-axis expansion upon lithiation, resulting in excellent battery durability.
Results and discussion
Engineering phases and planar defects of Nb2O5 by adjusting calcination temperature
Nb2O5 exists in different crystalline polymorphs depending on the synthesis methods, precursors and pyrolysis temperatures.23,35 Different combinations of octahedral linkages by either corner- or edge-sharing satisfying the requirement of O/Nb ratio of 2.5 produce structural diversity among Nb2O5. Crystal structures containing differently coordinated cations give additional complexity to Nb2O5 structures. Here commercial Nb2O5 hydrates were used as the starting materials to study the calcination temperature effect on its structural properties (Fig. S2, ESI†). Thermogravimetric analysis (TGA) shows that the weight of Nb2O5 hydrate becomes stable at around 200 °C (Fig. 1a). Differential scanning calorimetry (DSC) shows that an endothermic peak ends at this point corresponding to the end of the loss of crystallized water. The first obvious exothermic peak at ∼570 °C in the heat flow corresponds to the transition from amorphous Nb2O5 to T-Nb2O5. The second phase transition at around 950 °C is assigned to the transformation from T-Nb2O5 to H-Nb2O5. The broad exothermic peak indicates that the second transition is a kinetically sluggish process. Based on the TGA results, we set a series of temperature points that is higher than 700 °C to prepare different Nb2O5 samples. X-ray diffraction (XRD) reveals that Nb2O5 crystallizes in the T-phase at a temperature lower than 850 °C (Fig. 1b and Fig. S3a, ESI†). The second phase appears at 900 °C and the transition is completed above 950 °C, which is consistent with the DSC analysis. The samples in the temperature ranging from 950 to 1100 °C show a similar characteristic diffraction pattern of the H-phase. However, compared with pure H-Nb2O5 observed at 1300 °C (Fig. S3c, ESI†), notable difference in XRD of 950 °C sample was retained even after considering the particle size and preferred orientation of crystal growth. Their similar high-angle diffraction region in XRD patterns indicate nearly identical short-range motifs while distinct low-angle diffraction region suggests different atomic arrangements on the long-range scale. As shown in Fig. 1c, the broader Raman band for T-Nb2O5 is due to the coexistence of two different types of niobium coordination (NbO6 and NbO7) in the crystal structure.36 Except for the bending mode of Nb–O–Nb linkage at 200–300 cm−1 and the stretching mode of NbOx at ∼690 cm−1, a new peak appears for H-Nb2O5 corresponding to the stretching mode of a higher-order bond (Nb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O terminal bond) with a shorter bond distance.36,37 The Raman features for H-Nb2O5 prepared at different temperatures (e.g. 950 °C and 1300 °C) are highly similar indicating that their local bonding is almost the same, consistent with XRD results. The Raman spectra of the samples calcined at 900 °C show characteristic peaks of either H-phase or T-phase as the laser focused on different regions, confirming that the phase transition starts in this temperature range. SEM images show that starting from the nano-sized hydrate, the particle size of Nb2O5 increased with increasing the calcination temperature (Fig. 1d–f and Fig. S4, ESI†). The material starts to sinter at 900 °C and finally becomes bulk at 1300 °C, resulting in different surface areas, as shown in BET measurements (Table S1, ESI†). Growth and intergrowth of the original nano-particles of amorphous Nb2O5 produce corresponding aggregates of H-Nb2O5 with the size of several microns to tens of microns (Fig. 1e and f).
O terminal bond) with a shorter bond distance.36,37 The Raman features for H-Nb2O5 prepared at different temperatures (e.g. 950 °C and 1300 °C) are highly similar indicating that their local bonding is almost the same, consistent with XRD results. The Raman spectra of the samples calcined at 900 °C show characteristic peaks of either H-phase or T-phase as the laser focused on different regions, confirming that the phase transition starts in this temperature range. SEM images show that starting from the nano-sized hydrate, the particle size of Nb2O5 increased with increasing the calcination temperature (Fig. 1d–f and Fig. S4, ESI†). The material starts to sinter at 900 °C and finally becomes bulk at 1300 °C, resulting in different surface areas, as shown in BET measurements (Table S1, ESI†). Growth and intergrowth of the original nano-particles of amorphous Nb2O5 produce corresponding aggregates of H-Nb2O5 with the size of several microns to tens of microns (Fig. 1e and f).
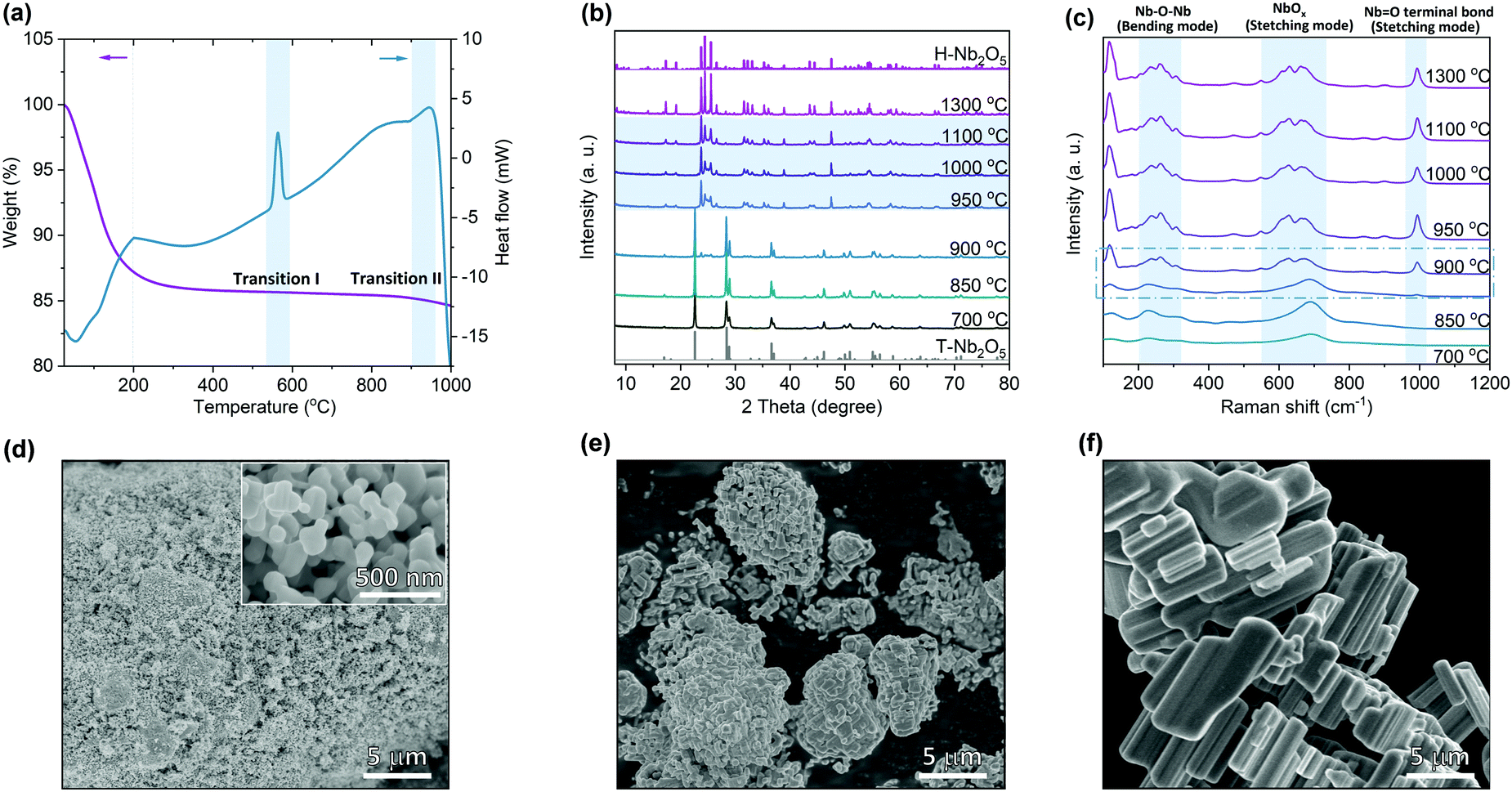 | ||
| Fig. 1 The temperature effect on the phase structures of Nb2O5. (a) Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) of Nb2O5 hydrate heated from 25 to 1000 °C at 5 °C min−1 under flowing air at 20 mL min−1. (b) XRD patterns of phases observed upon heating Nb2O5 hydrate in air at different temperatures. The standard PDF pattern for T-(orthorhombic) and H-(monoclinic) phase are inserted at the bottom and top of the graph, respectively. (c) Raman spectra of phases observed upon heating Nb2O5 hydrate in air. Three typical characteristic Raman active mode for Nb2O5 are highlighted by brilliant blue background. SEM images of Nb2O5 prepared at (d) 700 °C, (e) 950 °C and (f) 1300 °C. | ||
Considering the notable difference observed for H-Nb2O5 prepared at different temperatures, TEM analysis was performed to examine the local structure explicitly. Fig. 2a and b show the crystal model of ideal H-Nb2O5 along b axis and its corresponding simulated HRTEM, respectively. The bright dots in simulated image correspond to the empty area in between the corner shared oxygen octahedra. The sample obtained at 1300 °C shows well-defined micro-sized single crystals, as revealed by selected area electron diffraction (SAED) pattern of area D (Fig. 2c and d) while a few twin bands occur from time to time, as revealed by the streaks in TEM images. Compared with the simulated SAED pattern from [010] axis, these twins occur parallel to [101] direction thus streaking SAED spots along the [101] direction (Fig. S5, ESI†). As shown in Fig. 2e, the block structure is revealed by 2 × 3 and 2 × 4 bright dots stacking along the [100] direction, consistent with the modelling H-Nb2O5 pattern (inset picture). The crystal is long-range ordered on each side of the twin boundary and shows mirror symmetry. Overall, the grains obtained at 1300 °C show low density of planar defects and only obvious twin boundary was observed in TEM images (Fig. S5, ESI†). For the Nb2O5 sample calcined at 950 °C, TEM images captured from [010] orientation suggest the presence of abundant Wadsley defects in the sample (Fig. 2f and Fig. S6, ESI†). Short-range ordered regions and highly disordered regions were separated either by single or continuous twin bands in the crystals. As shown in Fig. 2g, splitting spots of SAED patterns were frequently observed. The diffuse scattering indicates atomic arrangement in the structure is an intermediate state of order. Splitting of spots as square indicate that two twin planes appear perpendicular to each other in one grain, as shown in Fig. 2f and Fig. S6 (ESI†).38,39 However, the blocks in these microdomains are still dominated by 3 × 4 and 3 × 5 blocks, which are the building units of H-Nb2O5 although their combination is more complex than pure H-Nb2O5 (Fig. 2h). There are two kinds of sub-twin bands, formed by rotating the longer side of either 3 × 4 or 3 × 5 block of host lattice by 90 degrees resulting in a V-type structure (Fig. S7, ESI†).38 As shown in Fig. 2h, these two micro-twin bands occur together as twin boundary changing the orientation of all blocks or appear by themselves forming two new Nb2O5 polymorphs (Nb28O70 and Nb56O140) without affecting the stoichiometry (enclosed area in Fig. 2h and Fig. S8, ESI†). The consecutive sub-twin or their multiple combinations give rich diverse stacking faults, which are the common defects in this sample. The array of 3 × 4, 3 × 5 blocks, and tetrahedral niobium site are packed, weaving a two-dimensional plane. Each block shape in the disordered region could still be clearly distinguishable indicating that the blocks are arranged coherently along the b axis though heavily disordered in the a–c plane (Fig. 2h, Fig. S8 and S9, ESI†). TEM results clarify the difference we found in both XRD and Raman spectra. Plenty of nano-scale Wadsley planar defects could be successfully formed in H-Nb2O5 by optimizing the calcination temperature. In the following discussion, the Nb2O5 prepared at 950 °C was denoted as d-H-Nb2O5 (i.e. defective H-Nb2O5) for clarity, to emphasize the planar defects or disordered structure of the sample.
 | ||
| Fig. 2 (a) Schematic of H-Nb2O5 crystallographic shear structure along b axis and (b) the corresponding simulated HRTEM image. Simulation conditions: defocus = 53 nm, thickness = 8.41 nm. The empty area in between corner shared NbO6 is enclosed with atomic columns for reference. (c) Dark-field TEM images of Nb2O5 prepared at 1300 °C. (d) Electron diffraction pattern from the crystal imaged in area D in (c) along [010] direction. (e) HR-TEM image showing the twin boundary along [101] direction, which is labeled as white dash line in (c). Simulated HR-TEM images inserted. (f) Dark-field TEM images of Nb2O5 prepared at 950 °C. (g) Electron diffraction pattern from short-range ordered region of crystal in (f) along [010] direction. (h) High-resolution TEM images of Nb2O5 prepared at 950 °C viewed down from the b axis. Twins and stacking faults were labeled as solid lines. The enclosed red model shown in (h) represents unit cell of H-Nb2O5 and two other Nb2O5 formed by consecutive sub-twins in the microdomains. | ||
Investigation into the electrochemical properties of different Nb2O5
The electrochemical performance of as-prepared d-H-Nb2O5 calcined at 950 °C was investigated using half-cells (Fig. 3 and Fig. S10–S18, ESI†). The T-Nb2O5 calcined at 700 °C and H-Nb2O5 without defects prepared at 1300 °C were also studied for comparison. As shown in Fig. 3a, both H-Nb2O5 and d-H-Nb2O5 delivered a higher capacity than T-Nb2O5 at 0.05C (1C = 0.2 A g−1). The electrochemical reaction of lithium intercalation into the structure of Nb2O5 can be expressed as Nb2O5 + xLi+ + xe− ↔ LixNb2O5. For T-Nb2O5, the discharge voltage decreases smoothly with increasing lithium content (x). This close-to-linear sloping voltage profile suggests that a single-phase formation reaction takes place during lithiation (Fig. 3a).27 For both H-Nb2O5 phases, the voltage profiles show three distinct regions, including a solid solution reaction at the beginning and near the end as well as a two-phase plateau.12 The cell voltage decreased at lower x values and reached approximately 1.7 V at x = ∼0.5 (corresponding to 0.25 Li per niobium in Fig. 3a). The cell voltage remained almost constant until x = ∼1.2 (corresponding to 0.6 Li per niobium), suggesting a two-phase equilibrium in LixNb2O5 in this region. There is another smooth decrease in voltage occurring near the end of the intercalation, where d-H-Nb2O5 starts to exhibit different electrochemical behavior and shows higher capacity than H-Nb2O5.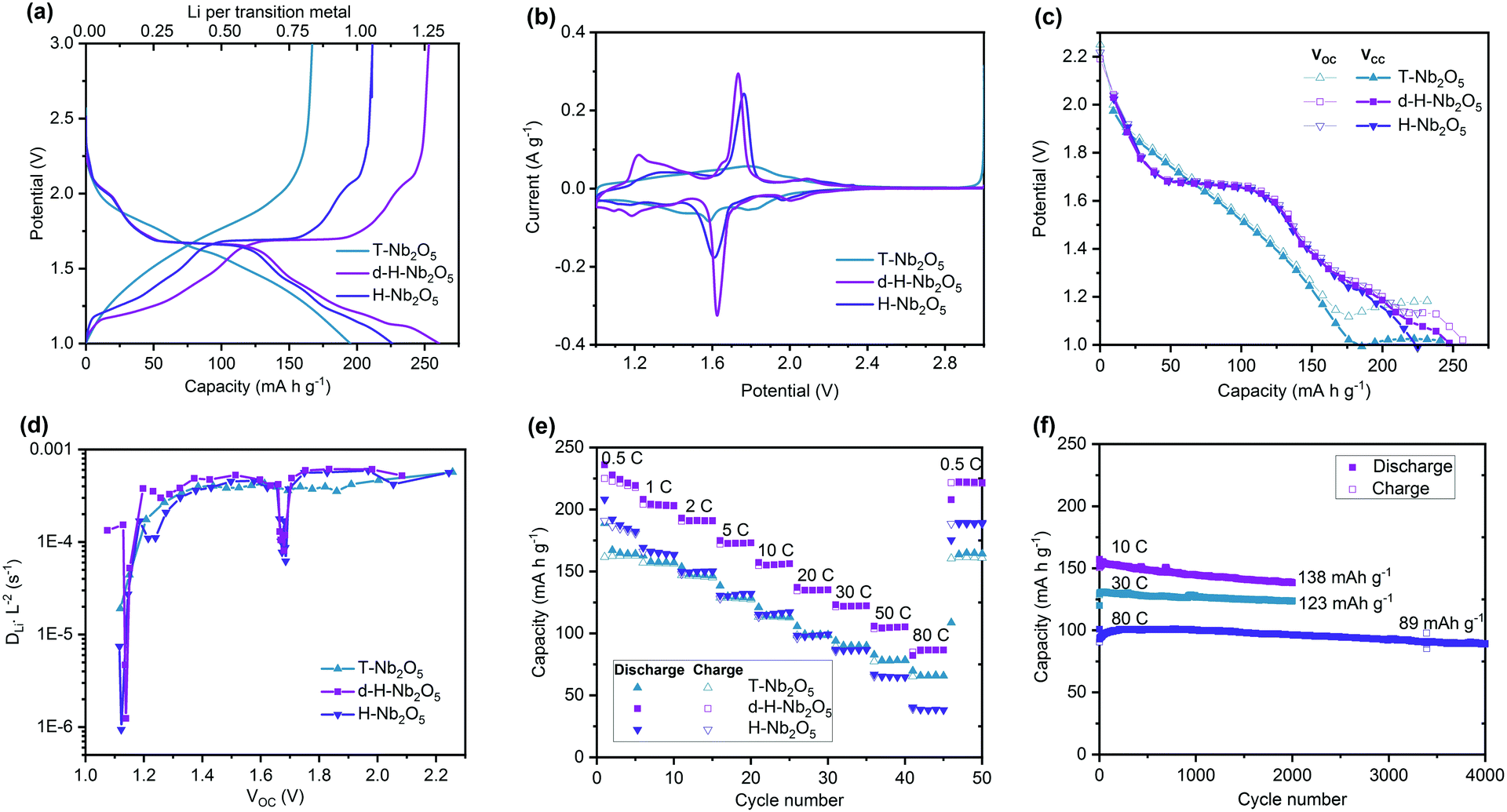 | ||
| Fig. 3 (a) Galvanostatic discharge–charge curves of Nb2O5 tested from 3.0 to 1.0 V at a rate of 0.05C (1C = 0.2 A g−1). (b) Cyclic voltammogram (CV) of the Nb2O5 samples at a sweep rate of 0.1 mV s−1. (c) Galvanostatic intermittent titration technique (GITT) curves. (d) DLiL−2 value, relative Li+ ions diffusion coefficients of Nb2O5, calculated from GITT curves (c). (e) Rate performance of the Nb2O5 samples at various current densities. (f) Cycling performance of d-H-Nb2O5 at 10C, 30C and 80C. | ||
The cyclic voltammetry (CV) curve of T-Nb2O5 shows a rectangular profile, a typical feature of pseudocapacitive behavior (Fig. 3b). Both H-Nb2O5 and d-H-Nb2O5 have a sharp redox peak at around 1.7 V, corresponding to a two-phase electrochemical process, while the intensity of redox peak for T-Nb2O5 is much weaker. The CV curves of d-H-Nb2O5 are slightly different from those of H-Nb2O5 in terms of peak position and intensity. The sharper and more symmetric redox peak with smaller polarization for the d-H-Nb2O5 sample indicates its favorable two-phase process kinetics for both lithium insertion and extraction.15 It has been reported that the cation disorder in the ternary niobium oxides such as TiNbO and NbWO could broaden the electrochemical feature relative to Nb2O5.4,14,16 Defects may also contribute to this phenomenon, as a “pseudo” redox peak at 1.2 V for the defective d-H-Nb2O5, which is also a typical feature for their dQ/dV plots (Fig. S10, ESI†). This peak further characterized by CV analysis did not show strong dependence on sweep rate while the other peaks exhibit either surface- or bulk-controlled electrochemical kinetics (Fig. S11, ESI†). For both H-Nb2O5 phases, the main redox peak related to the two-phase process is controlled by solid-state diffusion; their redox reaction at several shoulder peaks is limited by the surface process. For T-Nb2O5, the redox reaction is mainly associated with surface diffusion limitation.
Galvanostatic intermittent titration technique (GITT) was performed by minor current impulsion with long-time relaxation (Fig. 3c and Fig. S12, ESI†). The well overlapped Vcc (voltage at the beginning of relaxation) and Voc (voltage at the end of relaxation) represent good energy efficiency, which means that most of the lithium imposed could migrate from the surface to bulk to reach an equilibrium state internally by diffusion. For T-Nb2O5, the separation of the Vcc and Voc curves in the low potential window indicates decreased lithium diffusion kinetics (Fig. 3c and Fig. S12a, ESI†).40 The unchanged Voc and steady separated voltage trend suggests that T-Nb2O5 is overlithiated. For both H-Nb2O5 phases, hysteresis occurs between Vcc and Voc below 1.2 V and more lithium insertion could still be achieved by decreasing potential. The same discharge profile as in Fig. 3a was depicted by this impulsion relaxation operation as well; d-H-Nb2O5 still delivered higher intercalation capacity than H-Nb2O5. The different electrochemical behavior of H-Nb2O5 and d-H-Nb2O5 at near the equilibrium state (Fig. 3a–c) suggests this distinction comes from the intrinsic structure–property relationship rather than the effect of particle size or crystal orientation. Apparent diffusion coefficients (DLiL−2) of these materials were extracted from the GITT curve (Fig. 3d). There were no significant changes in DLiL−2 for T-Nb2O5 with lithium intercalation above 1.2 V. The DLiL−2 decreased rapidly below 1.2 V, indicating sluggish diffusion kinetics in the lithium-stuffed structure. Both H-Nb2O5 phases show a similar coefficient trend in the entire voltage region; the d-H-Nb2O5 shows a slightly higher DLiL−2 value than H-Nb2O5 at 1.2 V; their diffusion coefficients have not changed significantly besides rapid decay in the two-phase region and at around 1.2 V (Fig. S12d, ESI†). The reason for the latter decay is the same as that in T-Nb2O5; that is, an increasing lithium concentration hinders further continuous insertion. The decay in two-phase region however could not truly represent slower kinetics since Fick's law of diffusion for calculating DLiL−2 is not appropriate for a two-phase process.40
The higher intrinsic intercalation capacity and good ionic conductivity led to impressive rate performance of d-H-Nb2O5 (Fig. 3e). It exhibits high discharge capacity of 230 mA h g−1 at 0.5C, and 125 mA h g−1 at a rate as high as 50C, better than that nano-sized T-Nb2O5. In order to eliminate the effect of particle size on the electrochemical properties, H-Nb2O5 with the same particle size as d-H-Nb2O5 was prepared using a mild milling method (Fig. S13, ESI†). The phase purity and local structure remained the same after the downscaling process. The smaller particle size does not enhance the specific capacity of H-Nb2O5 significantly by shortening the lithium diffusion distance (Fig. S13f, ESI†). This result indicates that regardless of the particle size the extra amount of charge is stored in the Wadsley defects, which is a planar defect structure appearing not only on the surface but also in the bulk of d-H-Nb2O5. The d-H-Nb2O5 exhibits the electrochemical performance comparable to the state-of-the-art Nb-based compounds (Fig. S14, ESI†).4,14,15,41 As can be seen in Fig. 3f, d-H-Nb2O5 shows good cycling stability at a high rate. It maintained a specific capacity of 138 mA h g−1 at 10C (2 A g−1) and 123 mA h g−1 at 30C (6 A g−1) after 2000 cycles. Retention of almost 90% could be achieved for the cell running at 80C (a current density of 16 A g−1) after 4000 cycles, delivering capacity of 89 mA h g−1. The d-h-Nb2O5 retains the overall pattern of both XRD and XAS after 2000 cycles at 10C (Fig. S15, ESI†), indicating that the crystal structure is stable under the cycling conditions. In contrast, T-Nb2O5 degraded rapidly and lost 45% capacity after 300 cycles at 10C (Fig. S16, ESI†). This higher capacity without obvious stability loss is achieved by the cutoff voltage of 1.0 V (Fig. S17, ESI†), providing a higher energy density of the full cell when H-Nb2O5 is used as the anode. There is still a trade-off between mass loading and performance at a high rate, which is a common problem of both anodes and cathodes in the battery system (Fig. S18, ESI†). The appropriate design of the electrode could alleviate this problem of diffusion limitation in thicker electrodes in the future.42,43
The evolution of the valence of niobium and its local environment during the cycling
To get deep insight into the origin of high electrochemical performance of d-H-Nb2O5, operando X-ray absorption near edge structure (XANES) measurements were performed for d-H-Nb2O5 and T-Nb2O5 to measure the Nb average valence information and local environment variation during the charge–discharge process. First, operando Nb K-edge XANES was collected under two different C-rate conditions (0.25C and 10C) to elucidate the electronic structure change of both Nb2O5 phases during the redox reaction (Fig. S19, ESI†). As shown in Fig. 4a, b and Fig. S20 (ESI†), the absorption edge for all Nb2O5 phases exhibited a continuous chemical shift to lower values as Nb5+ is reduced during the lithiation process. The sharpened white-line peak during the discharge process suggests a less distorted Nb–O coordination environment.44 The d-H-Nb2O5 shows a wider variation range towards lower energies than T-Nb2O5 at both 0.25C (Fig. 4) and 10C (Fig. S20, ESI†). After delithiation at 3 V, T-Nb2O5 exhibited negatively shifted absorption edge compared with pristine spectra (Fig. 4a), resulting in a lower Coulombic efficiency. In contrast, better reversible redox behavior is observed for d-H-Nb2O5, which is interpreted by its fully overlapped edge after cycling (Fig. 4b). To elucidate real-time redox states during the electrochemical reaction, differentiated XANES spectra (Xi–Xpristine) were plotted in a 2D-contour map (Fig. 4c and d). In the main edge, more significant intensity change during lithiation was confirmed for d-H-Nb2O5. The disappearance of the pre-edge at approximately 18990 eV for both Nb2O5 phases during lithiation (Fig. 4a and b) correlates with a decreased intensity in the corresponding region in the 2D-contour map (Fig. 4c and d), implying that the local octahedral symmetry of Nb2O5 increases due to alleviation of the second-order Jahn–Teller (SOJT) distortion when lithium intercalates.4,15 In the case of 10C, pre-cycling activation of the electrode at 0.25C was performed followed by collection of operando XANES data at 10C. The changes during testing were similar to those of the case at 0.25C. The oxidation state changes of niobium during Li insertion–extraction was calibrated by the shift of the absorption edge of niobium reference (Fig. 4e, f and Fig. S21, ESI†). The pristine oxidation state of niobium in all Nb2O5 electrodes is slightly lower than Nb5+ due to self-discharge and the valence varies continuously on lithium intercalation. Both Nb2O5 phases show a linear relationship between the valence of the niobium and lithium contents. More than one lithium per niobium was inserted into the d-H-Nb2O5 at 0.25C, resulting in a capacity of over 200 mA h g−1, which is higher than that based on one electron-reduction. The accommodation of more lithium accounts for the higher intercalation capacity of d-H-Nb2O5 than that of T-Nb2O5 at both low and high rate (Fig. 4e and f). In addition, this lithium (de)intercalation in the shear structure of d-H-Nb2O5 occur more reversibly, as revealed by a closer niobium valence to that of the pristine oxidation state after cycling (Fig. 4e). The result here is consistent with our previous electrochemical discussion that d-H-Nb2O5 shows a higher capacity and better rate performance.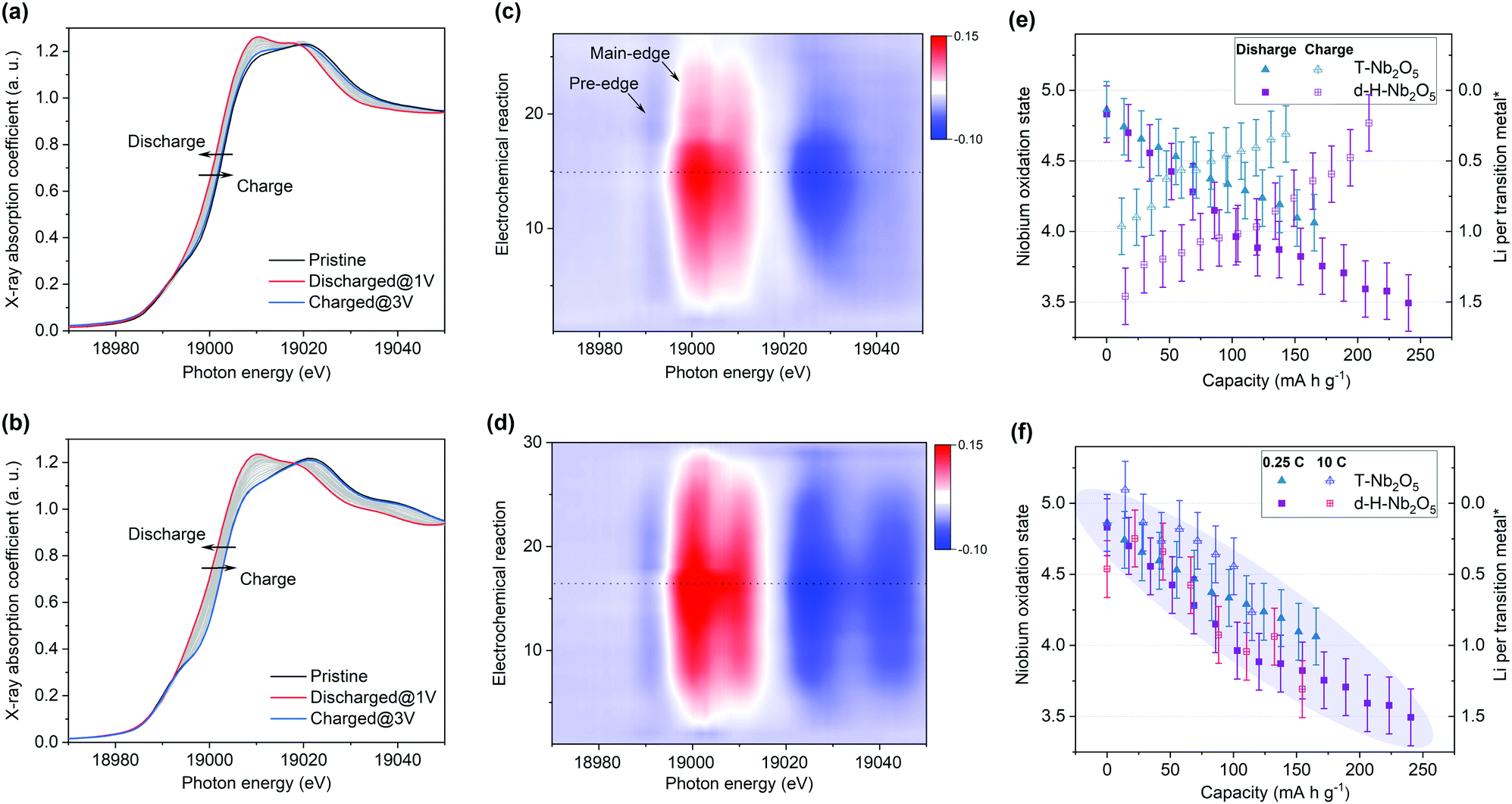 | ||
| Fig. 4 Operando Nb K-edge X-ray absorption near edge structure (XANES) profiles for (a) T-Nb2O5 and (b) d-H-Nb2O5 at 0.25C. 2D contour plot of operando XANES for (c) T-Nb2O5 and (d) d-H-Nb2O5 during cycling. The dashed line in the (c) and (d) is to distinguish discharge and charge process. Average Nb oxidation state (determined from the shift of XANES spectra) as a function of electrochemical reaction at (e) 0.25C rate and at (f) 10C rate for T-Nb2O5 and d-H-Nb2O5. Only the discharge process is shown in (f). Lithium per niobium atom on the right y-axis of (e and f) was calculated by niobium oxidation state (left y-axis) correspondingly. The error bars of ±0.2 here represents possible deviation due to XANES resolution. | ||
The local structure and coordination environment of Nb2O5 during lithiation was further analyzed by radial distributional functions (RDFs) which were obtained from operando EXAFS spectra via the Fourier transform method (Fig. 5 and Fig. S22, ESI†). Considering the complex coordination environment for the two Nb2O5 phases, only qualitative analysis of EXAFS without local structure fitting was discussed. The peaks at ∼1.75 Å and >3.00 Å (Fig. 5) correspond to the interatomic distances Nb–O and Nb–Nb or Nb–O–Nb, respectively. In the pristine state, the higher intensity of Nb–O peak with respect to Nb–Nb peak was observed in T-Nb2O5 while in contrast, the Nb–Nb peak intensity is much higher than Nb–O peak in the shear structure (Fig. 5a and c). Previous research explained the higher Nb–Nb peak intensity in M-Nb2O5 (tetragonal) to be a result of its two-dimensional (2D) planar arrangement of Nb atoms.27 However, the Nb atom arrangements of M-Nb2O5 and H-Nb2O5 perpendicular to the shear plane are quite different but with the same EXAFS phenomenon. The stronger Nb–Nb peak intensity in shear structure could be due to less shielding effect of oxygen on Nb–Nb interaction in the a–c plane. In other words, the oxygen in the dense-packed 4 h atomic layer of T-Nb2O5 has a larger shielding effect on the Nb–Nb scattering.18
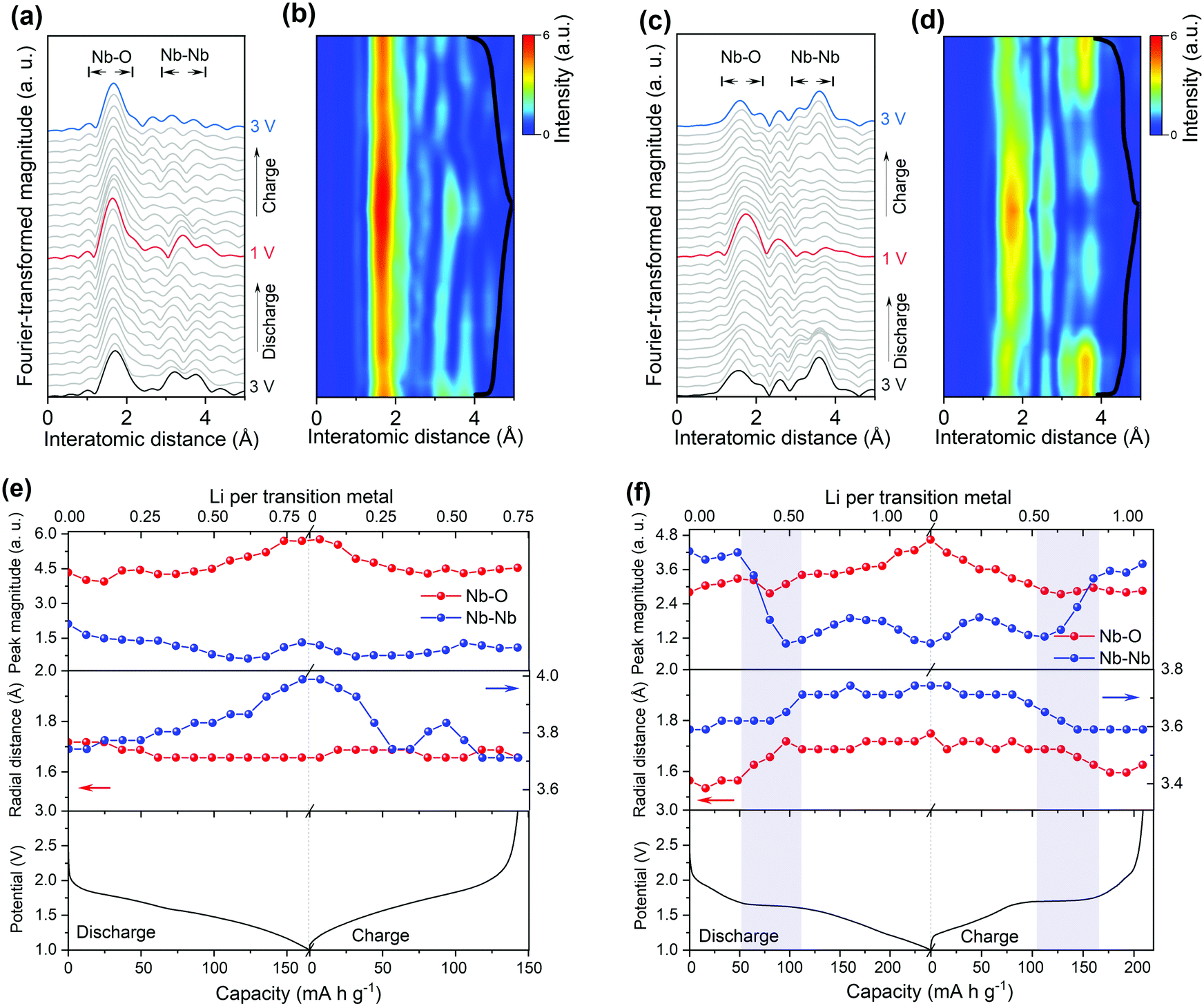 | ||
| Fig. 5 Radial distribution function (RDF) of Nb K-edges operando EXAFS spectra and their corresponding 2D contour plot for the electrode of (a and b) T-Nb2O5 and (c and d) d-H-Nb2O5 at 0.25C. The electrochemical discharge/charge profile (black curve) of operando cell was overlaid in the right side of contour plot. (e and f) Peak magnitudes and radial distance derived from RDF (a and c) as functions of electrochemical reaction. | ||
Upon lithium insertion, Nb–O peak intensity of both T-Nb2O5 and d-H-Nb2O5 increases (Fig. 5b, d–f). The intercalated lithium reduces the valence of Nb from Nb5+ to Nb4+ (or lower), alleviating the Jahn–Teller effect of distorted NbOx resulting in a less disordered Nb–O local structure.4,15 The uniform Nb–O distance leads to this increase in peak intensity.45–47 During the lithiation in T-Nb2O5, there is only change in the intensity of first Nb–O shell without being distance shifted (Fig. 5a), even at a high rate (Fig. S22, ESI†). A shoulder peak at 2.0 Å gradually appears at the same time and disappears after full delithiation. This is explained by the fact that 2D interplane lithium diffusion path of T-Nb2O5 has less effect on niobium coordination in the a–b plane but indeed enlarge the Nb–O distance along c axis due to Nb4+ cation. The results are consistent with in situ XRD of T-Nb2O5 that show a blue shift of (001) and (002) diffraction peaks upon lithiation due to the increase of the interplanar distance.30 On the other hand, its Nb–(O–)Nb distance increases upon Li intercalation (Fig. 5e). In particular, for Nb–Nb interaction of corner-shared NbOx along c axis, the corresponding FT intensity at 3.8 Å almost disappears due to the shielding effect of interlayer lithium (Fig. 5a).27 After one cycle, the Nb–Nb region shows notable difference with the pristine state. The weaker intensity of Nb–Nb peak suggests that lithium may remain in the interlayer which could also be responsible for the non-overlapping XANES spectra.
For the shear structure, the intensity of Nb–Nb dramatically decreases once the discharge reaches a plateau (Fig. 5d, f and Fig. S22, ESI†). Lithium is inserted into the Nb–Nb interlayer space between corner-shared NbO6 in the b axis direction coordinating with the oxygen site. From the view towards the a–c plane down the tunnels, the voids between corner-shared NbO6 in the blocks may be one-dimensional lithium transport pathways. Previous studies on TiNb2O7 also show that lithium diffusion is feasible down the tunnels and across the tunnels and is stored in the (001) lattice plane.16,48 Lithium insertion increases the shielding effect of both Nb–Nb corner-sharing interactions, causing the loss of their FT intensity. Simultaneously, both Nb–O and Nb–(O–)Nb distances shift to a higher distance in the plateau region and shift back in the beginning of the two-phase region of the charge process (Fig. 5f). The nonlinear increase of average interatomic distance and obvious variation of Nb–Nb peak intensity indicate the lithium insertion of the shear structure is quite different from the 2 D path in T-Nb2O5, suggesting that notable structure distortion occurs in the two-phase region. There could be a solid solution reaction before and after this phase-transition process that does not influence the local structure upon lithiation. Operando EXAFS measurements at 10C show that T-Nb2O5 and d-H-Nb2O5 show the same trend as the case at 0.25C in terms of peak shift and intensity variation (Fig. S22, ESI†). Despite the large deformation during discharge, the RDF returns to its original configuration for both low rate and high rate, indicating that the d-H-Nb2O5 structure is highly reversible. Our previous operando Raman validates 2D facile Li transport in T-Nb2O5.19 The operando Raman spectroscopic evolution of T-Nb2O5 upon lithiation showed intensity decrease of the NbOx stretching mode and a band splitting of the Nb–O–Nb bending mode though the latter could not be observed clearly in current ex situ experiments (Fig. S23a and b, ESI†). The ex situ Raman spectra show the structure–property relationship of the shear structure upon the incorporation of Li which is different from that in T-Nb2O5. The more complex Raman spectra of the shear structure reflect that its local structure and bond order are much more complex than in T-Nb2O5. In contrast to T-Nb2O5 in which the Raman spectra almost remain the same above 2.0 V, the stretching mode of H-Nb2O5 gradually decreases above 2.0 V and finally vanishes at 1.80 V. This shows that lithium is strongly bound in the oxygen site even in the dilute concentration, influencing the stretching energy between niobium and oxygen. The initial absorption may correspond to the first redox peak in CV curve. For the shear structure, the Nb–O–Nb bending mode varies through the discharge process and the change is more significant than in T-Nb2O5. In particular, at high lithium concentration, where the two-phase process occurs, the profile of pristine Nb–O–Nb bending mode becomes broader and a new peak located at 330 cm−1 appears at 1.5 V, corresponding to the bending mode of Nb–O–Li.15,49 The continued lithiation increases the intensity of this peak without further change in the Raman signal shape. The above EXAFS and Raman analysis show that the local environment of the shear structure changes significantly at the beginning of the two-phase reaction. At dilute lithium concentration, the interaction between lithium and oxygen is strong already. Afterwards high lithium concentration induces structure distortion in the two-phase region, which is characterized by the increase of Nb–O and Nb–Nb distances accompanied by the decrease of the Nb–Nb peak intensity in EXAFS and obvious variation of Nb–O–metal bending mode in the Raman spectrum.
Charge storage and lithium diffusion in different phases of Nb2O5
The above characterization of T-Nb2O5, d-H-Nb2O5 and H-Nb2O5 shows that the lithium storage capability and structural reversibility upon lithiation is highly different for the three phases. DFT-based computation was applied to examine the electrochemical performance of these niobium-based materials (Fig. 6 and Fig. S24, ESI†). Fig. 6 shows the computational models of lithiated Nb2O5, including T-Nb2O5, H-Nb2O5 and planar-defect d-H-Nb2O5. Two Nb2O5 polymorphs derived from two dominant defects as shown in Fig. 2h are studied, which are denoted as d1-H-Nb2O5 and d2-H-Nb2O5. Li ions preferentially occupy the sites between alternating oxygen to bridge them in the lattices. Accordingly, lithiated Li3Nb2O5 with Li sites fully filled are modeled for these charged samples. Chemical and physical properties of these niobium-based materials are extensively examined (Fig. 6c and Table 1). The ratio of Li/Nb (= 1.5) for the lithiated phases is the upper limit observed in the discharge–charge testing and XANES analysis (Fig. 3a and 4e).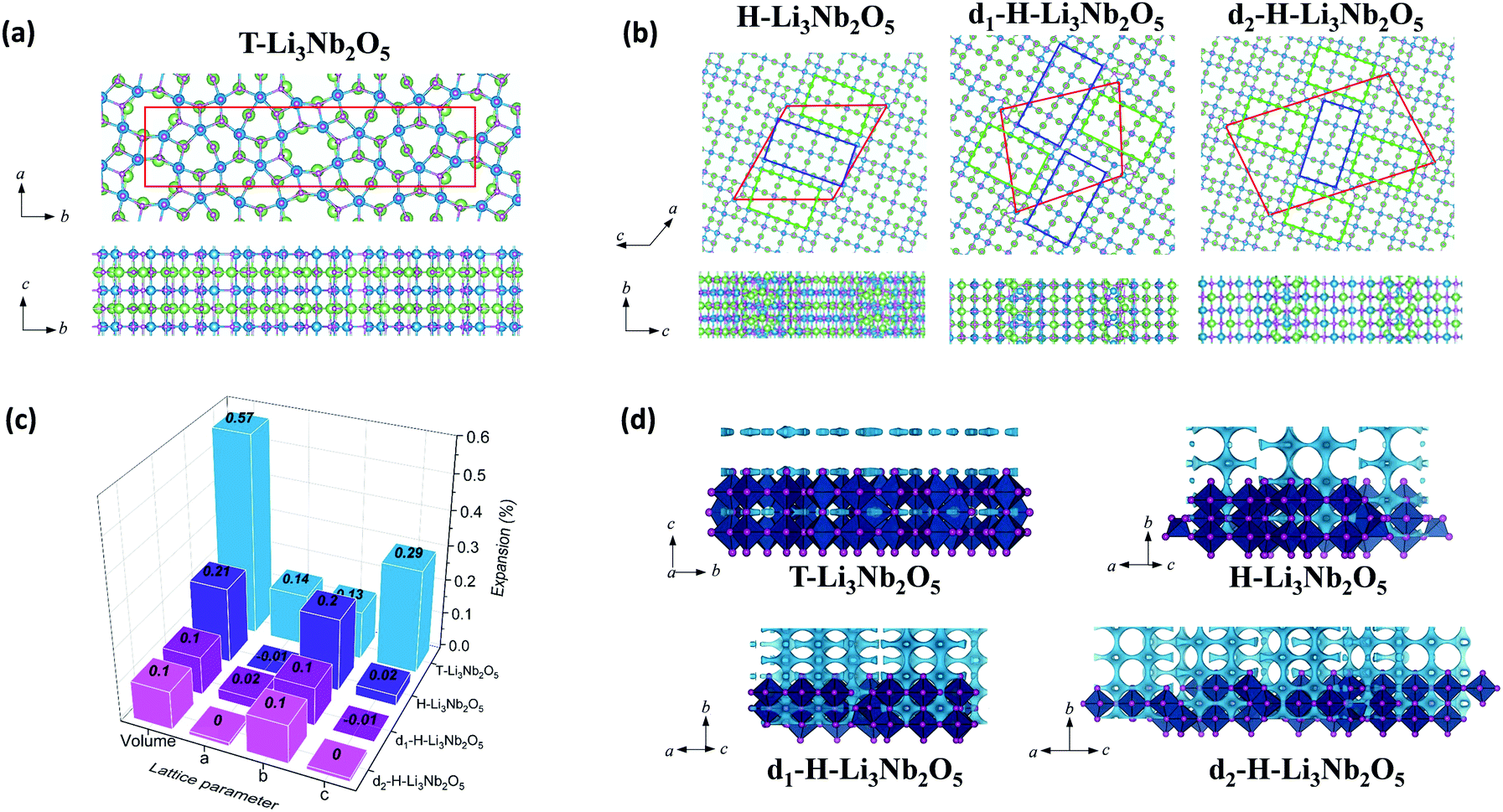 | ||
| Fig. 6 Top and side views of the lithiated phases, including (a) T-Nb2O5 and (b) H-Nb2O5, d1-H-Nb2O5 and d2-H-Nb2O5. The boxes with red lines indicate unit cells; green and blue rectangles indicate 3 × 4 and 3 × 5 blocks, observed in TEM (Fig. 2e and h); Nb, O and Li are represented with blue, purple and green balls, respectively. (c) The lattice variation of the lithiated phases per Nb2O5 unit. (d) BVS mapping of T-Nb2O5 and H-Nb2O5 with and without defects. The light blue isosurface illustrate the possible dimensionality and directions of lithium pathways in the structure. The isovalue of 1.5 eV above Emin was set for all the structure to put them on the same scale to visualize the lithium diffusion. | ||
| T-Nb2O5 | H-Nb2O5 | d1-H-Nb2O5 | d2-H-Nb2O5 | |
|---|---|---|---|---|
| Cell volume (Å3) | 92.53 | 102.49 | 102.40 | 102.44 |
| Density (g cm−3) | 4.79 | 4.32 | 4.33 | 4.32 |
| E ads(Li+) | −2.35 | −2.38 | −2.41 | −2.39 |
Among the unlithiated samples, T-Nb2O5 has the smallest cell volume per Nb2O5 and the highest density, indicating its densely packed structure. All H-Nb2O5 phases, including the defected derivatives, have similar volume and density (Table 1). Their larger volume compared to that of T-Nb2O5 is due to the less dense structure based on the corner-sharing tetrahedral structure, resulting in more space and easier accommodation for Li ions. In T-Li3Nb2O5, Li ions are located only in the 4 g layer, parallel to a–b plane whereas in H-Li3Nb2O5, d1-H-Li3Nb2O5 and d2-H-Li3Nb2O5 the ions intercalate into different layers, perpendicular to a–c plane (Fig. 6b). Electrochemical testing showed that the performance of T-Nb2O5 undergoes significant degradation at a high rate under deep discharge (Fig. S16, ESI†). Even though corner-shared NbOx along c axis performed like pillar hosting the 2D lithium channel, the increasing amount of intercalated lithium may impedes this facile diffusion, as evidenced by lower DLi+ under 1.2 V in GITT results (Fig. 3d and Fig. S12a, ESI†). The DFT results here also reveal that its layered distribution of Li ions leads to the larger volume expansion (per Li ion) for lithiated T-Li3Nb2O5 (0.57%). On the other hand, shear planes alleviate the expansion from lithiation and shows smaller expansion for lithiated H-Li3Nb2O5 (0.21%). Planar-defect d1-H-Li3Nb2O5 and d2-H-Li3Nb2O5 have staggered 3 × 4 and 3 × 5 blocks (green and blue rectangles, respectively, in Fig. 6b), perpendicular to each other, which further relieves the tension and results the smallest expansion (0.10%, Fig. 6c), concentrated along b axis. DFT results indicate that the volume expansion of both H-Nb2O5 phases shows strong anisotropy, i.e. strong expansion in the b direction and absent or slight contraction of the a–c plane. Ex situ XRD for d-H-Nb2O5 is consistent with this conclusion, as hinted by the red shift of the (405) and (402) planes, and the blue shift of (110) plane upon lithiation in the two-phase region (Fig. S23c and d, ESI†). This anisotropic lattice variation was also observed in the neutron diffraction analysis and recent SAED analysis.12,50,51 A similar block contraction was observed in XRD analysis of its ternary counterpart, Nb16W5O55, though the structural evolution is more complex.4 The shear plane with edge-shared NbO6 performed like the strap of a wooden bucket, alleviating structure expansion and helping the structure recover to the pristine state. The averaged adsorption energy of Li ions, Eads(Li+), was calculated for these lithiated materials. It was found that Li ions strongly adsorb on H-phase Nb2O5 and show the strongest adsorption on its defected derivatives (Table 1). The lithium diffusion transport in these Nb2O5 was semi-quantitatively analyzed by bond-valence sum (BVS) mapping (Fig. 6d). The light blue isosurface shows a 2D lithium pathway in T-Nb2O5, which is consistent with EXAFS analysis and previous results. For the shear structure, BVS mapping predicted the lithium migration in dilute lithium concentration before structure distortion. Regardless of disorder in the a–c plane, the lithium ions diffuse down the tunnels in every block along the b axis while the shear plane separates the lithium migration in the a–c plane. The anisotropy in the crystal growth of the shear structure, characterized by TEM confirms the long-range order in the b axis and disorder in the a–c plane, enabling its fast lithium transport. Furthermore, the electronic conductivity of both Nb2O5 is enhanced after lithium intercalation, as indicated by the change in color of the Nb2O5 pellet from white to dark during lithiation (Fig. S25, ESI†). DFT calculation of density of states (DOS) suggests that H-Nb2O5 and its derivatives have similar electronic conductivity, slightly higher than that of T-Nb2O5 (Fig. S26, ESI†). Upon lithiation, both Nb2O5 phases show enhanced conductivity, since the Fermi energy is located within the conduction band.
Based on the results by computation, planar-defect d1-H-Nb2O5 and d2-H-Nb2O5 have open structures and form the most stable lithiated d1-H-Li3Nb2O5 and d2-H-Li3Nb2O5 with the smallest volume expansion and strongest Eads(Li+), thus, resulting in optimal long-term stability and higher intercalation capacity. In contrast, T-Nb2O5 with a dense structure shows the weakest Eads(Li+) for its lithiated phase and the largest volume expansion, leading to smaller specific capacity and fast degradation upon cycling (Fig. 3e and Fig. S16, ESI†).
Conclusions
In summary, we have systematically studied the impact of planar defects on the electrochemistry of H-Nb2O5. In the defect region, stronger lithium adsorption and smaller volume change during lithiation due to the rearrangement of the building blocks of the shear structure increased lithium storage capability. By engineering the planar defects through optimization of the calcination temperature, micron-sized H-Nb2O5 achieved much better performance than nano-sized T-Nb2O5. The variation in the local environment of H-Nb2O5 upon lithiation was investigated and compared with that of T-Nb2O5. A stronger interaction between lithium and oxygen leads to an obvious distortion of the shear structure in the two-phase region during intercalation, as evidenced by the increase in Nb–O and Nb–Nb distance and the decrease in Nb–Nb peak intensity in EXAFS, and obvious variation in Nb–O–metal bending mode in the Raman spectra. DFT and ex situ XRD reveal that this distortion results in an anisotropic change in the cell volume, i.e. contraction in the a–c plane constrained by the shear plane, and expansion in the b direction along the lithium diffusion pathway. Compared with T-Nb2O5, the d-H-Nb2O5 structure is more reversible upon lithiation, which shows better stability at a high cycling rate. This work not only reports a highly promising anode for fast lithium storage, but also offers new insight into the lithium intercalation mechanism in the shear structure and proposes a new strategy to enhance battery performance that may be applicable to other similar structures with Wadsley planar defects for different applications.Author contributions
T. L. and G. N. contributed equally to this work. B. Z. and M. L. conceived the project. B. Z., Y. S. and M. L. supervised the research. T. L. performed most of the experiments and analyzed the experimental data with the help of L. S., Z. L., G. N., W. Z., T. Y. and P. J. Y. D. performed TEM and analyzed the data. G. N. and M. K. contributed to collecting and analyzing the XAFS data and co-wrote the paper. K. L., J. W. and M. A. performed computational studies and analyzed the computational data. All the authors discussed the results. T. L., J. W. and G. N. wrote the manuscript with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CBMM and US National Science Foundation (DMR-1742828). T. L. acknowledges the financial support of a scholarship from the China Scholarship Council (CSC).Notes and references
- Y. Liu, Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef.
- S. Ahmed, I. Bloom, A. N. Jansen, T. Tanim, E. J. Dufek, A. Pesaran, A. Burnham, R. B. Carlson, F. Dias, K. Hardy, M. Keyser, C. Kreuzer, A. Markel, A. Meintz, C. Michelbacher, M. Mohanpurkar, P. A. Nelson, D. C. Robertson, D. Scoffield, M. Shirk, T. Stephens, R. Vijayagopal and J. Zhang, J. Power Sources, 2017, 367, 250–262 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- K. J. Griffith, K. M. Wiaderek, G. Cibin, L. E. Marbella and C. P. Grey, Nature, 2018, 559, 556–563 CrossRef CAS PubMed.
- E. Ferg, R. J. Gummow, A. de Kock and M. M. Thackeray, J. Electrochem. Soc., 1994, 141, L147–L150 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- H. Liu, Z. Zhu, Q. Yan, S. Yu, X. He, Y. Chen, R. Zhang, L. Ma, T. Liu, M. Li, R. Lin, Y. Chen, Y. Li, X. Xing, Y. Choi, L. Gao, H. S.-y. Cho, K. An, J. Feng, R. Kostecki, K. Amine, T. Wu, J. Lu, H. L. Xin, S. P. Ong and P. Liu, Nature, 2020, 585, 63–67 CrossRef CAS PubMed.
- B. Zhao, R. Ran, M. Liu and Z. Shao, Mater. Sci. Eng., R, 2015, 98, 1–71 CrossRef.
- K. J. Griffith, A. C. Forse, J. M. Griffin and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 8888–8899 CrossRef CAS PubMed.
- J. Come, V. Augustyn, J. W. Kim, P. Rozier, P.-L. Taberna, P. Gogotsi, J. W. Long, B. Dunn and P. Simon, J. Electrochem. Soc., 2014, 161, A718–A725 CrossRef CAS.
- J. W. Kim, V. Augustyn and B. Dunn, Adv. Energy Mater., 2012, 2, 141–148 CrossRef CAS.
- Z. Song, H. Li, W. Liu, H. Zhang, J. Yan, Y. Tang, J. Huang, H. Zhang and X. Li, Adv. Mater., 2020, 32, 2001001 CrossRef CAS PubMed.
- Ö. Budak, M. Geißler, D. Becker, A. Kruth, A. Quade, R. Haberkorn, G. Kickelbick, B. J. M. Etzold and V. Presser, ACS Appl. Energy Mater., 2020, 3, 4275–4285 CrossRef.
- K. J. Griffith and C. P. Grey, Chem. Mater., 2020, 32, 3860–3868 CrossRef CAS.
- Y. Yang, H. Zhu, J. Xiao, H. Geng, Y. Zhang, J. Zhao, G. Li, X.-L. Wang, C. C. Li and Q. Liu, Adv. Mater., 2020, 32, 1905295 CrossRef CAS PubMed.
- K. J. Griffith, I. D. Seymour, M. A. Hope, M. M. Butala, L. K. Lamontagne, M. B. Preefer, C. P. Koçer, G. Henkelman, A. J. Morris, M. J. Cliffe, S. E. Dutton and C. P. Grey, J. Am. Chem. Soc., 2019, 141, 16706–16725 CrossRef CAS PubMed.
- L. Hu, L. Luo, L. Tang, C. Lin, R. Li and Y. Chen, J. Mater. Chem. A, 2018, 6, 9799–9815 RSC.
- T. Yuan, L. Soule, B. Zhao, J. Zou, J. Yang, M. Liu and S. Zheng, Energy Fuels, 2020, 34, 13321–13334 CrossRef CAS.
- D. Chen, J.-H. Wang, T.-F. Chou, B. Zhao, M. A. El-Sayed and M. Liu, J. Am. Chem. Soc., 2017, 139, 7071–7081 CrossRef CAS PubMed.
- C. P. Koçer, K. J. Griffith, C. P. Grey and A. J. Morris, J. Am. Chem. Soc., 2019, 141, 15121–15134 CrossRef PubMed.
- C. P. Koçer, K. J. Griffith, C. P. Grey and A. J. Morris, Chem. Mater., 2020, 32, 3980–3989 CrossRef PubMed.
- R. J. Cava, D. W. Murphy and S. M. Zahurak, J. Electrochem. Soc., 1983, 130, 2345–2351 CrossRef CAS.
- K. Kato and S. Tamura, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 673–677 CrossRef.
- B. M. Gatehouse and A. D. Wadsley, Acta Crystallogr., 1964, 17, 1545–1554 CrossRef CAS.
- N. Kumagai and K. Tanno, Denki Kagaku oyobi Kogyo Butsuri Kagaku, 1982, 50, 704–707 CrossRef CAS.
- N. Kumagai, K. Tanno, T. Nakajima and N. Watanabe, Electrochim. Acta, 1983, 28, 17–22 CrossRef CAS.
- R. Kodama, Y. Terada, I. Nakai, S. Komaba and N. Kumagai, J. Electrochem. Soc., 2006, 153, A583 CrossRef CAS.
- N. Kumagai, Y. Koishikawa, S. Komaba and N. Koshiba, J. Electrochem. Soc., 1999, 146, 3203–3210 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef CAS PubMed.
- A. A. Lubimtsev, P. R. C. Kent, B. G. Sumpter and P. Ganesh, J. Mater. Chem. A, 2013, 1, 14951–14956 RSC.
- C.-P. Liu, F. Zhou and V. Ozolins, http://meetings.aps.org/link/BAPS.2012.MAR.B26.3, 2012.
- K. J. Griffith, PhD thesis, University of Cambridge, 2018.
- S. Andersson, J. Solid State Chem., 1970, 1, iv–vi CrossRef.
- E. I. Ko and J. G. Weissman, Catal. Today, 1990, 8, 27–36 CrossRef CAS.
- J. M. Jehng and I. E. Wachs, Chem. Mater., 1991, 3, 100–107 CrossRef CAS.
- A. A. McConnell, J. S. Aderson and C. N. R. Rao, Spectrochim. Acta, Part A, 1976, 32, 1067–1076 CrossRef.
- S. Iijima, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 18–24 CrossRef CAS.
- F. E. Rohrer and A.-K. Larsson, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 780–784 CrossRef PubMed.
- K. J. Griffith, A. Senyshyn and C. P. Grey, Inorg. Chem., 2017, 56, 4002–4010 CrossRef CAS PubMed.
- M. B. Preefer, M. Saber, Q. Wei, N. H. Bashian, J. D. Bocarsly, W. Zhang, G. Lee, J. Milam-Guerrero, E. S. Howard, R. C. Vincent, B. C. Melot, A. Van der Ven, R. Seshadri and B. S. Dunn, Chem. Mater., 2020, 32, 4553–4563 CrossRef CAS.
- H. Sun, L. Mei, J. Liang, Z. Zhao, C. Lee, H. Fei, M. Ding, J. Lau, M. Li, C. Wang, X. Xu, G. Hao, B. Papandrea, I. Shakir, B. Dunn, Y. Huang and X. Duan, Science, 2017, 356, 599–604 CrossRef CAS PubMed.
- T. Tian, L.-L. Lu, Y.-C. Yin, F. Li, T.-W. Zhang, Y.-H. Song, Y.-H. Tan and H.-B. Yao, Adv. Funct. Mater., 2021, 31, 2007419 CrossRef CAS.
- B. Guo, X. Yu, X.-G. Sun, M. Chi, Z.-A. Qiao, J. Liu, Y.-S. Hu, X.-Q. Yang, J. B. Goodenough and S. Dai, Energy Environ. Sci., 2014, 7, 2220–2226 RSC.
- H. Koga, L. Croguennec, M. Ménétrier, P. Mannessiez, F. Weill, C. Delmas and S. Belin, J. Phys. Chem. C, 2014, 118, 5700–5709 CrossRef CAS.
- J. Sottmann, R. Homs-Regojo, D. S. Wragg, H. Fjellvag, S. Margadonna and H. Emerich, J. Appl. Crystallogr., 2016, 49, 1972–1981 CrossRef CAS.
- Y. Iwasawa, K. Asakura and M. Tada, XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, Springer, Cham, Switzerland, 2017 Search PubMed.
- X. Lu, Z. Jian, Z. Fang, L. Gu, Y.-S. Hu, W. Chen, Z. Wang and L. Chen, Energy Environ. Sci., 2011, 4, 2638–2644 RSC.
- L. J. Hardwick, M. Holzapfel, P. Novák, L. Dupont and E. Baudrin, Electrochim. Acta, 2007, 52, 5357–5367 CrossRef CAS.
- M. Catti and M. R. Ghaani, Phys. Chem. Chem. Phys., 2014, 16, 1385–1392 RSC.
- Y. Yang and J. Zhao, Adv. Sci., 2021, 8, 2004855 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, supplementary figures, supplementary tables, and supplementary references. See DOI: 10.1039/d1ee02664j |
| ‡ T. L. and G. N. contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |