
Selective methane electrosynthesis enabled by a hydrophobic carbon coated copper core–shell architecture†
Xin Yu
Zhang‡
a,
Wen Jing
Li‡
a,
Xue Feng
Wu
a,
Yuan Wei
Liu
a,
Jiacheng
Chen
b,
Minhui
Zhu
 b,
Hai Yang
Yuan
*a,
Sheng
Dai
c,
Hai Feng
Wang
d,
Zheng
Jiang
e,
Peng Fei
Liu
*a and
Hua Gui
Yang
*a
b,
Hai Yang
Yuan
*a,
Sheng
Dai
c,
Hai Feng
Wang
d,
Zheng
Jiang
e,
Peng Fei
Liu
*a and
Hua Gui
Yang
*a
aKey Laboratory for Ultrafine Materials of Ministry of Education, Shanghai Engineering Research Center of Hierarchical Nanomaterials, School of Materials Science and Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, China. E-mail: hgyang@ecust.edu.cn; pfliu@ecust.edu.cn; hyyuan@ecust.edu.cn
bState Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
cKey Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, China
dKey Laboratory for Advanced Materials, Centre for Computational Chemistry and Research Institute of Industrial Catalysis, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, China
eShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, 239 Zhangheng Road, Shanghai, 201204, China
First published on 1st December 2021
Abstract
The electrosynthesis of valuable chemicals via carbon dioxide reduction reaction (CO2RR) has provided a promising way to address global energy and sustainability problems. However, the selectivity and activity of deep-reduction products (DRPs) still remain as big challenges. Here, a copper–carbon-based catalyst with a hydrophobic core–shell architecture has been constructed and was found to exhibit excellent DRPs of methane generation with a faradaic efficiency of 81 ± 3% in a neutral medium and a maximum partial current density of −434 mA cm−2 in a flow cell configuration, which is among the best of CO2-to-CH4 electrocatalysts. Density functional theory calculations suggest that the hydrophobic structure decreasing the water coverage on the catalyst surface can promote the protonation of the *CO intermediate and block CO production, further favoring the generation of methane. These results provide a new insight into the electrosynthesis of DRPs via constructing a hydrophobic core–shell architecture for tuning the surface water coverage.
Broader contextFossil fuel has greatly driven the economic growth and improved the living standards of humans, but this has occurred at the expense of ever-increasing anthropogenic carbon dioxide (CO2) emissions, leading to irreversible climatic changes. For alleviating anthropogenic CO2 emissions, electrochemical conversion of CO2 into value-added fuels and chemicals utilizing renewable electricity is a practical and promising strategy. However, achieving high product selectivity and partial current density for desirable deep-reduction products (DPRs) like methane (CH4) is a big challenge. Due to the high susceptibility of the CO2 reduction reaction (CO2RR) to the local environment and mass transportation near the gas–solid–liquid interface, tuning interfacial properties by engineering the surface wettability of the catalysts holds great promise to regulate the CO2RR performance. In this work, we open up a new avenue for narrowing the selectivity for DRPs by controlling the hydrophobicity for tuning water availability on the core–shell architecture. The rationally designed and constructed hydrophobic core–shell Cu-based catalyst possesses remarkable CH4 formation activity and selectivity compared to the reported Cu-based catalysts. |
Introduction
The renewable-electricity-powered carbon dioxide reduction reaction (CO2RR) provides an effective strategy to utilize CO2 and mitigate CO2 emissions.1–3 Among the developed electrocatalytic material candidates, copper-based materials are well known as the group that can selectively convert CO2 to hydrocarbons and alcohols in an aqueous solution.4 Unfortunately, their poor selectivity and low partial current density, especially towards deep-reduction products (DRPs, e.g., eight-electron transfer for methane (CH4), and twelve-electron transfer for ethylene (C2H4) and ethanol (C2H5OH)), significantly limit the CO2 single-pass conversion, which is among the largest challenges for electrochemical CO2 utilization.5 Hitherto tremendous research efforts have been devoted to catalyst design via tuning the morphology,6,7 manipulating the oxidation states,8,9 or creating defects,10,11 with optimized adsorbate energies thereby promoting the selectivity of copper-based electrocatalysts.12–14Besides the modulation of the catalyst electronic structures, constraining the available CO2 reactant can also regulate product selectivity.15,16 Recent reports have exploited the idea that electrode design of triple-phase boundary construction can concentrate CO2 and accommodate the local alkali environment to enhance the CO2RR activity.17,18 For instance, increasing the hydrophobicity of the catalyst electrode for constructing abundant gas–liquid–solid interfaces has been proposed as the governing factor to boost DRP production.19 In parallel, co-feeding CO2 with other gases (e.g., CO, N2, and O2) to decrease the local CO2 concentration can also narrow the product distribution for DRPs.20,21 Moreover, it has been recently reported that the molar fraction of water (H2O) at the cathodic GDE/membrane interface does not change as a function of humidification of the CO2 feed.22 On the basis of the above results, we attempt to initially tune the availability of local H2O, which has a substantial effect not only on the competing hydrogen evolution reaction (HER) but also on the CO2RR, via the catalyst structure design. The elaborately designed hydrophobic structure is more sophisticated to form a gas–solid–liquid interface on the surface of each catalyst particle, and is conducive to the effective regulation of electron–proton transfer, significantly affecting the overall CO2RR performance.
Herein, we explore a new strategy to increase the DRP activity of CH4, the deepest C1 and the simplest hydrocarbon product, via controlling the local H2O availability on the surface of the copper-based materials over the hydrophobic core–shell architecture. A density functional theory (DFT) study illustrates the favourable pathway for CH4 generation on the Cu surface with low local H2O concentration compared with the competing *CO desorption and C–C coupling process. In experiments, we rationally crafted the hydrophobic carbon shell coated on the copper/cuprous oxide core (H-CuOx@C, although oxidized Cu would be reduced to metallic Cu during CO2RR, the obtained samples were all named according to their pristine structure) to push away the aqueous electrolyte for constraining the H2O coverage on the surface (Fig. 1a). The resultant H-CuOx@C can sustain the hydrophobicity under CO2RR conditions (aqueous solution and secure negative-potential biased) and deliver a partial current density of CH4 (jCH4) of −39 ± 3 mA cm−2 with a peak CH4 faradaic efficiency of 81 ± 3% at the potential of −1.60 V vs. reversible hydrogen electrode (vs. RHE, all potentials were referenced to RHE unless mentioned otherwise; the potentials were not iR-corrected if not mentioned) in the neutral medium. Through using a flow cell system in the alkaline medium, the crafted catalyst can reach a maximum FECH4 of 73.3 ± 5.5% at a j of −500 mA cm−2. Notably, jCH4 of H-CuOx@C is as high as −434 mA cm−2 at an applied j of −700 mA cm−2, reaching a maximum CO2-to-CH4 conversion rate of 0.56 μmol cm−2 s−1. These performances exceed those of the wettable CuOx (W-CuOx) controls and most-reported methane-selective catalysts. We highlight that controlling the hydrophobicity for tuning water availability on the core–shell architecture might open up a new avenue for narrowing the selectivity for DRPs.
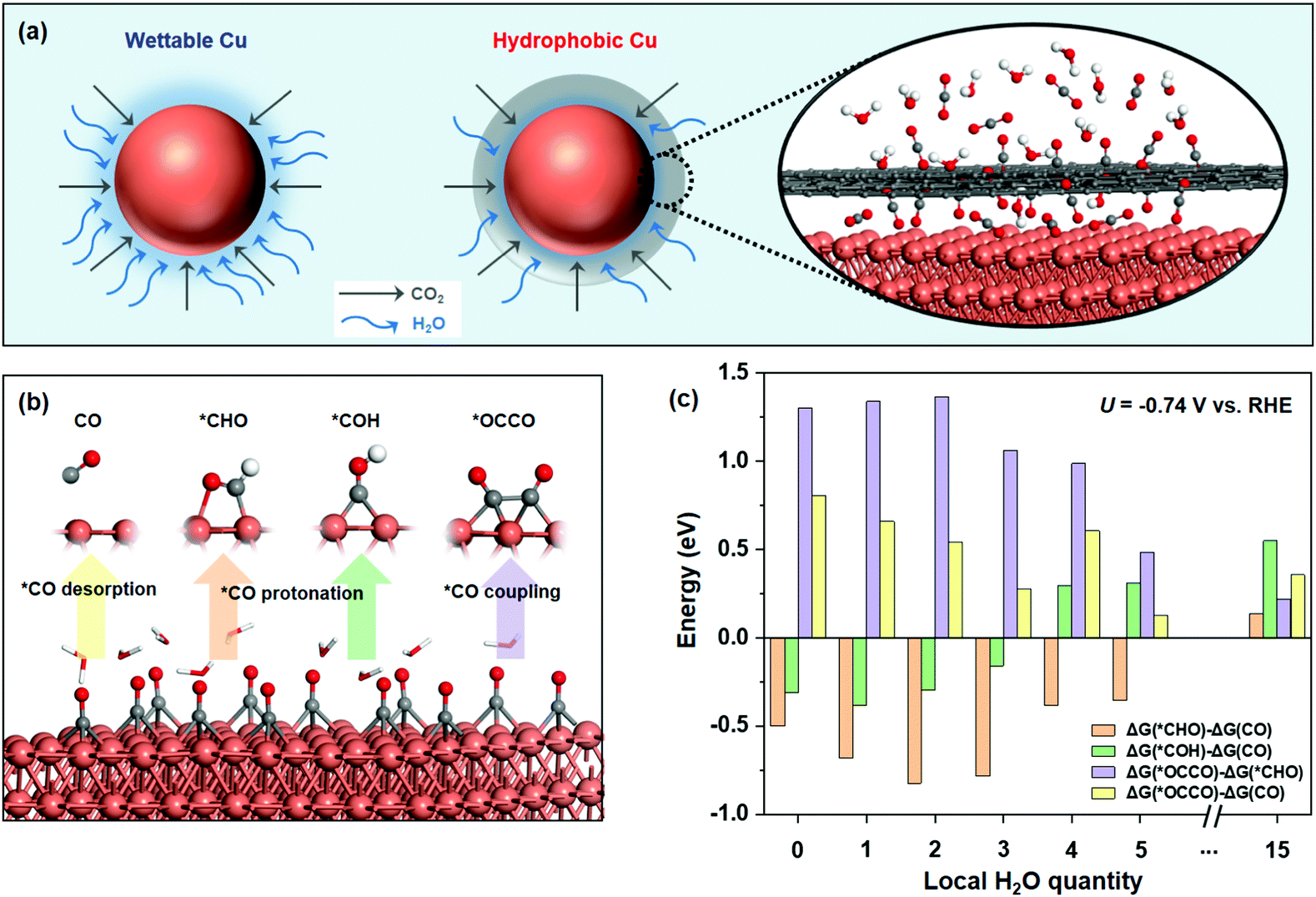 | ||
| Fig. 1 (a) Schematic illustration of the hydrophobic core–shell architecture, which can constrain H2O availability via hydrophobic carbon coating. (b) Three different pathways from *CO desorption to CO(g), *CO protonation to *CHO or *COH, and *CO coupling to *OCCO, with the active Cu site denoted by an asterisk (*). Orange, red, grey, and white balls represent copper, oxygen, carbon, and hydrogen atoms, respectively. (c) Reaction energies for subtraction of three competing reactions (*CO desorption, protonation of *CO to *CHO or *COH, and C–C coupling to *OCCO) under different H2O quantities considering the potential of −0.74 V vs. RHE. | ||
Experimental
Materials and chemicals
Copper nitrate trihydrate (Cu(NO3)2·3H2O) was purchased from Aladdin, and chitosan was obtained from Adamas Reagent Co., Ltd. Polyvinylpyrrolidone (PVP, M. W. 8000), carbon paper (TGP-H-060) and Nafion 115 proton exchange membrane were bought from Alfa Aesar. Ethylene glycol (AR, ≥99.0%) was obtained from Shanghai Titan Scientific Co., Ltd. Potassium bicarbonate (KHCO3) and isopropanol (AR, ≥99.0%) were obtained from Shanghai Chemical Reagent Co., Ltd. Nafion (5 wt% in a mixture of lower aliphatic alcohols and water) was obtained from Sigma-Aldrich. Carbon dioxide (CO2, 99.9999%) was bought from Shanghai Jiajie Special Gas Co., Ltd. All reagents were commercially available and of analytical grade. The water used was purified using a Millipore system (typically 18.2 MΩ cm resistivity).Materials characterization
The crystal structure was determined using X-ray diffraction (Bruker D8 Advanced Diffractometer with Cu Kα radiation). The morphology and structure were characterized using a scanning electron microscope (Hitachi S4800) and a transmission electron microscope (TEM, JEM 2100, 200 kV). Scanning transmission electron microscopy (STEM) characterization was performed using a ThermoFisher Talos F200X instrument. High-angle annular dark field (HAADF)-STEM images were recorded using a convergence semi angle of 11 mrad, and inner- and outer collection angles of 59 and 200 mrad, respectively. Energy dispersive X-ray spectroscopy (EDS) was carried out using 4 in-column Super-X detectors. Fourier transform infrared spectroscopy (FT-IR) was performed using a Nicolet 6700 spectrometer with a spectral range of 4000–400 cm−1. Volumetric CO2 adsorption measurement (Micromeritics ASAP 2020) was analyzed at 298 K and 1 atm. Brunauer–Emmett–Teller (BET, Micromeritics ASAP 2460) surface area measurement was performed at 77 K in the N2-adsorption mode. The contact angles were measured using a contact angle system (Shanghai Zhongchen, JC2000D3) at ambient temperature, with the probe liquid being 2 μL of water. The chemical state was analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Escalab 250), and the binding energy of the C 1s peak at 284.8 eV was taken as the internal standard. Raman analysis was carried out by using a Leica DMLM microscope (Renishaw) with a 514 nm laser. XAFS spectra at the Cu K-edge were performed on the 1W1B beamline station of the Beijing Synchrotron Radiation Facility (BSRF), China. Cu foil, Cu2O, and CuO were used as references.The in situ electrochemical attenuated total reflectance infrared spectroscopy (ATR-IR) was performed in a homemade cell attached to a PerkinElmer spectrum 100 spectrometer. Catalyst powders were deposited onto the carbon fiber paper electrode as the working electrode, tightly contacting the surface of the silica prism. A Pt counter electrode, an Ag/AgCl reference electrode, and a gas inlet to purge CO2 were also contained in the cell. Prior to electrochemical measurements, the electrolyte (0.1 M KHCO3) was injected into the cells and purged with high-purity CO2 (99.9999%). A CHI1242C potentiostat was employed to record the electrochemical response. The spectrum was collected every minute at −1.40 V vs. RHE with a dwell time of 30 min.
H-CuOx@C synthesis
The H-CuOx@C sample was constructed by a hydrothermal method. Firstly, 0.435 g of Cu(NO3)2·3H2O and 0.676 g of PVP were dissolved in 15 mL and 30 mL of ethylene glycol, respectively. Then, the Cu(NO3)2 solution was slowly dropped into the PVP solution under vigorous stirring to form homogeneous solutions, followed by adding 0.322 g chitosan. The autoclave was sealed and heated at 180 °C for 2 h. After the hydrothermal reaction, the obtained samples were thoroughly washed with ethanol and water three times each.W-CuOx synthesis
The preparation method of W-CuOx was the same as the above procedure except without the addition of chitosan.Preparation of electrodes
The sample ink was prepared by dispersing 5 mg of catalyst (H-CuOx@C or W-CuOx) and 50 μL of Nafion solution into 0.125 mL of H2O and 0.375 mL of isopropanol followed by sonication for more than 30 min. 25 μL of ink was dropped onto a carbon paper (CP, 1.0 × 1.0 cm2) and dried in the air 2 times. Then H-CuOx@C/CP and W-CuOx/CP were used for XRD, XPS, Raman, and XAFS measurements.A glassy carbon electrode (GCE) was used as the working electrode for electrochemical measurements in an H-type gas-tight reactor (H-Cell). The sample ink was prepared as mentioned above. 2.5 μL of the ink was dropped on the GCE (with a diameter of 3 mm) and dried in the air 2 times with the total catalyst loading amount of about 0.7 mg cm−2.
The gas diffusion electrodes (GDE) with the H-CuOx@C catalyst deposited on the Cu coated polytetrafluoroethylene (PTFE) gas diffusion layers were constructed and used as the working electrodes to evaluate CO2RR performance in a flow cell. To prepare Cu/PTFE, approximately 200 nm nominal thick Cu films were constructed by the vacuum evaporation method on the PTFE substrate (with an average pore diameter of 0.22 μm) using Cu targets (99.999%) at an evaporation rate of around 0.5 Å s−1 in an OMV FS300-S6 evaporating tool at a base pressure of <6 × 10−4 Torr. The sample ink was prepared by dispersing 5 mg of the catalyst and 20 μL of Nafion solution into 0.25 mL of H2O and 0.75 mL of ethanol followed by sonication for more than 1 h. Then the ink was sprayed onto a Cu/PTFE film electrode using an air-brush, with the total catalyst loading amount of about 0.5 mg cm−2.
Electrochemical measurements
All electrochemical studies were performed using an electrochemical station (CHI 660E). The H-Cell consists of two compartments which are separated by a Nafion 115 membrane. In the three-electrode system, a customized GCE with a surface area of 0.07 cm2 and a catalyst mass loading of 0.7 mg cm−2 was used as the working electrode. An Ag/AgCl electrode (3.5 M KCl) and platinum mesh were used as the reference electrode and the counter electrode, respectively. CO2-saturated 0.1 M KHCO3 (pH ≈ 6.75) was used as the electrolyte; CO2 was continuously purged through the electrolyte (both catholyte and anolyte were 30 mL in each compartment) at a rate of 20 sccm and was routed into the gas chromatograph. All potentials measured were calibrated to the RHE reference scale using ERHE = EAg/AgCl + 0.059 × pH + 0.205 (the potentials were not iR-corrected if not mentioned). The linear sweep voltammetry (LSV) at a scan rate of 1 mV s−1 was performed in Ar-saturated and CO2-saturated 0.1 M KHCO3 aqueous solution. The current density was calculated by normalizing the current to the corresponding geometric surface area.The large current density performance of H-CuOx@C was determined in a flow cell configuration. A Sustainion® membrane (Dioxide Materials) was activated in 1.0 M aqueous KOH solution for 24 h, washed with H2O, and used as the anion-exchange membrane. The anode consisted of IrTaOx supported on a Ti mesh was prepared by a dip coating and thermal decomposition method. 36 mg of IrCl3·was dissolved in 4.75 mL of H2O and 1.5 mL of concentrated HCl. 18.9 mg of TaCl5 was dissolved in 1.5 mL of methanol and added into the IrCl3 solution. A Ti mesh was dip coated with the mixed solution by sonication for 5 min, then dried at 120 °C for 0.5 h and calcinated at 500 °C for 1 h in air. The procedure was repeated ten times to increase the loading amount. The GDE was attached on the cathode using a copper tape at the edge of the electrode to electrically connect with the cathode, whose exposure area was 0.5 cm2. PTFE spacers were used for assembling the flow cell. 1.0 M KOH flowed (10 mL per min) in the chambers between the membrane and the working electrode, and the membrane and the anode as a liquid electrolyte. CO2 gas flowed behind the PTFE layer of GDE at a rate of 20 sccm. Chronopotentiometry experiments were performed at currents ranging from −300 to −800 mA cm−2, showing continuous gas product distributions.
Product analysis
The gas products of CO2 electroreduction were analyzed using a gas chromatography instrument (GC online test, RAMIN, GC2060), equipped with a flame ionization detector (FID for CO, CH4, and C2H4) and a thermal conductivity detector (TCD for H2). Ar was used as the carrier. Faradaic efficiencies were tested on-line and collected several times to obtain the average value. Liquid products were quantified by 1H nuclear magnetic resonance (NMR) (Varian 700 MHz spectrometer, 16.4T). In a typical analysis, after 5 hours’ CO2RR at an applied potential of −1.40 V vs. RHE, a mixture of 500 mg of the electrolyte and 100 mg of 4.8 ppm DMSO (used as internal standard) in D2O solution was used as the measured sample. The 1H spectra were obtained by using a pre-saturation method to suppress the water peak.Assuming that eight electrons are needed to produce one methane molecule, the FE and the partial current densities of CH4 formation were calculated as follows:
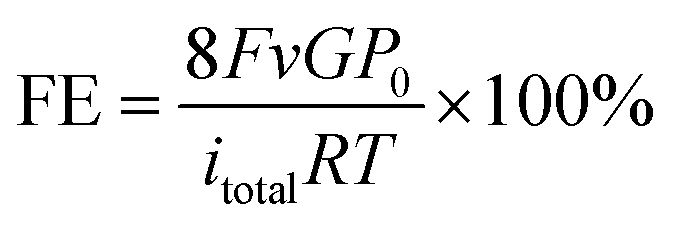
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1, and R = 8.314 J mol−1 K−1.then:
485 C mol−1, and R = 8.314 J mol−1 K−1.then:| jCH4 = FECH4 × itotal × (electrode area)−1 |
DFT calculation method
All the spin-polarized DFT computations were performed by using the Vienna ab initio simulation package (VASP)23 and the ion cores represented by the projector augmented wave (PAW) potentials. The generalized gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE) functional was used in this work.24 A cutoff energy of 450 eV for the plane-wave basis set was adopted. The convergence threshold for the structure relaxation was set to be 0.05 eV Å−1 in force and 10−5 eV in energy. A vacuum space exceeding 15 Å was employed. The computations on Cu(111) were performed in a p(2 × 2) periodic slab with four layers with a 2 × 2 × 1 Monkhorst–Pack k-point mesh for all the calculation models. The van der Waals (vdW) interactions were taken into consideration by the method of Grimme (DFT-D3).25Gibbs free energy calculation method
The reaction free energies of the four competing steps (*CO desorption, protonation of *CO to *CHO or *COH, and C–C coupling to *OCCO) were calculated using the following equations:| ΔG(CO) = E(*) + E(CO(g)) − E(*CO) + (ΔZPE − TΔS)CO |
| ΔG(*CHO) = E(*CHO) − E(H+ + e−) − E(*CO) + (ΔZPE − TΔS)*CHO + eU |
| ΔG(*COH) = E(*COH) − E(H+ + e−) − E(*CO) + (ΔZPE − TΔS)*CHO + eU |
| ΔG(*OCCO) = E(*OCCO) − 2E(*CO) + (ΔZPE − TΔS)*OCCO |
Results and discussion
We first sought to investigate the availability of the local H2O concentration on CO2RR selectivity over Cu catalysts with a relatively high CO2 concentration (corresponding to high *CO coverage15) using DFT. With *CO as an important intermediate in CO2RR, as shown in Fig. 1b, three competing conversion pathways occur: (i) *CO desorption to form the gaseous CO(g), (ii) *CO protonation into *CH4, *CO → *CHO (or *COH) →⋯→ *CH4, and (iii) the C–C bond coupling in two *CO to form multi-carbon products.28We studied the influence of local H2O quantities for CO2RR on common Cu(111) surface with high *CO coverage by the above competing pathways (Fig. S1, ESI†). The multi-point averaging molecular dynamics (MPA-MD) calculations29 were performed to reduce the energy difference originating from the different structures of water (see details in the ESI† and Fig. S2). After calculating the reaction free energy of *CO desorbing to CO(g) (ΔG(CO)), *CO to *CHO (ΔG(*CHO)), *CO to *COH (ΔG(*COH)), and C–C coupling to *OCCO (ΔG(*OCCO)) at different local H2O quantities (Fig. 1c and see the detailed data in Table S1, ESI†), we found that the formation of *CHO is more exothermic than that of *COH (ΔG(*CHO) − ΔG(*CO) < ΔG(*COH) − ΔG(*CO)), indicating that *CHO is the key intermediate to form CH4, which is similar to the reported results.28,30 Our calculation also shows that ΔG(*OCCO) − ΔG(CO) (or ΔG(*OCCO) − ΔG(*CHO)) is always positive, showing that ΔG(*OCCO) is larger than both ΔG(CO) and ΔG(*CHO). This result indicates that the C–C coupling into C2+ products is not favorable on the pristine Cu(111) surface. Furthermore, comparing ΔG(*CHO) and ΔG(CO), we found that ΔG(*CHO) − ΔG(CO) decreases with the increase in H2O content on the surface and gradually becomes more negative at the beginning. At a low H2O content with about 2 or 3 local molecules, ΔG(*CHO) − ΔG(CO) is the lowest, i.e., *CO protonation to form CH4 is more feasible, implying a higher CH4 selectivity vs. CO under these conditions. The whole energy change confirms what we proposed before (Fig. S3, ESI†). With further increase in the surface H2O content, ΔG(*CHO) − ΔG(CO) increases rapidly and becomes positive after the H2O content reaches a certain level (e.g., 15 molecules), indicating that CO desorption becomes favored instead of *CHO formation owing to the steric squeezing from the local H2O molecules (see the dynamic process in Fig. S4, ESI†), which seriously affects the product distribution of CH4 and CO. In this case, the selectivity of CO might be higher compared with that of CH4.
Taken together, we anticipate that controlling the local H2O content at a low coverage on Cu catalysts with high CO2 concentration could promote the selectivity to CH4 in CO2RR. Motivated by the DFT results, we rationally synthesized hydrophobic core–shell nanohybrids to reduce the local H2O coverage, containing a copper/cuprous oxide sphere core and a nanocarbon shell, via a modified one-step hydrothermal method with chitosan as the reductive agent and carbon source, which has abundant amino and hydroxyl functional groups for strong chelation of Cu ions. Carbon nanoshells stemmed from the hydrothermal carbonization of chitosan, which is consistent with past reports.31 In comparison, the cuprous oxide sphere without carbon coating was constructed as the control sample without the addition of chitosan. We investigated the crystal structure of the synthesized samples using X-ray diffraction (XRD) (Fig. 2a), which revealed that H-CuOx@C contains metallic Cu (JCPDS #04-0836), a small amount of Cu2O (JCPDS #65-3288), and a broad diffraction peak between 20° and 30° which is attributed to amorphous carbon.32 For the bare pristine W-CuOx control, all peaks could be indexed to Cu2O (JCPDS #65-3288, Fig. S5, ESI†), which is attributed to the lack of the reductive agent, chitosan.33 H-CuOx@C and W-CuOx were both reduced to metallic Cu with obvious Cu(111) diffraction peaks during the CO2RR process (Fig. S6, ESI†). Fig. S7a (ESI†) displays the scanning electron microscopy (SEM) image of H-CuOx@C in an enlarged view with the particle size ranging from 200 nm to 1.5 μm, and the corresponding energy-dispersive spectroscopy (EDS) mapping images suggest a sphere-like nanostructure with Cu, O, and C elements distributed universally over the sample (Fig. S7b–e, ESI†). In contrast, W-CuOx nanospheres show a relatively rougher surface, and only Cu and O elements exist in the sample (Fig. S8, ESI†).
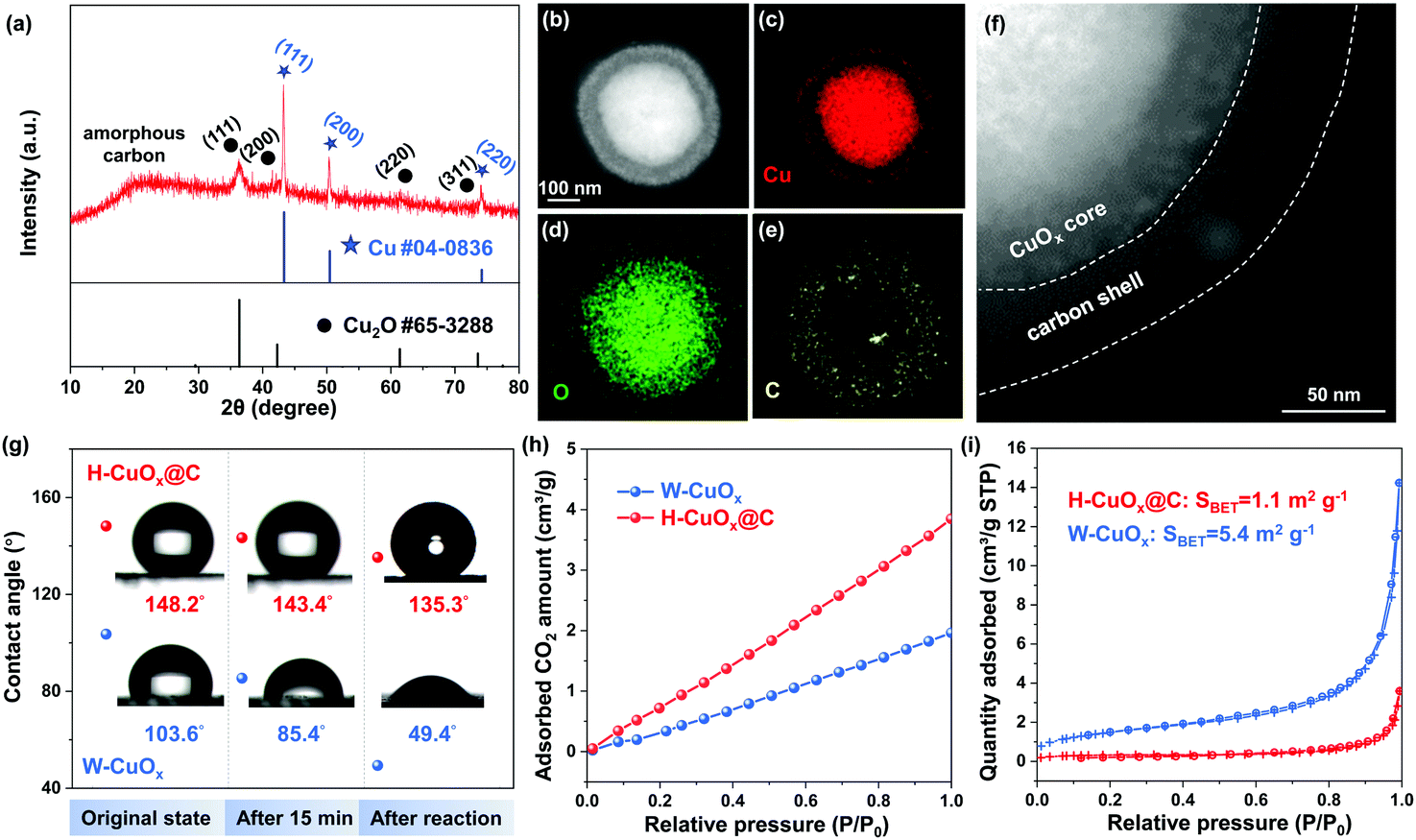 | ||
| Fig. 2 (a) XRD pattern of H-CuOx@C, indicating the coexistence of Cu, Cu2O, and amorphous carbon components. (b) HAADF-STEM image of H-CuOx@C and (c–e) the corresponding EDS elemental mapping images of (c) Cu, (d) O, and (e) C elements, respectively, displaying the carbon coating core–shell architecture. (f) Enlarged HAADF-STEM image of H-CuOx@C, showing the ∼50 nm thick carbon shell. (g) The CA measurements of the H-CuOx@C and W-CuOx electrodes in their original state, after being staged for 15 min, and after CO2RR at −1.40 V for 2 h, respectively. (h) CO2 adsorption isotherms of H-CuOx@C and W-CuOx, respectively, indicating the higher CO2 adsorption capability of H-CuOx@C. (i) N2 adsorption–desorption isotherm of H-CuOx@C and W-CuOx samples, respectively. | ||
Transmission electron microscopy (TEM) images of H-CuOx@C (Fig. S9 and S10, ESI†) confirm that carbon layers exist stably before and after 2 hours’ (h) electrolysis at −1.40 V. Please note that when the applied potential increased to −1.60 V, carbon layers were destroyed during the CO2RR process (Fig. S11, ESI†), indicating the tolerance of the carbon shell under the electrolyte with appropriate bias, which would also affect the hydrophobicity. No carbon shell could be found in the bare W-CuOx control (Fig. S12 and S13, ESI†). Both H-CuOx@C and W-CuOx samples were reduced to metallic Cu during the test with the obvious diffraction ring of Cu(111), which is consistent with the XRD patterns and demonstrates the validity of the DFT calculations. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was also conducted to gather detailed structure information about H-CuOx@C (Fig. 2b–f). The HAADF-STEM image of the typical H-CuOx@C particle (Fig. 2b) along with its corresponding EDS elemental maps demonstrate the core–shell architecture with a carbon shell. The enlarged HAADF-STEM image further confirms that the thickness of the carbon shell is about 50 nm (Fig. 2f). To characterize the cross-section structure and the crystal phase of H-CuOx@C, the sample was embedded in epoxy resin and then cut into ultrathin slices by ultramicrotomy. HRTEM imaging was acquired and the corresponding FFT was used for analysis (Fig. S14, ESI†), indicating the existence of an amorphous carbon shell and Cu2O component in the core. Furthermore, the FT-IR spectrum of H-CuOx@C identified the existence of oxy-hydrogenated amorphous carbon and Cu2O (Fig. S15, ESI†).34,35
Contact angle (CA) measurements were implemented to investigate the wettability of the samples. The pristine bare W-CuOx surface exhibits an external water CA of 103.6°, whereas the CA would be decreased under a solution environment, with CA of 85.4° staged for 15 min and 49.4° after 2 h of electrolysis at −1.40 V, which demonstrates its hydrophily under the real CO2RR conditions (Fig. 2g). In contrast, by introducing a carbon shell, the H-CuOx@C surface turns hydrophobic with a drastically increased CA of 148.2°, and could maintain the hydrophobicity both under the static state with surface water and CO2RR conditions biased with a secure negative potential applied on the electrode in the electrolyte (Fig. 2g). The detailed time dependencies of surface wettability of H-CuOx@C and W-CuOx were further studied, shown in Fig. S16 (ESI†), indicating the durable hydrophobicity of H-CuOx@C. Moreover, the CA of H-CuOx@C will decrease to 103.8° after which the carbon shell is destroyed, which demonstrates the importance of the carbon shell for hydrophobicity (Fig. S17, ESI†). We obtained in situ diffusion reflectance infrared Fourier transform spectra (DRIFTS) of H2O adsorption to detect the variation of H2O on the catalyst surface (measurement details are described in the ESI†). It has been reported that the more hydrophobic the material is, the more enhanced ordering and stronger H2O–H2O interactions via hydrogen-bonding can occur during sorption so that the maximum of band 3000–3500 cm−1 will shift to a lower wavenumber.36 These conclusions are consistent with our results shown in Fig. S18 (ESI†), which further proves that H-CuOx@C has stronger hydrophobicity. The hydrophilicity of the catalyst structure also allows the competition with adsorption of H2O, which affects their CO2 capture abilities significantly.37,38 In general, the hydrophobic surface is able to enhance the local CO2 concentration by keeping water away from the electrode surface and leaving water-free space to store gaseous CO2. In this work, the CO2 adsorption isotherm and N2 adsorption–desorption isotherm further confirm that the introduction of a carbon shell could enhance the CO2 adsorption capacity by 2 times and the CO2/N2 adsorption selectivity by 10 times, which might profit from the accelerated gas diffusion brought about by the hydrophobic carbon shell, thereby contributing to the enhanced generation of CH439 (Fig. 2h and i).
In order to identify the chemical composition and surface valence states of H-CuOx@C and W-CuOx samples, X-ray photoelectron spectroscopy (XPS) was conducted (Fig. 3a and Fig. S19, S20 (ESI†)). The peak of Cu 2p could be fitted into three constituent peaks (Fig. 3a), which are ascribed to Cu+/Cu0 (932.6 eV), Cu2+ (933.8 eV), and Cu(OH)2/CuCO3 (935.0 eV), and the surface Cu2+ species indicates that the Cu+/Cu0 components were partially oxidized, ascribed to slight surface oxidation in the air.40 In comparison, the W-CuOx sample exhibits the main component of oxidized Cu species, which results from the lack of a reductive agent (Fig. S20, ESI†), and these results are coincident with those of the XRD analysis. After CO2RR at a potential of −1.40 V for 2 h, the surface Cu2+ components of H-CuOx@C were reduced to Cu+/Cu0 (Fig. 3a). We also employed Raman spectroscopy to probe the existence of the carbon shell. In Fig. 3b and Fig. S21 (ESI†), Raman spectra show the Cu2O components in both H-CuOx@C and W-CuOx, probed by the characteristic vibrational fingerprints at 146, 217, 526, and 632 cm−1 (ref. 41). Additionally, two bands located at 1327 and 1593 cm−1 were only found in H-CuOx@C, which are indexed to the D band and the G band of carbon, respectively, demonstrating the specific carbon shell.42 After being applied at a cathodic potential of −1.40 V for 2 h, only carbon characteristic peaks could be observed in the Raman spectra, without obvious oxide Cu species (Fig. 3b). This also proves that Cu2O was reduced to metallic copper during the CO2RR, but the carbon shell was undamaged, featuring the ability to continuously tune the hydrophobicity.
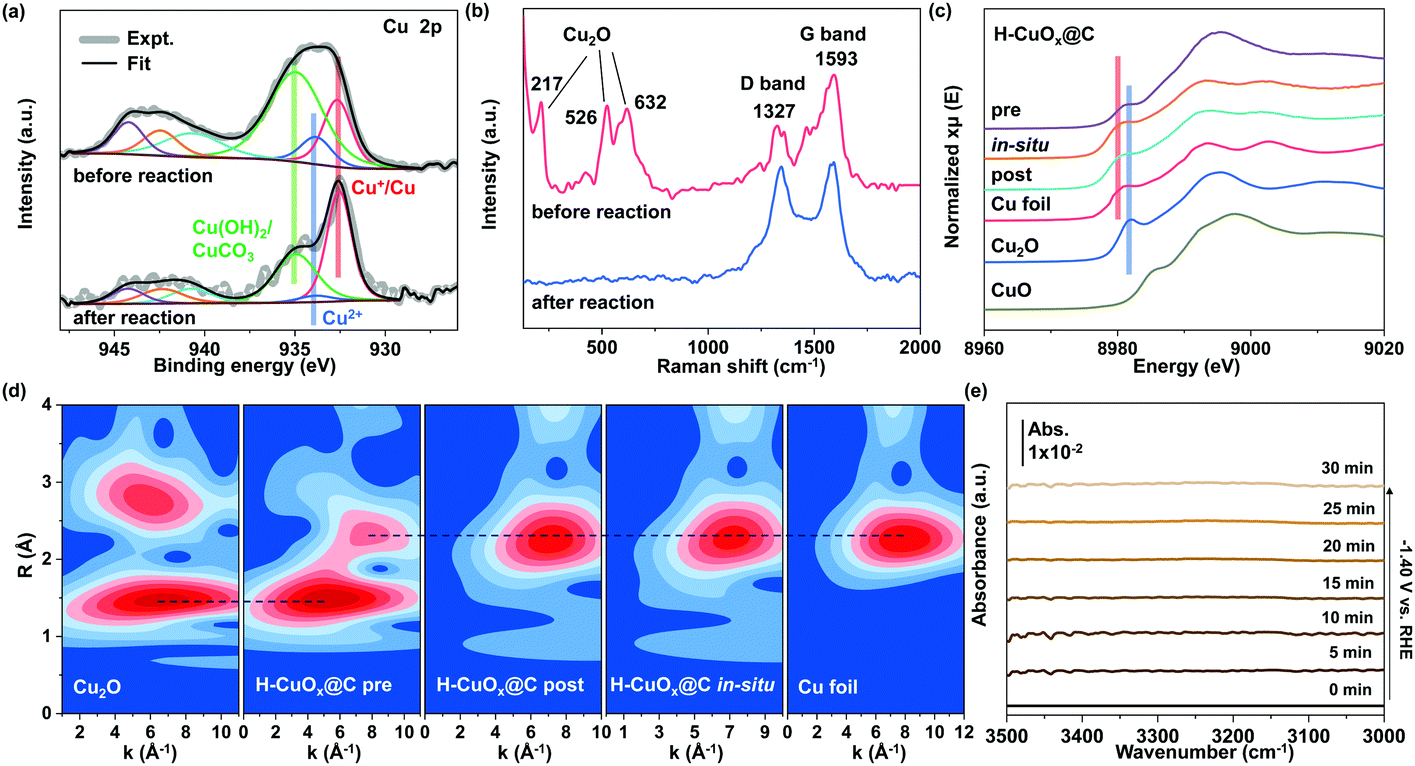 | ||
| Fig. 3 (a) XPS spectra of the Cu 2p region and (b) Raman spectra of H-CuOx@C collected before and after CO2RR at −1.40 V for 2 h. (c) Normalized Cu K-edge operando XANES spectra and (d) Morlet WT of the k3-weighted operando EXAFS data for H-CuOx@C with standard Cu foil, Cu2O, or CuO powders as controls. The XPS and Raman spectra along with operando XAFS analysis demonstrate the reductive process of oxide Cu species to the metallic Cu0 states. (e) Time-dependent in situ ATR-IR differential spectra of H-CuOx@C carried out at −1.40 V under the CO2RR in CO2-saturated 0.1 M KHCO3, indicating that the hydrophobicity of H-CuOx@C has a minor change over half an hour operation time. | ||
We further performed X-ray absorption fine structure spectroscopy (XAFS) measurements, which can provide further insights into the electronic structures and chemical bonding of bulk metal atoms in their local environment. The ex situ X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra indicate that H-CuOx@C is composed of Cu and Cu2O, while W-CuOx is consistent with those of Cu2O reference (Fig. S22, ESI†). To probe the oxidation state evolution of these electrocatalysts during the CO2RR, we carried out operando XANES measurements under the same electrochemical conditions. At −1.40 V, the operando XANES spectra clearly show that both samples of H-CuOx@C and W-CuOx exhibited metallic Cu0 during the CO2RR and kept their metallic Cu0 states after the test (Fig. 3c and Fig. S23, ESI†). The additional analysis of the Morlet wavelet transform (WT) was also performed to provide solid supports for the generation of metallic Cu0 during the CO2RR process. As shown in Fig. 3d, a WT maximum at 3–9 Å−1, which represents the existence of a Cu–O and Cu–Cu bond, is visible in H-CuOx@C, whereas it exhibits a distinct feature at 6–8 Å−1 during the CO2RR at a working potential of −1.40 V, corresponding to the Cu–Cu bond, which suggests that the metallic Cu0 species occupy the main composition in the bulk during the test,43 while for the W-CuOx catalyst, the pristine main component of Cu2O was also reduced to metallic Cu0 under the same reaction conditions (Fig. S24, ESI†). These XAFS results together with Raman analysis unambiguously prove the negligible changes in the valence states between H-CuOx@C and W-CuOx under the CO2RR conditions which could affect selectivity. We also performed in situ electrochemical attenuated total reflectance infrared spectroscopy (ATR-IR) to prove that the hydrophobicity of the carbon shell would persist over a significant operation time. A digital photograph of a homemade cell attached to a PerkinElmer spectrum 100 spectrometer for ATR-IR measurement is shown in Fig. S25 (ESI†). We can observe in Fig. 3e that the intensity of hydrogen-bonded water peak between 3100 and 3500 cm−1 remains almost the same with additional electrolysis time, which indicates that the amount of water bound on the H-CuOx@C catalyst surface would not increase gradually during the CO2RR process. This result also suggests that the electrowetting effect on the wettability of H-CuOx@C during the CO2RR experiment is minimal.
The activity of H-CuOx@C for electrocatalytic CO2RR was evaluated in a standard three-electrode configuration in a gastight two-compartment electrochemical cell (Fig. S26, ESI†). The electrodes were made by drop casting an ink solution containing a certain concentration of catalysts, isopropanol, H2O and Nafion onto the glassy carbon electrodes (GCE, for more details see the ESI†). The linear sweep voltammetry (LSV) curves of H-CuOx@C and W-CuOx were collected in a CO2- and Ar-saturated 0.1 M KHCO3 solution, respectively. As shown in Fig. 4a, the LSV curve of H-CuOx@C in a CO2-saturated solution shows a positive shift of the onset potential and significant increase in current density compared with that measured in an Ar-saturated solution, suggesting the occurrence of CO2RR.44 Thereafter, the H-CuOx@C catalyst was tested over a range of potentials for catalytic activity, and showed a marked selectivity for methane (FECH4) at potentials from −1.30 to −1.70 V, with a peak FECH4 reaching 81 ± 3% at −1.60 V (Fig. 4b). To further determine the selectivity of CO2RR, a nuclear magnetic resonance (NMR) test was performed, and no detectable liquid product was found in 1H NMR, which also suggests that the total FE of all the products is nearly 100% (Fig. S27, ESI†). However, at −1.60 V, the FEH2, FECO, FECH4 and FEC2H4 of W-CuOx are 44%, 40%, 12% and 3%, respectively (Fig. 4b). Notably, the partial current density towards CH4 (jCH4) of H-CuOx@C is obviously higher than that of the W-CuOx control from −1.30 to −1.70 V: its jCH4 is as high as −39 ± 3 mA cm−2 and 15-folder larger than that of W-CuOx at −1.60 V (Fig. S28, ESI†). As shown in Fig. 4c, H-CuOx@C produced nearly exclusively CH4 among the C1 products; in contrast, the C1 selectivity of W-CuOx shifted from hydrocarbon to CO over the course of cathodic potential increase. Moreover, the ratios of C2H4 and C1 products for H-CuOx@C and W-CuOx are fairly close (Fig. 4c), and these results also coincide with those of the DFT study. As illustrated in Fig. S29 (ESI†), H-CuOx@C features a Cdl of 0.69 mF cm−2, which is close to that of 0.52 mF cm−2 for W-CuOx, suggesting comparative active surface areas, which ambiguously demonstrates that the H-CuOx@C catalyst exhibits a higher intrinsic CH4 activity than the W-CuOx control. To identify the role of the hydrophobic core–shell structure without destroying this architecture or changing the particle size, we employed a hydrophilic organic modifier of cetyltrimethylammonium bromide (CTAB) to change the local hydrophilicity over the catalysts (details are described in the ESI†).45 As shown in Fig. S30a and b (ESI†), the CH4 selectivity will be decreased when the local hydrophobicity becomes weaker (without changing the morphology), with a maximum FECH4 of 38.9 ± 1.3% at −1.40 V. When the catalyst is converted to being hydrophilic, it shows a major selectivity for H2 at potentials from −1.20 to −1.70 V, with no detectable CH4 (Fig. S30c, ESI†). The above results further demonstrate the important role of local hydrophobicity of the core–shell structure in promoting CH4 selectivity.
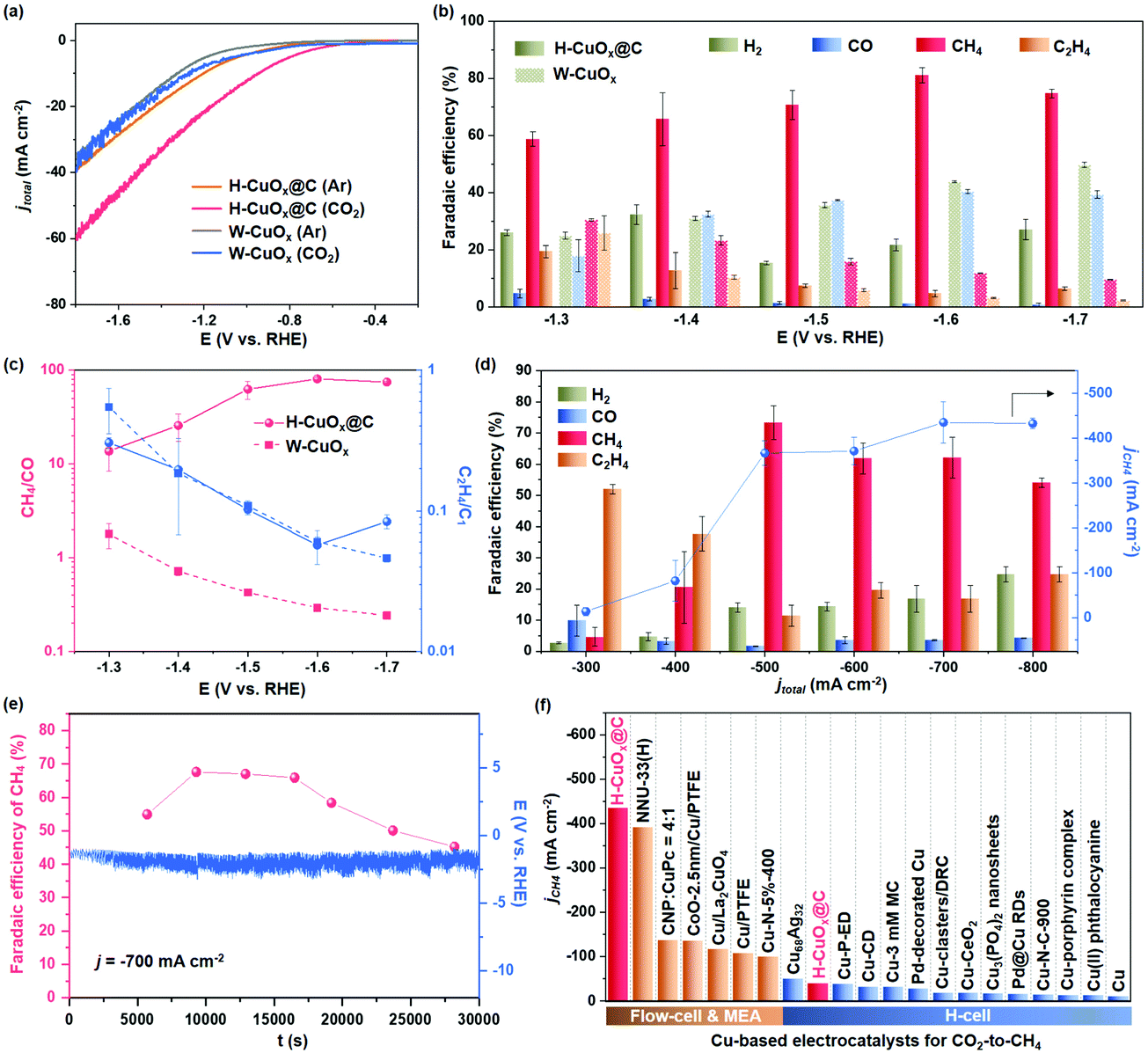 | ||
| Fig. 4 Electrochemical performance evaluation of H-CuOx@C and W-CuOx catalysts for CO2RR. (a) The LSV curves of H-CuOx@C and W-CuOx catalysts with respect to RHE at a scan rate of 1 mV s−1 in the CO2- or Ar-saturated 0.1 M KHCO3 electrolyte, indicating the occurrence of the CO2RR. (b) Faradaic efficiencies of gas products on H-CuOx@C and W-CuOx, exhibiting a higher CH4 selectivity for H-CuOx@C. (c) CH4/CO and C2H4/C1 ratios of H-CuOx@C and W-CuOx catalysts at different applied potentials, suggesting the superior CH4 selectivity for H-CuOx@C. (d) Faradaic efficiencies for gas products and jCH4 over H-CuOx@C at various applied current densities in a flow cell reactor with 1.0 M KOH as the electrolyte. Error bars were all calculated based on the standard deviation of three measurements under each test condition. (e) Stability test of electrochemical CO2 methanation during 30000 s of electrolysis under a j of −700 mA cm−2. Red dots represent FECH4, and the blue line represents the applied potential curve. Faradaic efficiencies of detailed gas products are shown in Table S2 (ESI†). (f) Maximum jCH4 of H-CuOx@C and recently reported Cu-based CO2RR catalysts (details are shown in Tables S3 and S4, ESI†). | ||
To further improve the activity of CO2-to-CH4, we evaluated CO2RR performance in a flow cell configuration. The sprayed gas diffusion electrode (GDE) and flow cell device are shown in Fig. S31 (ESI†). H-CuOx@C is evaluated at different j from −300 to −800 mA cm−2 and CH4 is detected as a major CO2RR product in a wide j range (Fig. 4d). The representative gas chromatography result of H-CuOx@C is shown in Fig. S32 (ESI†), showing the main CO2RR product of CH4. Notably, H-CuOx@C reaches a maximum FECH4 of 73.3 ± 5.5% at an applied j of −500 mA cm−2, which results in a jCH4 of −366.5 mA cm−2. In addition, the jCH4 of H-CuOx is as high as −434 mA cm−2 at an applied j of −700 mA cm−2, reaching a maximum CO2-to-CH4 conversion rate of 0.56 μmol cm−2 s−1 (Fig. 4d). The long-term stability test of H-CuOx@C electrode at the applied j of −700 mA cm−2 was also implemented in the flow cell configuration, which exhibits FECH4 of more than 50% over a time range of up to 23700 s, suggesting the jCH4 can be maintained over −350 mA cm−2 for more than 6 h (Fig. 4e and Table S2, ESI†). The attenuation of CO2RR performance might be influenced by the GDL fouling and the side reaction between the electrolyte and CO2 in flow cells. We also compared the maximum jCH4 for H-CuOx@C with that for other Cu/Cu-based catalysts (Fig. 4f and Tables S3, S4, ESI†), and H-CuOx@C with a jCH4 of −434 mA cm−2 was found to possess a remarkable CH4 formation activity compared to the reported Cu-based catalysts.
Conclusions
In summary, a hydrophobic core–shell architecture was designed and constructed to tune the H2O availability over the Cu-based catalyst surface, which could provide a high faradaic efficiency of 81 ± 3% towards CH4 in a neutral medium and a maximum jCH4 of −434 mA cm−2 in a flow cell configuration, which is among the best of the Cu-based CO2-to-CH4 electrocatalysts. The superior DRP selectivity originates from the improved CO2 capacity and decreased H2O coverage induced by the hydrophobic carbon shell, which is demonstrated using experimental data and computational techniques. We highlight that numerous Cu-based catalysts can be expected to benefit from this study, with appropriate H2O availability via catalyst architecture design to block CO generation, further improving the DRP selectivity along with other surface modulation strategies.Author contributions
H. G. Y. and P. F. L. directed the research. X. Y. Z. performed the experiments and data analyses. W. J. L. performed the DFT calculations. Z. J. and Y. W. L. performed the XAFS test and analyses. S. D. and X. F. W. provided assistance with the HAADF-STEM characterization. M. Z. and J. C. assisted with the in situ DRIFTS and ATR-IR tests and data analyses. H. F. W. and H. Y. Y. guided the DFT calculations. All the authors participated in writing and editing the manuscript, and contributed their efforts to the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Funds for Distinguished Young Scholars (51725201), the International (Regional) Cooperation and Exchange Projects of the National Natural Science Foundation of China (51920105003), the Innovation Program of Shanghai Municipal Education Commission (E00014), the National Natural Science Foundation of China (51902105, 21902048), the Fundamental Research Funds for the Central Universities (JKD01211519), the Shanghai Engineering Research Center of Hierarchical Nanomaterials (18DZ2252400) and the Shanghai Sailing Program (19YF1411600). The authors acknowledge the support by Shanghai Rising-star Program (20QA1402400). Additional support was provided by the Feringa Nobel Prize Scientist Joint Research Center. The authors also thank the Frontiers Science Center for Materiobiology and Dynamic Chemistry. The authors also thank the crew of the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF) and the 1W1B beamline of Beijing Synchrotron Radiation Facility (BSRF) for their constructive assistance with the XAFS measurements and data analyses.References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. M. Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- R.-B. Song, W. Zhu, J. Fu, Y. Chen, L. Liu, J.-R. Zhang, Y. Lin and J.-J. Zhu, Adv. Mater., 2019, 32, 1903796 CrossRef.
- R. G. Grim, Z. Huang, M. T. Guarnieri, J. R. Ferrell, L. Tao and J. A. Schaidle, Energy Environ. Sci., 2020, 13, 472–494 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- T. Möller, F. Scholten, T. N. Thanh, I. Sinev, J. Timoshenko, X. Wang, Z. Jovanov, M. Gliech, B. R. Cuenya, A. S. Varela and P. Strasser, Angew. Chem., Int. Ed., 2020, 59, 17974–17983 CrossRef.
- F. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC.
- M. G. Kibria, C.-T. Dinh, A. Seifitokaldani, P. D. Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. G. de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- D. Gao, I. Sinev, F. Scholten, R. M. Arán-Ais, N. J. Divins, K. Kvashnina, J. Timoshenko and B. R. Cuenya, Angew. Chem., Int. Ed., 2019, 58, 17047–17053 CrossRef CAS PubMed.
- T. Kim and G. T. R. Palmore, Nat. Commun., 2020, 11, 3622 CrossRef.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- X. Chang, T. Wang, Z.-J. Zhao, P. Yang, J. Greeley, R. Mu, G. Zhang, Z. Gong, Z. Luo, J. Chen, Y. Cui, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 15415–15419 CrossRef CAS.
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D.-H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C.-T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- X. Wang, A. Xu, F. Li, S.-F. Hung, D.-H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS.
- S. He, F. Ni, Y. Ji, L. Wang, Y. Wen, H. Bai, G. Liu, Y. Zhang, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2019, 577, 509–513 CrossRef PubMed.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- W. H. Lee, Y.-J. Ko, Y. Choi, S. Y. Lee, C. H. Choi, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, Nano Energy, 2020, 76, 105030 CrossRef CAS.
- Y. Xu, J. P. Edwards, J. Zhong, C. O'Brien, C. M. Gabardo, C. McCallum, J. Li, T. C. Dinh, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2020, 13, 554–561 RSC.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS.
- D. G. Wheeler, B. A. W. Mowbray, A. Reyes, F. Habibzadeh, J. He and C. P. Berlinguette, Energy Environ. Sci., 2020, 13, 5126–5134 RSC.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- M. W. J. Chase, NIST-JANAF Themochemical Tables, Fourth Edition, J. Phys. Chem. Ref. Data, 1998, 9, 1–1951 Search PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- T. Cheng, H. Xiao and W. A. Goddard III, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS.
- D. Wang, T. Sheng, J. Chen, H.-F. Wang and P. Hu, Nat. Catal., 2018, 1, 291–299 CrossRef CAS.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS.
- Y. Yang, X. Liang, F. Li, S. Li, X. Li, S.-P. Ng, C.-M. L. Wu and R. Li, ChemSusChem, 2018, 11, 376–388 CrossRef CAS PubMed.
- B. Priyanto, M. Saleh, S. Tunmee, C. Euaruksakul, Y. Cahyono, T. Triwikantoro and D. Darminto, Mater. Sci. Forum, 2019, 966, 95–99 Search PubMed.
- M. S. Almughamisi, Z. A. Khan, W. Alshitari and K. Z. Elwakeel, J. Polym. Environ., 2020, 28, 47–60 CrossRef CAS.
- V. Ţucureanu, A. Matei and A. M. Avram, Crit. Rev. Anal. Chem., 2016, 46, 502–520 CrossRef.
- A. O. Moghanlou, A. Bezaatpour, M. H. Sadr, M. Yosefi and F. Salimi, Mater. Sci. Semicond. Process., 2021, 130, 105838 CrossRef CAS.
- J.-L. Xu and A. A. Gowen, Spectrochim. Acta, Part A, 2021, 250, 119371 CrossRef CAS.
- J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560–4561 CrossRef CAS PubMed.
- N. Ding, H. Li, X. Feng, Q. Wang, S. Wang, L. Ma, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 10100–10103 CrossRef CAS PubMed.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- I. Platzman, R. Brener, H. Haick and R. Tannenbaum, J. Phys. Chem. C, 2008, 112, 1101–1108 CrossRef CAS.
- D. Ren, Y. L. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- J. Wu, D. Wang, S. Wan, H. Liu, C. Wang and X. Wang, Small, 2020, 16, 1900550 CrossRef CAS.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard III, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, TEM and SEM images, XRD, XPS, Raman, XAFS and performance comparison. See DOI: 10.1039/d1ee01493e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |