
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular-level insight into uptake of dimethylamine on hydrated nitric acid clusters†
Andriy
Pysanenko
a,
Karolína
Fárníková
a,
Jozef
Lengyel
 *b,
Eva
Pluhařová
*a and
Michal
Fárník
*a
*b,
Eva
Pluhařová
*a and
Michal
Fárník
*a
aJ. Heyrovský Institute of Physical Chemistry, v.v.i., The Czech Academy of Sciences, Dolejškova 2155/3, Prague 8 182 23, Czech Republic. E-mail: michal.farnik@jh-inst.cas.cz; eva.pluharova@jh-inst.cas.cz
bChair of Physical Chemistry, TUM School of Natural Sciences, Technical University of Munich, Garching 85748, Germany. E-mail: jozef.lengyel@tum.de
First published on 26th August 2022
Abstract
Mixed nitric acid/water clusters with dimethylamine (DMA) represent a suitable model system for understanding acid–base chemistry in atmospherically relevant clusters. We investigate these clusters in a detailed molecular-beam experiment accompanied by ab initio calculations. The (HNO3)M(H2O)N clusters are produced by supersonic expansion into vacuum and doped by DMA molecules in a pickup process. Two complementary mass spectrometry approaches are employed to analyze the resulting (DMA)K(HNO3)M(H2O)N clusters: (i) electron impact ionization at 70 eV to form positive cluster ions and (ii) low-energy electron attachment at 0–10 eV to form negative clusters. The positive ion spectra contain mainly protonated (DMA)k(HNO3)mH+ clusters with k = m + 1, whereas the negative ones are dominated by (DMA)k(HNO3)mNO−3 with m ≥ k, followed by (DMA)k(HNO3)mNO2− (m > k) ions with low abundances. These observations are rationalized by our calculations, which exhibit the tendency of the mixed clusters to maximize the number DMA·H+ and NO3− ions in the clusters. In the neutral clusters, this is fulfilled for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of DMA and HNO3, while the positively charged (DMA)k(HNO3)mH+ clusters satisfy this condition for k = m + 1. The protonated clusters always contain the DMA·H+ moiety. For the negatively charged cluster ions, thermochemistry supports the prevailing formation of NO−3 and m ≥ k ion composition. Furthermore, the NO−3-containing cluster ions can form when an electron attaches to the protonated moiety of the DMA·H+⋯NO−3 ion pair in the cluster, which leads to H atom evaporation. From the gas phase HNO3 molecule, where NO−2 is formed exclusively upon an electron attachment, the tendency to form NO−3 increases to hydrated HNO3 clusters, where both NO−2 and NO−3 ions are generated in approximately equal abundances, to the DMA doped clusters, where NO−3 strongly prevails NO−2.
:1 ratio of DMA and HNO3, while the positively charged (DMA)k(HNO3)mH+ clusters satisfy this condition for k = m + 1. The protonated clusters always contain the DMA·H+ moiety. For the negatively charged cluster ions, thermochemistry supports the prevailing formation of NO−3 and m ≥ k ion composition. Furthermore, the NO−3-containing cluster ions can form when an electron attaches to the protonated moiety of the DMA·H+⋯NO−3 ion pair in the cluster, which leads to H atom evaporation. From the gas phase HNO3 molecule, where NO−2 is formed exclusively upon an electron attachment, the tendency to form NO−3 increases to hydrated HNO3 clusters, where both NO−2 and NO−3 ions are generated in approximately equal abundances, to the DMA doped clusters, where NO−3 strongly prevails NO−2.
Environmental significanceNew particle formation (NPF) in atmospheric aerosol generation is still insufficiently understood. It is well-established that acid-base chemistry plays a key role in NPF, yet, it is difficult to obtain a truly molecular-level understanding from field and aerosol chamber investigations. Studies of isolated clusters in molecular beams can provide such picture. Nitric acid and dimethylamine (DMA) are ubiquitous throughout the atmosphere and are involved in NPF. We dope hydrated nitric acid clusters with DMA in a molecular beam experiment complemented by theoretical calculations, and provide a molecular-level insight into these species and their chemistry. Discussed relationship between neutral cluster structure and positively and negatively charged cluster ions is relevant for implementation of mass spectrometry in aerosol research. |
1 Introduction
Aerosol particles play a pivotal role in the atmosphere: they influence the Earth's energy balance by scattering and absorption of solar and terrestrial radiation and thus determine the global climate, provide environment for heterogeneous chemical reactions and multiphase processes and control the chemistry and composition of the atmosphere.1–4 Moreover, aerosol particles have pronounced effects on human health.2,5 Despite their significance and huge effort in their investigation, aerosols still belong to the least known component of our atmosphere.Significant fraction of atmospheric aerosol particles originates from molecular clusters formed from single molecules in the gas phase via gas-to-particle conversion known as new particle formation (NPF). They grow to larger sizes and eventually become stable against evaporation. However, the detection of the smallest particles in the nanometer size range directly in the atmosphere is difficult.6 Nevertheless, these sizes, where the transition from the gas phase clusters to stable particles occurs, are important.7 The laboratory alternative to investigate the clusters in the NPF process is offered by various aerosol chamber experiments such as CESAM in Paris8 or CLOUD in CERN9 and others,10 or various aerosol flow-tube experiments, see, e.g., ref. 11–13. A different perspective is offered by experiments with clusters in molecular beams in vacuum. They can provide a bottom-up approach and truly molecular-level insight.
It has been well established that the key components in the NPF process are acids, especially sulfuric acid, which have to be stabilized in the clusters by bases such as ammonia and the simple alkyl amines abundant in the atmosphere.2,14–17 Other species, such as highly oxygenated organic molecules (HOMs) might stabilize the clusters as well,18–21 and water vapour is indispensable in the NPF process too.2,14,15 However, dehydrated clusters are often detected in laboratory experiments,17,22–24 thus the exact role of water still has to be better understood.15
Though nitric acid (HNO3) is mainly considered in polar stratospheric cloud particles,3,25 it is ubiquitous throughout the entire atmosphere and its involvement in the particle growth in the atmosphere has been studied.26–29 Recent work has indicated a rapid NPF and growth by nitric acid and ammonia condensation.9 These species are more abundant in polluted areas and thus nitric acid can be responsible for urban smog. Although, there are no indications that clusters of nitric acid HNO3 with dimethylamine (CH3)2NH (DMA) are directly involved in NPF in atmosphere, they represent an excellent model system for understanding detailed acid–base chemistry in the NPF process. Nanoparticle formation and growth from the nitric acid HNO3 and dimethylamine (CH3)2NH (DMA) was investigated before in a flow tube experiment accompanied by theoretical modelling.13 The authors deduced acid:base ratio close to 1:1 in all their relative humidity conditions, which was different from the previous work on sulfuric acid/base particles, namely H2SO4–DMA.12,30,31 Here we approach similar system, DMA–HNO3–H2O clusters, from a complementary experimental and theoretical perspective.
Molecular beam studies of clusters with atmospheric relevance started from the early cluster experiments in 1970s (e.g., see recent reviews 32,33). The early experiments with the hydrated HNO3/H2O clusters performed in Castleman's group,34 revealed the acid dissociation and H3O+⋯NO−3 ion pair generation in these clusters. These early studies were extended by our experiments with Na-doping of the (HNO3)M(H2O)N clusters,35 and electron attachment to these clusters.36,37 Later on, we have investigated also uptake of various atmospherically relevant molecules by these clusters.33,38,39
Here, we dope the preexisting hydrated nitric acid clusters with the dimethylamine molecules: the small well defined (HNO3)M(H2O)N clusters fly isolated in a molecular beam in vacuum and adsorb individual DMA molecules in a pickup chamber.33 Due to the finite size of the clusters, this method allows us observing acid–base reactions and water evaporation directly in the small (DMA)K(HNO3)M(H2O)N clusters. The mass spectrometry of the positively as well as negatively charged clusters accompanied by ab initio quantum chemical calculations of these clusters offers an unprecedented detailed insight into the reaction and ionization processes in these clusters. This truly molecular-level insight provides a basis for detailed understanding of the NPF processes involving nitric acid and dimethylamine.
2 Experimental and theoretical methods
2.1 Experiment
The experiments have been performed with our versatile cluster beam apparatus CLUB described previously, e.g., in our recent reviews32,33 and references therein. The apparatus scheme is provided in accompanying ESI.† Briefly, the (HNO3)M(H2O)N clusters are produced by a continuous supersonic expansion of nitric acid vapour from HNO3-water solution (65% Lach-Ner) in buffer gas helium (99.9999%, Messer). The nitric acid solution was evaporated in a temperature controlled stainless steel reservoir placed directly in the source vacuum chamber with an attached conical nozzle (diameter 90 μm, length 2 mm, full opening angle 30 deg.). The nozzle was heated separately to a temperature usually 5–10 °C higher than the reservoir to prevent condensation in the nozzle. The clustering conditions were controlled by the reservoir temperature TR and He stagnation pressure P0, as exploited in our previous work.35–37 The present results were obtained at TR = 65 °C and P0 = 3 bar. Previous analysis35 revealed that under similar conditions neutral (HNO3)M(H2O)N clusters were generated with M ≈ 1–6 and N ≈ 1–15.The cluster beam was skimmed and passed to a pickup chamber, where dimethylamine (99.8% Sigma-Aldrich) was introduced via a needle valve with a controlled pressure ranging from pc = 2 × 10−4 mbar to 2 × 10−3 mbar. Mass spectra at different DMA pressures were recorded. It ought to be stressed that the individual clusters fly isolated without any interaction in high vacuum, and even at the highest pickup pressure pc = 2 × 10−3 mbar they undergo only several individual collisions with isolated molecules (note that this pickup pressure is still almost 6 orders of magnitude below the atmospheric pressure).
After flying another ≈ 1.5 m through two differentially pumped chambers in ultrahigh vacuum (≤10−7 mbar) the clusters entered the vacuum chamber with a perpendicularly mounted reflectron time-of-flight mass spectrometer (RTOF), which was operated either in the positive or negative ion mode.
In the positive mode, the clusters were ionized by a pulsed electron gun at a frequency of 10 kHz at 70 eV electron energy. A typical electron current was 10 μA. The ionization pulse width was 5 μs. After 0.5 μs delay, to avoid any free electron effects, the ions were extracted by a 4 kV pulse and further accelerated to 8 keV into the time-of-flight region. After ≈ 95 cm flight path, the ions were detected by an MCP detector.
In the negative ion mode, the voltages on the extraction plates were inverted. The clusters were ionized at low electron energies Ee 0–14 eV, and the spectra were recorded in the electron energy steps of 0.2 eV and integrated. The electron pulse was 5 μs long, and after 0.5 μs delay a 3 μs extraction pulse was applied to extract the negative ions. A typical electron current at the present experiment was 0.2 μA at 5 eV. The individual mass spectra recorded at different electron energies allow for the evaluation of the electron energy dependence of the negative ion yield for individual mass peaks.37 The energy spectra were normalized to the electron current. Since the current is very low at electron energies close to zero, below Ee ≈ 1.5 eV the spectra can be subject to a large error.37
2.2 Theory
In order to provide detailed interpretation of collected mass spectra, we characterized structures of dimethylamine with hydrated nitric acid clusters using ab initio calculations and ab initio molecular dynamics simulations. We focused on the following clusters: DMAK(HNO3)M(H2O)N (K = 1–3; M = 1–3; N = 2–6), DMAkHNO3(H2O)nH+ (k = 1–3; n = 2–6) and negatively charged DMAk(HNO3)m(H2O)nNO−3 and DMAk(HNO3)m(H2O)nNO−2 (k = 1–3; m = 1, 2; n = 0–4) which will be used to interpret the mass spectra.To sample the configurational space of the clusters, we used Born–Oppenheimer molecular dynamics40 using the CP2K program package.41 For treatment of the electronic structure we applied revPBE42 functional with the D3 dispersion correction.43 The core electrons were described by the Goedecker–Tetter–Hutter norm-conserving pseudopotentials,44 with Kohn–Sham orbitals being expanded in TZV2P MOLOPT basis set.45 For the auxiliary plane-basis set a cutoff of 400 Ry was used. The equations of motion were integrated with a time step of 0.5 fs. To keep the temperature constant a CSVR thermostat46 was used with a coupling constant of 50 fs. We generated at least one 20 ps trajectory at 300 K for each of the systems, specifically one for clusters containing one HNO3 unit, two for dehydrated clusters with two HNO3 units and three for larger clusters. Periodic boundary conditions were applied. Box length of the cubic simulation cell was 14 Å.
To calculate the most stable structure of each system, we optimized structures at first at the PM6 (ref. 47) level of theory which was then followed up by the M06-2X/aug-cc-pVDZ48,49 level of theory. The DFT functional has been chosen in order to be consistent with previous studies of systems containing hydrated nitric acid36,39 where it was benchmarked against CCSD(T). M06-2X has been shown to provide reliable results for atmospherically relevant clusters.50 We gathered these structures from snapshots of the previously calculated trajectories. Snapshots were spaced by 1 ps. We performed frequency analysis to confirm the local minimum. The energy span of the resulting structures was 20–90 kJ mol−1, the structure with the lowest energy was then used to evaluate the binding energy of either dimethylamine or water to the cluster at M06-2X/aug-cc-pVDZ level. The binding energy includes zero-point vibrational energy. We performed all quantum chemical calculations in the Gaussian 16.51
3 Experimental results
3.1 Positive spectra
(Fig. 1) summarizes the evolution of the positive ion mass spectra with the increasing pressure of DMA in the pickup chamber. Top panel (a) shows the spectrum of pure (HNO3)M(H2O)N clusters without any pickup. It exhibits the protonated mass peak series (HNO3)m(H2O)nH+, which are labeled by different symbols and colors for series with different number of nitric acid molecules m = 0–4. Some peaks are labeled by their acid-to-water m:n ratio and the corresponding m/z for easier orientation. The peculiar shape of the spectrum has been observed in previous experiments34,35,38,39 and it was explained by the acid dissociation and H3O+⋯NO−3 ion pair generation.34,35
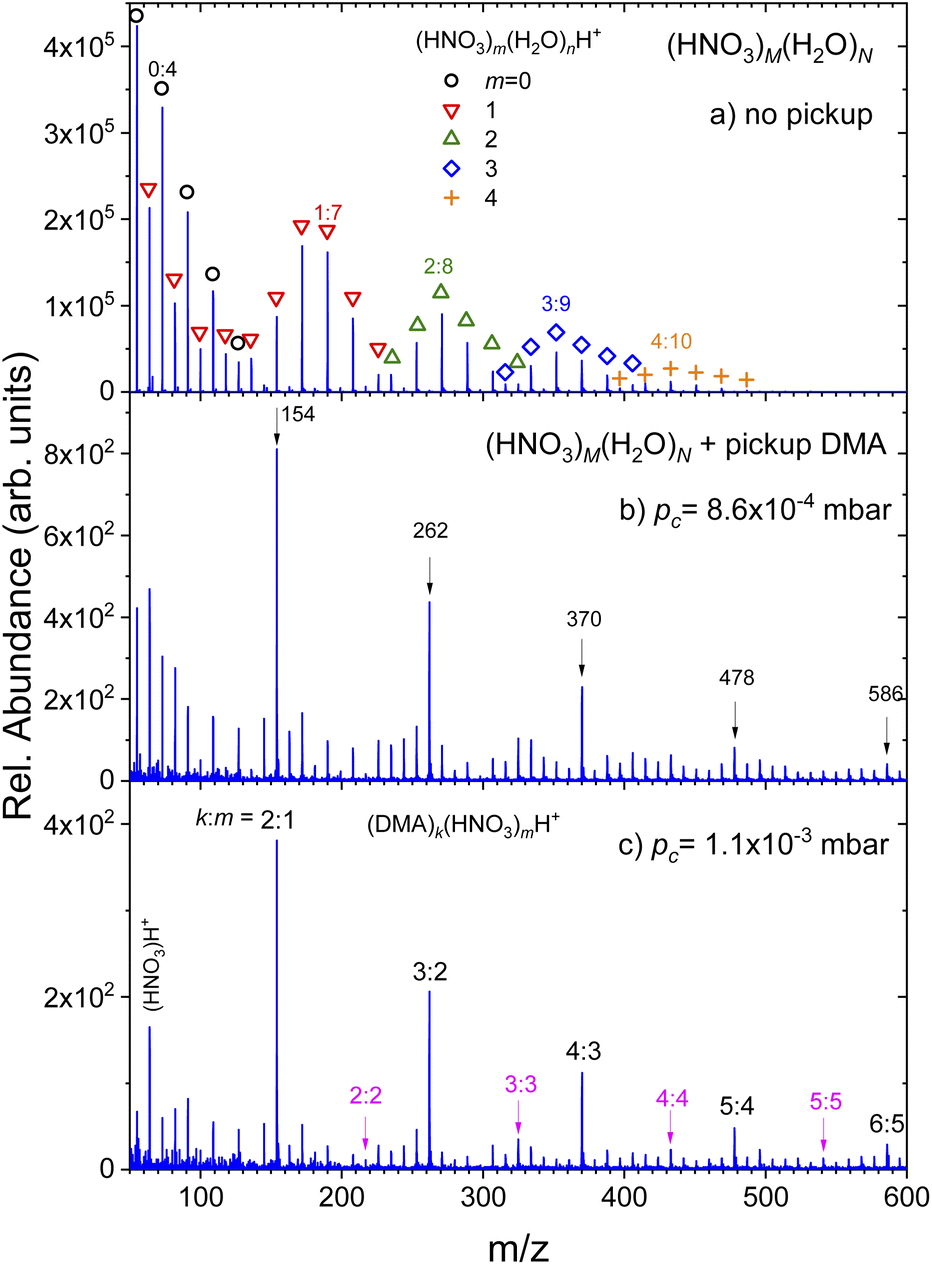 | ||
| Fig. 1 Mass spectra of (HNO3)M(H2O)N clusters after the pickup of DMA molecules. The top spectrum (a) shows the pure clusters without any pickup. Individual protonated mass peak series (HNO3)m(H2O)nH+ are labeled by symbols and some peaks are labeled by m:n ratio for easier orientation. Panels below show the spectra evolution with increasing pressure of DMA in the pickup chamber: (b) 8.6 × 10−4 mbar and (c) 1.1 × 10−3 mbar. The most pronounced peaks are labeled by their m/z in (b), and assigned k:m of (DMA)k(HNO3)mH+ peaks in (c). | ||
Panels below show the spectra evolution with increasing pressure of DMA in the pickup chamber (b) 8.6 × 10−4 mbar and (c) 1.1 × 10−3 mbar. Additional mass spectra measured at different pc pressures are shown in ESI.† The spectra were measured under the same conditions except for the pickup gas pressure pc. Thus, the differences between panels in the relative signal intensities reflect the decreasing cluster beam intensity with increasing DMA pressure due to the cluster scattering out of the beam. The relative increase of peak intensities at the higher mass side of the spectrum with respect to the lower mass side is simply the consequence of the easier scattering of smaller (lighter) clusters out of the beam.
The important difference between the spectra are the pronounced peaks that clearly stand out as the DMA pressure increases. They are labeled by their mass-to-charge ratio (m/z) in panel (b). Although there are mass coincidences with the original (HNO3)m(H2O)nH+ masses, these peaks must be due to the picked up DMA molecules. Therefore, they are assigned to (DMA)k(HNO3)mH+ ions with k = m + 1, as indicated in the last panel (c) where these peaks are labeled by k:m. Upon careful inspection, further mass peaks corresponding to the (DMA)k(HNO3)mH+ series with k = m could be unambiguously identified as indicated also in panel (c). However, this series is very weak compared to the k = m + 1 series. No hydrated species (DMA)k(HNO3)m(H2O)nH+ could be identified unambiguously. They would partly coincide with the original (HNO3)m(H2O)nH+ series, however, we have not observed any intensity anomalies which would point to the generation of the (DMA)k(HNO3)m(H2O)nH+ mass peaks upon the DMA uptake.
3.2 Negative spectra
(Fig. 2) shows the negative ion mass spectra after the electron attachment. Top panel (a) shows the spectrum of the pure (HNO3)M(H2O)N clusters. The expansion conditions and the clusters were the same as in (Fig. 1a). The major ion series (HNO3)m(H2O)nNO−2 (red circles) and (HNO3)m(H2O)nNO−3 (green triangles) are indicated. Similar spectra were published and discussed previously.36,37 We briefly recap: The dissociative electron attachment (DEA) to an isolated HNO3 molecule yielded essentially only NO−2 ions with the relative abundance of 96.5% and the rest were OH− ions. The NO−3 ions were observed with only negligible 0.03% abundance.36 On the other hand, the electron attachment to the hydrated HNO3 in the clusters changed the populations in favor of the NO−3 containing cluster ions. The (HNO3)m(H2O)nNO−3 ions with different m and n dominated the spectrum with integrated intensity of 56% over the (HNO3)m(H2O)nNO−2 ions with 32%. The rest corresponded to HNO−3 and OH− containing cluster ions with 8% and 2%, respectively.36,37 In our current spectrum in (Fig. 2a), the ratio of integrated intensities of the ion series containing NO−2 and NO−3 is 1.0 ± 0.1, which is slightly different from the previous spectra36,37 due to somewhat different expansion conditions as discussed in ESI.† Nevertheless, our main focus here is the qualitative change of the spectra with the addition of the DMA molecules.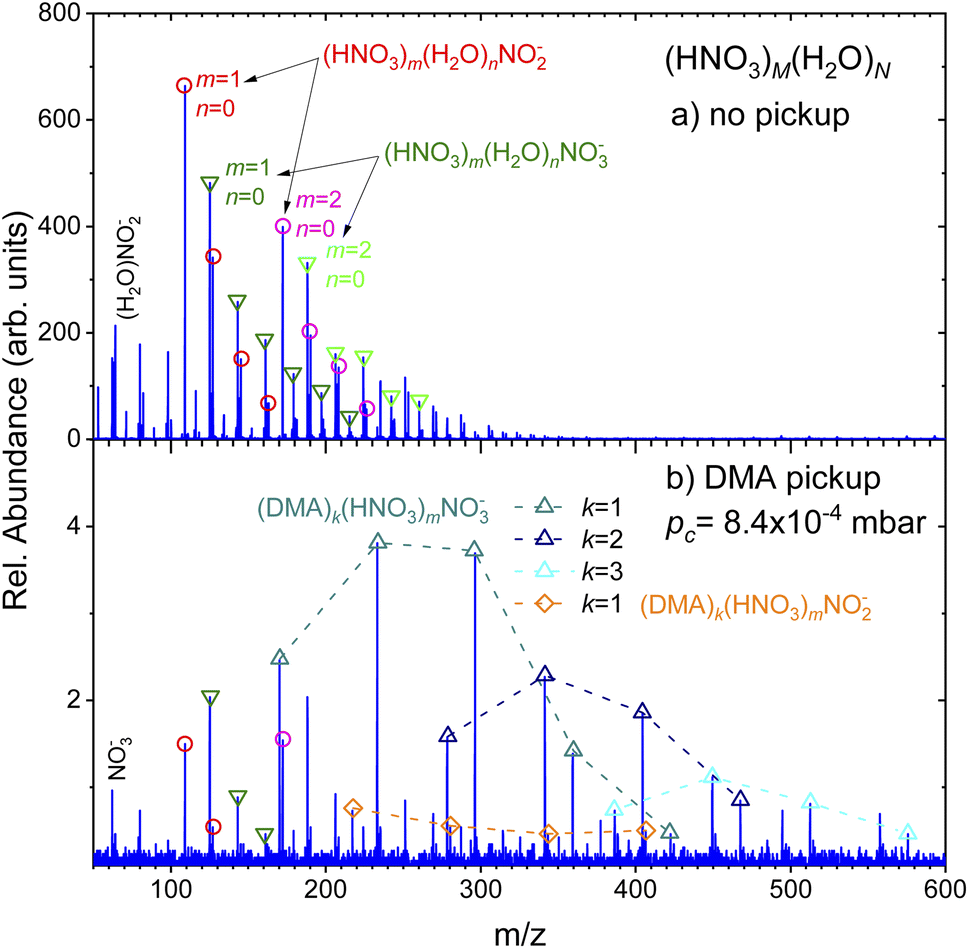 | ||
| Fig. 2 Top panel (a) shows the negative ion mass spectra after the electron attachment to the pure (HNO3)M(H2O)N clusters. Major ion series (HNO3)m(H2O)nNO−2 (circles) and (HNO3)m(H2O)nNO−3 (triangles down) are indicated. Bottom spectrum (b) shows the same clusters after pickup of DMA molecules (pressure in the pickup cell pc = 8.4 × 10−4 mbar. Here, the major series correspond to (DMA)k(HNO3)mNO−3 (triangles up). A minor (DMA)(HNO3)mNO−2 series is indicated (diamonds). | ||
The negative ion spectrum after the pickup is shown in the bottom panel (Fig. 2b). The change in the spectrum with pickup pressures was relatively abrupt and appeared just at higher pc. Therefore only the spectrum at pc = 8.4 × 10−4 mbar is shown in (Fig. 2b), which corresponds to the positive ion spectrum in (Fig. 1b). Due to the scattering of the clusters from the beam, the ion abundances are very low at higher pc pressures.
Clearly, new major series appear in the spectrum, which could be unambiguously assigned to (DMA)k(HNO3)mNO−3 ions (triangles up). The series distinguished by different colors and connected by dashed lines always start with k = m ion followed by ions with more HNO3 than DMA molecules m > k. Series with k = 1–3 and m up to k + 4 and k + 3 are indicated in (Fig. 2b). New series with DMA molecules and NO−2 ions, (DMA)k(HNO3)mNO−2, could be identified in the spectrum too, however, at very low intensities. The series indicated by the orange diamonds corresponds to the k = 1 case. The first member of this series corresponds to m = k + 1. A very weak k = 2 series could be found upon a closer inspection (not indicated to avoid congestion).
Traces of the series from the top spectrum can be found in (Fig. 2b) too, and are labeled by the same symbols as in the top panel (circles and triangles down). We note that the relative intensities of the original NO−2 and NO−3 series change upon the DMA pickup: while in the pure cluster spectrum (a), the NO−2 containing peaks (circles) are higher than the corresponding NO−3 containing peaks (triangles down), in the bottom spectrum with DMA pickup (b) the ratio is reversed. The total integrated intensities of the NO−3 ion containing series (including the ones with DMA) constitute over 80% of the mass spectrum, i.e., the ratio of the cluster ion series containing NO−3:NO−2 is about 4.3 ± 0.5 (the larger error bars are due to the lower intensity and ambiguity in the assignment of some less abundant peaks due to mass coincidences).
The positive ion mass spectra indicated no clear evidence for any mass peaks containing DMA, HNO3 and water. Since we start with strongly hydrated clusters that pick up DMA molecules, the question arises, if the water evaporates upon the DMA pickup and subsequent acid–base reaction, or if it is the result of the cluster ionization process eventually followed by ion–molecule reactions. Therefore, we seek for evidence of hydrated mixed clusters upon the electron attachment. The attachment might be a softer ionization leading less to water evaporation than the 70 eV electron impact ionization. There is a series of 3 peaks following the first (DMA)k(HNO3)mNO−3, k = m = 1, which are displaced by m/z = 18. They could correspond to the hydrated cluster ions (DMA)k(HNO3)m(H2O)nNO−3, n = 1–3. Due to mass coincidences we discuss these peaks in more detail in ESI.† Nevertheless, we can safely conclude that the abundance of hydrated cluster ions is strongly suppressed with respect to the non-hydrated ones.
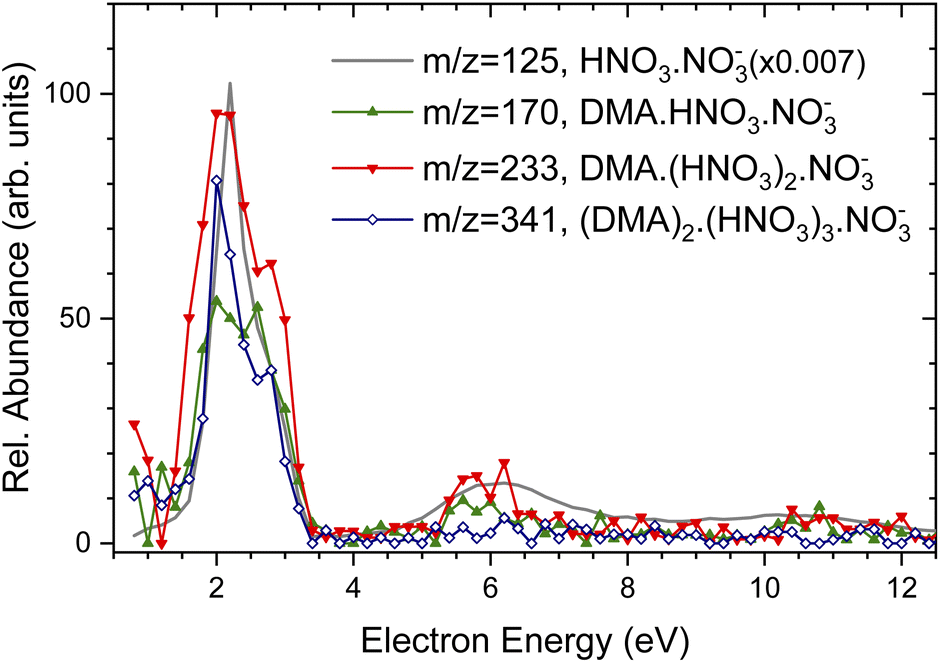 | ||
| Fig. 3 Negative ion yield dependence on the electron energy for different mass peaks: (DMA)1(HNO3)1NO−3, m/z = 170, green triangles up; (DMA)1(HNO3)2NO−3, m/z = 233, red triangles down; (DMA)2(HNO3)3NO−3, m/z = 341, blue opened diamonds. For comparison, the grey line shows the Ee dependence of (HNO3)NO−3, m/z = 125 mass peak in the pure spectrum without pickup (intensity multiplied by a factor of 0.007). | ||
For comparison, the grey line shows the Ee-dependence of (HNO3)NO−3, m/z = 125 mass peak in the pure spectrum without pickup (note, that its intensity was scaled down by more that two orders of magnitude). This spectrum is identical with the Ee-dependencies of electron attachment to the pure (HNO3)M(H2O)N clusters measured and analyzed in detail previously.37 Thus, Fig. 3 demonstrates that the energy spectra of the DMA-containing clusters exhibit essentially the same character as the spectra of the pure clusters without the DMA pickup, and the electron attachment proceeds via the same mechanism to the pure (HNO3)M(H2O)N as well as to the (DMA)K(HNO3)M(H2O)N clusters with the adsorbed DMA molecules. Due to the relatively low signal-to-noise ratio of the negative ion signal and mass coincidences, no further unambiguous conclusions could be drawn from the energy dependencies, and therefore we present somewhat extended discussion only in ESI† for interested reader.
4 Computational results
The calculations allow to disentangle the pickup of DMA on the hydrated acid clusters and subsequent positive or negative ionization.4.1 Pickup of DMA on neutral clusters
We start with pickup of DMA on (HNO3)(H2O)5 which corresponds to one of the more populated clusters in our experiment and is known to have acidically dissociated structure.36Fig. 4a and b show the binding energies of H2O and DMA molecules, respectively, as a function of hydration with n-water molecules. Note that, throughout the whole manuscript, we use enthalpies rather than free energies52 to allow for easier comparison with the experiment where the clusters have presumably a low temperature, though its exact value is challenging to determine experimentally. We use “binding strength” or “binding energy” without the negative sign, i.e., the enthalpy change associated with the processes listed in captions of Figs. 4, 5 and 7. The attachment of a DMA molecule to the (HNO3)(H2O)n cluster is followed by an energy release of about 100 kJ mol−1 for a structure containing n = 3 to 5 water molecules (Fig. 4b). This is due to the proton transfer from H3O+ to DMA in the clusters with ion pair already generated, n = 4 and 5. For n = 3 the proton transfers directly from the HNO3 molecule. The binding strength of the first attached DMA increases with the cluster size because of the favorable hydration of the charged species. The trend for the second (neutral) DMA is the opposite and the binding energy of the second DMA to the clusters that are more populated in our experiment (HNO3)(H2O)n, n = 3–5, is about 45 kJ mol−1.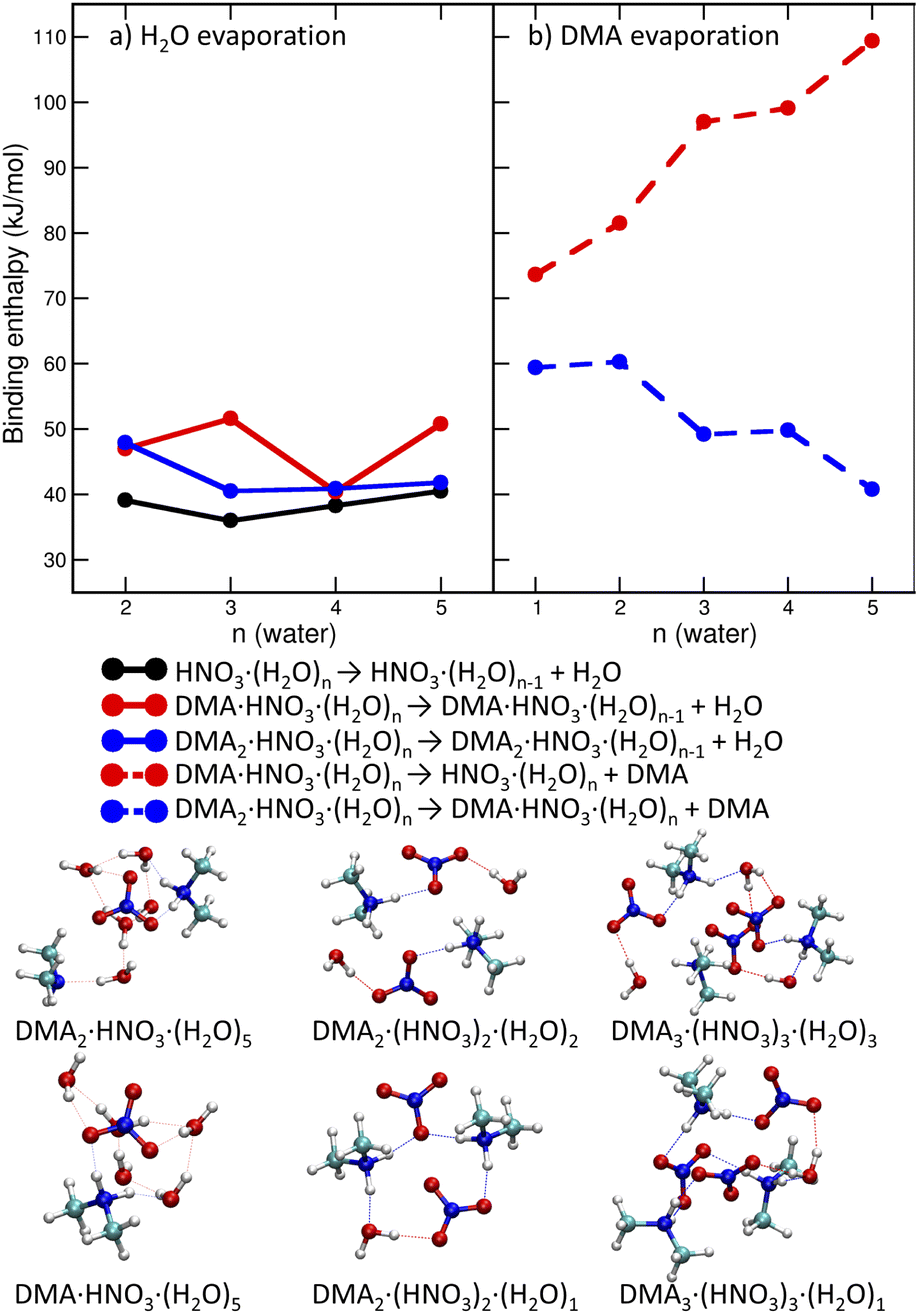 | ||
| Fig. 4 Binding enthalpies of water molecules (a, solid lines) and DMA molecules (b, dashed lines) in DMAK(HNO3)(H2O)N clusters as a function of number of water molecules. The bottom panel shows structures of selected neutral clusters. | ||
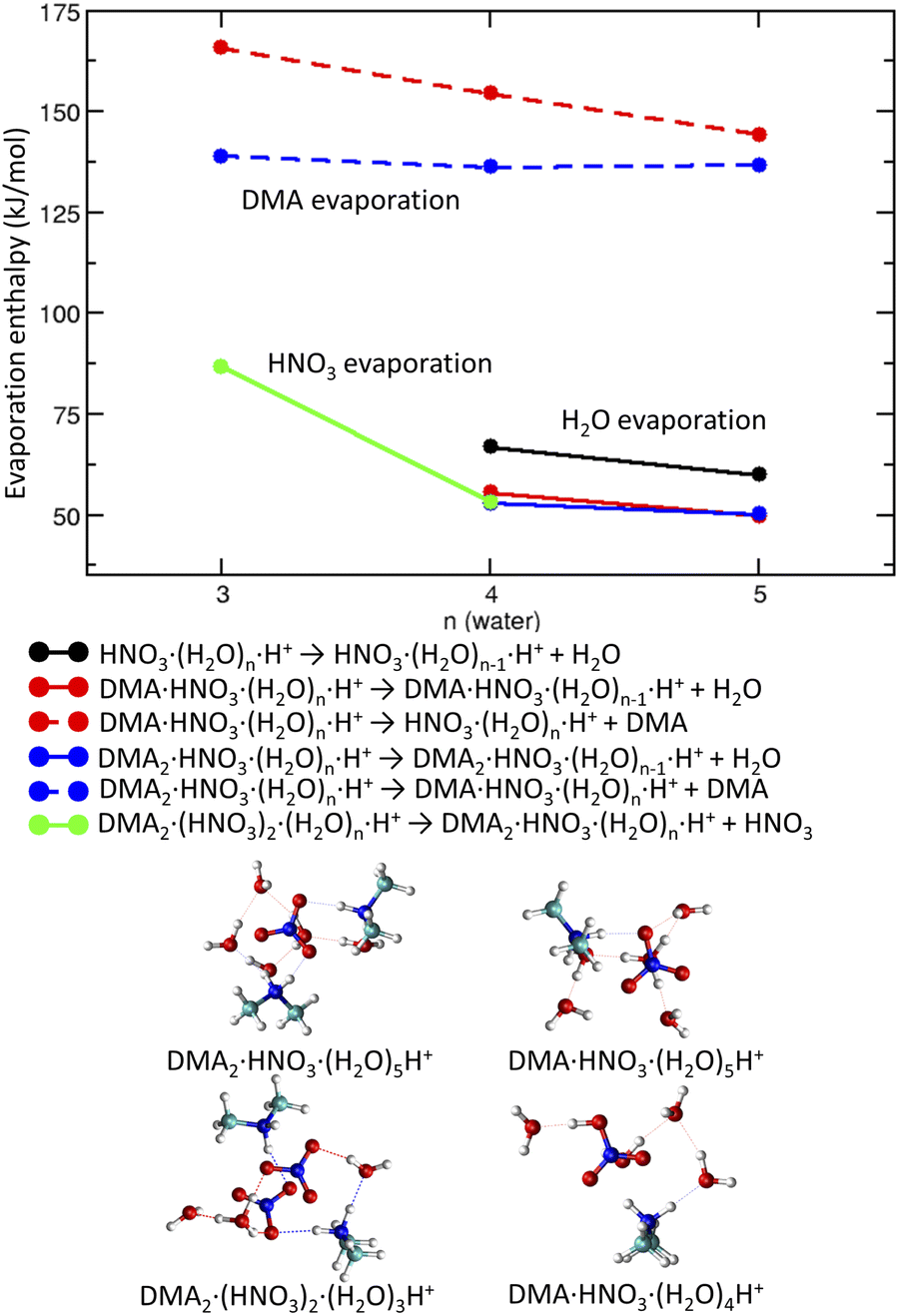 | ||
| Fig. 5 Binding enthalpies of water molecules (solid lines), DMA molecules (dashed lines) and HNO3 (green line) in DMAk(HNO3)(H2O)nH+ clusters as a function of number of water molecules (top). The bottom shows structures of selected protonated clusters. | ||
The left panel (Fig. 4a) illustrates that binding of a water molecule is about 45 kJ mol−1 nearly independently of the clusters size. Therefore, the pickup of DMA is sufficient to evaporate two water molecules. The pickup of the second DMA molecule is possible, but it releases less energy, because the molecule remains neutral and has a binding strength comparable to water.
DMA is a stronger base than NO−3 or water, thus clusters with n ≥ 1 contain the DMA·H+⋯NO−3 ion pair. This feature is preserved in larger structures (DMA:HNO3 2:2 or 3:3) which possess acidically dissociated structures even without any water (see Fig. 4 bottom).
4.2 Positively charged clusters
The second analyzed step was the ionization of clusters. The positive ionization of DMAK(HNO3)M(H2O)N by 70 eV electrons creates protonated clusters DMAk(HNO3)m(H2O)nH+. The structures always contain protonated DMA·H+. In the cluster ions DMA(HNO3)(H2O)nH+ with less than n ≤ 4 water molecules, the neutral HNO3 is formed (see Fig. 5 bottom, right). More hydrated clusters DMA(HNO3)(H2O)nH+ (n ≥ 5) possess structures with a DMA·H+⋯NO−3 ion pair and the additional proton resides on a water molecule forming H3O+. In case of (DMA)2(HNO3)2(H2O)nH+, structure containing H3O+ is preferred for n ≥ 3 and neutral HNO3 is more likely in case of n ≤ 2 (see ESI†).The experimental mass spectra contain predominantly the series (DMA)k(HNO3)mH+ ions with k = m + 1 and small signals corresponding to k = m. This can be explained by the binding energy of each of the molecules (Fig. 5 top). The most likely molecule to evaporate upon the ionization is water with binding energy of around 55 kJ mol−1 in protonated clusters of different composition. The binding of water to the protonated clusters containing DMA (red and blue circles, full lines) is slightly weaker than to those without DMA (black circles). The overall positive charge of the cluster strengthens water binding by about 10 kJ mol−1 in comparison with the neutral structure. The second weakest bound moiety is nitric acid with about 85 kJ mol−1 cost for evaporation from DMA2(HNO3)2(H2O)3H+ (green line). The most tightly bound species is DMA (about 150 kJ mol−1), because it is protonated in all clusters. The binding energy of the first DMA is by about 25 kJ mol−1 higher than of the second one for n = 3. This difference decreases with increasing number of water molecules. Thus, evaporation of HNO3 from DMA2(HNO3)2(H2O)nH+ giving DMA2(HNO3)(H2O)nH+ is plausible and corroborates with the dominant k = m + 1 series in the mass spectra.
The tight binding of both DMA molecules in the positively charged clusters corresponds to the fact that in order to evaporate them, the extra proton has to be accepted by a weaker base at first. From structural point of view, the clusters maximizing the number of DMA·H+ and NO−3 ions are the optimal ones. This is fulfilled for K = M in the neutral clusters and for k = m + 1 in the protonated ones (Fig. 5 left).
4.3 Negatively charged clusters
After a low energy electron attachment, the following chemical reactions releasing H, OH and NO2 radicals can occur:| DMAK(HNO3)M(H2O)N + e− → DMAK(HNO3)M−1(H2O)NNO3− + H | (1) |
| DMAK(HNO3)M(H2O)N + e− → DMAK(HNO3)M−1(H2O)NNO2− + OH | (2) |
| DMAK(HNO3)M(H2O)N + e− → DMAK(HNO3)M−2(H2O)N+1NO3− + NO2 | (3) |
Based on the reaction enthalpies (Fig. 6), the predominant pathway is the release of NO2 radical (∼−130 kJ mol−1) and formation of NO−3-containing ions. The reaction enthalpy does not depend significantly on the amount of DMA, HNO3 and H2O in the clusters. The second, but less likely possibility, is the formation of OH radical and generation of NO−2-containing ions which is slightly exothermic (∼−20 kJ mol−1) in our clusters. The hydrogen radical is unlikely to form, because the reactions are endothermic (∼40 kJ mol−1). Thus, the energetics suggests that the clusters containing NO−3 will be more populated than those with NO−2.
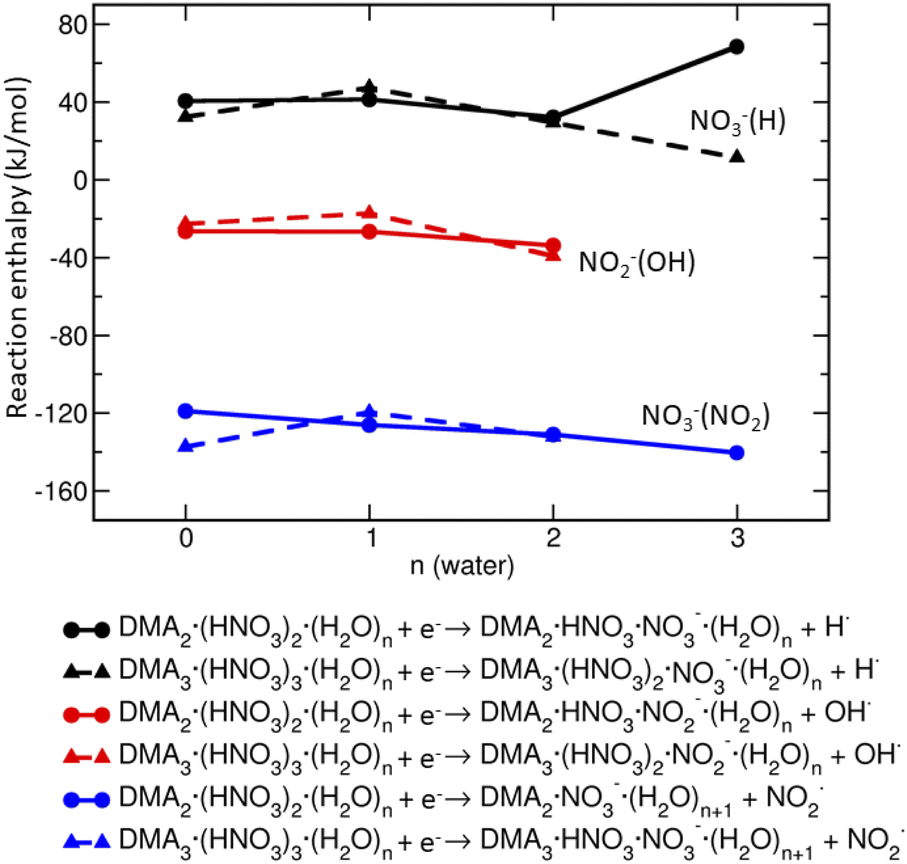 | ||
| Fig. 6 Reactions enthalpies of potential processes occurring after electron attachment to DMAK(HNO3)M(H2O)N: formation of H radical with NO−3 remaining in the cluster (black), OH radical with NO−2 remaining in the cluster (red), and NO2 radical with NO−3 remaining in the cluster (blue). Triangles correspond to the DMA3(HNO3)3(H2O)N, circles to the DMA2(HNO3)2(H2O)N clusters. | ||
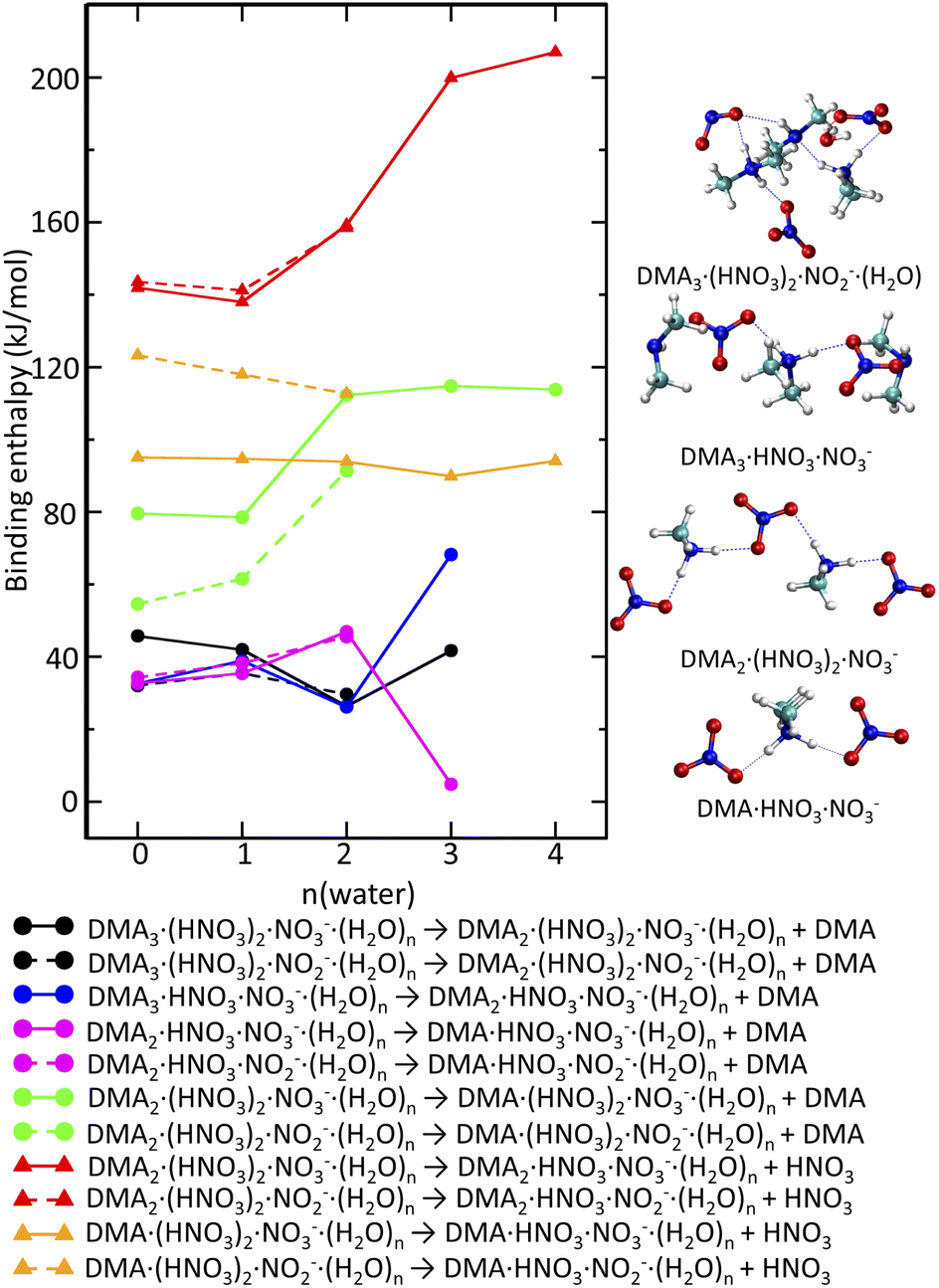 | ||
| Fig. 7 Binding enthalpies of DMA (circles) and HNO3 (triangles) in DMAk(HNO3)m(H2O)nNO−3 (solid lines) or DMAk(HNO3)m(H2O)nNO−2 (dashed lines) clusters as a function of number of water molecules. Different colours correspond to different cluster composition. The right panel shows structures of selected negative clusters. | ||
If just the reactions triggered by electron attachment to the DMAK(HNO3)M(H2O)N clusters containing the same number of DMA and HNO3 molecules K = M were considered, the resulting structures should contain more DMA than HNO3. This is not the case, since we observe DMAk(HNO3)mNO−3 cluster ions with m ≥ k. Therefore, evaporation of molecules from the clusters generated immediately after the electron attachment has to be quantified too. The binding strengths of DMA and H2O to the negatively charged clusters are similar to those in neutral clusters. The binding energy of a water molecule is about 50 kJ mol−1 (see ESI).† Binding of DMA in the intermediate clusters is on average by a few kJ mol−1 weaker (Fig. 7 black, blue and magenta lines). Clusters containing NO−3 or NO−2 do not differ in this aspect. It is likely that water molecules and DMA molecules which cannot be protonated evaporate leading to the cluster composition with m = k (see labels of the black, blue and magenta lines).
In contrast to that, HNO3 is tightly bound (at least 150 kJ mol−1) in the DMAk(HNO3)m(H2O)nNO−3 or DMAk(HNO3)m(H2O)nNO−2m = k clusters (Fig. 7 red). DMA binding to the m = k cluster (Fig. 7 green) is also stronger than to the above mentioned m ≤ k clusters, but the interaction is weaker than in case of HNO3. The corresponding values increase from 80 kJ mol−1 for less hydrated clusters containing NO−3 to 120 kJ mol−1 more hydrated clusters. The lower values for analogous process in DMAk(HNO3)m(H2O)nNO−2 can be rationalized by the presence of HONO (not NO−2) (see ESI).† Thus, the evaporation of DMA from the m = k clusters leading to the experimentally observed m ≥ k series is more likely than the hypothetical HNO3 evaporation. Even structures with m ≥ k bind HNO3 quite strongly, even though not as much as in the m = k case. The evaporation costs more than 90 kJ mol−1 (Fig. 7 orange).
Fig. 7 (right) shows some representative structures. The intermediate DMA3(HNO3)NO−3 formed after electron attachment to the neutral DMA3(HNO3)2 clusters has one DMA·H+ symmetrically surrounded by NO−3 and two neutral DMA molecules. The facile evaporation of the later leads to DMA(HNO3)NO−3 which is in fact DMA·H+(NO−3)2. The tendency to maximize the number of protonated DMA·H+ and anionic units is preserved in negatively charged clusters too. It occurs for k = m in DMAk(HNO3)mNO−3,2. In addition, the evaporation of DMA from the m = k clusters is more facile than evaporation of HNO3. The resulting m ≥ k structures observed in the experimental spectrum are still relatively stable with respect to the HNO3 evaporation.
5 Discussion
We start with the composition of the clusters focusing on the DMA:HNO3 ratio detected in the mass spectra under different types of ionization. The positive ion mass spectra in Fig. 1 show that upon pickup of DMA molecules on the hydrated (HNO3)M(H2O)N clusters and subsequent electron ionization, protonated clusters (DMA)k(HNO3)mH+ are generated with one more DMA molecule than HNO3, k = m + 1. Minor series with k = m is also discernible. On the other hand, electron attachment to the same neutral clusters yields mainly (DMA)k(HNO3)mNO−3 series with m ≥ k. Analogous series with NO−2 and m > k are also present at very small abundances.
The calculations suggest that the optimal structures are those maximizing the number of DMA·H+ and NO−3 ions. This occurs for K = M in neutral clusters, for k = m + 1 in protonated ones and for k = m in DMAk(HNO3)mNO−3,2. Fig. 7 rationalizes the experimental observation that the major peaks in the negative spectra correspond to m > k (specifically, m = k + 1 and m = k + 2), since additional DMA molecules can evaporate from the negatively charged clusters, while the evaporation of HNO3 is less likely, as discussed above. In addition, we have to explain why the clusters with m ≥ k + 2 are still observed experimentally. The experimental spectrum reflects the stability of the ions as well as the population of the original neutral species. If we start from neutral clusters with more HNO3 than DMA molecules M > K, it is easier to end up with the m > k negatively charged ionic clusters. It ought to be mentioned that the neutral (DMA)K(HNO3)M(H2O)N clusters are generated by the pickup of K molecules of DMA on (HNO3)M(H2O)N, and thus the initial K:M composition can be different from the optimal K = M neutral cluster structure, and the clusters with M > K can be more abundant. Upon the positive ionization, there is enough energy delivered in the ionization process to reach the optimal k = m + 1 structure of the protonated (DMA)k(HNO3)mH+ cluster ions, while the clusters with m ≥ k + 2 are still observed upon the electron attachment.
Second point to discuss is the dehydration of clusters. We start the experiment with highly hydrated clusters before the DMA pickup. In the positive (HNO3)m(H2O)nH+ spectrum of pure clusters without DMA pickup, the average cluster ion contains six to three H2O molecules per one HNO3 molecule. In addition, at least OH and probably also some water molecules are evaporated upon the electron ionization at 70 eV. Therefore, the number of H2O molecules in the neutral cluster is even larger than in the observed cluster ion fragment, N > n. Upon the DMA pickup mostly cluster ions without any water are observed in the mass spectrum. Does the water evaporate from the neutral cluster due to the acid–base reaction, or upon the subsequent ionization? The comparison of the binding energies suggest that the uptake of one DMA and subsequent acid–base reaction is sufficient to evaporate about two water molecules. In addition, the average collision energy of DMA molecules with clusters in our experiments corresponds to about 58 kJ mol−1, which could lead to another single water molecule evaporation. However, this is not quite sufficient for the complete dehydration of the clusters, thus, the remaining water molecules are evaporated by both, positive and negative ionization processes.
Finally, we concentrate on the effect of cluster medium observed in the negative ion spectra. Let us remind that the DEA of an isolated HNO3 molecule yielded essentially only NO−2 ions with the relative abundance of 96.5% with almost no traces of NO−3.36 Going from the gas phase HNO3 to the hydrated HNO3 clusters, our previous investigations36,37 showed that the relative population of NO−3 ions upon the electron attachment substantially increased. In the present experiment, the ratio of integrated intensities of the ion series containing NO−3 and NO−2 is 1.0 ± 0.1. Upon further addition of the DMA molecule, the propensity for the NO−3 ion based clusters is even stronger with the ratio of the cluster ion series containing NO−3:NO−2 of about 4.3 ± 0.5.
The change of the NO−3:NO−2 ratio upon the uptake of DMA can be discussed in terms of a simple mechanism of electron attachment to an ion pair formed in cluster. If a neutral cluster contains an ion pair – either H3O+⋯NO−3 or DMA·H+⋯NO−3 – the electron is likely to attach to the protonated species leading to the hydrogen atom ejection from the cluster and the NO−3-containing cluster ion formation. For HNO3(H2O)N clusters with single HNO3 molecule, the H3O+⋯NO−3 ion pair formation occurs at n ≥ 4.34–36,52,53 Our present calculations indicate that the DMA·H+⋯NO−3 ion pair is already formed in clusters with a single water molecule because of DMA higher basicity. With addition of second HNO3 molecule, the number of water molecules required to form an ion pair is reduced even further. For example, with two HNO3 molecules only three water molecules are needed for the ion pair to be energetically preferred in (HNO3)2(H2O)3 cluster, and the presence of DMA in the cluster induces spontaneous proton transfer even without any water. Thus, the relative abundance of ion-pair containing clusters increases upon the DMA addition. Since they lead to the NO−3-containing cluster ions upon the electron attachment, the relative abundance of these ions increases upon the DMA addition, as observed experimentally.
However, this simple mechanism cannot explain the observation of the NO−2 containing cluster ions, which are present in the spectra too, albeit at low intensities. Therefore, we present the argumentation based on the reaction energetics summarized in Fig. 6: the formation of NO−3 is more exothermic than NO−2, but NO−2-containing clusters can be formed as well.
6 Conclusions
We have investigated the formation of atmospherically relevant (DMA)K(HNO3)M(H2O)N clusters by uptake of DMA on (HNO3)M(H2O)N and subsequent mass spectrometry analysis using both positive and negative ionization. Our experimental results aided by theoretical calculations indicate that:• Optimal neutral cluster composition corresponds to DMA and HNO3 in 1:1 ratio. This is rationalized by the tendency to maximize the number DMA·H+ and NO−3 ions in the clusters and by binding energies of cluster constituents.
• The dominant series in the positive mass spectra is k = m + 1 which is nicely supported by calculated structures where the additional H+ resides on the extra DMA and prevents its evaporation.
• Upon an electron attachment, a formation of NO−3 is more likely than NO−2 which is due to the presence of the DMA·H+⋯NO−3 ion pair structures and reaction energetics (shown in Fig. 6). In addition, calculations give k = m as the optimal composition. The observation m > k is due to the fact that the negatively charged cluster evaporate DMA more easily than HNO3 according to the calculated evaporation enthalpies, and also due to the experimental neutral cluster composition.
An interesting point is the effect of cluster environment on the product ions observed in the electron attachment experiments. The population of NO−3 and NO−2 ions depends strongly on the medium. The dissociative electron attachment (DEA) to an isolated HNO3 molecule yields essentially exclusively NO−2 ions, while the NO−3 ion abundance increases with hydration: in the present pure HNO3/H2O clusters the ratio of integrated intensities of the ion series containing NO−3 and NO−2 ions is 1.0 ± 0.1. With addition of the strong base DMA, this ratio increased to about 4.3 ± 0.5. The propensity for NO−3-containing cluster ions can be explained by a simple model of electron attachment to the protonated species in the neutral clusters containing H3O+⋯NO−3 or DMA·H+⋯NO−3 ion pairs, which are generated due to the proton transfer in the acid–base reaction. Nevertheless, to justify the generation of both NO−3 and NO−2-containing cluster ions, we calculate the reaction energetics, which explains the observed tendencies qualitatively.
We also demonstrate that the observed ionic species strongly depend on the ionization procedure. While the positive ion spectrum suggests prevalence of DMA molecules in the clusters, the negative spectrum clearly points to more HNO3 molecules in clusters. In addition, the observed major cluster ions do not contain any water, while the spectra of pure clusters without DMA pickup clearly exhibit a high level of hydration in the neutral clusters. Therefore, extreme caution has to be exercised when conclusions about neutral cluster composition are made from mass spectra, which should be considered in any implementation of mass spectrometry in aerosol research.
Author contributions
A. Pysanenko: performed experiment and data evaluation; K. Fárníková: theoretical calculations and visualization; J. Lengyel: proposed idea for the experiment, helped data acquisition, interpretation and writing; E. Pluhařová: supervised and performed theory, interpretation and writing; M Fárník: supervised and designed experiment, interpretation, original draft and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support of the Czech Science Foundation (GAČR) project no.: 21-07062S. J. Lengyel acknowledges support of Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; Project LE 4583/1-1). Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA CZ LM2018140) supported by the Ministry of Education, Youth and Sports of the Czech Republic.Notes and references
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the upper and lower atmosphere, Academic Press, San Diego, 2000 Search PubMed.
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- C. E. Kolb and D. R. Wornsnop, Annu. Rev. Phys. Chem., 2012, 63, 471–491 CrossRef CAS PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons Inc., Hoboken, NJ, 2016, p. 3rd ed Search PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts, Science, 1997, 276, 1045–1051 CrossRef CAS PubMed.
- F. Bianchi, J. Tröstl, H. Junninen, C. Frege, S. Henne, C. R. Hoyle, U. Molteni, E. Herrmann, A. Adamov, N. Bukowiecki, X. Chen, J. Duplissy, M. Gysel, M. Hutterli, J. Kangasluoma, J. Kontkanen, A. Kürten, H. E. Manninen, S. Münch, O. Peräkylä, T. Petäjä, L. Rondo, C. Williamson, E. Weingartner, J. Curtius, D. R. Worsnop, M. Kulmala, J. Dommen and U. Baltensperger, Science, 2016, 352, 1109–1112 CrossRef CAS PubMed.
- M. Kulmala, J. Kontkanen, H. Junninen, K. Lehtipalo, H. E. Manninen, T. Nieminen, T. Petäjä, M. Sipilä, S. Schobesberger, P. Rantala, A. Franchin, T. Jokinen, E. Järvinen, M. Äijälä, J. Kangasluoma, J. Hakala, P. P. Aalto, P. Paasonen, J. Mikkilä, J. Vanhanen, J. Aalto, H. Hakola, U. Makkonen, T. Ruuskanen, R. L. Mauldin, J. Duplissy, H. Vehkamäki, J. Bäck, A. Kortelainen, I. Riipinen, T. Kurtén, M. V. Johnston, J. N. Smith, M. Ehn, T. F. Mentel, K. E. J. Lehtinen, A. Laaksonen, V.-M. Kerminen and D. R. Worsnop, Science, 2013, 339, 943–946 CrossRef CAS.
- J. Wang, J. F. Doussin, S. Perrier, E. Perraudin, Y. Katrib, E. Pangui and B. Picquet-Varrault, Atmos. Meas. Tech., 2011, 4, 2465–2494 CrossRef CAS.
- M. Wang, W. Kong, R. Marten, X.-C. He, D. Chen, J. Pfeifer, A. Heitto, J. Kontkanen, L. Dada, A. Kürten, T. Yli-Juuti, H. E. Manninen, S. Amanatidis, A. Amorim, R. Baalbaki, A. Baccarini, D. M. Bell, B. Bertozzi, S. Bräkling, S. Brilke, L. Caudillo Murillo, R. Chiu, B. Chu, L.-P. De Menezes, J. Duplissy5, H. Finkenzeller, L. Gonzalez Carracedo, M. Granzin, R. Guida, A. Hansel, V. Hofbauer, J. Krechmer, K. Lehtipalo, H. Lamkaddam, M. Lampimäki, C. Ping Lee, V. Makhmutov, G. Marie, S. Mathot, R. L. Mauldin, B. Mentler, T. Müller, A. Onnela, E. Partoll, T. Petäjä, M. Philippov, V. Pospisilova, A. Ranjithkumar, M. Rissanen, B. Rörup, W. Scholz, J. Shen, M. Simon, M. Sipilä, G. Steiner, D. Stolzenburg, Y. Jun Tham, A. Tomé, A. C. Wagner, D. S. Wang, Y. Wang, S. K. Weber, P. M. Winkler, P. J. Wlasits, Y. Wu, M. Xiao, Q. Ye, M. Zauner-Wieczorek, X. Zhou, R. Volkamer, I. Riipinen, J. Dommen, J. Curtius, U. Baltensperger, M. Kulmala, D. R. Worsnop, J. Kirkby, J. H. Seinfeld, I. El-Haddad, R. C. Flagan and N. M. Donahue, Nature, 2020, 581, 184–189 CrossRef CAS PubMed.
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Chem. Rev., 2015, 115, 4259–4334 CrossRef CAS PubMed.
- Y. Huang, M. M. Coggon, R. Zhao, H. Lingell, M. U. Bauer, C. Flagan and J. H. Seinfeld, Atmos. Meas. Tech., 2017, 10, 839–867 CrossRef CAS.
- H. Chen, S. Chee, M. J. Lawler, K. C. Barsanti, B. M. Wong and J. N. Smith, Aerosol Sci. Technol., 2018, 4, 1120–1133 CrossRef.
- S. Chee, N. Myllys, K. C. Barsanti, B. M. Wong and J. N. Smith, J. Phys. Chem. A, 2019, 123, 5640–5648 CrossRef CAS PubMed.
- R. Zhang, A. Khalizov, L. Wang, M. Hu and W. Xu, Chem. Rev., 2012, 112, 1957–2011 CrossRef CAS PubMed.
- J. Elm, J. Phys. Chem. A, 2021, 125, 895–902 CrossRef CAS.
- T. Olenius, O. Kupiainen-Määttä, I. K. Ortega, T. Kurtén and H. Vehkamäki, J. Chem. Phys., 2013, 139, 084312 CrossRef CAS PubMed.
- A. Kürten, T. Jokinenb, M. Simona, M. Sipilä, N. Sarnel, H. Junninen, A. Adamov, J. Almeida, A. Amorime, F. Bianchif, M. Breitenlechner, J. Dommenf, N. M. Donahuei, J. Duplissy, S. Ehrharta, R. C. Flagan, A. Franchin, J. Hakala, A. Hansel, M. Heinritzi, M. Hutterli, J. Kangasluoma, J. Kirkby, A. Laaksonen, K. Lehtipalo, M. Leiminger, V. Makhmutovo, S. Mathot, A. Onnelad, T. Petäjä, A. P. Praplan, F. Riccobono, M. P. Rissanen, L. Rondo, S. Schobesberger, J. H. Seinfeld, G. Steiner, A. Tomé, J. Tröstl, P. M. Winkler, C. Williamson, D. Wimmer, P. Ye, U. Baltensperger, K. S. Carslaw, M. Kulmala, D. R. Worsnop and J. Curtius, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15019–15024 CrossRef.
- J. Elm, T. Kurtén, M. Bilde and K. V. Mikkelsen, J. Phys. Chem. A, 2014, 118, 7892–7900 CrossRef CAS.
- J. Elm, N. Myllys, T. Olenius, R. Halonen, T. Kurtén and H. Vehkamäki, Phys. Chem. Chem. Phys., 2017, 19, 4877–4886 RSC.
- J. Elm, N. Myllys and T. Kurtén, J. Phys. Chem. A, 2017, 121, 4578–4587 CrossRef CAS PubMed.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS.
- J. Kirkby, J. Curtius, J. Almeida, E. Dunne, J. Duplissy, S. Ehrhart, A. Franchin, S. Gagne, L. Ickes, A. Kürten, A. Kupc, A. Metzger, F. Riccobono, L. Rondo, S. Schobesberger, G. Tsagkogeorgas, D. Wimmer, A. Amorim, F. Bianchi, M. Breitenlechner, A. David, J. Dommen, A. Downard, M. Ehn, R. C. Flagan, S. Haider, A. Hansel, D. Hauser, W. Jud, H. Junninen, F. Kreissl, A. Kvashin, A. Laaksonen, K. Lehtipalo, J. Lima, E. R. Lovejoy, V. Makhmutov, S. Mathot, J. Mikkilä, P. Minginette, S. Mogo, T. Nieminen, A. Onnela, P. Pereira, T. Petäjä, R. Schnitzhofer, J. H. Seinfeld, M. Sipilä, Y. Stozhkov, F. Stratmann, A. Tomé, J. Vanhanen, Y. Viisanen, A. Vrtala, P. E. Wagner, H. Walther, E. Weingartner, H. Wex, P. M. Winkler, K. S. Carslaw, D. R. Worsnop, U. Baltensperger and M. Kulmala, Nature, 2011, 476, 429–433 CrossRef CAS PubMed.
- K. Lehtipalo, L. Rondo, J. Kontkanen, S. Schobesberger, T. Jokinen, N. Sarnela, A. Kürten, S. Ehrhart, A. Franchin, T. Nieminen, F. Riccobono, M. Sipilä, T. Yli-Juuti, J. Duplissy, A. Adamov, L. Ahlm, J. Almeida, A. Amorim, F. Bianchi, M. Breitenlechner, J. Dommen, A. J. Downard, E. M. Dunne, R. C. Flagan, R. Guida, J. Hakala, A. Hansel, W. Jud, J. Kangasluoma, V.-M. Kerminen, H. Keskinen, J. Kim, J. Kirkby, A. Kupc, O. Kupiainen-Määttä, A. Laaksonen, M. J. Lawler, M. Leiminger, S. Mathot, T. Olenius, I. K. Ortega, A. Onnela, T. Petäjä, A. Praplan, M. P. Rissanen, T. Ruuskanen, F. D. Santos, S. Schallhart, R. Schnitzhofer, M. Simon, J. N. Smith, J. Tröstl, G. Tsagkogeorgas, A. Tomé, P. Vaattovaara, H. Vehkamäki, A. E. Vrtala, P. E. Wagner, C. Williamson, D. Wimmer, P. M. Winkler, A. Virtanen, N. M. Donahue, K. S. Carslaw, U. Baltensperger, I. Riipinen, J. Curtius, D. R. Worsnop and M. Kulmala, Nature, 2016, 7, 11594 CAS.
- J. Lengyel, A. Pysanenko and M. Fárník, Atmos. Chem. Phys., 2017, 17, 14171–14180 CrossRef CAS.
- T. Peter, Annu. Rev. Phys. Chem., 1997, 48, 785–822 CrossRef CAS.
- G. Huang, X. Zhou, G. Deng, H. Qiao and K. Civerolo, Atmos. Environ., 2002, 36, 2225–2235 CrossRef CAS.
- S. M. Murphy, A. Sorooshian, J. H. Kroll, N. L. Ng, P. Chhabra, C. Tong, J. D. Surratt, E. Knipping, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2007, 7, 2313–2337 CrossRef CAS.
- A. Sorooshian, S. M. Murphy, S. Hersey, H. Gates, L. T. Padro, A. Nenes, F. J. Brechtel, H. Jonsson, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2008, 8, 5489–5520 CrossRef CAS.
- B. R. Bzdek, D. P. Ridge and M. V. Johnston, Atmos. Chem. Phys., 2010, 10, 3495–3503 CrossRef CAS.
- J. Kim, L. Ahlm, T. Yli-Juuti, M. Lawler, H. Keskinen, J. Tröstl, S. Schobesberger, J. Duplissy, A. Amorim, F. Bianchi, N. M. Donahue, R. C. Flagan, J. Hakala, M. Heinritzi, T. Jokinen, A. Kürten, A. Laaksonen, K. Lehtipalo, P. Miettinen, T. Petäjä, M. P. Rissanen, L. Rondo, K. Sengupta, M. Simon, A. Tomé, C. Williamson, D. Wimmer, P. M. Winkler, S. Ehrhart, P. Ye, J. Kirkby, J. Curtius, U. Baltensperger, M. Kulmala, K. E. J. Lehtinen, J. N. Smith, I. Riipinen and A. Virtanen, Atmos. Chem. Phys., 2016, 16, 293–304 CrossRef CAS.
- M. J. Lawler, P. M. Winkler, J. Kim, L. Ahlm, J. Tröstl, A. P. Praplan, S. Schobesberger, A. Kürten, J. Kirkby, F. Bianchi, J. Duplissy, A. Hansel, T. Jokinen, H. Keskinen, K. Lehtipalo, M. Leiminger, T. Petäjä, M. Rissanen, L. Rondo, M. Simon, M. Sipilä, C. Williamson, D. Wimmer, I. Riipinen, A. Virtanen and J. N. Smith, Atmos. Chem. Phys., 2016, 16, 13601–13618 CrossRef CAS.
- M. Fárník and J. Lengyel, Mass Spectrom. Rev., 2018, 37, 630–651 CrossRef.
- M. Fárník, J. Fedor, J. Kočišek, J. Lengyel, E. Pluhařová, V. Poterya and A. Pysanenko, Phys. Chem. Chem. Phys., 2021, 23, 3195–3213 RSC.
- B. D. Kay, V. Hermann and A. W. Castleman, Chem. Phys. Lett., 1981, 80, 469–474 CrossRef CAS.
- J. Lengyel, A. Pysanenko, J. Kočišek, V. Poterya, C. Pradzynski, T. Zeuch, P. Slavíček and M. Fárník, J. Phys. Chem. Lett., 2012, 3, 3096–3109 CrossRef CAS PubMed.
- J. Lengyel, M. Ončák, J. Fedor, J. Kočišek, A. Pysanenko, M. K. Beyer and M. Fárník, Phys. Chem. Chem. Phys., 2017, 19, 11753–11758 RSC.
- J. Lengyel, J. Fedor and M. Fárník, Phys. Chem. Chem. Phys., 2019, 21, 8691–8697 RSC.
- A. Pysanenko, J. Lengyel and M. Fárník, J. Chem. Phys., 2017, 148, 154301 CrossRef.
- J. Lengyel, A. Pysanenko, K. Fárníková, E. Pluhařová and M. Fárník, J. Phys. Chem. Lett., 2020, 11, 2101–2105 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. D. Kühne, M. Iannuzzi, M. D. Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef.
- Y. Zhang and W. Yang, J. Chem. Phys., 1998, 109, 2604 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B, 1996, 54, 1703–1710 CrossRef CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- J. Elm, J. Kubečka, V. Besel, M. J. Jääskeläinen, R. Halonen, T. Kurtén and H. Vehkamäki, J. Aerosol Sci., 2020, 149, 105621 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian ∼16 Revision C.01, 2016, Gaussian Inc. Wallingford CT Search PubMed.
- P. R. McCurdy, W. P. Hess and S. S. Xantheas, J. Phys. Chem. A, 2002, 106, 7628–7635 CrossRef CAS.
- K. R. Leopold, Annu. Rev. Phys. Chem., 2011, 62, 327–349 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional mass spectra and their discussion, further computational results, cluster structures and coordinates. See https://doi.org/10.1039/d2ea00094f |
| This journal is © The Royal Society of Chemistry 2022 |