
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Seasonality of isoprene emissions and oxidation products above the remote Amazon†
B.
Langford
 *a,
E.
House
b,
A.
Valach‡
b,
C. N.
Hewitt
b,
P.
Artaxo
c,
M. P.
Barkley
de,
J.
Brito
j,
E.
Carnell
a,
B.
Davison
b,
A. R.
MacKenzie§
b,
E. A.
Marais
f,
M. J.
Newland
g,
A. R.
Rickard
gh,
M. D.
Shaw¶
b,
A. M.
Yáñez-Serrano||
i and
E.
Nemitz
a
*a,
E.
House
b,
A.
Valach‡
b,
C. N.
Hewitt
b,
P.
Artaxo
c,
M. P.
Barkley
de,
J.
Brito
j,
E.
Carnell
a,
B.
Davison
b,
A. R.
MacKenzie§
b,
E. A.
Marais
f,
M. J.
Newland
g,
A. R.
Rickard
gh,
M. D.
Shaw¶
b,
A. M.
Yáñez-Serrano||
i and
E.
Nemitz
a
aUK Centre for Ecology & Hydrology, Bush Estate, Penicuik, EH26 0QB, UK. E-mail: benngf@ceh.ac.uk
bLancaster Environment Centre, Lancaster University, Lancaster, LA1 4YQ, UK
cInstitute of Physics, University of São Paulo, São Paulo, Brazil
dEarth Observation Science Group, Department of Physics & Astronomy, University of Leicester, UK
eLeicester Institute for Space and Earth Observation (LISEO), University of Leicester, UK
fDepartment of Physics and Astronomy, University of Leicester, Leicester, UK
gWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, York, YO10 5DD, UK
hNational Centre for Atmospheric Science, University of York, York, YO10 5DD, UK
iAir Chemistry, Biogeochemistry and Multiphase Departments, Max Planck Institute for Chemistry, Mainz, Germany
jIMT Nord Europe, Institut Mines-Télécom, Université de Lille, Centre for Energy and Environment, 59000 Lille, France
First published on 17th January 2022
Abstract
The Amazon rainforest is the largest source of isoprene emissions to the atmosphere globally. Under low nitric oxide (NO) conditions (i.e. at NO mixing ratios less than about 40 pptv), isoprene reacts rapidly with hydroxyl (OH) to form isoprene-derived peroxy radicals (ISOPOO), which subsequently react with the hydroperoxyl radical (HO2) to form isoprene epoxydiols (IEPOX). IEPOX compounds are efficient precursors to the formation of secondary organic aerosols (SOA). Natural isoprene emissions, therefore, have the potential to influence cloudiness, rainfall, radiation balance and climate. Here, we present the first seasonal analysis of isoprene emissions and concentrations above the Amazon based on eddy covariance flux measurements made at a remote forest location. We reveal the forest to maintain a constant emission potential of isoprene throughout the year (6.9 mg m−2 h−1). The emission potential of isoprene is calculated by normalising the measured fluxes to a set of standard conditions (303 K and 1500 μmol m−2 s−1). During the wet season a factor of two reduction in absolute emissions was observed but this is explained entirely on the basis of meteorology and leaf area index, not by a change in isoprene emissions potential. Using an innovative analysis of the isoprene fluxes, in combination with measurements of its oxidation products and detailed chemical box-modelling, we explore whether concentrations of IEPOX follow the same seasonal cycle as the isoprene precursor. Our analysis implies that during the dry season (Sep–Jan) air pollution from regional biomass burning provides a modest increase in NO concentrations (indirectly inferred from a combination of other anthropogenic tracer measurements and box-modelling) which creates a competing oxidation pathway for ISOPOO; rather than forming IEPOX, alternative products are formed with less propensity to produce aerosol. This competition decreases IEPOX formation rates by a factor of two in the dry season compared with a scenario with no anthropogenic NO pollution, and by 30% throughout the year. The abundance of biogenic SOA precursors in the Amazon appears not to be dictated by the seasonality of natural isoprene emissions as previously thought, but is instead driven by regional anthropogenic pollution which modifies the atmospheric chemistry of isoprene.
Environmental significanceWe demonstrate the Amazon rainforest to maintain a constant isoprene emission potential, with the factor of two reduction in emission observed during the wet season explained entirely by meteorology and leaf area. Although isoprene emission rates are twice as large in the dry season, we suggest that widespread biomass burning at this time leads to modest increases in NO, which serves to suppress the formation of isoprene epoxydiol (IEPOX) secondary organic aerosol precursors by up to a factor of two. Rather than reacting with HO2 to form IEPOX, the isoprene peroxy radicals have an increased opportunity to react with NO. Therefore, the seasonal cycle in aerosol precursors in Amazonia is regulated, not by the availability of isoprene, but by anthropogenic biomass burning. |
1. Introduction
The Amazon rainforest is a globally important source of isoprene,1–3 yet only a few, short-term emission measurements of this reactive compound exist. These provide a tantalizing but narrow insight into the diurnal and seasonal dynamics of isoprene emission from the rainforest canopy, and its impact on atmospheric chemistry.4–9 Even less is known about how isoprene emissions might change as the forest transitions between the wet and dry seasons and what impact this might have on isoprene-derived SOA precursors such as IEPOX. Satellite observations of formaldehyde (HCHO), a second generation oxidation product of isoprene, have previously been used to probe seasonal isoprene emission dynamics indirectly.10 Barkley et al. (2009)11 showed that HCHO column density measured above the Amazon rainforest exhibits a clear and robust seasonal cycle, with lower HCHO amounts detected in the wet season and higher amounts in the dry season. This pattern reflects the metabolic processes that control the production of isoprene synthase, which are closely linked to photosynthesis and hence to light and temperature.12 However, this analysis also showed a marked dip in HCHO amounts in the transitional period between the wet and dry seasons, alluding to further controls. They postulated that the near absence of HCHO above background levels at this time was either due to a shutdown in isoprene emissions, caused by a wide-scale seasonal leaf flushing event, or to a dramatic decrease in the concentrations of the hydroxyl radical (OH), the primary chemical isoprene oxidant in the atmosphere.Understanding of isoprene oxidation chemistry has evolved rapidly in the last decade, with numerous laboratory and theoretical studies exploring in detail the key degradation pathways and branching ratios involved.13–17 Under pristine conditions (i.e. concentrations of NO < ∼40 pptv), isoprene reacts with OH to form a range of peroxy radical species (ISOPOO) which can react with HO2 radicals to produce isoprene hydroxyhydroperoxides (ISOPOOH).14 ISOPOOH can be further oxidised by OH to form lower volatility epoxydiols (IEPOX), which have been shown to efficiently condense onto pre-existing aerosol and so contribute mass to secondary organic aerosols (SOA)17–20 (see Section 9 of ESI†).
In more polluted environments, NO emitted from anthropogenic combustion processes (e.g. fossil fuel combustion and biomass burning) provides a competing reaction pathway for the ISOPOO radicals,21 resulting in the formation of greater amounts of methyl vinyl ketone (MVK) and methacrolein (MACR), as well as HCHO, glyoxal and methacrylic acid epoxide (MAE). These all give lower yields of SOA precursors than IEPOX22 under the temperature regime of the tropical forest.23 Previous field measurements of these species/processes in the Amazon rainforest have only been conducted downwind of the city of Manaus (ca. 2.1 million inhabitants) and are limited to the wet season.24,25 A detailed long-term study of isoprene emissions and its oxidation under remote conditions and through different seasons is still lacking.
To improve understanding of the sources and fate of isoprene in Amazonia, we measured the surface-atmosphere exchange of isoprene and the sum of its oxidation products (MVK + MACR + ISOPOOH, collectively termed Isoox) with hourly resolution using a proton transfer reaction mass spectrometer (PTR-MS)26 for an eleven month period at a remote site in the central region of the Amazon rainforest. We use this unique data set to reveal the seasonal dynamics of both isoprene emissions and its oxidation chemistry above the Amazon biome.
2. Results
Monthly average daytime (09:00–17:00 local time) isoprene concentrations and fluxes are shown in Fig. 1A and B respectively. A seasonal cycle is evident with both concentrations and fluxes largest during the dry season and up to a factor of two lower during the wet season. Fig. 1C shows the monthly and annual average isoprene emission potentials, which are calculated by normalising the measured fluxes to a set of standard conditions (303 K and 1500 μmol m−2 s−1) (see Section 2 of ESI†).27 Monthly values (except November and March, when data were limited and uncertainty large) deviate only very slightly and are statistically not significantly different from the annual average of 6.9 mg m−2 h−1. This demonstrates that the rainforest maintains a constant isoprene emission potential throughout the year and that the seasonal cycle in observed emission rates and ambient concentrations can be explained by fluctuations in light, temperature and leaf area index. It is, therefore, not necessary to invoke changes in the emission potential caused by phenomena such as leaf flushing or mass flowering events or changes in phenology8 to explain the observed seasonality in isoprene emission rates at this site. These measurements also provide, for the first time, direct unequivocal evidence that isoprene fluxes in Amazonia do not substantially decrease during the transitional period between wet and dry seasons.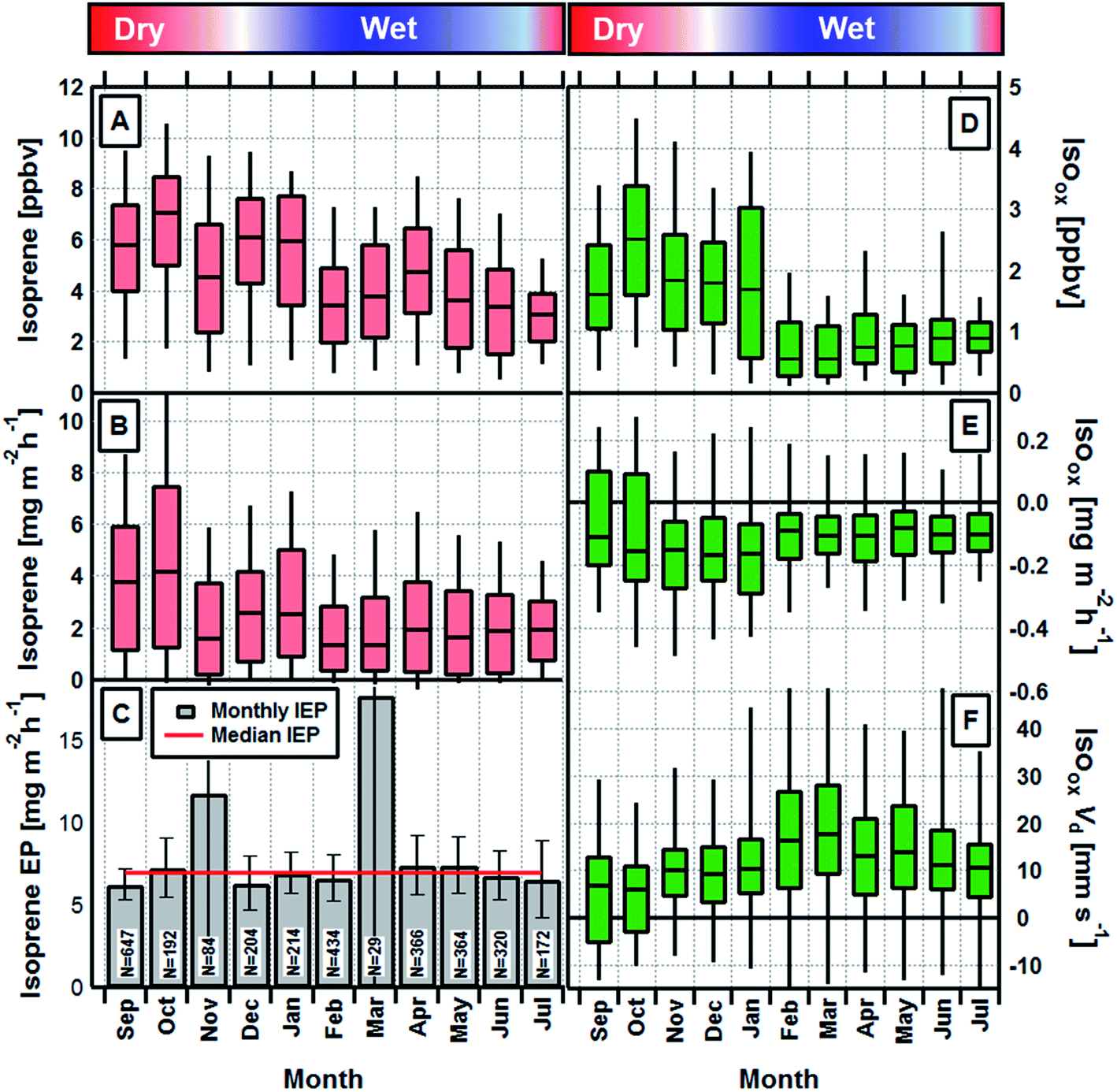 | ||
| Fig. 1 Box and whisker plots (centre line, median; box limits, upper and lower quartiles; whiskers, 10th and 90th percentiles) showing the monthly mean daytime (09:00 to 17:00 local) isoprene concentrations (A) and fluxes (B) measured above the rainforest, in relation to the wet–dry seasonality. Panel (C) shows the isoprene fluxes normalised to standard conditions (see text) to give the isoprene emission potential (IEP) of the forest. N = number of observations. Error bars show ±1σ of calculated emission potentials. Panels (D–F) show the concentration, flux and deposition velocities, respectively, for the isoprene oxidation products MVK, MACR and ISOPOOH, which we collectively term Isoox. | ||
In order to investigate potential seasonality in isoprene photochemistry we derived the relative abundance of the major oxidation products of isoprene, MVK + MACR and ISOPOOH (Isoox). ISOPOOH reacts on the metal surfaces within the PTR-MS sampling and analysis system (e.g. inlet lines, capillary and drift tube) and is converted to MVK. As a result, Isoox can be measured directly as the sum of species detected at m/z 71 by the PTR-MS.28 The exact conversion efficiency within our instrument is not known, but the long inlet line (>50 m) and steel capillary combined with a lack of signal at the ISOPOOH parent m/z suggest conversion was at or close to 100%.
Isoox concentrations and fluxes and its derived deposition velocity (Vd = −flux/concentration) are shown in Fig. 1D–F, respectively. The seasonal cycle in monthly average concentrations of Isoox (Fig. 1D) broadly follows that of isoprene but is enhanced further due to changes in the deposition process, as implied by the larger deposition velocity, between February and May (Fig. 1F).
Resistance analysis (see Methods) allows the total deposition resistance (Rt = 1/Vd) to be decomposed into the component resistances describing atmospheric transport (aerodynamic resistance, Ra and boundary layer resistance, Rb) and the interaction with the canopy (Rc). Fig. 2 shows a clear seasonal cycle in Rt (and thus Vd), which is driven almost entirely by variations in the canopy resistance, with Ra and Rb relatively constant throughout the year. This can be related to a change in the relative abundance of the three components of Isoox. Previous deposition measurements of ISOPOOH + IEPOX above a temperate forest suggest a near zero Rc.29 This is due to the rapid conversion of ISOPOOH to MVK upon contact with leaf surfaces and subsequent conversion to methyl ethyl ketone (MEK).30 In contrast, MVK + MACR deposits with an Rc value of at least 50 s m−1, as implied by the January measurements in Fig. 2. Utilizing the strongly contrasting canopy resistances of these compounds, we can estimate the fraction of ISOPOOH detected at m/z 71 as:
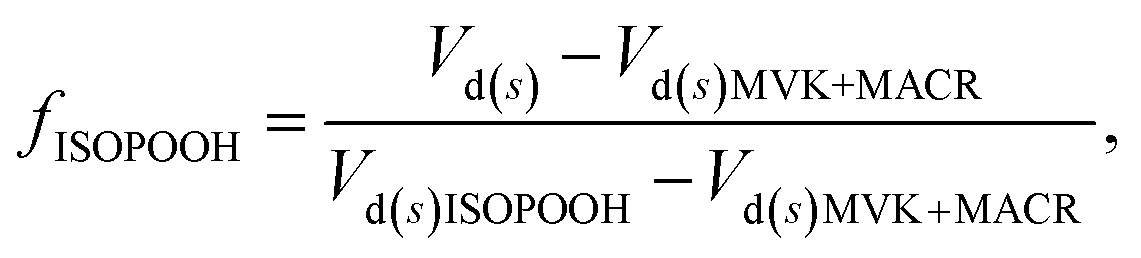 | (1) |
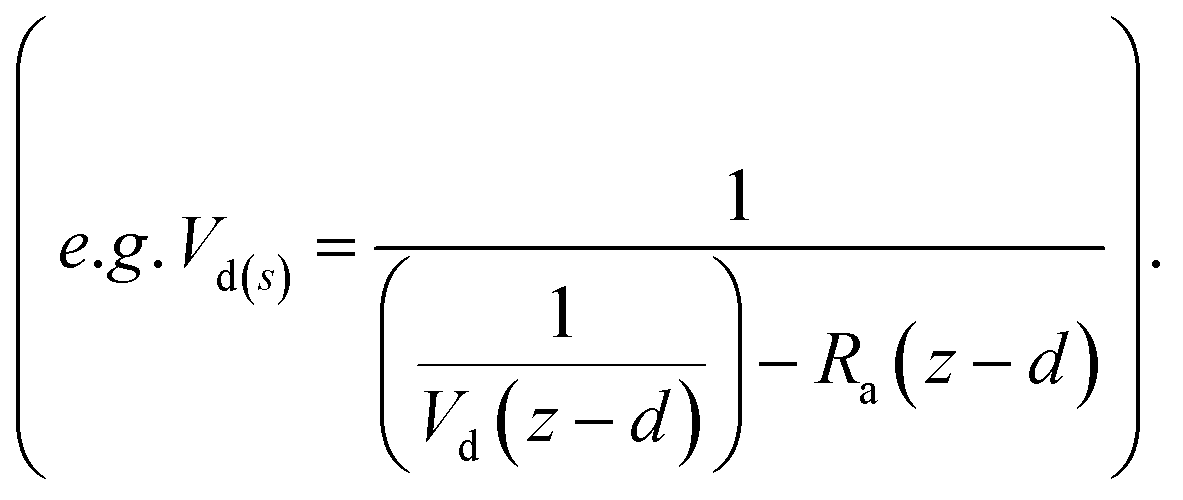 For ISOPOOH, where the value of Rc is expected to be at, or close to, zero, the Vd(s) can be given as 1/Rb, equal to 4.5 cm s−1. Determining the deposition velocity for MVK + MACR is more challenging due to a lack of previous MVK + MACR flux measurements that do not suffer from the ISOPOOH artefact. Here, we use a value of 0.45 cm s−1, which is the midpoint of the values used for MVK and MACR (0.3–0.6 cm s−1) in the dry deposition scheme presented by Zhang et al.31
For ISOPOOH, where the value of Rc is expected to be at, or close to, zero, the Vd(s) can be given as 1/Rb, equal to 4.5 cm s−1. Determining the deposition velocity for MVK + MACR is more challenging due to a lack of previous MVK + MACR flux measurements that do not suffer from the ISOPOOH artefact. Here, we use a value of 0.45 cm s−1, which is the midpoint of the values used for MVK and MACR (0.3–0.6 cm s−1) in the dry deposition scheme presented by Zhang et al.31
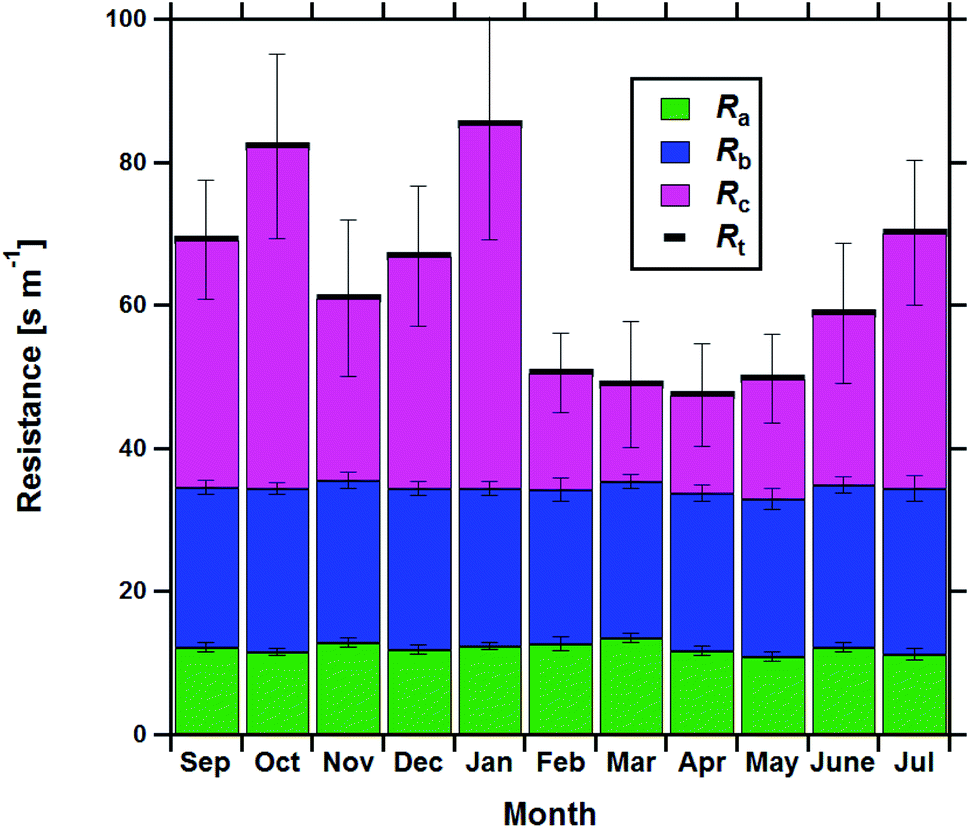 | ||
| Fig. 2 Monthly average total Isoox deposition resistance (Rt) broken down into the three major rate-limiting stages, the aerodynamic resistance (Ra), the boundary layer resistance (Rb) and the canopy resistance (Rc), on a monthly basis. Only data where the friction velocity (u*) exceeded 0.3 m s−1 and the Isoox deposition velocity was positive were included in this analysis. Error bars show ±1 standard error. | ||
Once the fraction of ISOPOOH is known on a concentration basis, it is possible to work back to the production ratio of ISOPOOH to MVK + MACR, χ, which accounts for the shorter chemical lifetime and faster deposition velocities of ISOPOOH relative to that of MVK + MACR, following the approach of Liu et al. (2016)24
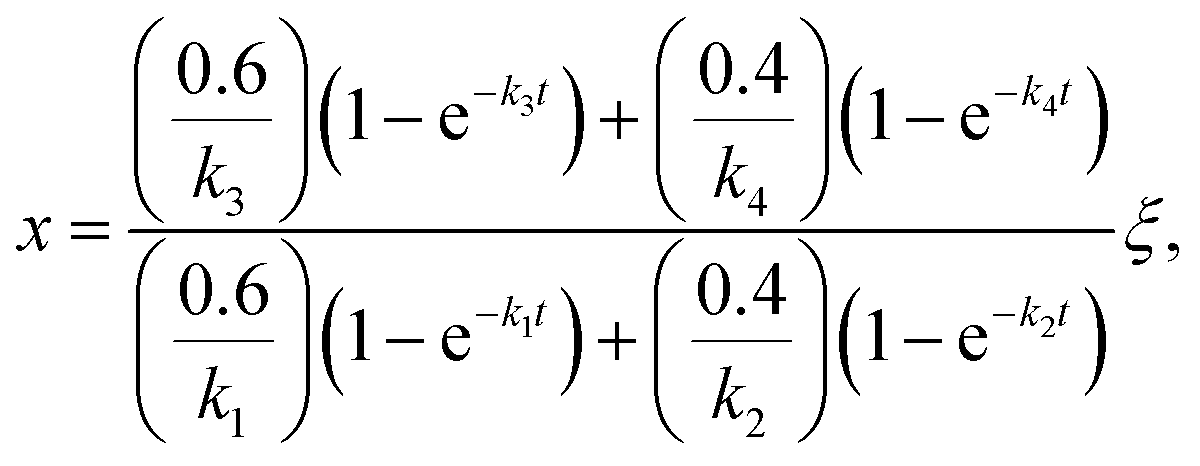 | (2) |
Fig. 3 shows a clear exponential relationship between the production ratio estimated using our deposition velocity approach and ambient concentrations of black carbon, which we take to be a proxy of anthropogenic pollution and possible marker of increased NO concentration (see Section 4). A similar exponential relationship was observed with measurements of acetonitrile, a commonly used tracer for biomass burning. At acetonitrile concentrations above ∼0.4 ppbv, the ISOPOOH to MVK + MACR production ratio is low (<0.2), consistent with NO concentrations of ∼200 pptv or greater. At lower pollutant concentrations, the production ratio increases, indicating a significant fraction of the isoprene peroxy radicals (ISOPOO) react with HO2, rather than NO, to form ISOPOOH.
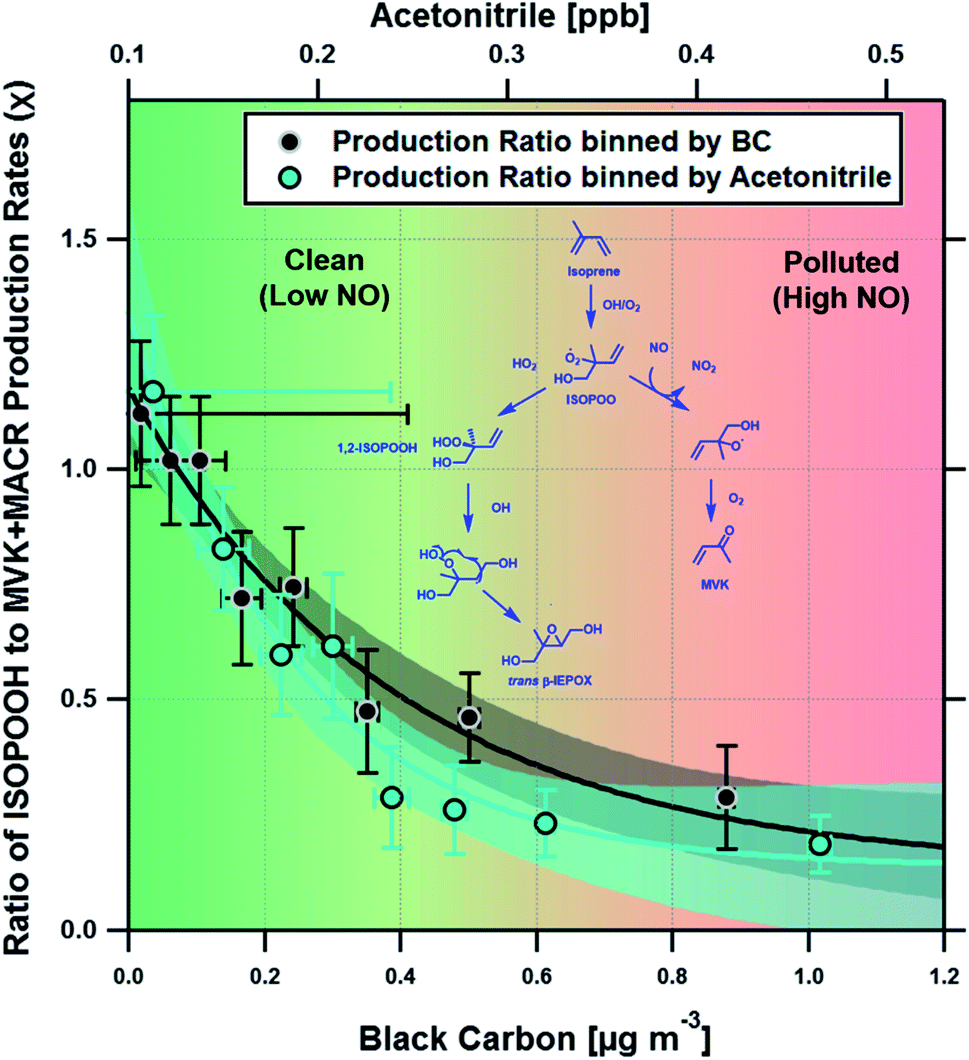 | ||
| Fig. 3 Scatter plot showing the measured relationship between the ratio of ISOPOOH to MVK + MACR production rates and both black carbon (BC) and acetonitrile (ACN) concentrations, two proxies for biomass burning and hence anthropogenic pollution. Data were stratified for each point to represent the average of 200 measurements and error bars show ±1 standard error. Solid lines show an exponential fit to the data and the shaded areas indicate the 95% confidence interval of the fit. The simplified chemical schematic shown highlights two of the possible loss routes for one of the primary ISOPOO species formed, which predominately follows either the HO2 (left hand side) or NO (right hand side) pathways to form either (1,2)-ISOPOOH isomer or MVK + HCHO respectively. An analogous pathway, not shown, would yield the (4,3)-ISOPOOH isomer or MACR + HCHO via HO2 and NO reactions, respectively. | ||
Having established the production ratios of ISOPOOH to MVK + MACR with monthly time resolution, we now combine our findings with detailed chemical box model simulations incorporating the Master Chemical Mechanism (MCM22) (see Methods and ESI† for details). Fig. 4, panel (a), shows the relationship between the ISOPOOH to MVK + MACR production ratio (χ) for a given NO concentration as predicted by the MCM box model. In the example shown, the model was initialized using the average daytime above-canopy isoprene concentration (5.8 ppbv) measured for the month of September. Panel (b) shows the fractional contribution of the four reaction pathways for ISOPOO, fHO2, fNO, fRO2 and fISOM over the same range of NO concentrations. Using an equivalent of these two plots for each month as a lookup table, we estimated the fraction of ISOPOO that reacts via the main HO2 and NO pathways as well as the contributions from peroxy radical (RO2) cross and self-reactions32 and isomerisation pathways16,33,34 over the 11 months measurement period. The results of this exercise are shown in Fig. 5a together with the average concentrations of BC and acetonitrile.
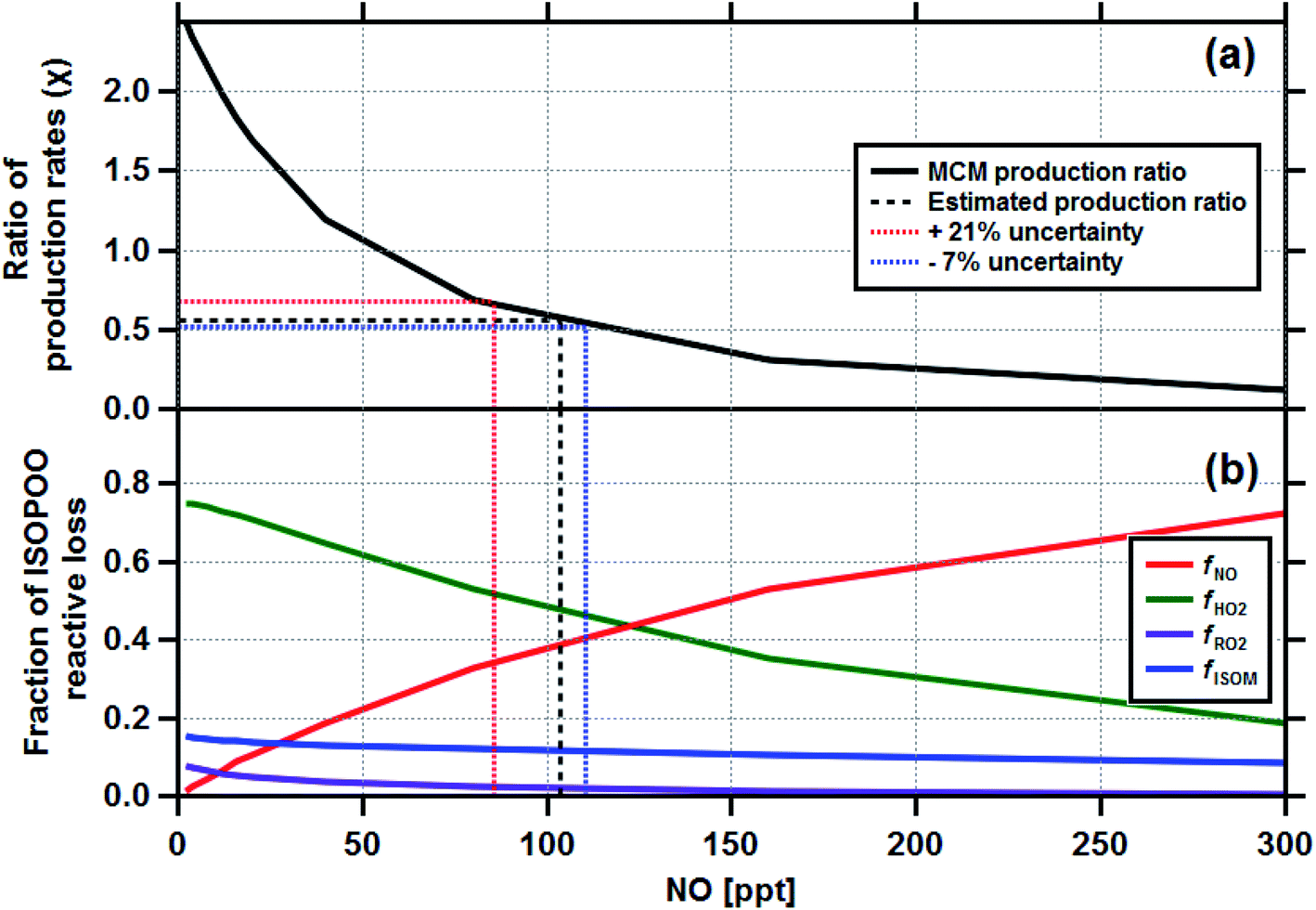 | ||
| Fig. 4 The output from the Master Chemical Mechanism box modelling study which calculated the ratio of ISOPOOH to MVK + MACR production rates (χ) for a range of NO concentrations (a) and the corresponding reactive loss to be expected to each of the four degradation pathways fNO, fHO2, fRO2 and fISOM (b). The box model was initialised for the average conditions of the month of September, where the measured isoprene concentration was 5.8 ppbv. Equivalent plots were made for the other 10 months of the study. The production ratio uncertainties are discussed in Section 4 and the ESI (Section 6.2†). | ||
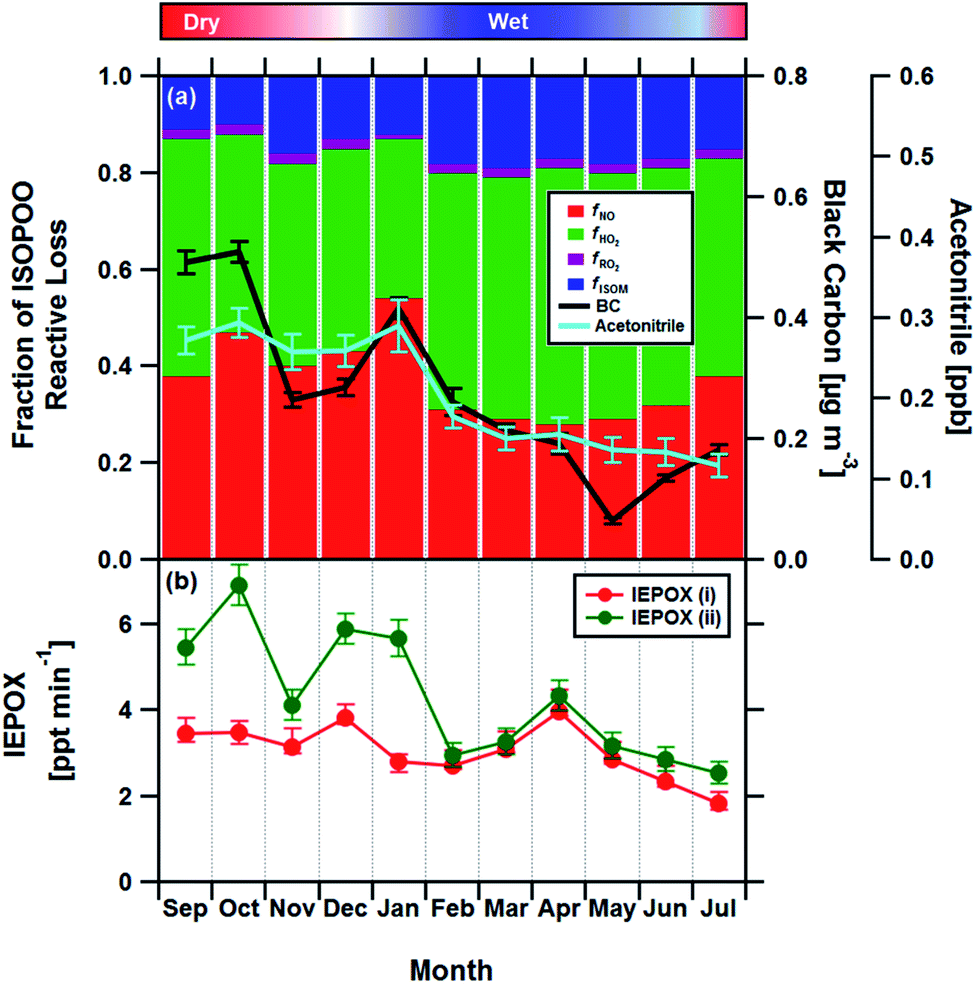 | ||
| Fig. 5 (a) Stacked bar chart showing the relative fractions of ISOPOO lost via the four major chemical pathways fNO, fHO2, fRO2 and fISOM for each month compared to the measured black carbon (BC) and acetonitrile concentrations (error bars show ±1 standard error), two markers of biomass burning, used here as a proxies for NO. Panel (b) shows the formation rate of the SOA precursor IEPOX from the MCM box model for the chemical regimes in panel (a) (IEPOX (i)), and for a regime assuming no anthropogenic influence, where NO is kept constant at 35 pptv throughout the year (IEPOX (ii)). IEPOX error bars reflect the uncertainties in the derived ISOPOOH to MVK + MACR production ratios (see Section 4†). | ||
The box model results suggest that HO2 accounts for 46% of the reactive loss of ISOPOO over the year, consistent with the view that HO2 is the principal ISOPOO sink in the Amazon, especially under the clean, low NO, conditions of the wet season when its contribution peaked at 53%.14,34 By comparison, the NO pathway accounts for between ∼28 and 54% of the ISOPOO reactive loss, with an average contribution of 37% over the year. The NO pathway is largest during September and January (the dry season) when increased deforestation activities and agricultural practices result in biomass burning plumes containing elevated concentrations of NO. These plumes are advected throughout the Amazon rainforest.35,36 Rainforest soils also act as a minor source of NO and these emissions may exhibit some seasonal cycle.37 However, the strong association between the NO pathway and both black carbon and acetonitrile, two tracers of biomass burning that are independent of soil emissions, suggests that locally produced NO cannot be the principal driver for the observed pattern. An analysis of terrestrial fire counts and brightness retrieved by the Terra MODIS satellite along calculated back trajectories confirms that biomass burning is the most likely dominant source of the measured BC and acetonitrile concentrations (see Section 8 of the ESI†).
To relate our results to current understanding, we additionally ran the GEOS-Chem global chemical transport model over the same period for the grid square coincident with our measurement site (see Section 5.3 of the ESI†). Concentrations of NO in the model were relatively constant (28 pptv) across the 11 months period, with an even split between anthropogenic (EDGAR v4.2) and natural emissions (GFED Version 4). The resulting fNO/fHO2 ratio was approximately unity throughout the year, which suggests the seasonal influence from biomass burning is not well captured within the model.
3. Discussion
The isoprene emissions dataset presented here shows, for the first time, that the seasonal cycle in isoprene emissions in Amazonia is controlled purely by variations in meteorology and leaf area, and that the emission potential of the forest remains rather constant throughout the seasons. This implies that the inability of chemical transport models to reconcile isoprene concentrations during the wet-to-dry transition points towards uncertainty in chemistry rather than in the modelled isoprene emission rates. This is consistent with the findings of Wells et al. (2020) who attribute the discrepancies in satellite-derived isoprene concentrations with model estimates to underestimates in NO concentrations, which bias modelled OH concentrations.38 Our measurements further support this conclusion, with NO concentrations ranging between 40 and 145 pptv required to obtain our derived production ratios in the chemical box model simulations. These are 30% higher than that predicted by GEOS-Chem for the wet season and a factor of four larger than in the dry season.While we have provided a robust isoprene emission potential for a single specific measurement site, recent airborne flux measurements of isoprene made above the rainforest of Amazonia have shown spatial variability in isoprene emission potentials, particularly across topographic gradients where species distributions change markedly.7 Accounting for spatial heterogeneity over the vast expanse of the Amazon rainforest can therefore only be achieved through Earth observations. New tools are now enabling direct observations of isoprene concentrations using these platforms, which should help to reveal the dynamics of isoprene emissions throughout the region.11,39,40
Our analysis of isoprene oxidation products point to seasonal changes in oxidation chemistry between the wet and dry seasons, driven by anthropogenic pollution, that are not currently captured by chemical transport models. We find that, under the most clean conditions (February to May), isoprene oxidation favours reaction with HO2 (fHO2 = 51%) which leads to the production of isoprene epoxydiols (IEPOX),17 a precursor to isoprene SOA. However, as anthropogenic NOx pollution increases, the NO oxidation pathway becomes progressively more important, especially in the dry season when regional biomass burning is at its peak. Such a shift in chemistry suppresses the formation of IEPOX via the HO2 pathway, despite isoprene emissions and concentrations being a factor of two larger than during the wet season.
In an attempt to quantify the magnitude of this effect, we repeated the box modelling for a scenario where we assume no anthropogenic influence and held NO concentration constant at values observed for the wet season in the remote Amazon (∼35 pptv of NO24). The results, shown in Fig. 5b, show that the seasonal variation in isoprene chemistry implied by our analysis would reduce the formation rate of IEPOX by up to a factor of two in the dry season and by 30% across the year. IEPOX are important precursors to the formation of isoprene-derived SOA17 and, therefore, identifying any seasonal pattern imposed by man-made pollution represents a key first step to understanding how cloud formation, rainfall and climate might be influenced by the increasing anthropogenic activity in this region. Our measurements were made during 2013, which had the second lowest number of forest fires ever recorded. Fires are predicted to increase by 20-100% in the coming decades41 and, therefore, the observed seasonality in isoprene chemistry and resulting IEPOX suppression reported here should be viewed as a lower limit.
The suppression of IEPOX formation due to NO pollution might appear at odds with the findings of Shirvastava et al. (2019)25, who posit urban NOx pollution to greatly enhance the formation of natural aerosol above the Amazon. However, at this remote site, our combined measurement and modelling approach imply only a modest increase in NO concentrations (<140 pptv of NO) when regional biomass burning events impact the site and is well below the levels seen within the fresh plume of urban pollution emitted from the city of Manaus. The fact that NOx pollution can both suppress and enhance the formation of IEPOX at different concentrations serves only to highlight the sensitivity of the atmospheric chemistry occurring in this region in response to anthropogenic perturbations.
4. Assumptions and uncertainties
Our analysis is based on several key assumptions, and therefore the absolute values of the chemical pathways reported are uncertain. The results are very dependent on the deposition velocities of MVK + MACR and ISOPOOH used in eqn (1). A sensitivity analysis, where the Vd(s) of ISOPOOH and MVK + MACR were varied, was used to assess the range of uncertainties associated with our estimates. The Vd(s) for ISOPOOH was already set to the maximum theoretically possible (e.g. 1/Rb) and, therefore, it was varied by −1 cm s−1, equivalent to an Rc of 6.5 s m−1. For MVK + MACR the Vd(s) was varied by ±0.15 cm s−1 to reflect the range of values used for these compounds in dry deposition schemes.31 Taking the two extreme scenarios, we derive an uncertainty estimate for the derived production ratios of plus 21% (ISOPOOH Vd(s) = 3.5 cm s−1; MVK + MACR Vd(s) = 0.3 cm s−1) and minus 7% (ISOPOOH Vd(s) = 4.5 cm s−1; MVK + MACR Vd(s) = 0.6 cm s−1). The uncertainties shown in Fig. 4 indicate how this uncertainty propagates through to the fraction of ISOPOO reactive loss for the month of September, with details of the other 10 months shown in Section 6.2 of the ESI.† Although the absolute production ratio is uncertain, any error is applied systematically to each point and, therefore, the seasonal pattern is unaffected. In contrast, changes in OH concentration across the seasons could directly affect the production ratio calculated for each month (e.g. ki,OH). A sensitivity analysis, where ki,OH was varied following the seasonal pattern of OH implied by isoprene flux to concentration ratios, shows changing OH concentrations to have a relatively minor impact on the estimated production rates (6% increase overall), with the largest difference noted for September, where the production rate increased by 15%. Even then, when accounting for variability in the OH concentrations, the seasonal pattern in ISOPOO reactive loss remained consistent (see Section 6.1 of the ESI†).Liu et al. (2016)24 reported direct observations of ISOPOOH to MVK + MACR production ratios at a different site (T3) within the Amazon basin during March 2014, using a more direct measurement approach, with observed values ranging between 0.6 and 0.9. Their site was closer to Manaus and experiences, at least at times, higher pollution (i.e. higher [NO]) than the ZF2 site used in this study, which would lower production ratios in favour of MVK + MACR. Our production ratio for March of the same year was 0.98, (0.91 to 1.18), falling just outside the range reported by Liu et al., providing a useful ground-truthing for our approach.
A further limitation of our study is the absence of NO measurements, which would provide confirmation that the changes in calculated production ratios reflect the availability of NO. A comparison of BC and NO measurements made between February 2014 and June 2015 at the T3 site, showed a clear relationship between the two species on a timescale of days to months (see Section 1.4 of the ESI†). This confirms that black carbon concentrations can offer a qualitative assessment of the anthropogenic trend in NO in Amazonia despite the very different chemical lifetimes of these two species. The fact that Fig. 3 shows such a close relationship between derived production ratios and two well-known markers of biomass burning provides confidence that our approach can broadly represent the trends in isoprene oxidation chemistry at this site. In addition, the average monthly NO concentrations implied from the ISOPOOH to MVK + MACR production ratios, ranged between 41 pptv (March) and 139 pptv (October), which compare well with previous background NO concentrations reported above the Amazon (15 to 80 pptv (ref. 24, 42 and 43)) providing a further sanity check for our results. Nonetheless, future work is required to better quantify the complex role that anthropogenic pollution plays in mediating isoprene chemistry and SOA formation in the remote Amazon. Recent advances in online mass spectrometry mean it is now possible to measure the major isoprene oxidation products, including both ISOPOOH and IEPOX, directly. Combining long-term ground based measurements of these species together with direct observations of NO should now become a priority, so the true impact that human pollution has in this remote part of the world can be further established.
5. Conclusions
Our findings shed light on the seasonal dynamics of both isoprene emissions from the tropical rainforest of Amazonia and its chemical fate. They confirm the tropical rainforest to maintain a constant isoprene emission potential throughout the year, with seasonal changes in emission rates explained by the latest isoprene emission algorithms on the basis of light temperature and leaf area index.The deposition rates of isoprene oxidation products, when explored to their fullest, hint at broad trends in the chemical fate of isoprene that are driven by anthropogenic pollution, specifically by nitric oxide. They indicate that reaction with HO2 is the dominant mechanistic pathway for isoprene peroxy radical oxidation in the remote Amazon, accounting for an average of 46% of the reactive loss of ISOPOO throughout the year, but that ISOPOO chemistry is sensitive to the level of NO present, with its oxidation pathways shifting towards reaction with NO during the dry season, when regional biomass burning is at its highest. This competition decreases IEPOX formation rates by a factor of two in the dry season compared with a scenario with no anthropogenic NO pollution, and by 30% throughout the year, despite absolute emissions of isoprene being a factor of two larger at this time. We conclude that the abundance of this important isoprene SOA precursor in the Amazon is, therefore, not dictated by the seasonality of natural isoprene emissions as previously thought, but instead, is strongly mediated by regional anthropogenic pollution.
6. Materials and methods
6.1 Flux measurements
Sample air was drawn from an inlet mounted at a height of 38 m on the triangular tower TT34 at the LBA ZF2 measurement site (2.594637° S, 60.209519° W, altitude 67 m) located in the Reserva Biologica do Cueiras in Central Amazonia, Brazil (see Section 1 of ESI†). Flux measurements were made using the virtual disjunct eddy covariance (vDEC) technique, in which a covariance function between vertical wind velocity measurements (w) and VOC mixing ratios (c) is used to determine the flux for each selected compound (Fc), such that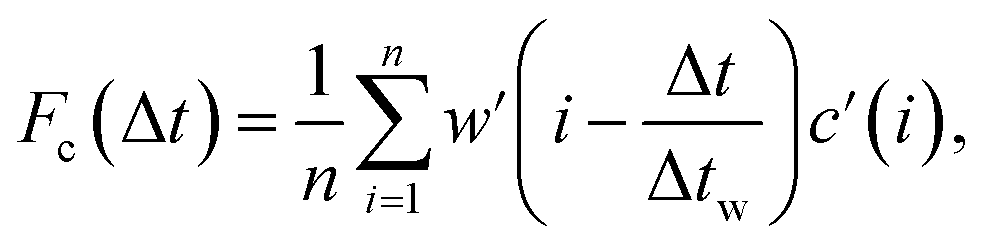 | (3) |
The flux limit of detection for each averaging period was calculated following the method outlined by Langford et al. (2015).44 A prescribed time lag of 8.5 s was applied to the isoprene data and 9 s was used for Isoox data. This time-lag (Δt) was determined from analysis of the histogram of time-lags determined from the maximum in the cross-covariance of the isoprene concentration and vertical wind velocity (MAX method) following the recommendations of Langford et al. (2015).44
To identify non-stationary periods the covariance of w′ and c′ was divided into nine intervals of five minutes and the average of these periods was compared with the covariance for the whole 45 min period. When the difference between these two values was greater than 60% the data were considered non-stationary and were rejected; this was the case for 17% of otherwise unfiltered fluxes.
Flux measurements were also corrected for high frequency flux losses using the approach of Horst (1997) and assuming a system response time of ∼0.2 s. This analysis showed high frequency flux contributions to be relatively minor at this tall forest site with a median flux loss of just 1.3%.
At night, a stable nocturnal boundary layer may develop potentially decoupling the forest canopy from our measurement location above. Emissions from the forest may accumulate and be seen as a burst in the morning as the boundary layer breaks up. This process of storage of emissions below the measurement height can be corrected for using the following equation
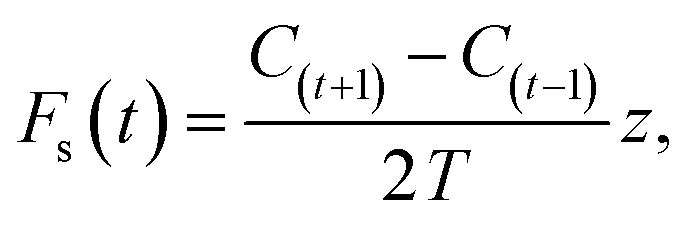 | (4) |
6.2 Resistance analysis
The deposition process is governed by three rate-limiting steps in series, the aerodynamic (Ra), boundary layer (Rb) and canopy (Rc) resistances which together form the total resistance (Rt). The deposition velocity (Vd) is therefore calculated as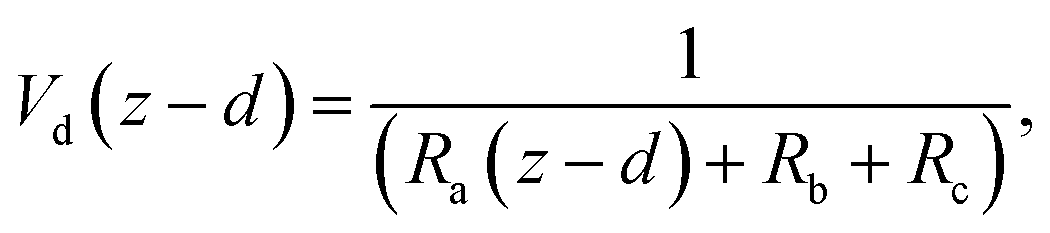 | (5) |
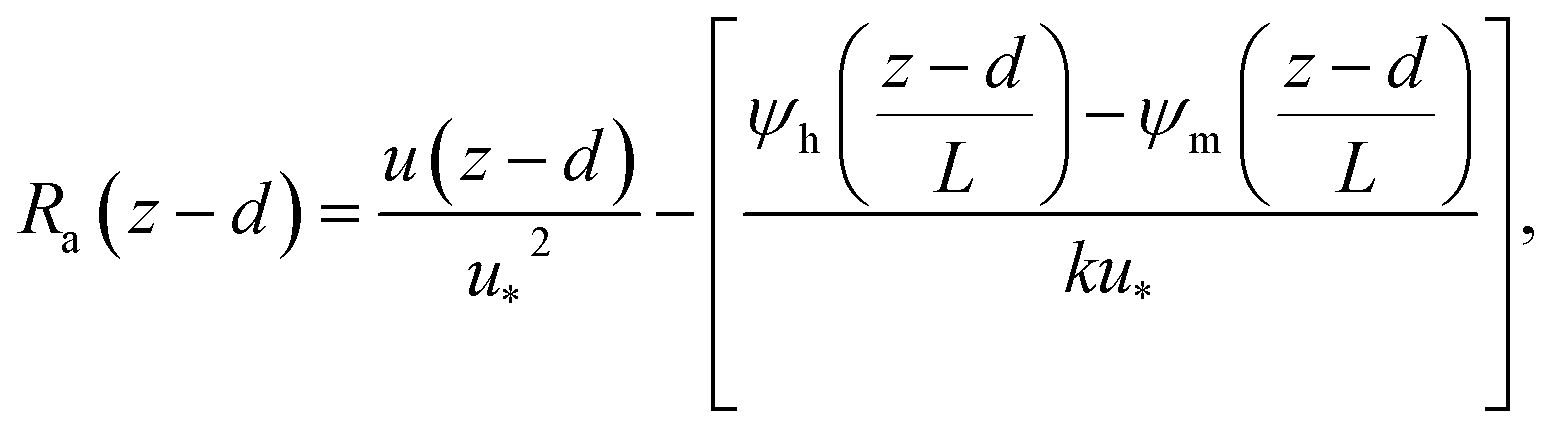 | (6) |
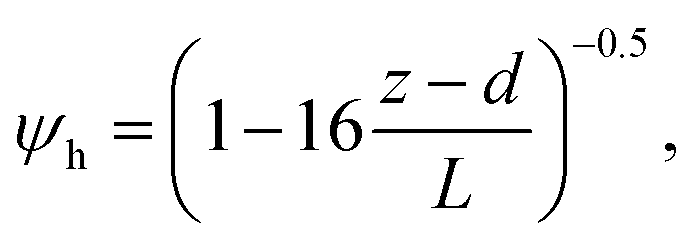 | (7) |
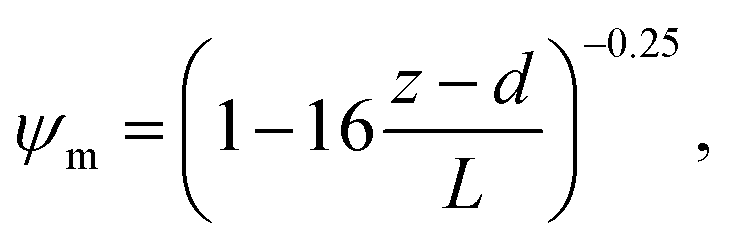 | (8) |
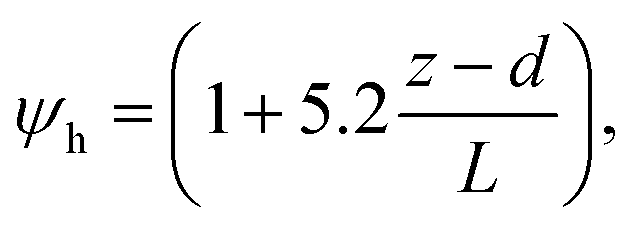 | (9) |
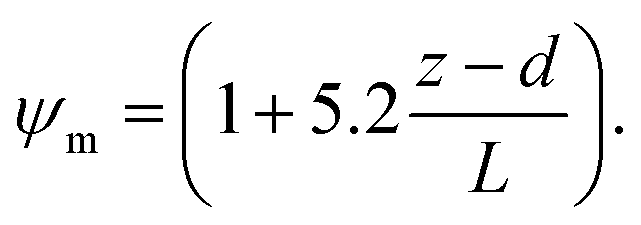 | (10) |
R b, describes the resistance created by the thin layer of laminar air that covers all surface elements and is inversely related to turbulence. Here, we calculate Rb following Jensen and Hummelshøj47,48 as
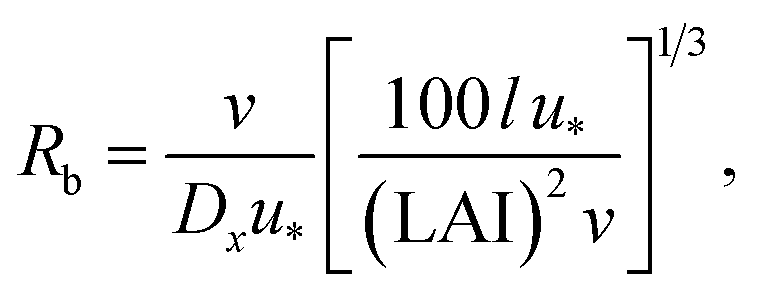 | (11) |
The final resistance, Rc, represents the canopy resistance and can be inferred from direct micrometeorological measurements of the deposition velocity which is calculated as
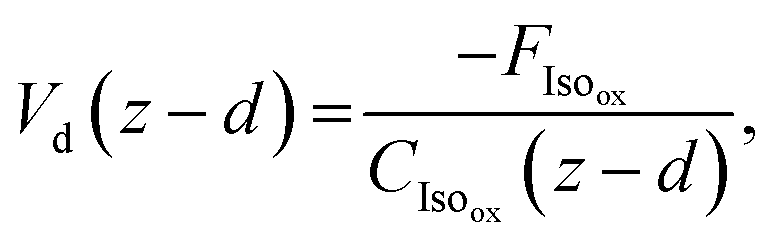 | (12) |
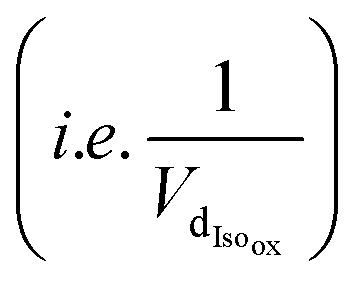 and the sum of Ra and Rb as shown in eqn (13):
and the sum of Ra and Rb as shown in eqn (13):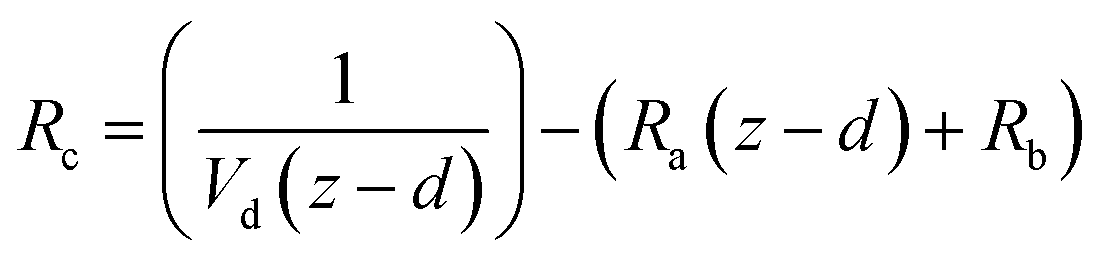 | (13) |
6.3 Box modelling
The Dynamically Simple Model of Atmospheric Chemical Complexity (DSMACC) box model49 was used, together with the near explicit chemical mechanism the Master Chemical Mechanism (MCM) v3.3.1 (ref. 22) (http://mcm.york.ac.uk)50 to (a) determine what was the fractional loss of ISOPOO to each of the four degradation pathways: reaction with NO, reaction with HO2, reaction with RO2, and isomerization, and (b) to investigate the sensitivity of HCHO and IEPOX production from isoprene photooxidation to the NO concentration, under a range of conditions representative of the observations.The complete isoprene degradation mechanism in MCM v3.3.1 consists of 1926 reactions of 602 closed shell and free radical species, which treat the chemistry initiated by reaction with OH radicals, NO3 radicals and ozone. It contains much of the isoprene HOX recycling chemistry identified as important in recent years under “low NO” conditions, including the peroxy radical 1,5 and 1,6 H-shift chemistry described in the LIM1 mechanism,16,51 as summarized in Wennberg et al. (2018)13 Model photolysis rates were calculated using the Tropospheric Ultraviolet and Visible Radiation Model (TUV v5.2).52
Author contributions
EN, CNH, ARM, BL and PA devised the project. EH, AV, BD, AMYS, JB, BL and MDS conducted the field measurements. BL and EH analysed the flux data. MJN, ARR and EM conducted the modelling work. MPB and EC analysed the satellite data and AV and BL analysed back trajectories. BL, EN, CNH, ARR and NJM wrote the manuscript with contributions from all co-authors.Conflicts of interest
The authors declare no competing interestsAcknowledgements
This work was funded by the UK Natural Environmental Research Council (NERC) via grant NE/I012036/1. BL and EN acknowledge further support from UK-SCAPE (NE/R016429/1). AV greatfully acknowledges a NERC studenship and CASE partnership with CEH Edinburgh. MJN and ARR acknowledge support provided by NERC via grant NE/M013448/1. ARR and MDS acknowledge support provided by the UK National Centre for Atmospheric Science (NCAS) Air Pollution Science Programme. PA acknowledge funding from FAPESP grants 2017-17047-0, and also the INCT MC Phase 2 under CNPq Grant 465501/2014-1, FAPESP Grant 2014/50848-9 and CAPES Grant 16/2014. The team acknowledge the support from the LBA central office coordinated by INPA (Instituto Nacional de Pesquisas da Amazonia). We thank James Cash for assistance in preparing Fig. 3 and Mark Sutton for helpful discussion.References
- D. J. Jacob and S. C. Wofsy, Photochemistry of biogenic emissions over the Amazon forest, J. Geophys. Res.: Atmos., 1988, 93(D2), 1477–1486 CrossRef CAS.
- J. Kesselmeier, et al., Atmospheric volatile organic compounds (VOC) at a remote tropical forest site in central Amazonia, Atmos. Environ., 2000, 34(24), 4063–4072 CrossRef CAS.
- A. Guenther, et al., A global-model of natural volatile organic-compound emissions, J. Geophys. Res.: Atmos., 1995, 100(D5), 8873–8892 CrossRef CAS.
- T. Karl, et al., The tropical forest and fire emissions experiment: Emission, chemistry, and transport of biogenic volatile organic compounds in the lower atmosphere over Amazonia, J. Geophys. Res., 2007, 112(D18), D18302 CrossRef.
- U. Kuhn, et al., Isoprene and monoterpene fluxes from Central Amazonian rainforest inferred from tower-based and airborne measurements, and implications on the atmospheric chemistry and the local carbon budget, Atmos. Chem. Phys., 2007, 7(11), 2855–2879 CrossRef CAS.
- U. Kuhn, et al., Exchange of short-chain monocarboxylic acids by vegetation at a remote tropical forest site in Amazonia, J. Geophys. Res., 2002, 107(D20), 8069 CrossRef.
- D. Gu, A. Guenther and J. Shilling, et al., Airborne observations reveal elevational gradient in tropical forest isoprene emissions, Nat. Commun., 2017, 8, 15541 CrossRef CAS PubMed.
- E. G. Alves, et al., Leaf phenology as one important driver of seasonal changes in isoprene emissions in central Amazonia, Biogeosciences, 2018, 15(13), 4019–4032 CrossRef CAS.
- H. J. I. Rinne, A. B. Guenther, J. P. Greenberg and P. C. Harley, Isoprene and monoterpene fluxes measured above Amazonian rainforest and their dependence on light and temperature, Atmos. Environ., 2002, 36(14), 2421–2426 CrossRef CAS.
- P. I. Palmer, et al., Mapping isoprene emissions over North America using formaldehyde column observations from space, J. Geophys. Res., 2003, 108(D6), 4180 CrossRef.
- M. P. Barkley, et al., Regulated large-scale annual shutdown of Amazonian isoprene emissions?, Geophys. Res. Lett., 2009, 36, L04803 CrossRef.
- R. K. Monson, et al., Relationships among isoprene emission rate, photosynthesis, and isoprene synthase activity as influenced by temperature, Plant Physiol., 1992, 98(3), 1175–1180 CrossRef CAS PubMed.
- P. O. Wennberg, et al., Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 2018, 118(7), 3337–3390 CrossRef CAS PubMed.
- Y. J. Liu, I. Herdlinger-Blatt, K. A. McKinney and S. T. Martin, Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation, Atmos. Chem. Phys., 2013, 13(11), 5715–5730 CrossRef.
- A. G. Carlton, C. Wiedinmyer and J. H. Kroll, A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 2009, 9(14), 4987–5005 CrossRef CAS.
- J. Peeters, T. L. Nguyen and L. Vereecken, HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 2009, 11(28), 5935–5939 RSC.
- F. Paulot, et al., Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 2009, 325(5941), 730–733 CrossRef CAS PubMed.
- T. B. Nguyen, et al., Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 2014, 14(7), 3497–3510 CrossRef.
- J. E. Krechmer, et al., Formation of low volatility organic compounds and secondary organic aerosol from isoprene hydroxyhydroperoxide low-no oxidation, Environ. Sci. Technol., 2015, 49(17), 10330–10339 CrossRef CAS PubMed.
- K. H. Bates, et al., Gas Phase Production and Loss of Isoprene Epoxydiols, J. Phys. Chem. A, 2014, 118(7), 1237–1246 CrossRef CAS PubMed.
- E. C. Tuazon and R. Atkinson, A product study of the gas-phase reaction of isoprene with the oh radical in the presence of nox, Int. J. Chem. Kinet., 1990, 22(12), 1221–1236 CrossRef CAS.
- M. E. Jenkin, J. C. Young and A. R. Rickard, The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 2015, 15(20), 11433–11459 CrossRef CAS.
- D. R. Worton, et al., Observational Insights into Aerosol Formation from Isoprene, Environ. Sci. Technol., 2013, 47(20), 11403–11413 CrossRef CAS PubMed.
- Y. J. Liu, et al., Isoprene photochemistry over the Amazon rainforest (vol 113, pg 6125, 2016), Proc. Natl. Acad. Sci. U. S. A., 2016, 113(51), E8358 CrossRef PubMed.
- M. Shrivastava, et al., Urban pollution greatly enhances formation of natural aerosols over the Amazon rainforest, Nat. Commun., 2019, 10(1), 1046 CrossRef PubMed.
- W. Lindinger, A. Hansel and A. Jordan, On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) - Medical applications, food control and environmental research, Int. J. Mass Spectrom., 1998, 173(3), 191–241 CrossRef CAS.
- B. Langford, et al., Isoprene emission potentials from European oak forests derived from canopy flux measurements: an assessment of uncertainties and inter-algorithm variability, Biogeosciences, 2017, 14(23), 5571–5594 CrossRef.
- J. C. Rivera-Rios, et al., Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 2014, 41(23), 8645–8651 CrossRef CAS.
- T. B. Nguyen, et al., Rapid deposition of oxidized biogenic compounds to a temperate forest, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(5), E392–E401 CrossRef CAS PubMed.
- E. Canaval, et al., Rapid conversion of isoprene photooxidation products in terrestrial plants, Commun. Earth Environ., 2020, 1(1), 44 CrossRef PubMed.
- L. Zhang, J. R. Brook and R. Vet, A revised parameterization for gaseous dry deposition in air-quality models, Atmos. Chem. Phys., 2003, 3(6), 2067–2082 CrossRef CAS.
- M. E. Jenkin, A. A. Boyd and R. Lesclaux, Peroxy radical kinetics resulting from the OH-Initiated oxidation of 1,3-butadiene, 2,3-dimethyl-1,3-butadiene and isoprene, J. Atmos. Chem., 1998, 29(3), 267–298 CrossRef CAS.
- G. Da Silva, C. Graham and Z. F. Wang, Unimolecular beta-Hydroxyperoxy Radical Decomposition with OH Recycling in the Photochemical Oxidation of Isoprene, Environ. Sci. Technol., 2010, 44(1), 250–256 CrossRef CAS PubMed.
- J. D. Crounse, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 2011, 13(30), 13607–13613 RSC.
- N. de Oliveira Alves, et al., Biomass burning in the Amazon region: Aerosol source apportionment and associated health risk assessment, Atmos. Environ., 2015, 120, 277–285 CrossRef.
- P. Artaxo, et al., Atmospheric aerosols in Amazonia and land use change: from natural biogenic to biomass burning conditions, Faraday Discuss., 2013, 165, 203–235 RSC.
- M. Keller, et al., Soil-atmosphere exchange of nitrous oxide, nitric oxide, methane, and carbon dioxide in logged and undisturbed forest in the Tapajos National Forest, Brazil, Earth Interact., 2005, 9, 23 Search PubMed.
- K. C. Wells, et al., Satellite isoprene retrievals constrain emissions and atmospheric oxidation, Nature, 2020, 585(7824), 225–233 CrossRef CAS PubMed.
- C. Shim, et al., Constraining global isoprene emissions with Global Ozone Monitoring Experiment (GOME) formaldehyde column measurements, J. Geophys. Res., 2005, 110(D24), D24301 CrossRef.
- M. P. Barkley, et al., Top-down isoprene emissions over tropical South America inferred from SCIAMACHY and OMI formaldehyde columns, J. Geophys. Res.: Atmos., 2013, 118(12), 6849–6868 CAS.
- M. G. Fonseca, et al., Effects of climate and land-use change scenarios on fire probability during the 21st century in the Brazilian Amazon, Global Change Biology, 2019, 25(9), 2931–2946 CrossRef PubMed.
- A. L. Torres and H. Buchan, Tropospheric nitric oxide measurements over the Amazon Basin, J. Geophys. Res.: Atmos., 1988, 93(D2), 1396–1406 CrossRef CAS.
- P. S. Bakwin, et al., Emission of nitric-oxide (no) from tropical forest soils and exchange of no between the forest canopy and atmospheric boundary-layers, J. Geophys. Res.: Atmos., 1990, 95(D10), 16755–16764 CrossRef CAS.
- B. Langford, W. Acton, C. Ammann, A. Valach and E. Nemitz, Eddy-covariance data with low signal-to-noise ratio: time-lag determination, uncertainties and limit of detection, Atmos. Meas. Tech., 2015, 8(10), 4197–4213 CrossRef.
- A. J. Dyer and B. B. Hicks, Flux-gradient relationships in the constant flux layer, Q. J. R. Metereol. Soc., 1970, 96(410), 715–721 CrossRef.
- E. K. Webb, Profile relationships: The log-linear range, and extension to strong stability, Q. J. R. Metereol. Soc., 1970, 96(407), 67–90 CrossRef.
- N. O. Jensen and P. Hummelshøj, Derivation of canopy resistance for water vapour fluxes over a spruce forest, using a new technique for the viscous sublayer resistance, Agric. For. Meteorol., 1995, 73(3), 339–352 CrossRef.
- N. O. Jensen and P. Hummelshøj, Derivation of canopy resistance for water vapor fluxes over a spruce forest, using a new technique for the viscous sublayer resistance (correction to vol. 73), p. 339, 1995), Agric. For. Meteorol., 1997, 85(3–4), 289 CrossRef.
- K. M. Emmerson and M. J. Evans, Comparison of tropospheric gas-phase chemistry schemes for use within global models, Atmos. Chem. Phys., 2009, 9(5), 1831–1845 CrossRef CAS.
- M. E. Jenkin, The CRI v2.2 reduced degradation scheme for isoprene, Atmos. Environ., 2019, 212, 172–182 CrossRef CAS.
- J. Peeters, J.-F. Müller, T. Stavrakou and V. S. Nguyen, Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: the upgraded lim1 mechanism, J. Phys. Chem. A, 2014, 118(38), 8625–8643 CrossRef CAS PubMed.
- S. Madronich, The Atmosphere and UV-B Radiation at Ground Level. Environmental UV Photobiology, ed. A. R. Young, J. Moan, L. O. Björn and W. Nultsch, Springer US, Boston, MA, 1993, pp. 1–39 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00057h |
| ‡ Now at: Climate and Agriculture Group, Agroscope, Switzerland. |
| § Now at: Birmingham Institute of Forest Research, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. |
| ¶ Now at: Wolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, York, YO10 5DD, UK. |
| || Now at: Center for Ecological Research and Forestry Applications (CREAF), Universitat Autònoma de Barcelona, 08193 Bellaterra (Barcelona), Spain. |
| This journal is © The Royal Society of Chemistry 2022 |