
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe crystal structure and luminescence properties of the first lithium oxonitridolithosilicate Li3SiNO2:Eu2+†
Daniel S.
Wimmer‡
a,
Kilian M.
Rießbeck‡
 a,
Markus
Seibald
b,
Dominik
Baumann
b,
Klaus
Wurst
a,
Gunter
Heymann
a and
Hubert
Huppertz
*a
a,
Markus
Seibald
b,
Dominik
Baumann
b,
Klaus
Wurst
a,
Gunter
Heymann
a and
Hubert
Huppertz
*a
aInstitut für Allgemeine, Anorganische und Theoretische Chemie, Universität Innsbruck, Innrain 80-82, A-6020 Innsbruck, Austria. E-mail: Hubert.Huppertz@uibk.ac.at; Web: https://www.uibk.ac.at/aatc/mitarbeiter/hub/
bams-OSRAM International GmbH, Mittelstetter Weg 2, D-86830 Schwabmünchen, Germany
First published on 12th October 2022
Abstract
The compound Li3SiNO2:Eu2+ was synthesized in high temperature solid-state reactions in weld shut tantalum ampules and the crystal structure of Li3SiNO2 has been determined by single-crystal X-ray diffraction. It crystallizes in the monoclinic space group C2/c (no. 15) with the lattice parameters a = 1049.01(3), b = 1103.42(3), c = 511.86(2) pm, β = 116.14(1)°, and a volume of V = 0.53187(2) nm3. This compound is built up from two different layers, which are arranged alternately along the crystallographic a-axis. The results from single-crystal diffraction were confirmed by the Rietveld analysis of bulk samples. Moreover, Li3SiNO2 could be successfully doped with the activator ion Eu2+ and the luminescence spectroscopy of single-crystals revealed broad band emission at λmax = 601 nm (fwhm = 90 nm).
Introduction
Although the field of lithium oxonitridosilicates has been known for quite a long time and the first compound Li4Si0.8N2.4(Li2O)1.6 was published by Juza, Weber, and Meyer-Simon in 1953,1 only a few more representatives are known today. A few years later (1981), the well-known LiSiON was synthesized by Laurent, Guyader, and Roult and analyzed by time-of-flight neutron diffraction.2 Ma et al. were able to successfully dope the compound LiSiON with the activator ion Eu2+ in 2012, which resulted in emission with a maximum of λmax = 426–478 nm (fwhm = 142–193 nm) depending on the activator ion concentration.3 Based on X-ray phase analysis and classical quantitative analysis, compounds such as Li4SiN2O, Li3SiNO2 or Li7SiN3O were predicted by Kraśnicka and Podsiadlo at the same time.4,5 The structure of the last compound, Li7SiN3O, was then elucidated by Casas-Cabanas and Palacín in 2014 using Rietveld refinement.6 All lithium oxonitridosilicates, whose structures are known to date, contain lithium ions exclusively in tetrahedral coordination. This results in structures that consist only of the combined network of tetrahedrally coordinated silicon and lithium ions. In the following, lithium oxonitridolithosilicate compounds shall be referred to as structures, which are built up by the aforementioned (Li/Si)–(O/N) tetrahedral network but additionally with lithium as a completing cation which occupies the cavities. Although no luminescent lithium oxonitridolithosilicate is known yet, there are already some established phosphors in the field of alkali lithosilicates.7–10 The best common examples are Na[Li3SiO4]:Eu2+ (NLSO:Eu2+) (λmax = 469 nm; fwhm = 32 nm), K[Li3SiO4]:Eu2+ (KLSO:Eu2+) (λmax = 604 nm; fwhm = 150 nm), NaK7[Li3SiO4]8:Eu2+ (NKLSO:Eu2+) (λmax = 515 nm and 598; fwhm = 49 nm and 138 nm),11–14 RbNa3[Li3SiO4]4:Eu2+ (λmax = 471 nm; fwhm = 22.4 nm),8,15,16 RbNa[Li3SiO4]2:Eu2+ (λmax = 523 nm; fwhm = 41 nm),17 RbLi[Li3SiO4]2:Eu2+ (RLSO:Eu2+) (λmax = 530 nm; fwhm = 42 nm),18 RbNa2K[Li3SiO4]4:Eu2+ (λmax = 480 nm; fwhm = 26 nm),19 CsNa2K[Li3SiO4]4:Eu2+ (λmax = 485 nm; fwhm = 26 nm),19 LiK7[Li3SiO4]8:Eu2+ (LKLSO:Eu2+) (λmax = 511 nm; fwhm = 45 nm),20 Na2K2[Li3SiO4]4:Eu2+ (λmax = 486 nm; fwhm = 20.7 nm),21,22 RbKLi2[Li3SiO4]4:Eu2+ (λmax = 474 nm and 532; fwhm = 24.8 nm and 43.5 nm),7,23 and Cs4−x−y−zRbxNayLiz[Li3SiO4]:Eu2+ (CRNLLSO:Eu2+) (λmax = 473 nm and 531 nm; fwhm = 25.2 nm and 58 nm).24 These compounds exhibit interesting luminescence properties and some of them are characterized by their narrow single band emission in the blue and green spectral range.In this article, we present the structure of Li3SiNO2 based on single-crystal X-ray data refinement. Additionally, the title phase was doped with the activator ion Eu2+ and, in comparison with the well-known alkali lithosilicates, nitrogen was successfully incorporated into the crystal structure resulting in a shift of the emission band to higher wavelengths compared to the aforementioned oxosilicates.25–27 The here presented compound can be seen as the first example in the field of lithium oxonitridolithosilicates and additionally it exhibits single band emission in the orange spectral region, which is a novelty even for the substance class of oxonitridolithosilicates.
Results and discussion
Crystal structure
The compound Li3SiNO2 crystallizes in the monoclinic space group C2/c with the lattice parameters a = 1049.01(3), b = 1103.42(3), c = 511.86(2) pm, β = 116.14(1)°, and a volume of V = 0.53187(2) nm3. Details on the crystal-structure refinement are given in Table 1. Atomic coordinates, displacement parameters, and interatomic distances are listed in Tables 2–4.| Empirical formula | Li3SiNO2 |
| Molar mass/g mol−1 | 94.92 |
| Crystal system | Monoclinic |
| Space group | C2/c |
| Powder data | |
| Powder diffractometer | STOE Stadi P |
| Radiation | Mo-Kα1 (λ = 70.93 pm) |
| a/pm | 1054.82(8) |
| b/pm | 1102.96(7) |
| c/pm | 512.56(4) |
| β/° | 116.66(1) |
| V/nm3 | 0.53293(6) |
| Single-crystal data | |
| Single-crystal diffractometer | Bruker D8 Quest Kappa |
| Radiation | Mo-Kα (λ = 71.073 pm) |
| a/pm | 1049.01(3) |
| b/pm | 1103.42(3) |
| c/pm | 511.86(2) |
| β/° | 116.14(1) |
| V/nm3 | 0.53187(2) |
| Formula units per cell Z | 8 |
| Calculated density/g cm−3 | 2.371 |
| Crystal size/mm3 | 0.200 × 0.070 × 0.070 |
| Temperature/K | 297(2) |
| Detector distance/mm | 40 |
| Exposure time | 0.5° per frame; 40 s per frame |
| Absorption coefficient/mm−1 | 0.604 |
| F(000)/e | 368 |
| θ-Range/° | 2.84–40.25 |
| Range in hkl | ±19, ±20, ±9 |
| Reflections total/independent | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 474/1622 474/1622 |
| R int | 0.0318 |
| Reflections with I ≥ 2σ(I) | 1579 |
| Data/ref. parameters | 1622/66 |
| Absorption correction | Multi-scan |
| Final R1/wR2 [I ≥ 2σ(I)] | 0.0222/0.0591 |
| Final R1/wR2 (all data) | 0.0226/0.0594 |
| Goodness of fit on F2 | 1.147 |
| Largest diff. peak/hole/e Å−3 | 0.516/−0.640 |
| Atom | Wyckoff-position | x | y | z | U eq | Occ. |
|---|---|---|---|---|---|---|
| Si1 | 8f | 0.22086(2) | 0.43007(2) | 0.25806(3) | 0.00621(6) | 1 |
| Li1 | 4e | ½ | 0.2978(2) | ¼ | 0.0101(2) | 1 |
| Li2 | 4e | ½ | 0.4746(2) | ¾ | 0.0153(3) | 1 |
| Li3 | 4e | 0 | 0.4338(2) | ¾ | 0.0205(4) | 1 |
| Li4 | 4e | 0 | 0.3190(3) | ¼ | 0.0188(4) | 0.76 |
| Li4A | 4e | 0 | 0.275(2) | ¼ | 0.019(2) | 0.24 |
| Li5 | 8f | 0.2769(2) | 0.1976(2) | 0.2176(3) | 0.0198(3) | 1 |
| O1 | 8f | 0.12887(4) | 0.31224(3) | 0.05472(8) | 0.00791(8) | 1 |
| O2 | 8f | 0.38906(4) | 0.39292(5) | 0.3760(2) | 0.01470(9) | 1 |
| N1 | 8f | 0.17022(5) | 0.44522(4) | 0.5324(2) | 0.00872(8) | 1 |
| Atom | U 11 | U 22 | U 33 | U 23 | U 13 | U 12 |
|---|---|---|---|---|---|---|
| Si1 | 0.00646(8) | 0.00652(8) | 0.00580(7) | −0.00072(3) | 0.00283(5) | 0.00006(4) |
| Li1 | 0.0103(6) | 0.0084(5) | 0.0133(6) | 0 | 0.0068(5) | 0 |
| Li2 | 0.0161(7) | 0.0172(7) | 0.0098(6) | 0 | 0.0031(5) | 0 |
| Li3 | 0.0180(8) | 0.0129(7) | 0.0179(8) | 0 | −0.0039(7) | 0 |
| Li5 | 0.0255(6) | 0.0191(6) | 0.0205(6) | 0.0106(4) | 0.0154(5) | 0.0119(5) |
| O1 | 0.0082(2) | 0.0068(2) | 0.0081(2) | −0.00118(9) | 0.0031(2) | −0.0003(2) |
| O2 | 0.0077(2) | 0.0199(2) | 0.0150(2) | −0.0062(2) | 0.0035(2) | 0.0019(2) |
| N1 | 0.0129(2) | 0.0072(2) | 0.0076(2) | 0.0002(2) | 0.0059(2) | 0.0008(2) |
| Si1–O2 | 1.6453(4) | Li3–O1 | 2.050(2) 2× | Li4A–O1 | 2.047(2) 2× |
| Si1–O1 | 1.6792(4) | ∅ Li3–O | 2.050(2) | Li4A–O2 | 2.544(8) 2× |
| ∅ Si1–O | 1.6668(4) | ∅ Li4A–O | 2.296(8) | ||
| Li3–N1 | 2.189(2) 2× | ||||
| Si1–N1 | 1.7146(5) | Li3–N1 | 2.4894(5) 2× | Li4A–N1 | 2.558(8) 2× |
| Si1–N1 | 1.7231(5) | ∅ Li3–N | 2.339(2) | ∅ Li4A–N | 2.558(8) |
| ∅ Si1–N | 1.7189(5) | ||||
| Li4–O1 | 2.0058(4) 2× | Li5–O1 | 1.889(2) | ||
| Li1–O2 | 1.880(2) | ∅ Li4–O | 2.0058(4) | Li5–O1 | 2.036(2) |
| Li1–O1 | 1.969(2) 3× | Li5–O2 | 2.418(2) | ||
| ∅ Li1–O | 1.947(2) | Li4–N1 | 2.223(2) 2× | ∅ Li5–O | 2.114(2) |
| ∅ Li4–N | 2.223(2) | ||||
| Li2–O2 | 1.967(2) 2× | Li5–N1 | 1.950(2) | ||
| Li2–O2 | 2.138(2) 2× | ∅ Li5–N | 1.950(2) | ||
| ∅ Li2–O | 2.053(2) |
The three-dimensional framework in the structure of Li3SiNO2 is built up from two different layers, which are stacked alternately along the crystallographic a-axis. As shown in Fig. 1, the first layer, named layer A, consists of condensed SiO2N2 and LiO3N tetrahedra, where the vertex-sharing SiO2N2 tetrahedra form unbranched zweier single chains28 along the crystallographic c-axis via bridging N[2]3− anions. The structural feature of such unbranched zweier single chains was found for the first time in nature in the mineral diopside CaMgSi2O6, which belongs to the mineral group of pyroxenes and, according to the silicate classification by Liebau, it is an inosilicate.28–30 SiO2N2 tetrahedra can also be found in compounds such as Ca3SiO4N2,31 where these tetrahedra are also connected to each other via N3− anions. The Si–O distances, which vary from 164.53(4) to 167.92(4) pm, and the Si–N distances with values from 171.46(5) to 172.31(5) pm are in good agreement with the sum of the ionic radii.23,24,32,33 The occupation of the N1 site with O instead of N leads to a significant under-occupation, indicating the presence of N at this position. These unbranched zweier single chains are then connected to each other via LiO3N tetrahedra, which form Li2O4N2 dimer units, when only layer A is considered.34 Hereby two LiO3N tetrahedra share a common edge via the O2− anions. Although the dimer units are not connected to each other, they connect the unbranched zweier single chains in such a way that they are corner-sharing via the N3− anions and edge-sharing via two O2− anions. In the LiO3N tetrahedra, the Li–O distances range from 188.9(2) to 241.8(2) pm and the Li–N distance is 195.0(2) pm. Since one Li–O distance is quite elongated, the corresponding coordination polyhedra can also be described as elongated trigonal pyramids exhibiting a 3 + 1 coordination, where the Li+ cations are slightly displaced from the centers (see Fig. 1).12,33,35
 | ||
Fig. 1 Layer A of Li3SiNO2. In the upper left, the LiO3N tetrahedra (orange) with the Li–N and Li–O distances and the dimer units formed from them are shown. In the upper right, the SiO2N2 tetrahedron (blue) is illustrated, including the Si–N and Si–O distances, and the unbranched zweier single chains built up from it. In the lower half, layer A, which is formed from LiO3N and SiO2N2 tetrahedra, is represented along [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 00]. 00]. | ||
The connectivity within layer A in the structure of Li3SiNO2 is related to that in the compounds Na2Mn2S336 and Na6In2S6,37 where the SiO2N2 tetrahedra correspond to the MnS4 tetrahedra (Mn1 site) and NaS4 tetrahedra, respectively, and the LiO3N tetrahedra correspond to the MnS4 tetrahedra (Mn2 site) and InS4 tetrahedra, respectively (see Fig. 2).
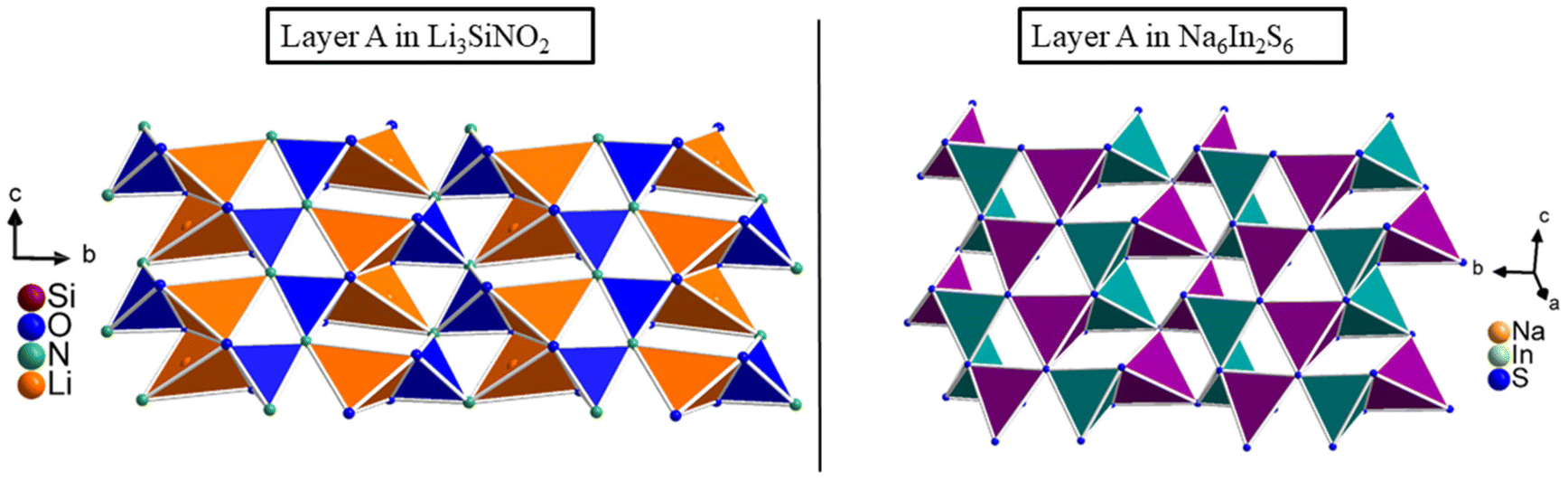 | ||
| Fig. 2 On the left, layer A of Li3SiNO2 is shown compared to layer A of the compound Na6In2S6, which is represented on the right. The LiO3N tetrahedra are displayed in orange, SiO2N2 tetrahedra are displayed in blue, the NaS4 tetrahedra are displayed in violet and the InS4 tetrahedra are displayed in teal. | ||
The second layer, named layer B, is built up from three different coordination polyhedra consisting of LiO4 tetrahedra, LiO2N4 octahedra, and LiO4N2 octahedra, whereby within the latter dynamic positional disorder occurs. In this layer, there are five crystallographically distinguishable lithium sites. The Li1 and Li2 sites form LiO4 tetrahedra with Li1–O and Li2–O distances ranging from 188.0(2) to 196.9(2) pm and 196.7(2) to 213.8(2) pm, respectively, which are in good agreement with the sum of the ionic radii.20,23 The Li3 site is coordinated by four N3− and two O2− anions and constitutes a distorted octahedral arrangement, where the Li3–O distances are 205.0(2) pm and the Li3–N distances are between 218.9(2) and 248.94(5) pm. Since two of the Li3–N distances are slightly longer and are located opposite to each other, this can also be described as an elongated octahedron. For Li4/4A, dynamic positional disorder occurs in the LiO4N2 octahedra, where the blurred electron density can be described by two partially occupied Li+ positions (Li4 and Li4A). The two sites are coordinated by four O2− anions and two N3− anions, where the Li4–O distances range from 200.58(4) to 292.0(3) pm and the Li4–N distance is 222.3(2) pm. The Li4A–O distances range from 204.7(2) to 254.4(8) pm and the Li4A–N distance is 255.8(8) pm, implying a highly distorted octahedron. Due to the spatial proximity, only one of the two positions (Li4 and Li4A) can be occupied at the same time and due to a strong correlation between the two sites, only an isotropic refinement of the displacement parameters in the structure solution was applied. For comparison, the displacement ellipsoids of the asymmetric unit of Li3SiNO2 are shown in the ESI (see Fig. S1†). The asymmetric units are illustrated for the cases where the Li4 site is described by only one fully occupied Li site and by two partially occupied Li sites, respectively. The occupation of the two sites was approximated in such a way that the two isotropic displacement parameters were approximately equal in size and then fixed manually at this value. The crystal structure refinement results in occupations of Li4 = 0.76 and Li4A = 0.24 implying that the Li4 site is statistically preferred. To confirm the occupations of these sites, five crystals from four different syntheses were measured by single-crystal X-ray diffraction at room temperature. All crystal structure refinements showed the same results with minimal deviations. Single-crystal X-ray measurements at low temperatures (−90 °C) have shown that the occupation of the Li4 and Li4A sites changes by 8% (Occ. of Li4 = 0.84 and Li4A = 0.16) and the Li4 site is more preferred. This indicates dynamic positional disorder between the two sites, which is temperature dependent. The results of the single-crystal measurements at low temperature are shown in the ESI,† listed in Tables S2–S5.† One reason for the dynamic positional disorder could be that the volume of the LiO4N2 octahedron (V = 16.73 Å3) is larger than that of the LiO2N4 octahedron (V = 14.19 Å3) and thus the Li+ cation has more space to displace, which becomes higher with increasing temperature (see Fig. 3). The program Vesta 338 was used to calculate the polyhedron sizes.
 | ||
| Fig. 3 The different coordination polyhedra of layer B are shown. On the upper half, the LiO4 tetrahedra of the Li1 and Li2 sites are displayed in orange including the Li–O distances. On the lower left, the LiO2N4 octahedron is displayed in green including the Li–O and Li–N distances and the polyhedron size, respectively. On the lower right, the LiO4N2 octahedron with the two partially occupied Li+ positions is represented in bright green including the Li–O and Li–N distances and the polyhedron size. The Li–O and Li–N distances of the Li4 site are shown in red. | ||
The four different coordination polyhedra around the four Li sites, as mentioned before, form an interconnected tetrahedra and octahedra band spanning the condensed layer B (see Fig. 4). The LiO2N4 and LiO4N2 octahedra built up the octahedra band, named band X, along the crystallographic c-axis, where the LiO2N4 octahedra are double edge-sharing to each other by two common N3− anions on each edge. The LiO4N2 octahedra, on the other hand, are not connected to each other, but share common edges with the LiO2N4 octahedra within the octahedra band X. The LiO2N4 and LiO4N2 octahedra are connected in such a way that one LiO4N2 octahedron shares common edges with three LiO2N4 octahedra once via two N3− anions and twice via one O2− and one N3− anions. Considering the cb-plane, the LiO4N2 octahedra are arranged alternately to the right and to the left of the LiO2N4 octahedra. A similar connection pattern is formed by the LiO4 tetrahedra within the tetrahedra band, named band Y. The LiO4 tetrahedra of the Li2 site are connected to each other along the crystallographic c-axis by double edge-sharing via two common O2− anions on each edge. The LiO4 tetrahedra of the Li1 site, which are not connected to each other within the tetrahedral band Y, always arrange themselves alternately to the left and right of the LiO4 tetrahedra of the Li2 site from the viewpoint perpendicular to the cb-plane. The LiO4 tetrahedra of the Li1 and Li2 sites share a common edge via two O2− anions. Along the crystallographic b-axis, the bands X and Y are arranged alternately. The bands X and Y are connected in such a way that the LiO4 tetrahedra of the Li1 site share a common edge with the LiO2N4 octahedra and two common edges with the LiO4N2 octahedra, each via two O2− anions. Additionally, the LiO4N2 octahedra and the LiO4 tetrahedra of the Li2 site are edge-sharing via two O2− anions. Furthermore, the c glide plane, derived from the space group C2/c, is visible when the mirror plane is placed perpendicular to the crystallographic b-axis through the center of the octahedral band X (see Fig. 4). Since the Li+ cations are not only part of the tetrahedral substructure, we assign this compound to the substance class of lithium oxonitridolithosilicates.
 | ||
| Fig. 4 The highly condensed layer B. In the upper half, the construction of the two different bands (X and Y) are presented. In the lower half, the arrangement and interconnection of the two different bands (X and Y), that build up layer B, are shown. The LiO4 tetrahedra of the Li1 and Li2 sites are displayed in orange, the LiO2N4 octahedra are displayed in green, and the LiO4N2 octahedra including the dynamic positional disorder of the Li4 and Li4A sites are represented in bright green. | ||
The entire structure of Li3SiNO2 is built up from the two layers A and B by stacking them alternately along the crystallographic a-axis with the characteristic pattern BAB′A′ (see Fig. 5). Although layers A and B are equivalent in construction to layers A′ and B′, they are differently orientated due to the most favorable arrangement in terms of energy. Layers A and A′ are related via a two-fold rotation axis that runs along the crystallographic b-axis through layer B. Layers B and B′ are connected via a 21 screw axis, which is additionally caused by the space group C2/c and runs along the crystallographic b-axis through layers A and A′. The two layers A and B are connected in such a way that the LiO3N tetrahedra of the Li5 site share a common edge with the LiO4 tetrahedra of the Li1 site via two O2− anions, and are edge-shared to the LiO4N2 octahedra and the LiO2N4 octahedra via an O2− and N3− anion. In addition, the LiO4 tetrahedra of the Li5 site are also corner-shared to the LiO4 tetrahedra of the Li2 site via an O2− anion. The SiO2N2 tetrahedra of the unbranched zweier single chains show a similar connection pattern. The SiO2N2 tetrahedra share common corners with the LiO4 tetrahedra of the Li2 sites via O2− anions. They share a common edge with the LiO2N4 octahedra via two N3− anions as well as via one N3− and one O2− anion. In addition, it is edge-shared to the LiO4N2 octahedra via an O2− and N3− anion.
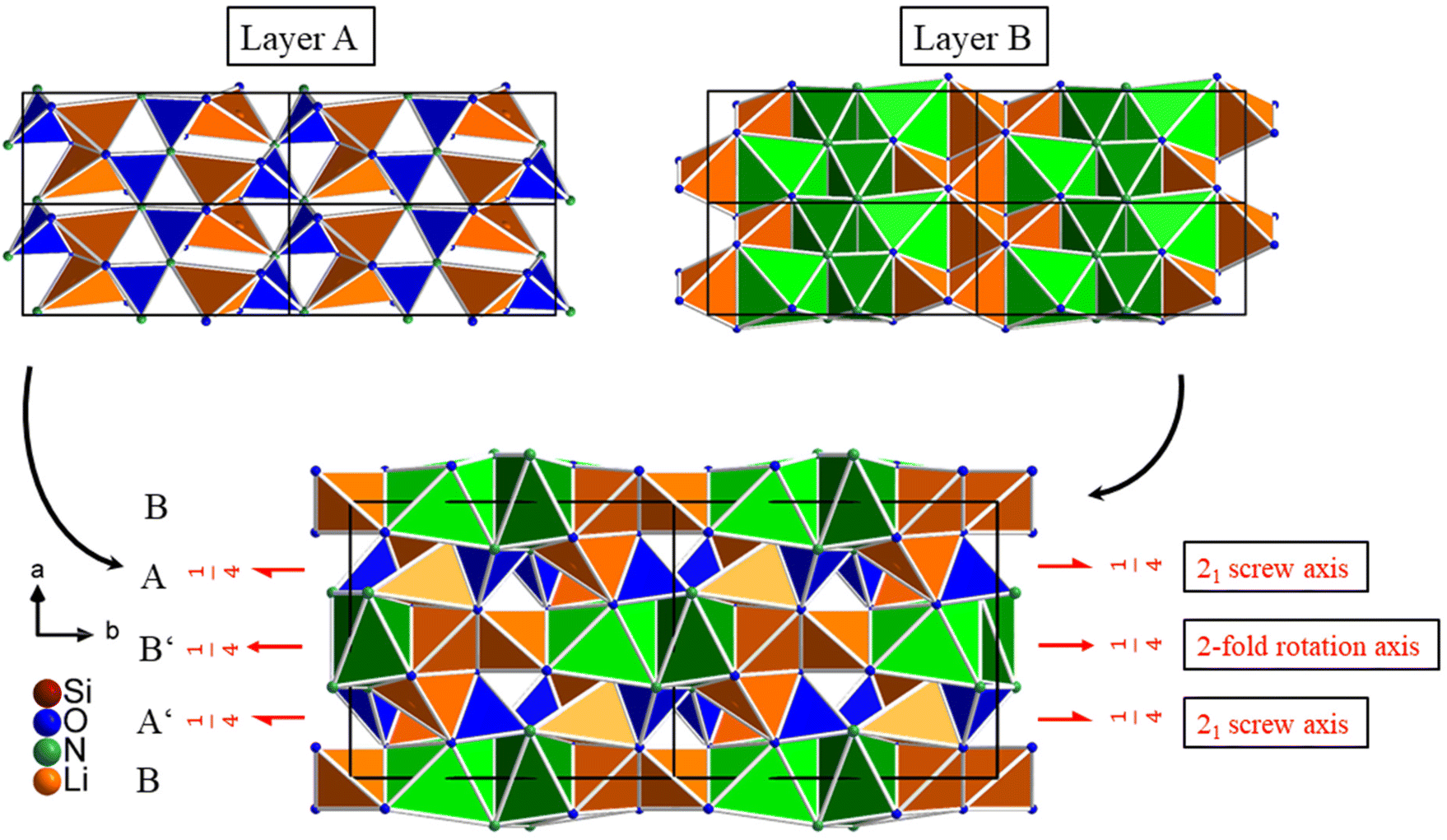 | ||
| Fig. 5 The entire structure of Li3SiNO2 built up from layers A and B. The LiO4 tetrahedra of the Li1 and Li2 sites and the LiO3N tetrahedra are displayed in orange, the SiO2N2 tetrahedra are displayed in blue, the LiO2N4 octahedra are displayed in green and the LiO4N2 octahedra including the dynamic positional disorder of the Li4 and Li4A sites are represented in bright green. | ||
The compound Li3SiNO2 could be successfully doped with the activator ion Eu2+ and the resulting luminescence properties are discussed in the Luminescence section.
MAPLE, CHARDI, and BLBS
The electrostatic consistency of the crystal structure is proved by MAPLE (Madelung Part of Lattice Energy),39–41 BLBS (bond length–bond strength),42,43 and CHARDI (Charge Distribution method)44,45 calculations. In the latter method, the charge distribution is calculated using the concept of effective coordination numbers in combination with bond strength based on Pauling's concept. The BLBS calculations predict the formal oxidation state of an atom by correlating the bond strengths (bond valences) and bond lengths, where the sum of all bond valences should add up to the oxidation state of the respective atom. The MAPLE and CHARDI calculations were performed for the cases where either the Li4 or the Li4A site is fully occupied. The results of the CHARDI and BLBS calculations are shown in Table 5. The partial MAPLE values and MAPLE sums of Li3SiNO2 are listed in Table 6.| Si1 | Li1 | Li2 | Li3 | Li4 | Li4A | Li5 | O1 | O2 | N1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ∑V | 4.10 | 1.10 | 1.00 | 1.02 | 0.89 | 0.68 | 1.01 | −2.09 | −1.85 | −3.07 |
| ∑Q1 | 4.15 | 0.92 | 1.01 | 0.98 | 0.93 | — | 0.93 | −2.50 | −1.97 | −2.52 |
| ∑Q2 | 4.21 | 0.98 | 0.99 | 0.99 | — | 0.83 | 0.94 | −2.59 | −2.02 | −2.40 |
| Li3SiNO2 | Li4 fully occupied | Li4A fully occupied | Model | |
|---|---|---|---|---|
|
a Typical partial MAPLE values (kJ mol−1): Si4+ 9000–10200; Li+ 600–860; O2− 2000–2800; N3− 5000–6200.47 The MAPLE sums of LiSiON and Li2O were calculated from the published single-crystal data.
|
||||
| Si1 | 9200 | 9261 | ||
| Li1 | 808 | 780 | ||
| Li2 | 678 | 635 | ||
| Li3 | 637 | 696 | ||
| Li4 | 667 | — | ||
| Li4A | — | 644 | ||
| Li5 | 653 | 648 | ||
| O1 | 2573 | 2549 | +1 | LiSiON2 |
| O2 | 2529 | 2592 | +1 | Li2O46 |
| N1 | 5783 | 5646 | ||
|
Σ = 22133 |
Σ = 22106 |
Σ = 22191 |
||
| Δ = 0.26% | Δ = 0.38% | |||
The results from the BLBS method show the expected charge for the atoms with the exception of the Li4A atom and are in good accordance with the oxidation states based on crystallographic criteria. The outcome of the BLBS method is supported by the CHARDI calculations, where only the atoms O1 and N1 show larger deviations. The deviations of the N1 and O1 atoms could be explained by the fact that they are spatially very close to the dynamic positional disordered Li4 and Li4A sites and thus interact with them more strongly than the O2 atom. The deviation of the Li4A atom with respect to the BLBS calculation could be due to the fact that the position could describe the blurred electron density very well, but does not correspond to a “typical” Li position in relation to the coordination and the partially longer Li–O/N distances, so the description of a dynamic process with a static model is limited. Furthermore, the partial MAPLE values and the MAPLE sums are compared to the reference values to verify the refined crystal structure. The partial MAPLE values of all individual atoms are in good agreement with the reference values known from the literature and confirm the assignment of the different atoms and the incorporation of N and O into the structure. The results of the calculations are consistent with those from the SCXRD refinement, and the assignment of the N and O atoms to the corresponding sites seems plausible. Comparing the MAPLE sums of LiSiON and Li2O with the calculated MAPLE sums of Li3SiNO2, the values were found to deviate by only 0.26 and 0.38% depending on the full occupation of the Li4 or Li4A site, which indicates the validity of the crystal structure.
Luminescence
The luminescence properties of Li3SiNO2:Eu2+ were investigated on single-crystals and powder samples, where the latter was used to determine the excitation spectrum and the thermal quenching behavior.The powder sample of Li3SiNO2 could not be synthesized in the pure phase and contains 3 wt% of LiSi2N3. The results of the Rietveld refinement can be found in the ESI (see Fig. S2 and Table S1†). Li3SiNO2 with a nominal activator concentration of 2 mol% Eu2+ based on the content of lithium can be excited with near-UV to blue light (e.g. λexc = 448 nm) and the single-crystals exhibit broad band emission at λmax = 601 nm with a full width at half maximum (fwhm) of 90 nm (0.32 eV, 2597 cm−1). In comparison, the powder sample (λexc = 400 nm) exhibits broad band emission at λmax = 606 nm with a full width at half maximum (fwhm) of 100 nm (0.33 eV, 2695 cm−1). In the CIE-xy color space, this corresponds to the values x = 0.564(1) and y = 0.433(1) for the measured single-crystal emission band and to x = 0.562(1) and y = 0.436(1) for the powder sample. Furthermore, an excitation spectrum of the powder sample was recorded exhibiting a maximum at 376 nm (see Fig. 6a and b). The first lab samples without any systematic engineering regarding the emission properties already yielded quantum efficiencies (QE) above 30%. The compound has a brown to gray body color under daylight and is stable under atmospheric conditions for several months.
 | ||
| Fig. 6 (a) The red line represents the luminescence spectrum of a Li3SiNO2:Eu2+ crystal recorded at an excitation wavelength of 448 nm. The green line shows the luminescence spectrum of a powder sample of Li3SiNO2:Eu2+ recorded at an excitation wavelength of 400 nm. The blue line represents the excitation spectrum of the powder sample of Li3SiNO2:Eu2+ monitored at λem = 590 nm (based on raw data maximum). (b) It shows the color point of the Li3SiNO2:Eu2+ single-crystals in the CIE-xy color space.48 | ||
The thermal quenching behavior (TQ) of Li3SiNO2:Eu2+ was examined on a powder sample (see Fig. 7).
 | ||
| Fig. 7 Representation of the thermal quenching behavior of Li3SiNO2:Eu2+ as the relative integral photoluminescence intensity to that at 25 °C measured in steps of 25 °C up to 225 °C. | ||
As can be seen in Fig. 7, the relative integral photoluminescence intensity decreases with increasing temperature and drops to 66% of the initial value at a temperature of 225 °C. At a typical operating temperature of 125 °C for a pc-LED, the relative integral intensity is still above 85%. Nevertheless, QE and TQ would have to be improved for any industrial use.
Since the compound shows only one emission band, it would indicate that only one position in the structure is suitable for incorporating the activator ion Eu2+. One possible position would be the Li3 site, which forms an octahedral coordination to two O2− anions with Li–O distances of 205.0(2) pm and to four N3− anions with Li–N distances between 218.9(2) and 248.94(5) pm resulting in a polyhedron size of 14.19 Å3. Although the distances are too short regarding typical Eu–O/N coordination (Eu–O distances range from 240 to 300 pm and Eu–N distances range from 260 to 325 pm),49–51 local spatial distortion or enlargement of the octahedron could occur in contrast to the average structure. To maintain charge neutrality, the inclusion of a divalent Eu2+ cation may be compensated by Li-vacancies in the surroundings or by local O/N substitution. In comparison with the LiO2N4 octahedron of the Li3 site, the LiO4N2 octahedron including the dynamic positional disordered Li4 and Li4A sites is slightly larger and would also be a possible position to host the Eu2+ cation. The Li4/Li4A site is coordinated octahedrally by four O2− anions with Li–O distances between 200.58(4) and 292.0(3) pm and two N3− anions with Li–N distances between 222.3(2) and 255.8(8) pm resulting in a polyhedron size of 16.73 Å3. Due to the two short Li–O distances, this would also lead to an enlargement or spatial distortion of the octahedron. Compared to compounds such as Li2CaSi2N4:Eu2+,52 Mg3GaN3:Eu2+,53 and Li-α-SiAlON:Eu2+,54 the polyhedron size and Li–O/N distances of the Li3 and Li4/Li4A sites in Li3SiNO2:Eu2+ are in a similar range and it seems possible that one of them hosts the activator ion Eu2+. In the former compound, there are two crystallographically distinguishable Ca sites, which are octahedrally coordinated by six N3− anions and the compound exhibits an emission maximum at λmax = 583–585 nm. The Ca1 site has Ca1–N distances between 248.9(3) and 250.9(3) pm resulting in a polyhedron size of 16.54 Å3 and the Ca2 site with a Ca2–N distances of 258.6(3) pm has a polyhedron size of 18.95 Å3, whereby the author supposed that both Ca sites are suitable to host the activator ion Eu2+. In Mg3GaN3:Eu2+, it was assumed that the activator ion Eu2+ is located in octahedral voids, where the possible observed Eu–N distances are between 232.0 and 248.9 pm resulting in an estimated polyhedron sizes between 14.5 and 16.5 Å3 exhibiting an emission maximum at 578 nm with a fwhm of 132 nm. In Eu2+ doped Li-α-SiAlON, it was supposed that the activator ion Eu2+ occupies a Li site, which is sevenfold coordinated by N3−/O2− anions with Li–O/N distances ranging from 205.2 to 276.7 pm. Depending on the Eu2+ concentration, the compound Li-α-SiAlON exhibits an emission maximum in the range λmax = 563–586 nm.
It would also be possible that the activator ion Eu2+ is situated on the Li2 site. As illustrated in Fig. 8, a lithium channel is formed in the structure of Li3SiNO2 along the crystallographic c-axis, which consists of an edge-shared LiO4 tetrahedral band (Li2 site) and corresponds to band Y. This band Y is connected to two unbranched zweier single chains of SiO2N2 tetrahedra by corner-sharing via the O2− anion, respectively. Due to this special arrangement of the unbranched zweier single chains of SiO2N2 tetrahedra, there would also be two N3− anions in close spatial proximity to the Li2 site, whose Li–N distance would be 314.9 pm. Although this Li–N distance is too large for a Li+ cation's first coordination sphere, it would be a permissible Eu–N distance for the activator ion Eu2+. Considering these two N3− anions, an octahedral coordination would also result for the Eu2+ cation on the Li2 site and is consequently a possible position hosting the Eu2+ cation. Due to the additional tetrahedral coordination of the O2− anions of the Li2 site, this would form a distorted octahedron with a polyhedron size of 12.52 Å3 and, due to the short Li–O distances, there would also be an enlargement or spatial distortion of the octahedron. The incorporation of the activator ion Eu2+ into lithium channels or domains has already been observed in other compounds known from the literature, such as RbKLi2[Li3SiO4]4:Eu2+,7,23 Cs4−x−y−zRbxNayLiz[Li3SiO4]:Eu2+,24 and LiK7[Li3SiO4]8:Eu2+.20 However, it should be considered that in these compounds the Li+ cations form an approximately square planar coordination to four O2− anions instead of a tetrahedral one.
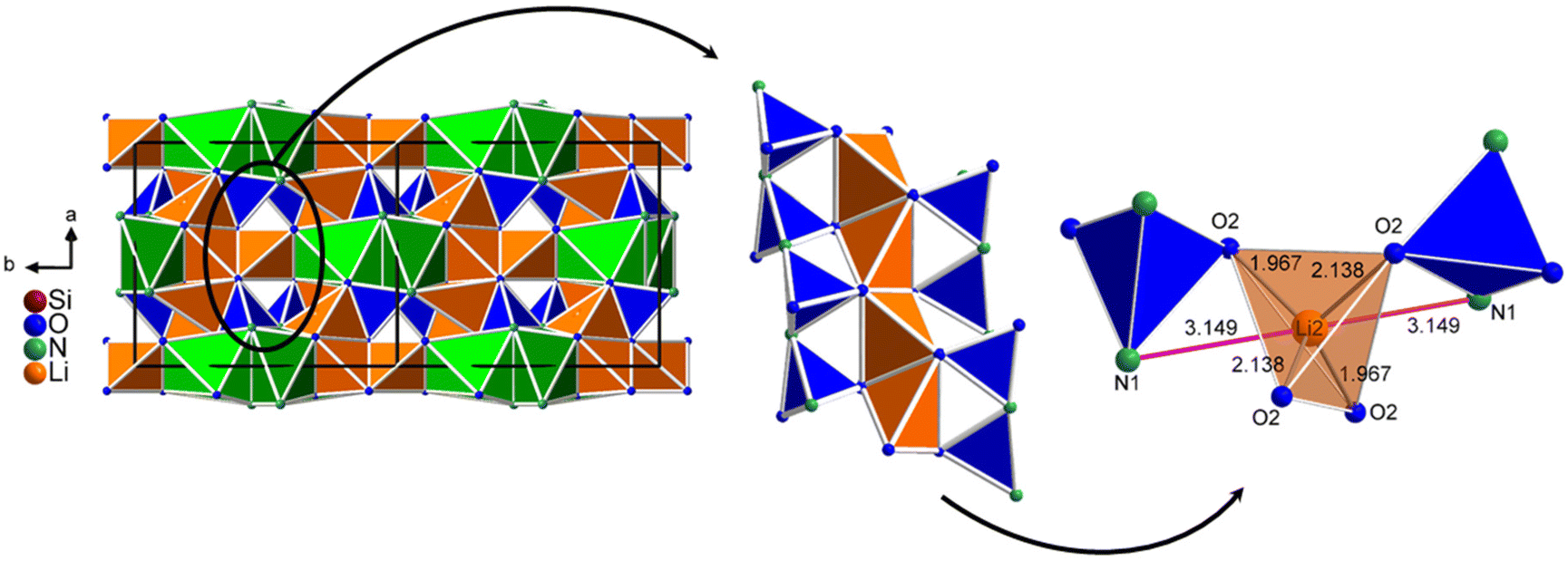 | ||
| Fig. 8 On the left, the position of one of the lithium channels in the structure of Li3SiNO2 is shown. In the middle, the connection of two unbranched zweier single chains of SiO2N2 tetrahedra (blue) with the LiO4 tetrahedra of the Li2 site (orange, band Y) is represented. On the right, the Li2 site including the possible octahedral coordination to four O2− and two N3− anions is illustrated. | ||
Due to the special arrangement of the various polyhedra, unoccupied distorted octahedral voids are formed in the structure, which could also be a possible position hosting the activator ion Eu2+. A similar behavior was observed in the compound Mg3GaN3:Eu2+, as mentioned before. The unoccupied distorted octahedral void is coordinated by five O2− anions and one N3− anion, resulting in a MO5N octahedron with an O1–O2 distance of 465.1, an O2–O2 distance of 511.9, and a N1–O2 distance of 430.6 pm (see Fig. 9). Comparing the anion–anion distances with those of the LiO2N4 octahedron (two times a N1–O1 distances of 421.1, and a N1–N1 distance of 497.2 pm) and the dynamic positional disordered LiO4N2 octahedron ( two times a O2–N1 distances of 501.8, and an O1–O1 distance of 400.9 pm), it is found to be similar in size to the last one and the MO5N octahedron would result in a polyhedron size of 16.46 Å3. The polyhedron size of the MO5N octahedron was calculated by placing a dummy atom in the center of the octahedral void without considering any structural relaxation. Considering these anion–anion distances of the MO5N octahedron, a spatial distortion or enlargement of the octahedral void could occur, when an Eu2+ cation is incorporated.
 | ||
| Fig. 9 On the left, the unoccupied distorted octahedral void (OV) in the structure of Li3SiNO2 is shown by the labeled O2− anions and the N3− anion, which is constructed by the special arrangement of the various polyhedra. On the right, the corresponding anions of the unoccupied MO5N octahedron are shown including the specific anion–anion distances. In the middle, the resulting MO5N octahedron (gray) with hypothetical M–O/N distances including the polyhedron size is illustrated. The LiO4 and LiO3N tetrahedra are displayed in orange, the LiO2N4 octahedron is displayed in green, the SiO2N2 tetrahedra are displayed in blue and the LiO4N2 octahedron including the dynamic positional disordered Li4 and Li4A sites is displayed in bright green. | ||
All four positions could act as doping sites for the activator ion Eu2+ that explain the observed single band emission. Presumably, however, it is assumable that only one of these sites is preferred, otherwise a split or broad emission profile should be observed. It seems most likely that the activator ion Eu2+ is incorporated into the Li2 site building up the lithium channel or in the center of the LiO4N2 octahedron including the dynamic positional disordered Li4 and Li4A sites. One possible reason for the latter is that, compared to the octahedrally coordinated Li3 site, it has larger cation–anion distances, which partially correspond to the observed Eu–N and Eu–O distances, therefore resulting in a larger polyhedron size. In the case of the Li2 site, an increased electron density could be observed at this site in the Eu2+ doped crystals, which roughly corresponds to the 2 mol% activator ion Eu2+ concentration used in the syntheses. In addition, the residual electron density near this site was also found to be aligned towards the N3− anions and the resulting distance to the N3− anions is around 250 pm, which is approximately in the range of Eu–N distances. Considering the unoccupied distorted octahedral void as a possible position hosting the Eu2+ cation, it should be possible to detect low electron density at this position in the single-crystal data. However, no electron density could be observed around this position and consequently the unoccupied distorted octahedral void is presumably not occupied by the activator ion Eu2+. Based on SCXRD, an unambiguous localization of the luminescence center in Li3SiNO2:Eu2+ was not possible.
Compared to other luminescent lithium (oxo)nitride silicates such as LiSi2N3:Eu2+ (ref. 55) or LiSiON:Eu2+,3 the Li–O/N distances of Li3SiNO2:Eu2+ are in a similar range. LiSi2N3:Eu2+ crystallizes in the orthorhombic space group Cmc21 and has one Li site tetrahedrally coordinated by four N3− anions with Li–N distances between 202(2) and 264(2) pm. The compound exhibits broadband emission with a maximum at λmax = 572–584 nm. The compound LiSiON:Eu2+, which crystallizes in the orthorhombic space group Pca21, also has one Li site. The Li+ cation has a coordination sphere to three O2− and one N3− anions, whose Li–O distances are between 191.8 and 206.8 pm and the Li–N distance is 225.2 pm resulting in an emission maximum at λmax = 426–478 nm (fwhm = 142–193 nm). In the publication of LiSi2N3:Eu2+ and LiSiON:Eu2+, no possible positions for the activator ion Eu2+ were discussed. In relation to LiSi2N3:Eu2+ and LiSiON:Eu2+, a blue shift in luminescence due to the partial substitution of N3− anions by O2− anions cannot be observed for Li3SiNO2:Eu2+. Since the structures have fundamental differences regarding the coordination of the Li+ cations, a comparison of these compounds is not possible. Other compounds such as Sr[LiAl3N4]:Eu2+ (SLA:Eu2+) (λmax = 654 nm; fwhm = 50 nm)56 and Sr[Li2Al2O2N2]:Eu2+ (SALON:Eu2+) (λmax = 614 nm; fwhm = 48 nm),57 for example, have comparable structures, where a partial substitution of N3− anions by O2− anions leads to a blue shift in the observed emission bands. The nephelauxetic effect is stronger for the activator ion Eu2+ when it is coordinated by N3− anions rather than by O2− anions due to the higher formal charge of N3− anions. Consequently it leads to a lowering of the 5d energy levels resulting in a red-shift of the Eu2+ emissions induced from the d to f transitions.58
Conclusion
In this study, a new orange-emitting phosphor Li3SiNO2:Eu2+ has been successfully prepared by a solid-state reaction in tantalum ampules. The compound can be excited effectively by near-UV to blue light and the single-crystals of Li3SiNO2:Eu2+ exhibit an emission maximum at λmax = 601 nm with a full width at half maximum (fwhm) of 90 nm (0.32 eV, 2597 cm−1). The crystal structure with its 3D-network can be described as a condensed layered structure consisting of two different layers A and B, which arrange alternately along the crystallographic a-axis. Although layer A has structural similarities to other compounds such as Na2Mn2S3 and Na6In2S6 and zweier single chains can basically also be found in phosphors such as MSi2O2N2 (M = Sr, Ba, Eu),26,59–73 the structural motif and connection pattern in layer B are still unknown. The latter consists of pure Li+ cations, which build up a dense network of corner- and edge-shared tetrahedra and octahedra with the O2− and N3− anions. Furthermore, we discussed four possible positions in the structure hosting the activator ion Eu2+, which include two different octahedral coordination spheres, one octahedral void spanned by a special arrangement of different polyhedra and a tetrahedrally coordinated Li-channel. In comparison with other compounds known from the literature such as α- and β-SiALON,74–76 we see that heavy cations are not necessarily required as counterions in the structure that can be substituted by the activator ion Eu2+ resulting in interesting luminescence properties. Although the new phosphor can be synthesized by low-cost educts and shows relatively good stability against moisture and atmospheric conditions, a further optimization of the material's optical properties must be achieved for possible applications as a phosphor material for example in pc-LEDs.Experimental section and theoretical methods
Synthesis
Since it is crucial to maintain control over the amount of oxygen for synthesizing oxonitrides, all preparations were carried out using an inert gas (Ar 5.0, Messer Austria GmbH) filled glovebox (MBraun, O2 < 1 ppm, H2O < 1 ppm). The starting materials Si3N4 (UBE SN-E10, >99%) and Li2O (Sigma Aldrich, >97%) with a stoichiometric ratio of 1:4 were weighed and mixed with 5 wt% LiF (Sigma Aldrich, >99.99%) as a flux and 2mol% EuF2 (Alfa Aesar, >99.9%) using an agate mortar. Afterwards, the powder mixture was filled into a tantalum ampule and sealed via arc welding under an argon atmosphere. The reaction vessel was then placed inside a silica-glass ampule containing a 400 mbar inert gas atmosphere (Ar 5.0, Messer Austria GmbH). The temperature profile of the synthesis consists of a heating ramp with 4 °C min−1 up to 960 °C, a dwelling time of 4 h, and subsequently a cooling ramp of −0.3 °C min−1 to 300 °C. Once cooled down to room temperature, the ampules could be opened and the samples were further examined under atmospheric conditions without detectable decomposition within several months. The product shows intense yellow to orange luminescence under UV-light (see Fig. 10).
 | ||
| Fig. 10 Left: optical appearance of the 2 mol% Eu2+-doped compound Li3SiNO2 under daylight. Right: optical appearance of the 2 mol% Eu2+-doped compound Li3SiNO2 under UV-light (365 nm). | ||
Single-crystal X-ray diffraction
Under a polarization microscope, suitable single-crystals of Li3SiNO2 were isolated and subsequently investigated with a Bruker D8 Quest diffractometer (Mo-Kα radiation, λ = 0.7107 Å) equipped with a Photon 300 CMOS detector. The programs Saint77 and Sadabs78 were used for data processing and multi-scan absorption correction. For the structure solution and parameter refinement, ShelXS79 using Direct Methods and ShelXL80 using the least squares method, both implemented in the program WinGX,81 were applied. The structural data were standardized employing Structure Tidy82 as implemented in Platon (version 170613).83 Further details on the crystal structure investigation of the single-crystal of Li3SiNO2 can be obtained from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures/under the deposition number 2208257.†Powder X-ray diffraction
Powder X-ray diffraction (PXRD), equipped with a STOE Stadi P powder diffractometer with Ge(111)-monochromatized Mo-Kα1 (λ = 70.93 pm) radiation and a Mythen 1K detector operated in Debye–Scherrer geometry across a 2θ range of 2°–40°, was used for phase analysis. The experimental powder pattern was compared with the calculated powder pattern derived from the single-crystal data of Li3SiNO2. The secondary phase identification was carried out with the ICDD PDF-2 database and the program Topas 4.284 was applied for Rietveld refinements.Luminescence
The emission spectra of representatives from Li3SiNO2 were recorded by a setup of a blue laser diode (λ = 448 nm, THORLABS, Newton, NJ, USA) in combination with a CCD detector (AVA AvaSpec 2048, AVANTES, Apeldoorn, The Netherlands). A tungsten–halogen calibration lamp was used for the previous spectral radiance calibration of the setup. The software AVA AvaSoft (version 7) was used for the data preparation. The analysis of the luminescence properties of powder samples was carried out using a Fluoromax 4 spectrophotometer (Horiba). The emission spectrum was recorded in the wavelength range between 470 and 750 nm (step size 1 nm) using an excitation wavelength of 400 nm. The same method was used to record the excitation spectra monitored at the corresponding maximum intensity and for the thermal-quenching analysis.Author contributions
All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.Conflicts of interest
The authors declare no conflict of interest regarding this article.Acknowledgements
A part of this work is funded by the German Federal Ministry of Economic Affairs and Climate Action (Bundesministerium für Wirtschaft und Klimaschutz) in the frame of the “Important Project of Common European Interest (IPCEI) on microelectronics”. The authors would like to thank Christiane Stoll for her support in the single-grain and powder luminescence as well as single-crystal X-ray diffraction measurements (ams-OSRAM International GmbH).References
- R. Juza, H. H. Weber and E. Meyer-Simon, Z. Anorg. Allg. Chem., 1953, 273, 48–64 CrossRef CAS.
- Y. Laurent, J. Guyader and G. Roult, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, B37, 911–913 CrossRef CAS.
- Y. Ma, F. Xiao, S. Ye, Q. Zhang, Z. Jiang and Y. Qian, ECS J. Solid State Sci. Technol., 2012, 1, R1 CrossRef CAS.
- A. Kraśnicka and S. Podsiadlo, J. Therm. Anal. Calorim., 1988, 34, 305–310 CrossRef.
- S. Podsiadło, J. Therm. Anal., 1987, 32, 771–775 CrossRef.
- M. Casas-Cabanas, H. Santner and M. Palacín, J. Solid State Chem., 2014, 213, 152–157 CrossRef CAS.
- K. Bernet and R. Hoppe, Z. Anorg. Allg. Chem., 1991, 592, 93–105 CrossRef CAS.
- J. Hoffmann, R. Brandes and R. Hoppe, Z. Anorg. Allg. Chem., 1994, 620, 1495–1508 CrossRef.
- B. Nowitzki and R. Hoppe, Chem. Inf., 1986, 17, 217–230 Search PubMed.
- D. Baumann, M. Seibald, T. Fiedler, S. Lange, H. Huppertz, D. Dutzler, T. Schröder, D. Bichler, G. Plundrich, S. Peschke, G. Hoerder, G. Achreiner and K. Wurst, WO/2018/029304, 2018.
- R. Werthmann and R. Hoppe, Z. Anorg. Allg. Chem., 1984, 509, 7–22 CrossRef CAS.
- D. Dutzler, M. Seibald, D. Baumann and H. Huppertz, Angew. Chem., Int. Ed., 2018, 57, 13676–13680 CrossRef CAS PubMed.
- M. Iwaki, S. Kumagai, S. Konishi, A. Koizumi, T. Hasegawa, K. Uematsu, A. Itadani, K. Toda and M. Sato, J. Alloys Compd., 2019, 776, 1016–1024 CrossRef CAS.
- B. Nowitzki and R. Hoppe, Rev. Chim. Minér., 1986, 23, 217–230 CAS.
- H. Liao, M. Zhao, M. S. Molokeev, Q. Liu and Z. Xia, Angew. Chem., Int. Ed., 2018, 57, 11728–11731 CrossRef CAS PubMed.
- H. Liao, M. Zhao, M. S. Molokeev, Q. Liu and Z. Xia, Angew. Chem., 2018, 130, 11902–11905 CrossRef.
- H. Liao, M. Zhao, Y. Zhou, M. S. Molokeev, Q. Liu, Q. Zhang and Z. Xia, Adv. Funct. Mater., 2019, 29, 1901988 CrossRef.
- M. Zhao, H. Liao, L. Ning, Q. Zhang, Q. Liu and Z. Xia, Adv. Mater., 2018, 30, 1802489 CrossRef PubMed.
- M. Zhao, Y. Zhou, M. S. Molokeev, Q. Zhang, Q. Liu and Z. Xia, Adv. Opt. Mater., 2019, 7, 1801631 CrossRef.
- D. S. Wimmer, M. Seibald, D. Baumann, S. Peschke, K. Wurst, G. Heymann, D. Dutzler, A. Garcia-Fuente, W. Urland and H. Huppertz, Eur. J. Inorg. Chem., 2021, 2021, 4470–4481 CrossRef CAS.
- M. Zhao, H. Liao, M. S. Molokeev, Y. Zhou, Q. Zhang, Q. Liu and Z. Xia, Light: Sci. Appl., 2019, 8, 1–9 CrossRef CAS PubMed.
- F. Ruegenberg, A. García-Fuente, M. Seibald, D. Baumann, S. Peschke, W. Urland, A. Meijerink, H. Huppertz and M. Suta, Adv. Opt. Mater., 2021, 9, 2101643 CrossRef CAS.
- D. Dutzler, M. Seibald, D. Baumann, F. Philipp, S. Peschke and H. Huppertz, Z. Naturforsch., B: J. Chem. Sci., 2019, 74, 535–546 CrossRef CAS.
- F. Ruegenberg, M. Seibald, D. Baumann, S. Peschke, P. C. Schmid and H. Huppertz, Chem. – Eur. J., 2020, 26, 2204–2210 CrossRef CAS PubMed.
- H.-W. Wei, X.-M. Wang, H. Jiao and X.-P. Jing, J. Alloys Compd., 2017, 726, 22–29 CrossRef CAS.
- V. Bachmann, C. Ronda, O. Oeckler, W. Schnick and A. Meijerink, Chem. Mater., 2009, 21, 316–325 CrossRef CAS.
- H. A. Höppe, H. Lutz, P. Morys, W. Schnick and A. Seilmeier, J. Phys. Chem. Solids, 2000, 61, 2001–2006 CrossRef.
- According to F. Liebau, polysilicate units (e.g. chains according to the term “dimensionality” = 1) are classified according to the lowest number of SiO4 tetrahedra per identity period (“periodicity” of the chain) as the chains of zwei, drei, vier etc. tetrahedra. The terms derive from German numerals zwei (2, two), drei (3, three), vier (4, four), etc. by adding the suffix “er” to the numeral. F. Liebau, Structural Chemistry of Silicates, Springer, Berlin, Heidelberg, 1985. Furthermore, according to the number of polysilicate chains connected to each other via Si–O–Si bridges to form bands (“multiplicity” of the chains), a distinction is made between single, double, triple, etc. chains.
- J. Zussman, Earth-Sci. Rev., 1968, 4, 39–67 CrossRef CAS.
- B. Warren and W. L. Bragg, Z. Kristallogr.-Cryst. Mater., 1929, 69, 168–193 CrossRef.
- X.-M. Wang, C.-H. Wang, M. Wu, Y. Wang and X.-P. Jing, J. Mater. Chem., 2012, 22, 3388–3394 RSC.
- S. Schmiechen, P. Strobel, C. Hecht, T. Reith, M. Siegert, P. J. Schmidt, P. Huppertz, D. Wiechert and W. Schnick, Chem. Mater., 2015, 27, 1780–1785 CrossRef CAS.
- P. Strobel, V. Weiler, C. Hecht, P. J. Schmidt and W. Schnick, Chem. Mater., 2017, 29, 1377–1383 CrossRef CAS.
- P. Strobel, S. Schmiechen, M. Siegert, A. Tücks, P. J. Schmidt and W. Schnick, Chem. Mater., 2015, 27, 6109–6115 CrossRef CAS.
- D. Wilhelm, D. Baumann, M. Seibald, K. Wurst, G. Heymann and H. Huppertz, Chem. Mater., 2017, 29, 1204–1209 CrossRef CAS.
- K. Klepp, P. Böttcher and W. Bronger, J. Solid State Chem., 1983, 47, 301–306 CrossRef CAS.
- B. Eisenmann and A. Hofmann, Z. Kristallogr., 1991, 197, 151–152 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1966, 5, 95–106 CrossRef CAS.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1970, 9, 25–34 CrossRef CAS.
- M. v. R. Hübenthal, Maple, version 4, University of Gießen, Gießen (Germany), 1993 Search PubMed.
- I. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- N. Brese and M. O'keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- R. Hoppe, Z. Kristallogr.-Cryst. Mater., 1979, 150, 23–52 CrossRef CAS.
- R. Hoppe, S. Voigt, H. Glaum, J. Kissel, H. P. Müller and K. Bernet, J. Less-Common Met., 1989, 156, 105–122 CrossRef CAS.
- T. Farley, W. Hayes, S. Hull, M. Hutchings and M. Vrtis, J. Phys.: Condens. Matter, 1991, 3, 4761 CrossRef CAS.
- M. Zeuner, S. Pagano and W. Schnick, Angew. Chem., Int. Ed., 2011, 50, 7754–7775 CrossRef CAS PubMed.
- CIE-Diagram: https://de.wikipedia.org/wiki/Datei:Cie_chromaticity_diagram_wavelength.png#/media/Datei:CIExy1931.png (04.08.2021).
- C. Funk, J. r. Köhler, I. Lazar, D. Kajewski, K. Roleder, J. r. Nuss, A. Bussmann-Holder, H. Bamberger, J. van Slageren and D. Enseling, Cryst. Growth Des., 2018, 18, 6316–6325 CrossRef CAS.
- M. Zeuner, S. Pagano, P. Matthes, D. Bichler, D. Johrendt, T. Harmening, R. Pöttgen and W. Schnick, J. Am. Chem. Soc., 2009, 131, 11242–11248 CrossRef CAS PubMed.
- J. Felsche, Sci. Nat., 1971, 58, 218–219 CrossRef CAS.
- M. Zeuner, S. Pagano, S. Hug, P. Pust, S. Schmiechen, C. Scheu and W. Schnick, Eur. J. Inorg. Chem., 2010, 2010, 4945–4951 CrossRef.
- F. Hintze, N. W. Johnson, M. Seibald, D. Muir, A. Moewes and W. Schnick, Chem. Mater., 2013, 25, 4044–4052 CrossRef CAS.
- R.-J. Xie, N. Hirosaki, M. Mitomo, K. Sakuma and N. Kimura, Appl. Phys. Lett., 2006, 89, 241103 CrossRef.
- Y. Li, N. Hirosaki, R. Xie, T. Takeka and M. Mitomo, J. Solid State Chem., 2009, 182, 301–311 CrossRef CAS.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS PubMed.
- G. J. Hoerder, M. Seibald, D. Baumann, T. Schröder, S. Peschke, P. C. Schmid, T. Tyborski, P. Pust, I. Stoll, M. Bergler, C. Patzig, S. Reißaus, M. Krause, L. Berthold, T. Höche, D. Johrendt and H. Huppertz, Nat. Commun., 2019, 10, 1824 CrossRef PubMed.
- J. L. Leaño, M.-H. Fang and R.-S. Liu, ECS J. Solid State Sci. Technol., 2017, 7, R3111 CrossRef.
- O. Oeckler, F. Stadler, T. Rosenthal and W. Schnick, Solid State Sci., 2007, 9, 205–212 CrossRef CAS.
- F. Stadler, O. Oeckler, H. A. Höppe, M. H. Möller, R. Pöttgen, B. D. Mosel, P. Schmidt, V. Duppel, A. Simon and W. Schnick, Chem. – Eur. J., 2006, 12, 6984–6990 CrossRef CAS PubMed.
- K. Park, D. Kim, Y. Jeong, J. Kim and T. Kim, J. Nanosci. Nanotechnol., 2016, 16, 1700–1702 CrossRef CAS PubMed.
- V. Bachmann, T. Jüstel, A. Meijerink, C. Ronda and P. J. Schmidt, J. Lumin., 2006, 121, 441–449 CrossRef CAS.
- X. Song, H. He, R. Fu, D. Wang, X. Zhao and Z. Pan, J. Phys. D: Appl. Phys., 2009, 42, 065409 CrossRef.
- M. Seibald, T. Rosenthal, O. Oeckler, C. Maak, A. Tücks, P. J. Schmidt, D. Wiechert and W. Schnick, Chem. Mater., 2013, 25, 1852–1857 CrossRef CAS.
- J. Botterman, K. Van den Eeckhout, I. De Baere, D. Poelman and P. F. Smet, Acta Mater., 2012, 60, 5494–5500 CrossRef CAS.
- X. Song, R. Fu, S. Agathopoulos, H. He, X. Zhao and X. Yu, J. Am. Ceram. Soc., 2011, 94, 501–507 CrossRef CAS.
- J. A. Kechele, O. Oeckler, F. Stadler and W. Schnick, Solid State Sci., 2009, 11, 537–543 CrossRef CAS.
- C. H. Hsu, B. M. Cheng and C. H. Lu, J. Am. Ceram. Soc., 2011, 94, 2878–2883 CrossRef CAS.
- R. Fu, S. Agathopoulos, X. Song, X. Zhao, H. He and X. Yu, Opt. Mater., 2010, 33, 99–102 CrossRef CAS.
- H. A. Höppe, F. Stadler, O. Oeckler and W. Schnick, Angew. Chem., Int. Ed., 2004, 43, 5540–5542 CrossRef PubMed.
- J. Botterman, K. Van den Eeckhout, A. J. Bos, P. Dorenbos and P. F. Smet, Opt. Mater. Express, 2012, 2, 341–349 CrossRef CAS.
- M. Seibald, T. Rosenthal, O. Oeckler and W. Schnick, Crit. Rev. Solid State Mater. Sci., 2014, 39, 215–229 CrossRef CAS.
- M. Seibald, T. Rosenthal, O. Oeckler, F. Fahrnbauer, A. Tücks, P. J. Schmidt and W. Schnick, Chem. – Eur. J., 2012, 18, 13446–13452 CrossRef CAS PubMed.
- K. Kimoto, R.-J. Xie, Y. Matsui, K. Ishizuka and N. Hirosaki, Appl. Phys. Lett., 2009, 94, 041908 CrossRef.
- N. Hirosaki, R.-J. Xie, K. Kimoto, T. Sekiguchi, Y. Yamamoto, T. Suehiro and M. Mitomo, Appl. Phys. Lett., 2005, 86, 211905 CrossRef.
- R.-J. Xie, N. Hirosaki, M. Mitomo, Y. Yamamoto, T. Suehiro and K. Sakuma, J. Phys. Chem. Lett., 2004, 108, 12027–12031 CrossRef CAS.
- Saint, version 8.34a, Bruker AXS Inc., Madison, Wisconsin (USA), 2014 Search PubMed.
- Sadabs, version 2014/5, Bruker AXS Inc., Madison, Wisconsin (USA), 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- L. Gelato and E. Parthé, J. Appl. Crystallogr., 1987, 20, 139–143 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- Topas, version 4.2, Bruker AXS Inc., Madison, Wisconsin (USA), 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2208257. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03064k |
| ‡ D. W. and K. R. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |