
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoparticle-based supramolecular approach for dye-sensitized H2-evolving photocathodes†
Noémie
Lalaoui‡
 ab,
Mohamed
Abdellah‡
ac,
Kelly L.
Materna
a,
Bo
Xu
a,
Haining
Tian
a,
Anders
Thapper
a,
Jacinto
Sa
ad,
Leif
Hammarström
*a and
Sascha
Ott
*a
ab,
Mohamed
Abdellah‡
ac,
Kelly L.
Materna
a,
Bo
Xu
a,
Haining
Tian
a,
Anders
Thapper
a,
Jacinto
Sa
ad,
Leif
Hammarström
*a and
Sascha
Ott
*a
aDepartment of Chemistry-Ångström Laboratories, Uppsala University, Box 523, SE75120 Uppsala, Sweden. E-mail: leif.hammarstrom@kemi.uu.se; sascha.ott@kemi.uu.se
bUniv. Grenoble Alpes, UMR CNRS 5250, Département de Chimie Moléculaire, 38000 Grenoble, France
cDepartment of Chemistry, Qena Faculty of Science, South Valley University, 83523 Qena, Egypt
dInstitute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Poland
First published on 23rd September 2022
Abstract
Solar conversion of water into the storable energy carrier H2 can be achieved through photoelectrochemical water splitting using light adsorbing anodes and cathodes bearing O2 and H2 evolving catalysts, respectively. Herein a novel photocathode nanohybrid system is reported. This photocathode consists of a dye-sensitized p-type nickel oxide (NiO) with a perylene-based chromophore (PCA) and a tetra-adamantane modified cobaloxime reduction catalyst (Co) that photo-reduces aqueous protons to H2. An original supramolecular approach was employed, using β-cyclodextrin functionalized gold nanoparticles (β-CD-AuNPs) to link the alkane chain of the PCA dye to the adamantane moieties of the cobaloxime catalyst (Co). This new architecture was investigated by photoelectrochemical measurements and via femtosecond-transient absorption spectroscopy. The results show that irradiation of the complete NiO|PCA|β-CD-AuNPs|Co electrode leads to ultrafast hole injection into NiO (π = 3 ps) from the excited dye, followed by rapid reduction of the catalyst, and finally H2 evolution.
Introduction
The development of sustainable energy technologies that allows us to decrease our use of fossil fuels, with their negative environmental impact, is one of the greatest challenges of our times. Inspired by natural photosynthesis, the conversion of solar energy into fuels (e.g. H2, CH4, CO) from abundant substrates (H2O, CO2) by photoelectrochemical cells (PECs) is a promising strategy to address this challenge.1–4 The electrodes in dye-sensitized photoelectrochemical cells (DS-PECs) are typically based on a wide band gap semiconductor that is sensitized with molecular dye molecules as light harvesting compounds5–8 and catalysts for solar fuel production.9,10 In a complete tandem cell, water oxidation occurs at the photoanode, while H2 is produced at the photocathode.Significant work has been done to improve the photoanodes, which can now achieve relatively high photocurrents and efficiencies.5,7,8 In comparison, the photocathodes have been less explored, and their performances are currently limiting the efficiency of complete DS-PECs.6,11–13 In dye-sensitized photocathodes (DS-PCs), light absorption by the surface bound dye is followed by hole injection into the valence band of the semiconductor, creating a reduced dye that can transfer an electron to the co-immobilized catalyst. Two reducing equivalents as well as two protons are needed by the catalyst to form H2. Rapid charge recombination relative to the rate of proton reduction limits the overall efficiency of DS-PCs. To generate higher photocurrents, the dominant recombination mechanisms between holes in the p-type semiconductor and (1) the reduced dye or (2) the reduced catalyst must be prevented.12
The nature of the molecular components, the semiconductor itself, and the way by which photosensitizer and catalyst are arranged on the electrode surface greatly impact the performance of DS-PCs. Several DS-PCs photocathodes have been described with the majority relying on NiO substrates.6,14–28 However, problems associated with the high density of traps and low hole mobility have been identified,29–32 and other p-type metal oxide semiconductors such as LaFeO3 and CuCrO2 have been recently proposed as alternative materials for the construction of DS-PCs for solar-driven H2 generation.33–35 As far as the dye is concerned, organic chromophores and ruthenium based complexes are the two families of light-harvesting units that are frequently used. The best photosensitizers have a donor–bridge–acceptor structure, where the HOMO is positioned close to the anchoring group and the LUMO is positioned away from the NiO surface.13,36–38 This push–pull structure is thought to promote charge-separation and slow down charge recombination by keeping the electron on the reduced dye physically distant to the holes in the NiO. Among organic chromophores, perylene-based dyes are renowned for their high stability and intense visible light absorption. PMI based photocathodes have been assembled with heterogeneous catalysts,37 molecular catalysts in solution39 and immobilized Ni(P2N2)2 – based catalysts,26,35 but to our knowledge, never with a cobaloxime type catalysts.
To improve PC performance, many PC designs have focused on optimizing the electrode assembly. The different strategies are DS-PCs combined with a catalyst in solution,16,36,39 co-grafted dye/catalyst systems,6,19,22,40 layer by layer assemblies of the dye and the catalyst,17,18,20 and supramolecular or covalent dye/catalyst assemblies.14,15,21,23
In this study, we report the design and synthesis of a new dye/catalyst supramolecular assembly based on β-cyclodextrin functionalized gold nanoparticles (β-CD-AuNPs) that connect a perylene-based dye (PCA) to a cobaloxime catalyst (Co). A rational and simple procedure of sequential immersion of NiO films into solutions of PCA, β-CD-AuNPs and Co was employed to produce electrodes of the type NiO|PCA|β-CD-AuNPs|Co (Fig. 1A) where the catalyst is spatially separated from the semiconductor electrode to reduce inefficiency from charge recombination. β-CD binds strongly to the alkane chain of the dye and the adamantane moiety of the catalyst to form the supramolecular assembly. Cyclodextrin has been previously used to form host–guest interactions in order to immobilized molecular catalysts on electrode surfaces.41,42 In the present architecture, AuNPs are proposed to act as electron transfer bridges by accepting electrons from PCA and transporting them to Co. This function of the AuNPs is motivated by previous studies in which such constructs were shown to enhance electron transfer rates at protein/electrode interfaces in bioelectrochemical systems.43,44 Other studies have shown that small gold nanoparticles (2–3 nm) significantly increase charge separation in chromophore-gold nano-assemblies, where the charge separation is sustained for several microseconds before undergoing recombination.45,46 However, to the best of our knowledge, this strategy has never been explored to develop operative photocathodes for H2 evolution.
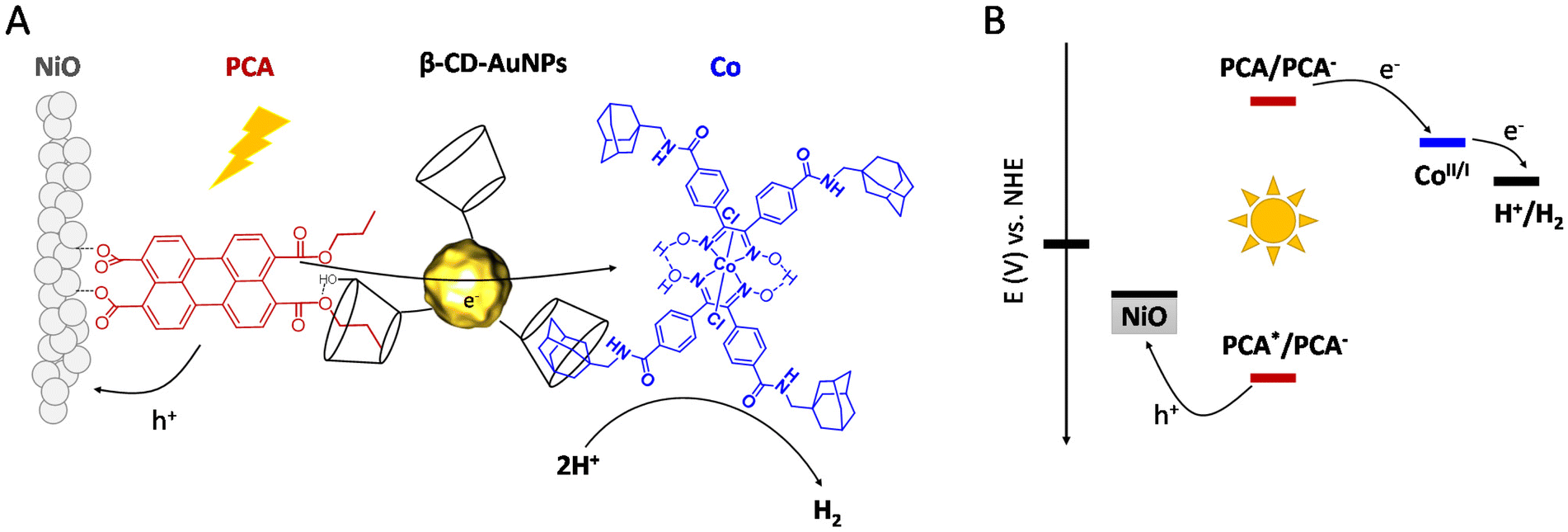 | ||
| Fig. 1 (A) Representation of the PCA|β-CD-AuNPs|Co supramolecular assembly anchored onto a mesoporous NiO thin film, (B) energy diagram of the photocathode. | ||
Results and discussion
Synthesis and characterization
PCA,47 β-CD-AuNPs48,49 and [Co(dcpgH)(dcpgH2)]Cl2501 were synthesized as previously reported. The adamantane moiety was added to 1via peptide coupling (see ESI†) to afford Co. The target complex was characterized by 1H NMR, Fourier transform infrared (FT-IR) spectroscopy, high-resolution mass spectrometry (HR-MS) and electrochemistry (Fig. S1–S3†). Fig. S11† shows a transmission electron microscopy (TEM) image of the functionalized AuNPs and reveals an average AuNP size of 3.2 ± 0.7 nm, which is in good agreement with previous studies.49 This type of AuNPs was selected to form a supramolecular assembly due to the cyclodextrin moieties which are able to interact with a variety of guests.51 The PCA dye was selected for its ability to interact with both NiO and β-CD. The monoanhydride precursor of PCA opens to form the dicarboxylate when exposed to metal oxide semiconductors for convenient electrode functionalization,52 while the hydrophobic alkane chain can interact with the β-CD moiety of the AuNPs, as illustrated in Fig. 1A. For short alkane chain, a host : guest ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 is expected.53 The optical and electrochemical properties of PCA are display in Table 1. The dye anion exhibits sufficient driving force (EPCA/PCA− = −0.91 V vs. NHE) to reduce Co to a catalytically active state (Eco,inset = −0.38 V vs. NHE).54 Hole injection into the valence band of NiO (E ≈ 0.46 V vs. NHE) is facilitated by the highly anodic excited state reduction potentials (EPCA*/PCA− = +1.73 V vs. NHE). PCA displays a broad absorption from 450 to 550 nm with a maximum at 503 nm and a shoulder at 476 nm (Fig. S6†). The intense visible light absorption and favourable electrochemical properties of PCA are well suited for the application of the dye in DS-PCs with Co as the catalyst (Fig. 1B). To confirm this ability to reduce the immobilized catalyst, PCs were assembled for photoelectrochemical analysis.
:1 is expected.53 The optical and electrochemical properties of PCA are display in Table 1. The dye anion exhibits sufficient driving force (EPCA/PCA− = −0.91 V vs. NHE) to reduce Co to a catalytically active state (Eco,inset = −0.38 V vs. NHE).54 Hole injection into the valence band of NiO (E ≈ 0.46 V vs. NHE) is facilitated by the highly anodic excited state reduction potentials (EPCA*/PCA− = +1.73 V vs. NHE). PCA displays a broad absorption from 450 to 550 nm with a maximum at 503 nm and a shoulder at 476 nm (Fig. S6†). The intense visible light absorption and favourable electrochemical properties of PCA are well suited for the application of the dye in DS-PCs with Co as the catalyst (Fig. 1B). To confirm this ability to reduce the immobilized catalyst, PCs were assembled for photoelectrochemical analysis.
|
λ
maxa |
εa (M−1 cm−1) |
E red (V vs. Fc+/Fc) | E ox (V vs. Fc+/Fc) | E red (V vs. NHE)e | E ox (V vs. NHE)e | |
|---|---|---|---|---|---|---|
| a Adsorption spectra in CH2Cl2. b Redox potentials obtained in a DMF solution. c Redox potentials obtained in a CH3CN solution. d Redox potentials obtained for surface bound molecules. e Redox potentials vs. NHE were calculated from the values vs. Fc+/0 by addition of 730 mV.55 | ||||||
| PCAc,d | 503 | 36000 |
−1.64 | 1 | −0.91 | 1.73 |
| 476 | 29300 |
|||||
| Co | — | — | −1.13 | −0.4 | −0.4 | 0.33 |
Electrode preparation and pre-catalysis characterization
PCA, β-CD-AuNPs, and Co were used to prepare a photocathode for H2 evolution based on FTO coated NiO films.26,56 NiO electrodes were prepared by immersion into a solution of PCA (0.2 mM) in a mixture of CH2Cl2 and methanol (1:1) for 24 hours to give NiO|PCA.
Co was immobilized on the NiO|PCA photocathodes in a layered assembly using β-CD-AuNPs to link the alkane chain of the dye to the adamantane moiety of the catalyst. Sequential loading of molecules was carried out by soaking the NiO|PCA films in an aqueous solution of β-CD-AuNPs (0.5 mg mL−1) for 1 h to give NiO|PCA|β-CD-AuNPs, followed by soaking in a Co solution (0.2 mM in MeOH-1%DMF) for 30 minutes to afford NiO|PCA|β-CD-AuNPs|Co electrodes.
Control electrodes without β-CD-AuNPs (denoted as NiO|PCA + Co) were obtained by soaking the NiO|PCA films in a Co solution following the same protocol. All immobilization steps were carried out at room temperature in the dark (see experimental part for details). The absorption spectra of the sensitized electrodes are depicted in Fig. 2B. UV-vis spectra of the films show the characteristic visible light absorption profiles of the dye. NiO|PCA electrodes display a blue shift and higher intensity for the peak at 448 nm than at 503 nm as opposed to the solution-based experiments (Fig. S6 and S9†). This is attributed to the anhydride opening and the binding of the carboxylates to NiO.57,58 β-CD-AuNPs binding was confirmed by UV-vis spectroscopy. In aqueous solution (Fig. 2A) a relatively weak plasmon resonance absorption maxima at 507 nm is observed.49The difference absorption spectrum of NiO|PCA and NiO|PCA|β-CD-AuNPs electrodes (Fig. 2C) exibits a peak at 538 nm attributed to the surface plasmon resonance of immobilized β-CD-AuNPs.
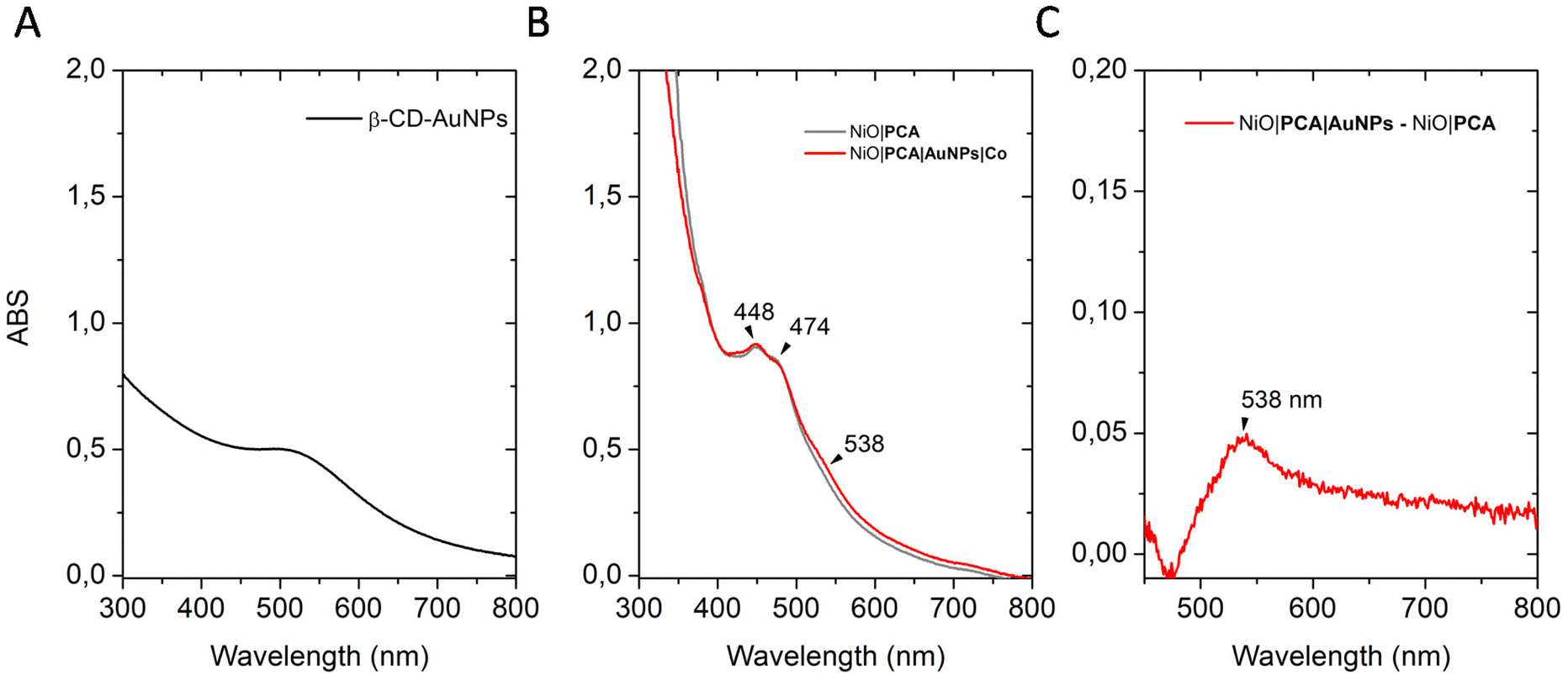 | ||
| Fig. 2 Absorption UV-vis spectrum of (A) an aqueous solution of β-CD-AuNPs, (B) NiO|PCA (grey) and NiO|PCA|β-CD-AuNPs|Co (red) electrodes (NiO electrodes were used to set the background), and (C) Difference in absorption of NiO|PCA|β-CD-AuNPs and NiO|PCA electrodes. | ||
The immobilization of the β-CD-AuNPs and the catalyst does not affect the NiO|PCA interface. The amount of dye loading on the photoelectrode was quantified by measuring the UV-vis spectra of PCA solution after sensitization of NiO films. A surface coverage of 20 ± 2 nmol cm−2 was found, which is in the same range as those determined in previous studies using similar electrodes.59 The amount of catalyst loading on the photoelectrodes was quantified by ICP-OES after acid digestion of the electrodes. A coverage of 0.5 ± 0.3 nmol cm−2 was found for NiO|PCA|β-CD-AuNPs|Co electrodes. No catalyst was observed for the NiO|PCA + Co electrodes confirming the crucial role of β-CD-AuNPs for the immobilization of Co catalysts.
The presence of PCA, β-CD-AuNPs and Co at the surface of NiO electrodes was further investigated by XPS analyses. The survey spectrum of the NiO electrodes (Fig. S12†) shows peaks attributed to Ni 2p and NiLMM Auger photoelectrons. After chemisorption of PCA onto NiO, the survey spectrum of the resulting material NiO|PCA shows an increase in the level of C and O compared to the NiO electrodes, consistent with the presence of PCA (Fig. S13†). The survey spectrum of NiO|PCA|β-CD-AuNPs|Co electrodes (Fig. S14†) shows the same features, but also additional information such as the presence of N and Au. A signal in the 398–402 eV range of the N 1s core level region (Fig. S13†) corresponds to N atoms present in grafted Co. The doublets for Au 4f7/2 (83.8 eV) and Au 4f5/2 (87.5 eV) shown in Fig. S14† are characteristic of Au0 of the β-CD-AuNPs.60,61 The absence of a signal at 84.9 eV as found for AuI in a gold octanethiol complex62 indicates that the gold atoms in the clusters are largely present as Au0. Further substantiation can be found on Au 4f peaks with slight asymmetry consistent with the presence of small nanoparticles/cluster.63 Au0 peaks due to metallic conduction (i.e. non-occupied states at the Fermi level) should be asymmetric with a tailing towards higher binding energy. XPS data clearly evidence the immobilization of PCA, β-CD-AuNPs and Co at the surface of NiO electrodes.
Photoelectrochemistry with NiO|PCA and NiO|PCA|β-CD-AuNPs
To ensure that NiO|PCA|β-CD-AuNPs electrodes could function without the kinetic limitations of sluggish catalyst reactions and charge recombination, photoelectrochemical measurements were performed in the presence of a soluble irreversible electron acceptor (IEA) in solution (Fig. 3A). Linear sweep voltammetry (LSV) was carried out in MES buffer electrolyte (0.1M, pH 5), at room temperature in an Ar-purged one compartment three electrodes electrochemical cell using a Pt counter electrode and a Ag/AgCl reference electrode. Irradiation of NiO|PCA and NiO|PCA|β-CD-AuNPs electrodes resulted in only minor photocurrents without a soluble acceptor (Fig. 3A). Addition of the IEA chloropentamminecobalt(III) chloride (10 mM) in the electrolyte solution allows for estimation of a maximal attainable photocurrent as [CoIII(NH3)5Cl2]Cl is known to be irreversibly reduced in solution.64 The IEA allows the photo-reduced dye to dispose its photoelectrons and to regenerate the ground state, thereby limiting the effects of reductive dye decomposition and charge recombination, thereby enhancing the photocurrent response for NiO|PCA and NiO|PCA|β-CD-AuNPs electrodes dramatically (Fig. 3A).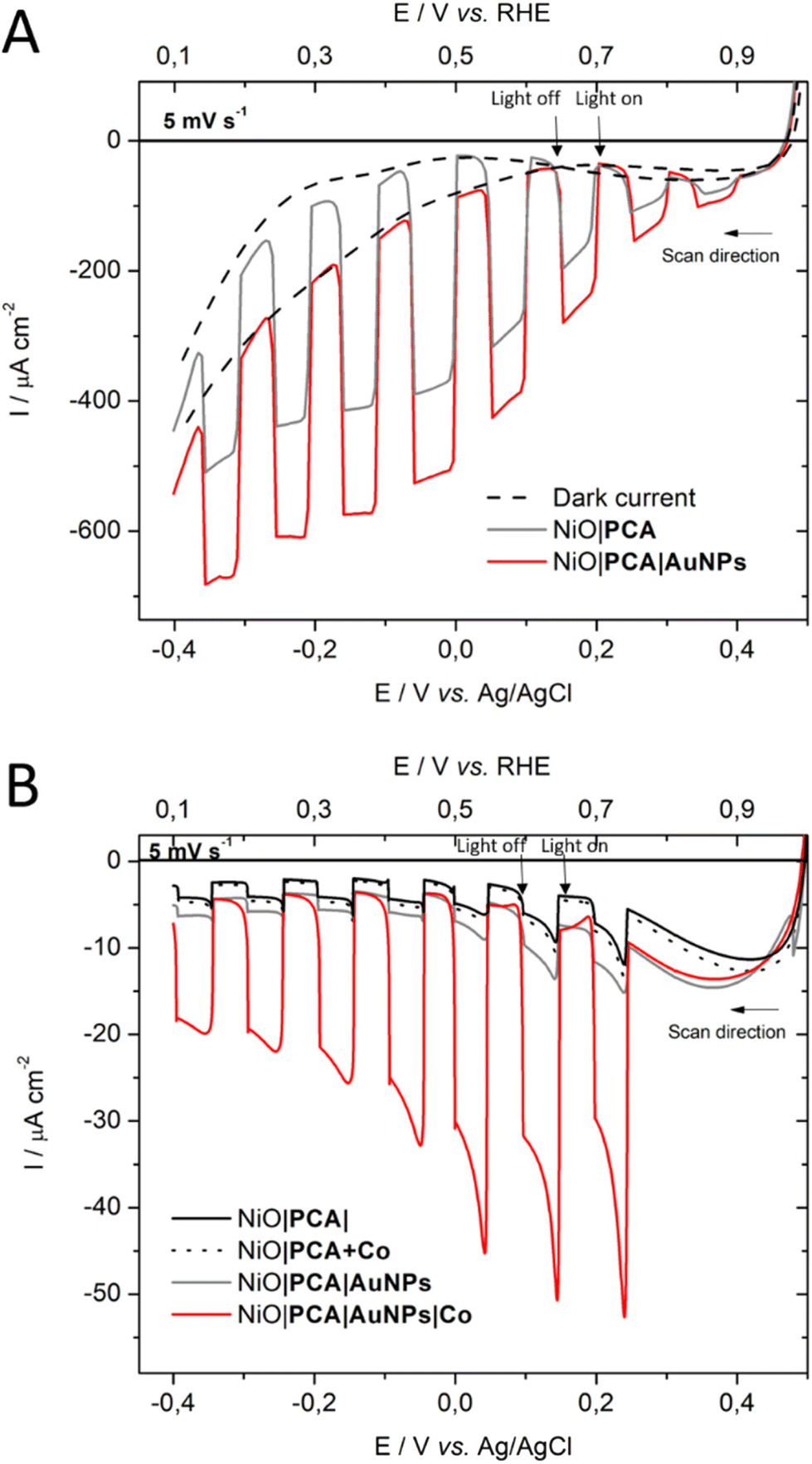 | ||
| Fig. 3 Linear sweep voltammograms (LSVs) under chopped light illumination of (A) NiO|PCA (grey) electrodes, and NiO|PCA|β-CD-AuNPs (red) electrodes with 10 mM of [CoIII(NH3)5Cl2]Cl acceptor in solution. (B) LSV scans of NiO|PCA, NiO|PCA|β-CD-AuNPs, NiO|PCA + Co and NiO|PCA|β-CD-AuNPs|Co. All experiments were performed in MES buffer (0.1 M, pH 5). Illumination with a white LED with 100 mW cm−2. An active electrode area of 1 cm−2 was used with a scan rate of 5 mVs−1. | ||
With NiO|PCA|β-CD-AuNPs electrodes, an absolute photocurrent response of |I| ≈ 420 μA cm−2 (0.0 V vs. Ag/AgCl) was observed, which indicates efficient light induced hole injection from the dye to the valence band of NiO with reduction of the IEA by PCA−. For comparison, NiO|PCA electrodes displayed a lower maximum photocurrent (|I| ≈ 344 μA cm−2, 0.0 V vs. Ag/AgCl), suggesting lower susceptibility to recombination between PCA− and holes in NiO when AuNPs are present. This finding supports that co-anchoring a catalyst could be a viable approach to exploit the reductive power of NiO|PCA|β-CD-AuNPs electrodes for H2 photo-production.
Photoelectrochemistry with NiO|PCA|β-CD-AuNPs|Co and H2 generation
The photoelectrochemical properties of the NiO|PCA|β-CD-AuNPs|Co photocathodes were evaluated in aqueous solution (0.1 M MES buffer, pH 5). Linear sweep voltammograms were recorded under chopped light irradiation (Fig. 3B). Immobilization of Co catalyst on the electrode resulted in a 10-fold photocurrent increase compared to that of NiO|PCA|β-CD-AuNPs electrodes (|j| = 15.8 μA cm−2, −0.2 V vs. Ag/AgCl). This enhanced response is attributed to the ability of PCA− to reduce Co and ultimately protons. The absence of current enhancement in the control experiment confirmed the crucial role of β-CD-AuNPs for the immobilization of Co catalysts.Chronoamperometric measurements were performed under chopped light irradiation at −0.1 V vs. Ag/AgCl over a period of 15 min showing steady-state cathodic photocurrent (Fig. 4) with a current density of ca. −11 μA cm−2. Photoelectrochemical H2 generation was confirmed by long term electrolysis over a period of 1 h (Fig. S16†), during which the magnitude of the current decreases slowly.
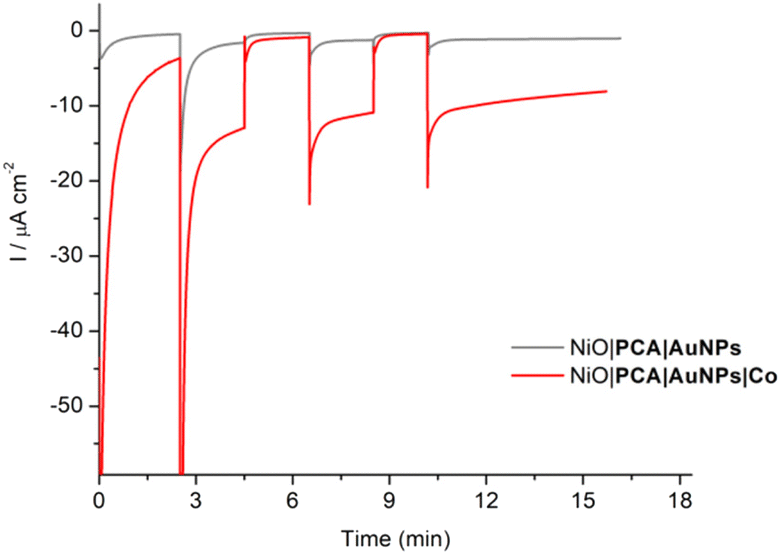 | ||
| Fig. 4 Chopped-light chronoamperometric measurements of NiO|PCA|β-CD-AuNPs and NiO|PCA|β-CD-AuNPs|Co photocathodes at an applied bias of −0.1 V vs. Ag/AgCl in pH 5 MES buffer. | ||
Clark electrode measurements confirmed the evolution of hydrogen in solution during the chronoamperometric measurements with a faradaic efficiency of 10 ± 2% and a TON of 8. Noteworthy is that these measurements do not take into account H2 present in the headspace.65 Moreover, no H2 generation was observed in the absence of catalyst or in the absence of light. This relatively low yield is within the current state-of-the-art in NiO-based dye-sensitized H2-evolving photocathodes.34 Attempts to increase the yield by increasing the catalyst loading on the electrode resulted in smaller photocurrents. The decrease in the photocurrent (from 9.8 to 4 μA cm−2) observed during long-term electrolysis can be attributed partly to some leaching or demetalation66 of the catalyst since we observed a decrease of catalyst loading by ICP measurements after photoelectrochemical measurements (Table S1†). Dye aggregation and competing reactions, such as NiO reduction15,67,68 may also be considered.
Transient absorption spectroscopy
Femtosecond transient UV/Vis absorption measurements were conducted on NiO|PCA, NiO|PCA|β-CD-AuNPs, and NiO|PCA|β-CD-AuNPs|Co photocathodes to understand the charge dynamics upon PCA excitation. Energetically, hole injection is possible from excited PCA dye to the NiO valence band.69 The excited state dynamics of PCA in NiO|PCA was compared to that of a ZrO2|PCA reference system, in which neither electrons nor holes can be injected due to the high-energy conduction band and low-energy valence band of ZrO2.70Fig. 5 shows the transient absorption spectra of the PCA dye attached to ZrO2 and NiO at different time delays after excitation. Both sets of spectra display the ground state bleach of the PCA dye at around 420 to 550 nm. The spectra of the ZrO2|PCA system shows an excited state absorption (ESA) from 550 to 750 nm with a distinct maximum around 580 nm, as expected.52 In NiO|PCA, the induced absorption is seen further to the red, without a distinct maximum. This difference agrees with the difference spectrum after spectroelectrochemical reduction of the NiO-bound dye (Fig. S10†). Fig. S18† presents the kinetic traces associated with ESA (probed at 580 nm) in ZrO2|PCA, and the reduced dye in NiO|PCA (probed at 625 nm), both of which were fitted with a with a double-exponential function.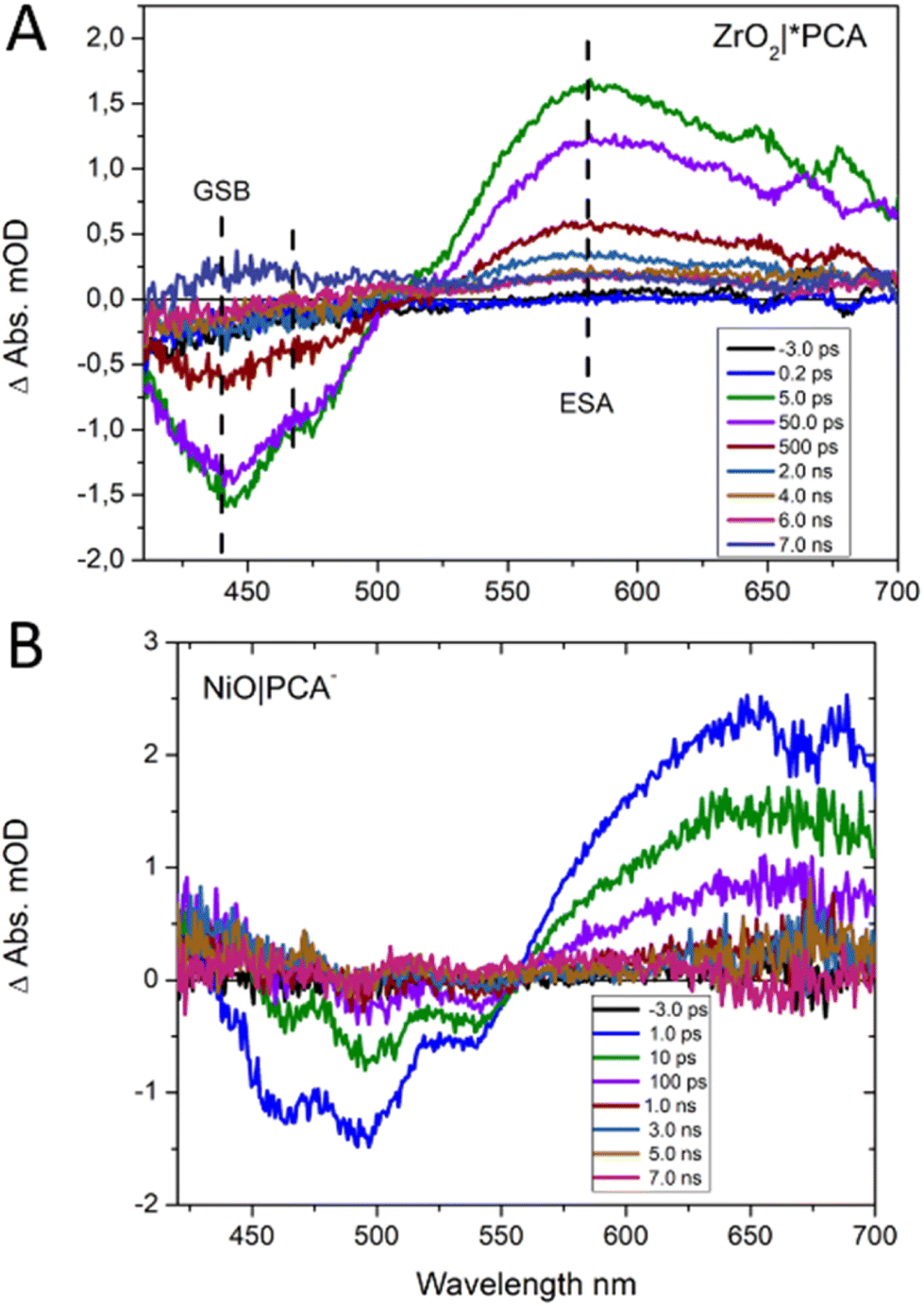 | ||
| Fig. 5 fs TA spectra of (A) ZrO2|PCA and (B) NiO|PCA, after excitation at 440 nm. | ||
Clearly, attaching PCA to NiO (τ1 = 15 ps (60%), τ2 = 300 ps (40%)) results in faster dynamics than having the dye on ZrO2 (τ1 = 30 ps (60%), τ2 = 500 ps (40%)).39 This is attributed to the photo driven hole injection from excited PCA into the NiO valence band.39 The thereby produced charge separated state NiO+|PCA− is however very short lived, and recombines to the ground state on the picosecond timescale, giving rise to the observed overall faster kinetics in NiO|PCA compared to that in ZrO2|PCA. The photoelectrochemical properties the NiO|PCA|β-CD-AuNPs|Co photocathodes (Fig. 3B) shows the role of the Au NPs as an important structural component in the supramolecular system. Fig. S17C† presents the TA spectra of the NiO|PCA|β-CD-AuNPs|Co photocathode which qualitatively contains the same features as those of NiO|PCA, but clearly with even shorter lifetimes of the signal that is attributed to the reduced PCA. Fig. 6 presents the kinetic traces of the decay of the reduced dye in NiO|PCA|β-CD-AuNPs|Co photocathodes, with a fit (τ1 = 3 ps (60%), τ2 = 40 ps (40%)), in comparison with the same features in NiO|PCA. In addition to the fast dynamic that is caused by attaching the dye to NiO (due to the hole injection and recombination), directed electron transfer from the reduced PCA via β-CD-AuNPs to the catalyst results in even faster dynamics of PCA− (Fig. 6). On one hand, inserting the β-CD-AuNPs will improve the loading of the catalyst and at the same time, it will guide the electron transfer from the reduced dye to the catalyst. It's important to note that the transient absorption data were acquired without applied bias, the presence of which can alter charge recombination rates.28,32,71 The main focus of the transient absorption study was however to demonstrate hole injection and find support for electron transfer to the AuNPs. Spectroscopically, it was not possible to detect the reduced form of the Co catalyst due to its comparably week spectroscopic features compare to the dye GSB.72
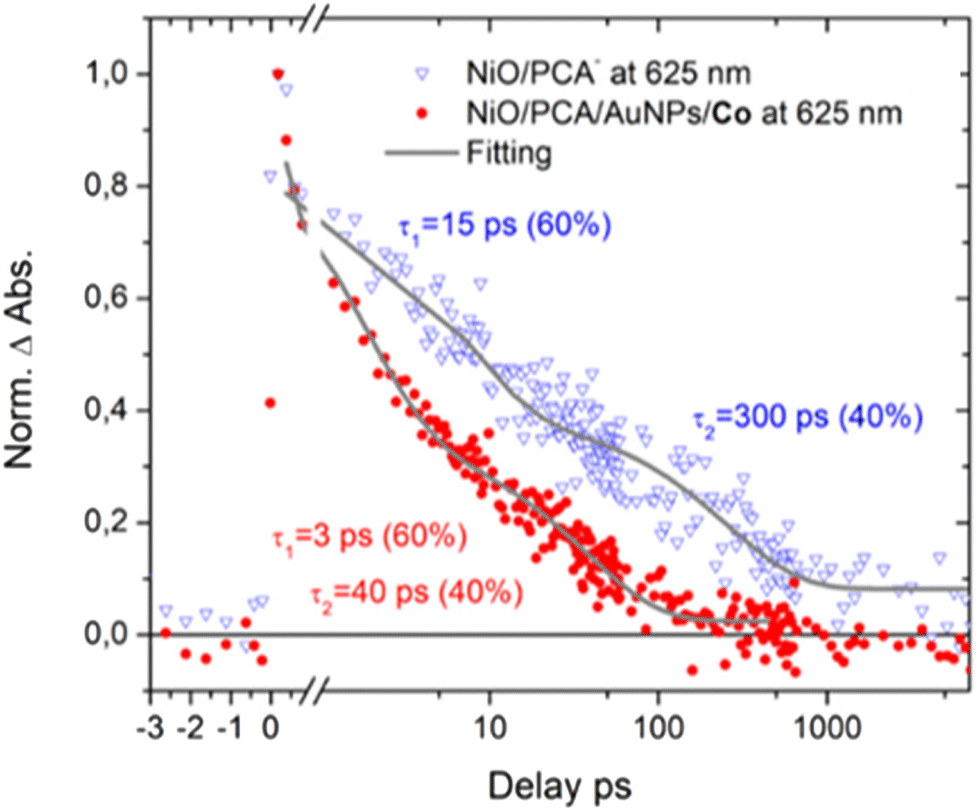 | ||
| Fig. 6 Time traces of NiO|PCA− (blue triangles) and NiO|PCA|AuNPs|Co (red dots) at 625 nm (reduced dye). | ||
Conclusions
Here, we report the first supramolecular assembly based on cyclodextrin functionalized gold nanoparticles for the construction of a NiO-based photocathode for H2 evolution from water. β-CD-AuNPs play a crucial role in the immobilization of Co catalysts and guide the electron transfer from the reduced dye to the catalyst. It was shown that light irradiation initiates an electron transfer from the NiO to the dye molecule, and subsequently from the reduced dye to the catalyst. The photocathode produces H2 with a faradaic efficiency of 10%, which is in the same range as those of previously reported molecular photocathodes based on cobaloxime catalysts.34 Further work to improve this system will target the use of novel p-type transparent materials such as CuCrO2, which have already proved promising.34,35Experimental
Materials
All chemicals were purchased from commercial suppliers (Sigma-Aldrich, VWR and Alfa-Aesar) and used without further purification, unless stated otherwise. Solvents were dried using drying agents and freshly distilled under argon before use. All organic reactions were performed under inert argon atmosphere.Instrumentation
1H NMR spectra were measured on a JEOL Eclipse 400 MHz spectrometer. High-resolution mass spectral analyses (HRMS) were performed on a pneumatically-assisted electrospray ionization Fourier transform (FTMS + pNSI) mass spectrometer (orbitrapXL). UV-vis spectroscopic measurements were collected using a Varian Cary 50 Bio UV-vis spectrophotometer. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopic measurements were performed using a PerkinElmer Spectrum One FTIR-Spectrometer with a PerkinElmer Universal ATR Sampling Accessory. Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) experiments were performed using a PerkinElmer Optical Emission Spectrometer. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a PHI Quantera II scanning XPS microprobe with monochromated Al Kα radiation (1486.6 eV). Survey spectra were captures at a pass energy of 224 eV, while for high-resolution spectra a pass energy of 55 eV was used. All spectra were referenced against an internal signature (C 1s, 284.8 eV). Transmission electron microscopy (TEM) images were obtained using JEOL JEM 1400 instrument. The particle size distribution was obtained from digitalized amplified micrographs by averaging the larger and smaller axis diameters measured in each particle.Electrochemical and photoelectrochemical measurements were acquired with an Autolab potentiostat (PGSTAT 302N). Electrochemical measurements were conducted in a traditional three-electrodes system with Ag/AgNO3 reference electrode, a platinum rod as counter electrode and either glassy carbon or FTO/NiO as working electrodes. Ferrocene was used as an internal standard and all the potentials converted into the Normal Hydrogen Electrode using EFc+/Fc = 0.64 V vs. NHE. LED lamp (cool white, 5000K) was used as light source. A Unisense Clark electrode H2 sensor was used for the detection and the quantification of H2. A Dektak150 Surface Profiler was used to measure the NiO film thicknesses.
TA spectroscopy
Femto-second transient absorption spectroscopy (fs-TAS) was performed probing in the UV-vis region using a Newport TAS. A Coherent Libra Ti:sapphire amplifier (1.5 mJ, 3 kHz, 800 nm, fwhm 40 fs) was used and split into pump and probe beams. An excitation wavelength of 440 nm was generated by directing the pump beam into the optical parametric amplifiers (TOPAS-Prime and NIRUVVIS, Light Conversion) and then focused and centered on the dry films with a pump power adjusted to 500 μW. The 800 nm fundamental of the amplifier was focused on a CaF2 crystal (Newport TAS), generating the white light supercontinuum probe. A silicon diode array (Newport custom made) was used to record the probe spectrum. A mechanical chopper blocked every other pump pulse, and the transient absorption at each time point was calculated for an average of 1000 ms chopped/un-chopped pulse pairs. To record the transient absorption spectra at different time points, an optical delay line was used to scan the delay of the probe beam relative to the pump beam from −5 ps to 7.5 ns. A total of 10–15 scans were collected and averaged for each sample. Prior to analysis, the data was corrected for the spectral chirp using Surface Xplorer 4.2, where single wavelength fits were also performed.Synthesis
[Co(dcpgH)(dcpgH2)]Cl254 (dcpgH2 = diphenylglyoxime-4,4′-dicarboxylic acid) 1, PCA dye47 and β-CD-AuNPs48,49 were synthesized according to literature method.Synthesis of Co (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP, 4 equiv., 0.51 mmol) was added to a solution of 1 (1 equiv., 0.13 mmol), diisopropylethylamine (7 equiv., 0.90 mmol) and 1-adamantanemethylamine (4 equiv., 0.51 mmol) dissolved in DMF (7 mL). Upon addition, 1-adamantanemethylamine became completely soluble. The reaction mixture was then stirred at R.T. overnight. DMF was removed under reduce pressure and the resulting oil was dissolved in 10 mL of CH2Cl2. The organic phase was washed with brine (2 × 5 mL), water (2 × 5 mL), dried (MgSO4), filtered and reduced to 2 mL. Then the crude orange product was precipitated with Et2O, filtered and washed with Et2O. The solid was purified by flash chromatography (0 to 7% methanol in CH2Cl2). The pure product was obtained after evaporation of the solvent in combine fractions to give 2 as an orange solid (83 mg, 46%). 1H NMR (CD2Cl2, 400 MHz): δ (ppm) 19.25 (Br, 2H, oxime), 7.51–7.26 (m, 16H, Ar), 7.10 (s, 4H, NH amide), 3.08 (s, 8H, CH2), 1.94 (s, 12H, CH adamantane), 1.68–1.52 (m, 48H, CH2 adamantane). HR-MS (ESI+): m/z, 1302.6 [M-H-2Cl]+.
Electrode preparation
First, FTO-coated glass slides were cleaned via sequential sonication steps using detergent, acetone, and isopropanol. Then, NiO was screen printed onto the clean FTO coated glass using a NiO paste as previously reported.56,73 After each layer of screen printing, the electrodes were annealed at 120 °C for 9 minutes on a hot plate. Three layers were screen printed and were finally annealed at 450 °C for 30 minutes in an oven, resulting in a thickness of ∼1.2 μm determined by profilometry.Photocathode assemblies
NiO electrodes were soaked in a 1:1 CH2Cl2:MeOH solution of PCA (0.2 mM) for 24 h at room temperature, then rinsed with CH2Cl2; MeOH and dried in air affording NiO|PCA electrodes. For the molecular assemblies PCA|β-CD-AuNP|Co, sequential loading of molecules was carried out by soaking the films in aqueous solution of β-CD-AuNPs (0.5 mg mL−1) for 1 h (giving NiO|PCA|β-CD-AuNPs electrodes), followed by soaking in methanol-1%DMF solution of the catalyst Co (0.2 mM) for 0.5 h. The electrodes were then rinsed with MeOH-1%DMF solution and dried in air. Water and methanol were used to rinse the films between the two loading solutions. NiO|PCA|Co electrodes were obtained following the same procedure without the soaking step in solution of β-CD-AuNPs.
Surface loading quantification
Surface coverage (Γ in mol cm−1) of PCA was evaluated by measuring the UV-vis spectra of PCA solution before and after sensitization of NiO films. A surface coverage of 20 ± 2 nmol cm−2 was found.Co was quantified by ICP-OES after digestion of NiO photocathodes in a mixture of conc. H2SO4 (1 mL) and HNO3 (1 mL) by heating in a Biotage (Uppsala Sweden) SPX microwave reactor for 10 min at 100 °C. The resulting clear solution was filtered with a micron filter syringe, then diluted using deionized water and analyzed by ICP-OES (pre-calibrated with known concentrations of each element (sulfur and cobalt) of interest). The geometric surface areas of the dissolved electrodes were measured prior to being digested in order to determine the surface loading.
Photoelectrochemistry measurement
All the PEC experiments were performed in 0.1 M MES buffer at pH 5 using an Ag/AgCl (sat'd NaCl) reference electrode and a Pt wire counter electrode. Linear sweep votammetric measurements were performed in a one-chamber cell degassed with Ar. A white LED was used to illuminate the photocathode, to generate a power density of approximatively 100 mW cm−2. Long term chronoamperometric experiments were performed in an H-cell in order to avoid leakage from the Pt counter electrode onto the working electrode. The photocathode and the reference were in one compartment, while the Pt counter electrode was in the other one. All samples were degassed under Ar prior to measurements.Clark electrode H2 measurements
H2 detection using a Clark electrode was performed based on a previously reported method.65 Prior to measurements, the Unisense Clark electrode was calibrated using known concentrations of dissolved H2 in 0.1 M MES buffer at pH 5 and measuring the Clark electrode response signal.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the Swedish Energy Agency (Grant no. 11674-8) for funding this research. We also thank Dr Souvik Roy for providing [Co(dcpgH)(dcpgH2)]Cl2 and Ashleigh Castner for the help with the ICP-OES experiments.References
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 RSC.
- E. A. Gibson, Chem. Soc. Rev., 2017, 46, 6194–6209 RSC.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- E. Benazzi, J. Mallows, G. H. Summers, F. A. Black and E. A. Gibson, J. Mater. Chem. C, 2019, 7, 10409–10445 RSC.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farré, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- L. Tong, A. Iwase, A. Nattestad, U. Bach, M. Weidelener, G. Götz, A. Mishra, P. Bäuerle, R. Amal, G. G. Wallace and A. J. Mozer, Energy Environ. Sci., 2012, 5, 9472–9475 RSC.
- P. B. Pati, L. Zhang, B. Philippe, R. Fernández-Terán, S. Ahmadi, L. Tian, H. Rensmo, L. Hammarström and H. Tian, ChemSusChem, 2017, 10, 2480–2495 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- B. Shan, B. D. Sherman, C. M. Klug, A. Nayak, S. L. Marquard, Q. Liu, R. M. Bullock and T. J. Meyer, J. Phys. Chem. Lett., 2017, 8, 4374–4379 CrossRef CAS.
- B. Shan, A. Nayak, M. K. Brennaman, M. Liu, S. L. Marquard, M. S. Eberhart and T. J. Meyer, J. Am. Chem. Soc., 2018, 140(20), 6493–6500 CrossRef CAS PubMed.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS PubMed.
- L. J. Antila, P. Ghamgosar, S. Maji, H. Tian, S. Ott and L. Hammarström, ACS Energy Lett., 2016, 1, 1106–1111 CrossRef CAS.
- S. Lyu, J. Massin, M. Pavone, A. B. Muñoz-García, C. Labrugère, T. Toupance, M. Chavarot-Kerlidou, V. Artero and C. Olivier, ACS Appl. Energy Mater., 2019, 2, 4971–4980 CrossRef CAS.
- B. van den Bosch, J. A. Rombouts, R. V. A. Orru, J. N. H. Reek and R. J. Detz, ChemCatChem, 2016, 8, 1392–1398 CrossRef CAS.
- B. Shan, A. K. Das, S. Marquard, B. H. Farnum, D. Wang, R. M. Bullock and T. J. Meyer, Energy Environ. Sci., 2016, 9, 3693–3697 RSC.
- K. L. Materna, N. Lalaoui, J. A. Laureanti, A. P. Walsh, B. P. Rimgard, R. Lomoth, A. Thapper, S. Ott, W. J. Shaw, H. Tian and L. Hammarström, ACS Appl. Mater. Interfaces, 2020, 12, 4501–4509 CrossRef CAS.
- J. Huang, J. Sun, Y. Wu and C. Turro, J. Am. Chem. Soc., 2021, 143, 1610–1617 CrossRef CAS PubMed.
- E. Giannoudis, S. Bold, C. Müller, A. Schwab, J. Bruhnke, N. Queyriaux, C. Gablin, D. Leonard, C. Saint-Pierre, D. Gasparutto, D. Aldakov, S. Kupfer, V. Artero, B. Dietzek and M. Chavarot-Kerlidou, ACS Appl. Mater. Interfaces, 2021, 13, 49802–49815 CrossRef CAS.
- L. D'Amario, J. Fohlinger, G. Boschloo and L. Hammarstrom, Chem. Sci., 2018, 9, 223–230 RSC.
- S. Mori, S. Fukuda, S. Sumikura, Y. Takeda, Y. Tamaki, E. Suzuki and T. Abe, J. Phys. Chem. C, 2008, 112, 16134–16139 CrossRef CAS.
- L. D'Amario, R. Jiang, U. B. Cappel, E. A. Gibson, G. Boschloo, H. Rensmo, L. Sun, L. Hammarström and H. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 33470–33477 CrossRef PubMed.
- L. D'Amario, L. J. Antila, B. Pettersson Rimgard, G. Boschloo and L. Hammarström, J. Phys. Chem. Lett., 2015, 6, 779–783 CrossRef PubMed.
- F. Li, R. Xu, C. Nie, X. Wu, P. Zhang, L. Duan and L. Sun, Chem. Commun., 2019, 55, 12940–12943 RSC.
- C. E. Creissen, J. Warnan and E. Reisner, Chem. Sci., 2018, 9, 1439–1447 RSC.
- C. E. Creissen, J. Warnan, D. Antón-García, Y. Farré, F. Odobel and E. Reisner, ACS Catal., 2019, 9, 9530–9538 CrossRef CAS.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- D. A. Hoogeveen, M. Fournier, S. A. Bonke, X.-Y. Fang, A. J. Mozer, A. Mishra, P. Bäuerle, A. N. Simonov and L. Spiccia, Electrochim. Acta, 2016, 219, 773–780 CrossRef CAS.
- S. Lyu, Y. Farré, L. Ducasse, Y. Pellegrin, T. Toupance, C. Olivier and F. Odobel, RSC Adv., 2016, 6, 19928–19936 RSC.
- R. J. Kamire, M. B. Majewski, W. L. Hoffeditz, B. T. Phelan, O. K. Farha, J. T. Hupp and M. R. Wasielewski, Chem. Sci., 2017, 8, 541–549 RSC.
- N. Kaeffer, C. D. Windle, R. Brisse, C. Gablin, D. Leonard, B. Jousselme, M. Chavarot-Kerlidou and V. Artero, Chem. Sci., 2018, 9, 6721–6738 RSC.
- H. Li, F. Li, B. Zhang, X. Zhou, F. Yu and L. Sun, J. Am. Chem. Soc., 2015, 137, 4332–4335 CrossRef CAS PubMed.
- L. Sévery, J. Szczerbiński, M. Taskin, I. Tuncay, F. Brandalise Nunes, C. Cignarella, G. Tocci, O. Blacque, J. Osterwalder, R. Zenobi, M. Iannuzzi and S. D. Tilley, Nat. Chem., 2021, 13, 523–529 CrossRef PubMed.
- N. Lalaoui, P. Rousselot-Pailley, V. Robert, Y. Mekmouche, R. Villalonga, M. Holzinger, S. Cosnier, T. Tron and A. Le Goff, ACS Catal., 2016, 6, 1894–1900 CrossRef CAS.
- C. Gutiérrez-Sánchez, M. Pita, C. Vaz-Domínguez, S. Shleev and A. L. De Lacey, J. Am. Chem. Soc., 2012, 134, 17212–17220 CrossRef PubMed.
- B. I. Ipe, K. G. Thomas, S. Barazzouk, S. Hotchandani and P. V. Kamat, J. Phys. Chem. B, 2002, 106, 18–21 CrossRef CAS.
- A. Kotiaho, R. M. Lahtinen, N. V. Tkachenko, A. Efimov, A. Kira, H. Imahori and H. Lemmetyinen, Langmuir, 2007, 23, 13117–13125 CrossRef CAS PubMed.
- R. K. Gupta and A. S. Achalkumar, J. Org. Chem., 2018, 83, 6290–6300 CrossRef CAS PubMed.
- M. T. Rojas, R. Koeniger, J. F. Stoddart and A. E. Kaifer, J. Am. Chem. Soc., 1995, 117, 336–343 CrossRef CAS.
- J. Liu, W. Ong, E. Román, M. J. Lynn and A. E. Kaifer, Langmuir, 2000, 16, 3000–3002 CrossRef CAS.
- S. Roy and E. Reisner, Angew. Chem., 2019, 131, 12308–12312 CrossRef.
- G. Chen and M. Jiang, Chem. Soc. Rev., 2011, 40, 2254–2266 RSC.
- R. J. Lindquist, B. T. Phelan, A. Reynal, E. A. Margulies, L. E. Shoer, J. R. Durrant and M. R. Wasielewski, J. Mater. Chem. A, 2016, 4, 2880–2893 RSC.
- C. Zhou, J. Huang and Y. Yan, Soft Matter, 2016, 12, 1579–1585 RSC.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS.
- J. R. Aranzaes, M.-C. Daniel and D. Astruc, Can. J. Chem., 2006, 84, 288–299 CrossRef.
- B. Xu, L. Tian, A. S. Etman, J. Sun and H. Tian, Nano Energy, 2019, 55, 59–64 CrossRef CAS.
- Y. Farré, F. Maschietto, J. Föhlinger, M. Wykes, A. Planchat, Y. Pellegrin, E. Blart, I. Ciofini, L. Hammarström and F. Odobel, ChemSusChem, 2020, 13, 1844–1855 CrossRef PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- F. Odobel, Y. Farré, F. Maschietto, J. Föhlinger, M. Wykes, A. Planchat, Y. Pellegrin, E. Blart, I. Ciofini and L. Hammarström, ChemSusChem, 2020, 13, 1844–1855 CrossRef PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- O. Swiech, R. Bilewicz and E. Megiel, RSC Adv., 2013, 3, 5979–5986 RSC.
- A. McNeillie, D. H. Brown, W. E. Smith, M. Gibson and L. Watson, J. Chem. Soc., Dalton Trans., 1980, 767–770 RSC.
- C. Paun, G. Słowik, E. Lewin and J. Sá, RSC Adv., 2016, 6, 87564–87568 RSC.
- N. Queyriaux, R. A. Wahyuono, J. Fize, C. Gablin, M. Wächtler, E. Martinez, D. Léonard, B. Dietzek, V. Artero and M. Chavarot-Kerlidou, J. Phys. Chem. C, 2017, 121, 5891–5904 CrossRef CAS PubMed.
- C. D. Windle, J. Massin, M. Chavarot-Kerlidou and V. Artero, Dalton Trans., 2018, 47, 10509–10516 RSC.
- K. L. Materna, A. M. Beiler, A. Thapper, S. Ott, H. Tian and L. Hammarström, ACS Appl. Mater. Interfaces, 2020, 12, 31372–31381 CrossRef CAS PubMed.
- D. A. Hoogeveen, M. Fournier, S. A. Bonke, A. Nattestad, A. Mishra, P. Bäuerle, L. Spiccia, A. J. Mozer and A. N. Simonov, J. Phys. Chem. C, 2017, 121, 25836–25846 CrossRef CAS.
- L. Tian, R. Tyburski, C. Wen, R. Sun, M. Abdellah, J. Huang, L. D'Amario, G. Boschloo, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2020, 142, 18668–18678 CrossRef CAS PubMed.
- Z. Liu, D. Xiong, X. Xu, Q. Arooj, H. Wang, L. Yin, W. Li, H. Wu, Z. Zhao, W. Chen, M. Wang, F. Wang, Y. B. Cheng and H. He, ACS Appl. Mater. Interfaces, 2014, 6, 3448–3454 CrossRef CAS PubMed.
- M. Abdellah, A. M. El-Zohry, L. J. Antila, C. D. Windle, E. Reisner and L. Hammarstrom, J. Am. Chem. Soc., 2017, 139, 1226–1232 CrossRef CAS PubMed.
- R. J. Dillon, L. Alibabaei, T. J. Meyer and J. M. Papanikolas, ACS Appl. Mater. Interfaces, 2017, 9, 26786–26796 CrossRef CAS PubMed.
- M. Abdellah, S. Zhang, M. Wang and L. Hammarström, ACS Energy Lett., 2017, 2, 2576–2580 CrossRef CAS.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y. B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2009, 9, 31 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt02798d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |