
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUranium oxide hydrate frameworks with Er(III) or Y(III) ions: revealing structural insights leading to the low symmetry†
Timothy A.
Ablott
,
Kimbal T.
Lu
,
Robert D.
Aughterson
and
Yingjie
Zhang
 *
*
Australian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia. E-mail: yzx@ansto.gov.au
First published on 29th September 2022
Abstract
Two new mixed-valence uranium oxide hydrate frameworks (UOFs), incorporating either Er3+ or Y3+ ions, were successfully synthesised under hydrothermal conditions and characterised with single-crystal X-ray diffraction and a variety of other structural and spectroscopic techniques. Both frameworks are isostructural and crystallise in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, consisting of β-U3O8 type layers pillared by additional uranyl centres, with the Er3+/Y3+ ions lying in the channels of the framework. SEM-EDS analysis found that both materials existed in plate-like morphologies, with a U:Er/Y ratio of 5.5. Bond valence sum analysis revealed the possible existence of pentavalent uranium centres, which was confirmed with diffuse reflectance spectroscopy. Being the first reported UOFs in this space group, this work highlights the complex and flexible nature of these materials, and the broader uranium oxide hydrate systems which exist in the surrounds of spent nuclear fuel disposal in the underground repository.
space group, consisting of β-U3O8 type layers pillared by additional uranyl centres, with the Er3+/Y3+ ions lying in the channels of the framework. SEM-EDS analysis found that both materials existed in plate-like morphologies, with a U:Er/Y ratio of 5.5. Bond valence sum analysis revealed the possible existence of pentavalent uranium centres, which was confirmed with diffuse reflectance spectroscopy. Being the first reported UOFs in this space group, this work highlights the complex and flexible nature of these materials, and the broader uranium oxide hydrate systems which exist in the surrounds of spent nuclear fuel disposal in the underground repository.
1. Introduction
The study of uranium oxide synthetic compounds has seen significant interest and substantial exploration due to their close relationship with the uranium oxide systems used as nuclear fuel in nuclear reactors around the world.1,2 With a push towards cleaner energy production to combat climate change, nuclear energy has again become a focus for many countries.1,2 One issue clearly identified with these fuel systems is the need for the safe management and disposal of the spent nuclear fuel (SNF) once removed from the nuclear reactor, in order to isolate these highly radioactive materials from the general public.3,4 With a variety of options available, changing from country to country, one of the most widely accepted approaches is the disposal of SNF in stable underground geological repositories, wherein the SNF is essentially safely isolated from the outside world.2 To ensure this isolated SNF, most commonly present as uranium oxide (UO2), is safe to store within these repositories for the long-term, studies into the behavior of these materials in these geological environments are critical.Both natural and synthetic uranium oxide compounds have proven extremely useful in replicating the conditions that these SNFs are exposed to in the repository environment. The study of these uranium oxide compounds has been driven by the knowledge of the natural weathering of uraninite (UO2+x).5,6 When exposed to oxidative conditions uraninite is known to undergo oxidation from U4+ to U6+, often existing in the form of uranyl [(UO2)2+] cations which further react with electron donors in the surrounding environment, forming a plethora of uranyl oxide compounds.7,8 These uranyl moieties typically contain two strongly bound oxygen atoms in the axial positions, with the equatorial positions regularly binding additional O2− or OH− ions, giving rise to typical coordination geometries of tetragonal, pentagonal, or hexagonal bipyramidal polyhedra.9,10 Extension of these polyhedra via corner- or edge-sharing results in the formation of layered (sheet) structures, which are commonly bridged by interlayer cations. Logically, given their compositions, these materials have been assigned the name uranium oxide hydrates (UOH).11,12 The study of uranium-containing minerals has identified several dozen UOH-based minerals,1,11 and with this knowledge more than a dozen synthetic UOH materials have since been reported.13–18 The distinctive features in these materials are the differing secondary metal ions which make up the interlayer cation layer, and the O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OH ratios of the uranyl oxide hydroxide sheets.
:OH ratios of the uranyl oxide hydroxide sheets.
A subset of UOH materials are those labelled uranium oxide frameworks, or UOFs. They differ from UOHs in that they form a framework-type structure, with uranyl oxide moieties linking two layered sheets, with the secondary cations lying within the channels formed by these linkers. A small but wide variety of secondary cations have been successfully incorporated into these UOF structures, including Cs+, Sr2+, Pb2+, Sm3+, Eu3+, Gd3+ and U4+.19–24 Given the evident diversity of what can be stabilised within the channels of these materials, further study is therefore needed in this field in order to truly capture the possible chemistry which can occur in the surrounds of the SNF disposed within a deep geological repository.
With lanthanides often found alongside uranium in the environment, as well as being present in SNF as fission products, they are therefore expected to be heavily involved in the modification of both natural UOH minerals and synthetic UOH compounds. In addition, they can also be applied as surrogates for minor actinides (i.e. Am3+ and Cm3+) which would also be present in the SNF environment,25,26 which makes Ln3+ species an ideal target of study for synthetic UOHs. Up to this point, no natural UOH minerals containing lanthanides have been identified, thus this area has been driven by synthetic UOH research. Early research into these synthetic compounds discovered both the uranium precursor as well as starting pH, which controls uranium hydrolysis, were crucial in their successful synthesis. Research using a schoepite precursor first led to the successful incorporation of a variety of Ln ions (Ln = Tb, Dy, Ho and Yb) between α-U3O8 uranyl oxide layers, with a U:Ln ratio of 2.27 Interestingly, in 2020 Lu et al. were able to synthesise a second UOH-Tb material with a U:Tb ratio of 6 and a different uranyl oxide hydroxide layer topology, now resembling a β-U3O8 uranyl oxide layer.28 This highlighted the potential for the interlayer cations to directly affect the chemistry of the uranyl oxide layers within these materials. Other UOH-Ln compounds have also been reported sans crystal structures, clearly demonstrating the challenge that exists in obtaining single crystals of UOH-Ln which are of high enough purity for single crystal analysis. These UOH-Ln compounds are listed in Table 1.
| UOH-Ln3+ | U/Ln ratio | Single crystal structure | Other characterization |
|---|---|---|---|
| UOH-Sm | 1 | No | XRD, SEM-EDS, TG |
| UOH-La/Pr/Nd/Tb/Dy/Ho/Yb | 2 | Yes | XRD, SEM-EDS, TEM, Raman, UV-vis, TG |
| UOH-Dy/Ho/Er/Tm/Tb/Yb/Lu | 2 | No | XRD, IR, TG |
| UOH-La/Ce/Pr/Nd/Sm | 3 | No | XRD, IR, TG |
| UOH-Sm | 4 | No | XRD, SEM-EDS |
| UOF-Sm/Eu/Gd | 5.5 | Yes | XRD, SEM-EDS, TEM, Raman, UV-vis-NIR, TG |
| UOH-Tb | 6 | Yes | — |
| UOH-Nd/Sm/Eu/Gd/Tb/Dy | 6 | No | XRD, IR, TG |
Intriguingly, as evidenced by Table 1, only Sm, Eu and Gd have been found to form the UOF sub-structure, with the Ln3+ ions found within the 3D channels of the framework.21,22 However, this work has also found that the pH of the synthesis is highly influential on the formation of either a UOF or UOH structure. For UOH/F-Sm, a synthesis pH < 4 gave rise to a UOF structure, whereas increasing the pH to 4–5 resulted in a layered UOH material in its place.21 This further highlights the complex synthetic chemistry of these materials, driving the need for further research. It has also been hypothesised as to whether the preferential formation of a Ln3+ containing UOF-type structure over a UOH-layered material is moderated by the ionic radius of the selected cation.12 Given, of the lanthanide structures reported, the three which all have very similar ionic radii are the only ones to form a framework structure, this suggestion has merit. Thus, erbium, having a smaller ionic radius to those of Sm, Eu and Gd, is a prime candidate to probe such a hypothesis. Only one synthesis of a UOH-Er material has been reported, however no single-crystal analysis was done on the material which prevented any structural insights from being explored.30 Yttrium was a second candidate that was identified for this study, with no previously reported structures containing Y having been found. Being chemically similar to Ln3+ ions and having an ionic radius close to that of erbium, it was selected as a possible UOH/F-Ln surrogate which could further aid in the understanding of the chemistry driving the formation of these materials. Herein, we report the hydrothermal synthesis of two novel synthetic UOF compounds, UOF-Er and UOF-Y, and their structural and spectroscopic analyses. The crystals isolated from the hydrothermal reaction of uranyl nitrate with either Er3+ or Y3+ ions were revealed to have an as of yet unseen 3D framework-type structure via synchrotron single crystal X-ray diffraction analysis, with the crystals subsequently explored using scanning and transmission electron microscopy alongside Raman and diffuse reflectance spectroscopy.
2. Experimental
2.1. Syntheses of materials
Uranyl nitrate hexahydrate with natural uranium was used in the synthesis of the materials. Compounds with uranium are radioactive and should be handled in the regulated laboratory. All other chemicals in A.R. grade were from Sigma-Aldrich (Merck).2.2. Characterizations
35 using the Olex2 graphical user interface.36 All atoms with ≥0.5 occupancies were located on the electron density maps and refined anisotropically. Hydrogen atoms on hydroxyl groups and water molecules were unable to be located and they were omitted in the structure refinements. Both compounds contain U as a strong X-ray absorber. In addition, the one-circle goniometer setup on the MX2 beamline provided less redundant data for absorption corrections. As such there were some Q-peaks around U atoms due to the ineffective absorption corrections.
3. Results and discussion
3.1. Material synthesis, microstructure and U to Er/Y ratios
Both UOF-Er and UOF-Y were successfully synthesised using uranyl nitrate as uranium precursor, with the solution pH directly controlled using NaOH. The formation of UOF-Er was achieved by dissolving equimolar amounts of UO2(NO3)2·6H2O and Er(NO3)3·5H2O in H2O and adjusting the starting pH to 6.18, with the solution then subsequently heated at 200 °C for 48 hours. Similarly, UOF-Y was synthesised using an almost identical procedure, with equimolar UO2(NO3)2·6H2O and Y(NO3)3·6H2O first dissolved in water, with the pH adjusted to 5.88. The solution was then heated at 240 °C for 72 hours.SEM analysis of UOF-Er revealed the presence of two distinct crystal morphologies. The major phase exists as large blocks/plates (Fig. 1a, left), with the minor phase consisting of thin plates, a very common crystal morphology for both synthetic UOH compounds20,27,28 and UOH minerals.42 SEM-EDS analysis of the major phase confirmed the presence of only U, Er and O in the material, with a U:Er ratio of ∼5.7 (Fig. 1a, right, Fig. S1 and Table S1, ESI†). The minor phase also contained only U, Er and O in the material, with a U:Er ratio of ∼2.5 (Fig. S2 and Table S1, ESI†), again consistent with other synthetic UOH materials.20,27,28 The EDS results are consistent with the major phase existing as a UOF with the minor phase that of a layered UOH material. Given these systems are known to be complex and can readily be influenced by reaction conditions (temperature, duration and solution pH), these findings are not entirely surprising. As such, the synthetic conditions reported for UOF-Er appear to allow for several phases to co-exist, with UOF-Er existing as the major phase.
 | ||
| Fig. 1 SEM analysis of (a) UOF-Er and (b) UOF-Y: secondary SEM images of the crystals on the left and their corresponding EDS spectra on the right confirming the presence of both U and the Er/Y in a ∼5.5:1 atomic ratio. | ||
UOF-Y was consistent with the findings of UOF-Er, with two phases immediately apparent upon examination with SEM. The major phase is present in the similar block/plate-like morphology (Fig. 1b, left), with SEM-EDS analysis indicating a U:Y ratio of ∼5.8 (Fig. 1b, Fig. S3 and Table S1, ESI†), also confirming the presence of only U, Y and O in the crystal. The minor phase gave a U:Y ratio of ∼3 (Table S1, ESI†) and appeared in the similar small plates, likely to exist as a layered UOH structure. Given the complexity of separating these two phases, and with the minor phase seemingly existing as a UOH structure, further characterization of this phase was not carried out.
3.2. Crystal structures and discussion
The crystal data and structural refinement details for compounds UOF-Er and UOF-Y are summarised in Table 2, with selected bond lengths (Å) and angles (°) in Table S2, ESI.† The calculated bond valence sums (BVSs)9 are presented in Tables S3 and S4, ESI,† with the parameters for U6+ taken from the literature.9,43| Compound | UOF-Er | UOF-Y |
|---|---|---|
| a R 1 = ∑||Fo|−|Fc||/|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | ||
| CSD | 2192944 | 2192943 |
| Empirical formula | ErO22.5U5.5 | YO22.5U5.5 |
| Formula weight | 1836.43 | 1758.08 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P |
P |
| a (Å) | 8.1060(16) | 8.0890(16) |
| b (Å) | 11.435(2) | 11.408(2) |
| c (Å) | 11.582(2) | 11.517(2) |
| α/(°) | 111.33(3) | 111.13(3) |
| β/(°) | 102.97(3) | 103.02(3) |
| γ/(°) | 106.48(3) | 106.40(3) |
| Volume (Å3) | 892.0(4) | 885.2(4) |
| Z/μ (mm−1) | 2/54.500 | 2/53.474 |
| Min./Max. θ [°] | 2.030/24.999 | 2.037/24.999 |
| d calcd (g cm−3) | 6.837 | 6.596 |
| GOF | 1.070 | 1.123 |
| Final R1a[I > 2σ(I)] |
0.0914 | 0.0468 |
| Final wR2b[I > 2σ(I)] |
0.2623 | 0.1402 |
Both compounds UOF-Er and UOF-Y were found to crystallise in the triclinic P space group, each containing six distinct U sites (U1–U5 in full occupancy and U6 modelled in half occupancies as it is on a centre of symmetry, Tables S3 and S4, ESI†) in the asymmetric unit. Two of these sites exist in an octahedral geometry (U1 and U6) and four in a pentagonal bipyramidal coordination geometry (U2–U5). In both structures, the Er/Y3+ species exists in an 8-coordinate, trigonal prismatic geometry, with both materials found to be isostructural.
Examination of the broader structure of UOF-Er and UOF-Y reveals that the material exists as a framework-type structure, with the secondary cations (Er3+/Y3+) lying within the channels of the framework (Fig. 2a). The backbone of the framework is composed of two distinct structural features. The first of these are uranyl polyhedra sheets connected through O–O equatorial edges of U3–U6 to form β-U3O8-type layers (Fig. 2b). These polyhedra sheets consist of two distinct chains of the pentagonal bipyramidal U3–U5 in a –(U3-U5-U4-U4-U5-U3)– motif, with these chains linked by U1 and U6 octahedra. A pair of U2 centers are connected by edge-shared O–O, which pillar the β-U3O8-type layers through U1, U3 and U4 (Fig. 2c). The axial oxygens of U4–U6 coordinate the 8-coordinate Er/Y, which are also coordinated to four water molecules and located in the channels of the framework (Fig. 2d and e).
 | ||
| Fig. 2 Crystal structure of compound UOF-Er/UOF-Y: a polyhedral crystal structure along the c-axis (a), a polyhedral view of a uranyl oxide hydroxide layer with a β-U3O8 topology (b), the uranyl oxide hydroxide layers linked by double U2 pentagonal bipyramids (c), with 8-fold coordinated Er/Y(III) interlayer cations in the framework channels [(d) and (e)], U in yellow and Er/Y in blue. | ||
The octahedral U1 consists of two axial U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds ranging from 1.821(14) to 1.939(14) Å, and four equatorial U–O bonds of 2.15(3) to 2.234(13) Å. These bond lengths, along with the near linear bond angle (176.0(15)°–176.5(6)°) of the two axial UO bonds, are consistent with similar octahedral uranyl units reported for other UOH/F materials in the literature.10,22,44,45 The four U sites existing in a pentagonal bipyramidal geometry (U2–U5), have two axial UO bonds of 1.72(3) to 2.001(13) Å and OUO angles of 173.3(6)° to 178.8(6)°, and five equatorial U–O bonds of 2.210(13) to 2.53(3) Å. As with the U1 site, these values are broadly consistent with those previously reported.10,22,44,45 The U6 site contains four O atoms at shorter U–O distances of 2.05(3) to 2.08(4) Å, arranged in a distorted square, and two axial oxygen atoms with slightly longer U–O bonds of 2.14(4) Å, which coordinate to the secondary metal cations.
O bonds ranging from 1.821(14) to 1.939(14) Å, and four equatorial U–O bonds of 2.15(3) to 2.234(13) Å. These bond lengths, along with the near linear bond angle (176.0(15)°–176.5(6)°) of the two axial UO bonds, are consistent with similar octahedral uranyl units reported for other UOH/F materials in the literature.10,22,44,45 The four U sites existing in a pentagonal bipyramidal geometry (U2–U5), have two axial UO bonds of 1.72(3) to 2.001(13) Å and OUO angles of 173.3(6)° to 178.8(6)°, and five equatorial U–O bonds of 2.210(13) to 2.53(3) Å. As with the U1 site, these values are broadly consistent with those previously reported.10,22,44,45 The U6 site contains four O atoms at shorter U–O distances of 2.05(3) to 2.08(4) Å, arranged in a distorted square, and two axial oxygen atoms with slightly longer U–O bonds of 2.14(4) Å, which coordinate to the secondary metal cations.
From the BVS calculations (Tables S3 and S4, ESI†), assuming the presence of U6+ (RU–O = 2.051; B = 0.519),9 it can be determined that five of the six U centers (U1–U5) in both UOF-Er and UOF-Y exist as U6+, as expected. Interestingly, the U6 site in both materials, having calculated BVS values of 5.58 and 5.54, suggests the presence of U5+ at this site in the structure. This is consistent with previously reported UOF structures, wherein U5+ sites have been observed at the octahedral U site which coordinates the secondary cations.21,22 The oxygen donors were found to be a majority O, with four H2O molecules coordinated to the Er/Y cations, a coordinated OH (O17 disordered in two positions) linking two U5 centers, and two more OH groups for O3 and O19. Based on the unit cell content and the types of U and O, the general formula is simplified to Z = 1: M2(H2O)8[(UO2)10UO14(OH)3], where M = Er for UOF-Er and Y for UOF-Y. As such, the structure complexity for the UOFs, measured by Ichem (bits/formula),11 is ∼114.5, quite complex given the average structure complexity of all known UOH minerals is ∼76.
Of significant interest is the identified P space group. Whilst layered UOH structures have been reported in the same space group,30,45 no triclinic UOF has been identified yet in the literature. Looking along the channels of the framework (Fig. 2e), it is immediately apparent that the (Er/Y)3+ ions don't lie in the center of the channel, instead lying closer to a corner of the space, with each Er/Y ion not connected to the next Er/Y ions lying in the channel. This is distinctly different from previously reported UOF structures, which are highlighted in Table 3. In these earlier structures, the interlayer cations form –(M–O–M)– chains along the channels, with these chains composed of two or more unique cation species.21,22 In these sequences, the interlayer cations are separated by distances of ∼3.8–4.0 Å, however for UOF-Er and UOF-Y this interaction distance is 5.8–6.2 Å, possibly explaining the lack of connectivity between the cations. Also, of interest is that in these previously reported materials, these cation species are disordered, existing across two sites in partial occupancies, allowing them to be aligned well inside the framework channels. However, in UOF-Er and UOF-Y these cation species have full occupancy preventing them alignment in the center of the channels. A second distinct, but related, difference is the chemical environment about the U1 and U3 centers. Each of these U centers contains axial O atoms projecting into the channel of the framework (O3 and O9, respectively) with relatively longer UO bonds leading to lower-than-normal BVS values for O3 and O9. Whereas in previously reported structures21,22 these oxygen species are involved in the coordination of the interlayer cation species, oxygens in the two new structures remain unbound, resulting in a unique pore environment yet unseen in UOF structures. This is further confirmed by the calculated crystal topologies (Table 3) for each of the UOF systems which show that the UOF-Er/Y system is distinct from those previously reported.
| Compound | Space group, formula and cell parameters | Asymmetric unita | Ln/Y ions | Topology |
|---|---|---|---|---|
| a Site occupancy in brackets. | ||||
| UOF-Eu/Gd22 | C2221: orthorhombic (Eu/Gd)2(OH)(H2O)5[(UO2)10O10(OH)2] [(UO4)(H2O)2] a = 11.629(2), b = 20.973(4), c = 14.170(3) Å | 4 U (1), 3 U (0.5), Eu/Gd (0.65, 0.35) | Eu/Gd disordered on 2 sites | 3^9,5,6^3,7^4,8-c net |
| UOF-Sm21 | C2: monoclinic Sm2(OH)(H2O)5[(UO2)10O10(OH)2] [(UO4)(H2O)2] a = 11.626(2), b = 20.975(4), c = 14.199(3) Å; β = 90.04(3)° | 11 U (1), 2 Sm (0.65, 0.35) | Sm disordered on 2 sites | 3^18,5^3,6^4,7^6,8^2-c net |
| UOF-Er/Y |
P: triclinic (Er/Y)2(H2O)8[(UO2)10UO14(OH)3] a = 8.1060(16), b = 11.435(2), c = 11.582(2) Å; α = 111.33(3)°, β = 102.97(3)°, γ = 106.48(3)° |
5 U (1), 1 U (1/2), Er/Y (1) | Er/Y on 1 site | 3^8,4,5^2,6^2,7^2-c net |
3.3. TEM characterization
Given their isostructural nature, only UOF-Er was further studied with TEM. A high-resolution TEM (HRTEM) image (Fig. 3) shows crystal lattice fringes. In Fig. 3, variations in the crystal fragment thickness, plus the presence of nano-domains highlight a complex structure. The selected area electron diffraction (SAED) pattern (inset in Fig. 3), viewed down the [3 5 9] zone axis, is indexed to the P space group. In addition, electron diffraction spots correlating to a 3× superstructure along the [1 −6 3] direction, marked with arrows, are also evident, indicating the presence of a modulated structure. The extra Bragg maxima fall around the 5.8 Å region, in real space, and also show splitting.
 | ||
| Fig. 3 HRTEM of UOF-Er: a high-resolution TEM image with an inset of a selected area electron diffraction pattern viewed down the [3 5 9] zone axis, also highlighting Bragg maxima from a modulated structure (arrows) along the (1 −6 3) direction indicating a 3× superstructure. | ||
3.4. Electronic structures and uranium valences
To further investigate the potential for U5+ being present in both UOF-Er and UOF-Y, the U valences were characterised using DRS. This technique has been employed in the past to analyse U valences in UOH/F materials,20,24 as it allows for the distinction between U4+, U5+ and U6+. The U4+, having a 5f2 electronic configuration, gives both sharp (zero-phonon lines) and broad (vibronic) absorptions across both the infrared and visible spectrum ranges.46 The presence of U5+ (5f1) is categorised by peaks limited to the near infrared. These peaks are due to the crystal-field splitting of 2F5/2–2F7/2, giving rise to distinct transitions in the range of 1538–833 nm (6500–12000 cm−1).46,47 Lastly U6+ (5f0), having no f electrons, has only charge-transfer bands which are visible in the UV and far UV regions of the spectrum.
The first feature of note in the spectra for both UOF-Er and UOF-Y (Fig. 4) is the broad, indistinct absorption between 250–550 nm. The two absorption maxima present in UOF-Y (Fig. 4a, top) at 350 nm and 450 nm can be attributed to the charge transfer of U6+, which is consistent with values found for other uranyl oxide compounds.48,49 The same feature is less visible in the UOF-Er spectrum, however given the location and broadness, is also attributed to U6+ charge transfer. Also located in this region, and possibly contributing to the broadness of the peak, are Er3+ f–f transitions (Fig. S4, ESI†). Examination of the NIR region (Fig. 4b) shows distinct peaks, strongly suggesting the presence of U5+ in both materials. For UOF-Er (Fig. 4b, bottom), this is signified by the strong, sharp peak at 910 nm/1490 nm. Two other peaks are also evident in this spectrum, which can be attributed to the Er3+ 4I15/2 → 4I11/2 (970 nm) and 4I15/2 → 4I13/2 (1556 nm) transitions.50 The first thing of note in the NIR spectrum for UOF-Y (Fig. 4b, top) is the absence of the peaks at 970 and 1556 nm, further evidence towards their assignment as Er3+ transitions. Importantly however is that the same peak attributed to U5+, whilst obfuscated, is evident in the spectrum of UOF-Y at 910 nm. Thus, the evidence of this peak, along with the BVS and crystal data, strongly suggests both UOF-Er and UOF-Y are mixed uranium valence compounds.
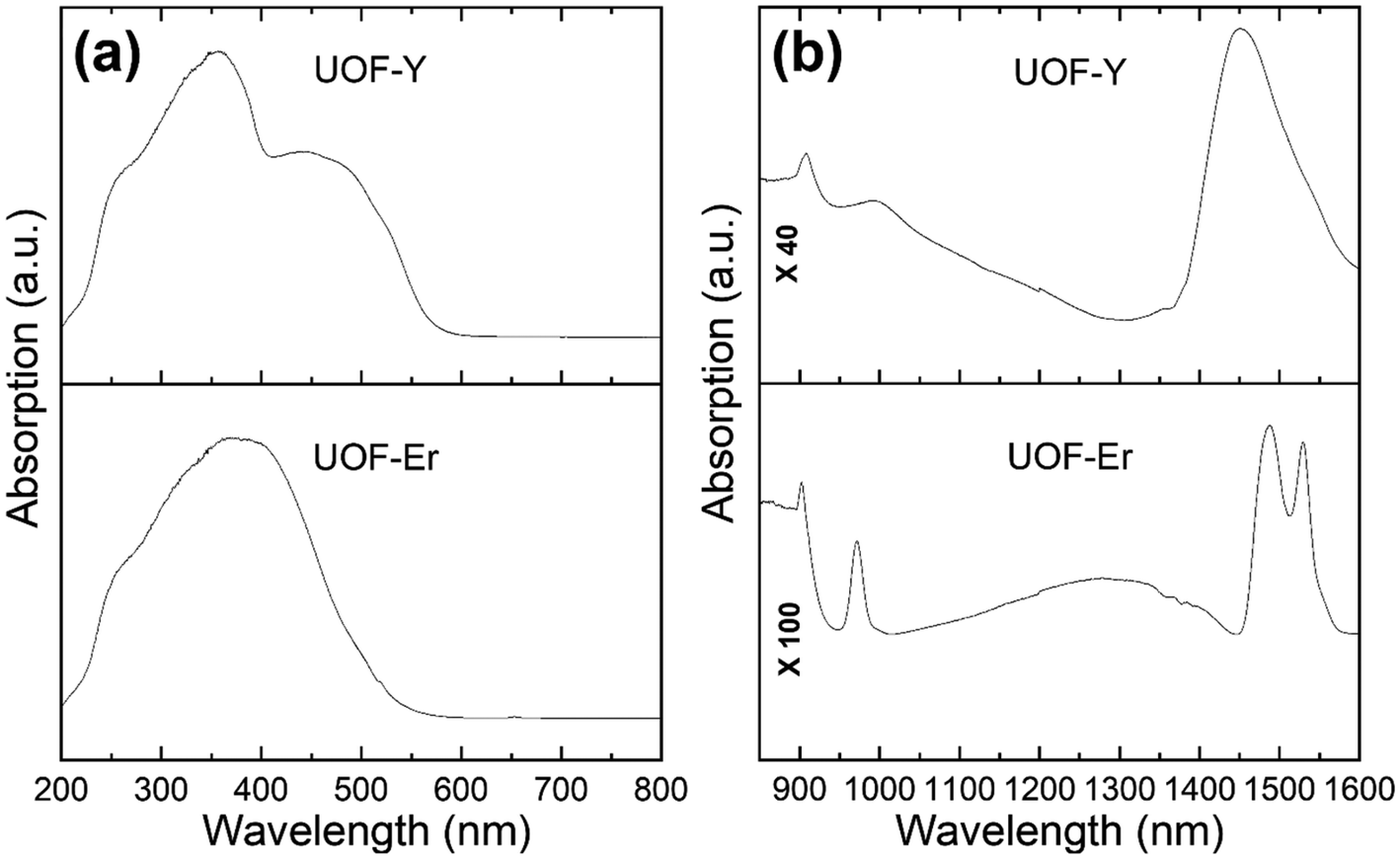 | ||
| Fig. 4 The DRS spectra of UOF-Er and UOF-Y in the (a) UV-vis and (b) the near infrared regions. | ||
3.5. Vibrational modes
The vibrational modes of both materials were examined using Raman spectroscopy, which is shown in Fig. 5. Previous studies on such UOH and UOF materials have identified the regions relating to ν2(δ)(UO2)2+ between 200–300 cm−1 with broad, weak peaks, ν(U3O) and γ[U3(OH)3] in the range of 300 to 600 cm−1 as broad, medium peaks and ν1(UO2)2+ between 700 and 900 cm−1 with sharp, strong peaks.49,51 In exploring the spectrum for UOF-Er (Fig. 5a), a sharp, strong peak is evident at ∼840 cm−1, corresponding to the UO bonds. The broader peaks between 150–550 cm−1 can be assigned to the various U–O bonds within the material. Despite a poorly resolved spectrum for UOF-Y (Fig. 5b), these same peaks are also visible, an expected result given the two materials are isostructural. Note the absence of Raman vibrations for the (UO2)+ uranyl ion is consistent with the crystallographic results that U5+ ions in these compounds are present in an octahedral coordination environment, similar to the U5+ ions in Y0.51U0.49Ti2O6 or Eu0.53U0.47Ti2O6 brannerite.52
 | ||
| Fig. 5 The Raman spectra of UOF-Er (a) and UOF-Y (b). | ||
3.6. Implications and perspectives
The herein reported synthesis of two isostructural, novel UOF structures clearly demonstrates that the scope of lanthanides which can be introduced into these framework-type structures is much broader than previously hypothesised with Er3+, Sm3+, Eu3+ and Gd3+, also including Y3+, now all reported in synthetic UOF materials. The structural parameters also appear to fit well to that of wyartite, the first pentavalent-uranium mineral.53,54 The use of higher solution pH and different hydrothermal conditions in this study compared to that previously reported is also of interest. The solution pH is one of the driving forces leading to the final products. While the initial hydrolysed uranyl species, [(UO2)(OH)]+, is less than 5% at the solution pH < 4, it increases to nearly 15% at the solution pH ∼ 4.5.55 Further hydrolysed uranyl species such as [(UO2)(OH)2], [(UO2)2(OH)2]2+ and [(UO2)3(OH)5]+ will be present at the solution pH above 5. These findings clearly suggest that the flexible β-U3O8 type layer which makes up the backbone of the framework tolerates subtle structural changes, which in turn allows for a variety of conditions and secondary metal ions to be used in their synthesis. This highlights that more work is required in this area to gain a better understanding of the diversity tolerated by these materials whilst maintaining UOF formation.However, given the hydrothermal conditions used in this study could sensibly be comparable to the conditions found in the geological repositories used for storing SNF, the understanding garnered from the synthesis UOF-Er and UOF-Y can be extended to better understanding the alteration chemistry of UO2 based species in these SNF environments. As the results in this study are examining the high temperature chemistry of these materials over a short time period (days), this work best captures the early stages of the alteration chemistry in these repositories. Further time under these high temperature conditions could exacerbate these changes, or even lead to further changes within these materials, and thus warrants further study.
The implication of the reported UOFs may extend to broader work on nuclear materials as well. Work has previously been performed on incorporating lanthanides as surrogates of minor actinides (i.e. Ce4+/Nd3+ in place of Pu4+/Cm3+) as the interlayer cations of UOH-based minerals.25,26 Given the ionic radii of 8-coordinate Er3+ (RCN = 8 = 1.004 Å) and Y3+ (RCN = 8 = 1.019 Å) closely match that of Cm3+ (RCN = 8 = 0.97 Å),56 it stands to reason that the incorporation of this cation into a similar UOF structure as the one reported here may be possible.
A recent study by Murphy and co-workers examined the intercalation of the anionic IO3− species within a UOH structure,57 which is the first such study incorporating an anionic species into these materials. The authors proposed that in capturing the radiolytic IO3− this slowed its release into the biosphere, and thus the presence of the UOH material in a geological repository could prove beneficial. It stands to reason that this work could be extended to other anions present in SNF such as pertechnetate (TcO4)−, with the framework-type structure of UOFs possibly offering additional benefits.
4. Conclusions
Two new UOFs with incorporated Er3+/Y3+ ions have been synthesised under hydrothermal conditions using uranyl nitrate as the U precursor. The U/(Er/Y) ratio of 5.5 matches those found for previously reported UOFs with other lanthanide ions, however the triclinic P space group is novel amongst previously reported framework-type structures. The pentagonal bipyramidal and octahedral uranium polyhedra extend to form β-U3O8-type sheets, which are pillared by an additional pentagonal bipyramidal uranium center to form the backbone of the framework, with the secondary Er3+/Y3+ cations found within the channels. Unlike previously reported UOFs, the secondary cations are not interconnected, instead existing as a single cation species. The presence of U5+ in the structure was confirmed by DRS, existing as pentavalent uranium at one of the octahedral coordination sites.
These compounds, in expanding the knowledge previously obtained from published UOF structures, clearly demonstrate the complex, flexible and highly sensitive chemistry which governs the formation and reactivity of uranium oxide hydrate-based systems which is dependent on, amongst other things, temperature, redox potential, and pH. Further work is still warranted to develop this understanding, with systematic laboratory studies controlling and exploring these conditions on such way to extra thus knowledge.
Author contributions
T. A. Ablott: Conceptualization, data curation, formal analysis, writing – original draft, writing – review & editing. K. T. Lu: Data curation, formal analysis, writing – review & editing. R. D. Aughterson: Data curation, formal analysis, writing – review & editing. Y. Zhang: Conceptualization, data curation, formal analysis, project administration, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There is no conflict of interest.Acknowledgements
We would like to thank I. Karatchevtseva for Raman measurement, the Nuclear Science and Technology (NST) at ANSTO for synthesis and characterization of materials. The crystallographic data for compounds UOF-Er and UOF-Y were collected on the MX2 beamline at the Australian Synchrotron, a part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.References
- J. Plásil, J. Geosci., 2014, 59, 99–114 CrossRef
.
- R. J. Baker, Coord. Chem. Rev., 2014, 266–267, 123–136 CrossRef CAS
- Y. Zhang, T. Wei, A. Xu, P. Dayal and D. J. Gregg, J. Am. Ceram. Soc., 2021, 104, 5981–5989 CrossRef CAS
- S. Alyokhina, Nucl. Eng. Technol., 2018, 50, 717–723 CrossRef CAS
- J. Janeczek and R. C. Ewing, J. Nucl. Mater., 1992, 190, 157–173 CrossRef CAS
- J. Janeczek and R. C. Ewing, J. Nucl. Mater., 1992, 190, 128–132 CrossRef CAS
- R. J. Finch and R. C. Ewing, J. Nucl. Mater., 1992, 190, 133–156 CrossRef CAS
- D. J. Wronkiewicz, J. K. Bates, S. F. Wolf and E. C. Buck, J. Nucl. Mater., 1996, 238, 78–95 CrossRef CAS
- P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551–1570 CAS
-
P. C. Burns, in Structural Chemistry of Inorganic Actinide Compounds, ed. S. V. Krivovichev, P. C. Burns and I. G. Tananaev, Elsevier, Amsterdam, 2007, pp. 1–30 Search PubMed
- J. Plásil, Eur. J. Mineral., 2018, 30, 237–251 CrossRef
- Y. Zhang, K. T. Lu and R. Zheng, Dalton Trans., 2022, 51, 2158–2169 RSC
- N. G. Chernorukov, O. V. Nipruk, G. N. Chernorukov, R. V. Abrazheev and K. A. Chaplieva, Russ. J. Gen. Chem., 2019, 89, 71–75 CrossRef CAS
- R. Vochten, L. van Haverbeke and R. Sobry, J. Mater. Chem., 1991, 1, 637–642 RSC
- Y. Li and P. C. Burns, Can. Mineral., 2000, 38, 737–749 CrossRef CAS
- R. E. Glatz, Y. Li, K.-A. Hughes, C. L. Cahill and P. C. Burns, Can. Mineral., 2002, 40, 217–224 CrossRef CAS
- P. C. Burns and F. C. Hill, Can. Mineral., 2000, 38, 163–173 CrossRef CAS
- Y. Zhang, J. Čejka, G. R. Lumpkin, T. T. Tran, I. Aharonovich, I. Karatchevtseva, J. R. Price, N. Scales and K. Lu, New J. Chem, 2016, 40, 5357 RSC
- K. A. Kubatko and P. C. Burns, Inorg. Chem., 2006, 45, 10277–10281 CrossRef CAS PubMed
- K. T. Lu, Y. Zhang, T. Wei, T. A. Ablott, T. H. Nguyen and R. Zheng, New J. Chem, 2022, 46, 1371 RSC
- K. T. Lu, Y. Zhang, T. Wei, Z. Wang, D. T. Oldfield and R. Zheng, Inorg. Chem., 2021, 60, 13233–13241 CrossRef CAS
- K. T. Lu, Y. Zhang, R. D. Aughterson and R. Zheng, Dalton Trans., 2020, 49, 15854–15863 RSC
- Y. Li and P. C. Burns, Can. Mineral., 2000, 38, 1433–1441 CrossRef CAS
- Y. Zhang, T. Wei, T. T. Tran, K. T. Lu, Z. Zhang, J. R. Price, I. Aharonovich and R. Zheng, Inorg. Chem., 2020, 59, 12166–12175 CrossRef CAS
- C. W. Kim, D. J. Wronkiewicz, R. J. Finch and E. C. Buck, J. Nucl. Mater., 2006, 353, 147–157 CrossRef CAS
- S. Biswas, R. Steudtner, M. Schmidt, C. McKenna, L. L. Vintró, B. Twamley and R. J. Baker, Dalton Trans., 2016, 45, 6383–6393 RSC
- Y. Zhang, R. Aughterson, I. Karatchevtseva, L. Kong, T. T. Tran, J. Čejka, I. Aharonovich and G. R. Lumpkin, New J. Chem., 2018, 42, 12386–12393 RSC
- K. T. Lu, Y. Zhang, T. Wei, J. Čejka and R. Zheng, Dalton Trans., 2020, 49, 5832–5841 RSC
- Y. Zhang, R. D. Aughterson, Z. Zhang, T. Wei, K. Lu, J. Čejka and I. Karatchevtseva, Inorg. Chem., 2019, 58, 10812–10821 CrossRef CAS
- L. N. G. Chernorukov, O. V. Nipruk, K. A. Klin, G. N. Chernorukov and O. N. Tumaeva, Radiochemistry, 2021, 63, 110–120 CrossRef
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed
-
G. M. Sheldrick, SADABS, Empirical Absorption and Correction Software, University of Göttingen, Göttingen, Germany, 1996 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed
- E. V. Alexandrov, V. A. Blatov, A. V. Kochetkov and D. M. Proserpio, CrystEngComm, 2011, 13, 3947–3958 RSC
- V. A. Blatov and A. P. Shevchenko, Struct. Chem., 2021, 32, 507–519 CrossRef
- E. V. Alexandrov, A. P. Shevchenko and V. A. Blatov, Cryst. Growth Des., 2019, 19, 2604–2614 CrossRef CAS
- M. K. Pagoaga, D. E. Appleman and J. M. Stewart, Am. Mineral., 1987, 72, 1230–1238 CAS
- F. Zocchi, J. Mol. Struct.: THEOCHEM, 2007, 805, 73–78 CrossRef CAS
- P. C. Burns, Can. Mineral., 2005, 43, 1839–1894 CrossRef CAS
- M. Rivenet, N. Vigier, P. Roussel and F. Abraham, J. Solid State Chem., 2009, 182, 905–912 CrossRef CAS
- K. S. Finnie, Z. Zhang, E. R. Vance and M. L. Carter, J. Nucl. Mater., 2003, 317, 46–53 CrossRef CAS
- Y. Zhang, T. Wei, Z. Zhang, L. Kong, P. Dayal and D. J. Gregg, J. Am. Ceram. Soc., 2019, 102, 7699–7709 CrossRef CAS
- Y. Zhang, J. K. Clegg, K. Lu, G. R. Lumpkin, T. T. Tran, I. Aharonovich, N. Scales and F. Li, ChemistrySelect, 2016, 1, 7–12 CrossRef CAS
- Y. Zhang, M. Bhadbhade, I. Karatchevtseva, J. R. Price, H. Liu, Z. Zhang, L. Kong, J. Čejka, K. Lu and G. R. Lumpkin, J. Solid State Chem., 2015, 226, 42–49 CrossRef CAS
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4424 CrossRef CAS
- R. L. Frost, J. Čejka and M. L. Weier, J. Raman Spectrosc., 2007, 38, 460–466 CrossRef CAS
- Y. Zhang, I. Karatchevtseva, L. Kong, T. Wei and Z. Zhang, J. Am. Ceram. Soc., 2018, 101, 5219–5228 CrossRef CAS
- F. C. Hawthorne, R. J. Finch and R. C. Ewing, Can. Mineral., 2006, 44, 1379–1385 CrossRef CAS
- P. C. Burns and R. J. Finch, Am. Mineral., 1999, 84, 1456–1460 CrossRef CAS
- C. Moulin and P. Decambox, Anal. Chem., 1995, 67, 348–353 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- G. L. Murphy, P. Kegler, M. Klinkenberg, A. Wilden, M. Henkes, D. Schneider and E. V. Alekseev, Dalton Trans., 2021, 50, 17257–17264 RSC
Footnote |
| † Electronic supplementary information (ESI) available: SEM-EDS. CCDC 2192943 (UOF-Y) and 2192944 (UOF-Er). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02763a |
| This journal is © The Royal Society of Chemistry 2022 |