
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the non-existence of a square-planar pentaiodide coordination complex I(I)4−†
Lars
Kloo

Applied Physical Chemistry, Department of Chemistry, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: Lakloo@kth.se
First published on 30th August 2022
Abstract
The properties of two conformers of the pentaiodide ion, a V-shaped and regularly observed I5− ion, and a so far undetected square-planar coordination complex of II4− composition, have been investigated by computational methods. The latter compound is indicated by the analogy to the coordination chemistry of gold with halide ligands, as well as isoelectronic main-group compounds. Static and dynamic simulations at density-functional and semi-empirical level including effects of solvent and counter ions indicate that the square-planar II4− indeed represents a well-defined local minimum on the pentaiodide potential energy surface, albeit less stable than the typically observed V-shaped I5−. No simple pathway of transformation between the two forms of the pentaiodide ion can be identified. Molecular dynamics simulations indicate that the presence of cations, unavoidable during the synthesis of polyiodide compounds, may trigger decomposition of the II4− coordination complex into smaller polyiodide building blocks and thus constitute the main reason why this conformer so far has not been identified in solid polyiodide compounds. However, its intrinsic stability indicates that the square-planar form should be possible to isolate in solid compounds given the right conditions of synthesis.
Introduction
Polyhalide compounds in general and polyiodides in particular offer a rich structural chemistry of catenated, low-dimensional compounds.1,2 The vast majority of polyiodide compounds can be visualized in terms of simple coordination chemistry, in which the ionic building blocks of I− and I3− can be regarded as solvated by the neutral solvent molecules I2. The sticky bonding properties of the relatively small and few building blocks generate chain-like structures, which can be attributed to topological electron densities of the building blocks (σ-holes).2,3 The predominant bonding contributions have been assigned to secondary interactions, such as dispersion and induction, complemented by weak covalency mediated by overlap of the large valence orbitals of iodine.4 However, this scheme has recently been challenged and the bonding been solely attributed to non-covalent interactions.5More specifically, a large number of compounds named as pentaiodides can be found in a search of the Cambridge Structural Database (CSD).6 The denotation pentaiodide refers primarily to the stoichiometry with respect to a suitable monocation, often a small organic cation. However, even such a simple stoichiometry offers a large diversity in terms of geometric structure. The stoichiometric pentaiodides can be found as isolated ions or as the repeating unit in typically chain-like structures, and they can be described as either of V-shape [(I−)·2I2] or of L-shape [(I3−)·I2], see Fig. 1.1
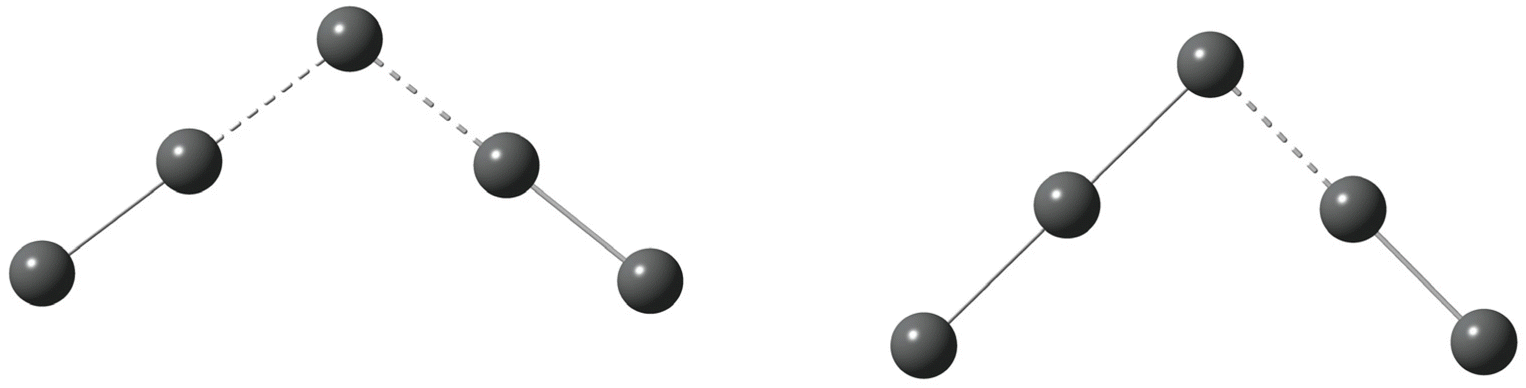 | ||
| Fig. 1 Pentaiodide ions in V-shaped (left) or L-shaped (right) forms; isolated or as part of an extended, chain-like structure; iodine atoms (grey). | ||
Alongside the catenated chemistry of polyiodides, there is an interesting analogy between the coordination chemistry of interhalogen compounds and gold-halide complexes. For instance, the well-known, linear gold(I) complexes AuCl2− and AuI2− can be compared to the interhalogen ions (complexes) ICl2− and II2− (i.e. the triiodide ion I3−) in the sense that they are structurally highly similar and the coordination centre can be regarded to be in a +I oxidation state. The view of iodine in a formal positive oxidation state as a coordination centre has recently been highlighted in comparison to that of silver(I), as well as to gold(I and III).7,8 In fact, the mixed anion, or as it might better be described as the iodide-bridged, gold(I)-iodide species Au2I3− is structurally analogous to the V-shaped pentaiodide ion.9 I analogy, the square-planar gold(III) complexes AuCl4− and AuI4− can be compared to ICl4− and II4− (i.e. another structural form of the pentaiodide ion I5−), see Fig. 2. However, here there is one clear discrepancy, the pentaiodide ion has to our knowledge so far not been found as a square-planar coordination complex, but rather in the forms shown in Fig. 1. Why not?
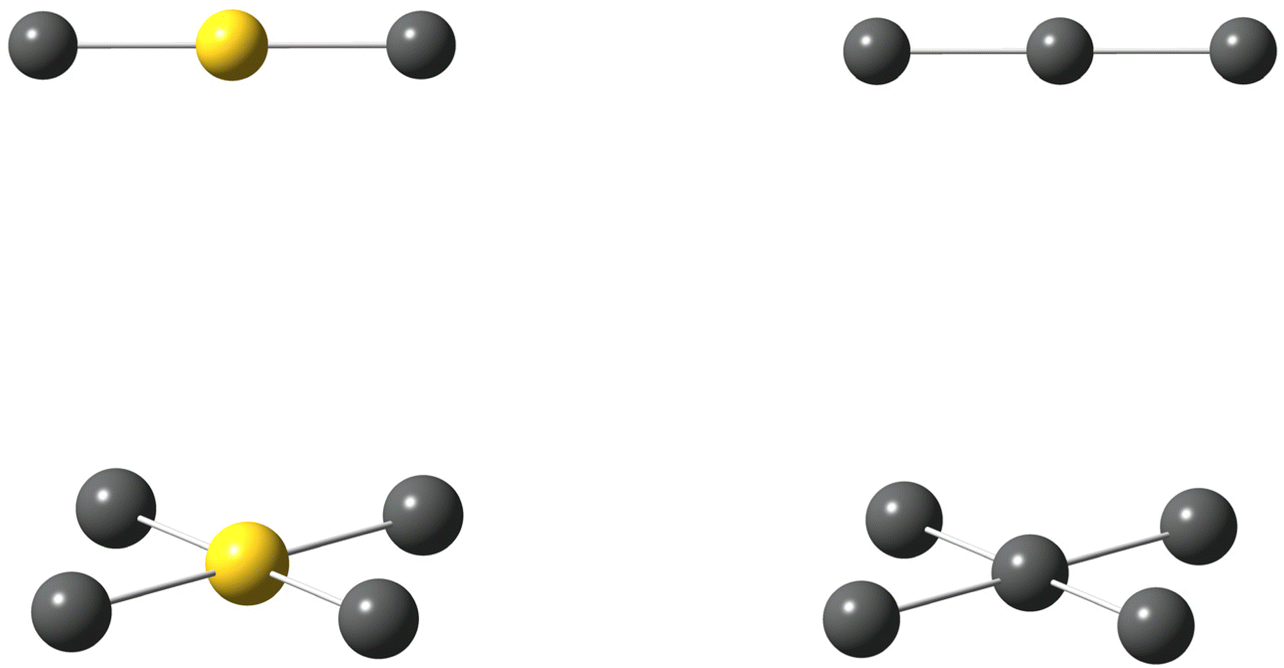 | ||
| Fig. 2 The well-known, linear AuI2− (top left), square-planar AuI4− complex (bottom left) and triiodide ion II2− (top right), as well as a hypothetical analogous II4− ion (bottom right); gold atoms (yellow), iodine atoms (grey). | ||
In this context, it is relevant to mention some analogies to similar and isoelectronic main-group compounds. Regarding valence electron numbers, a square-planar II4− can be thought of as a congener to the well-known, square-planar molecules SF42− and XeF4; compounds that essentially every undergraduate student in introductory chemistry has been exposed to when introduced to chemical bonding and Lewis electron structures. Among interhalogen compounds, the square-planar configuration is observed for a series of XF4− compounds (X = Cl, Br or I) and the above noted ICl4− ion; where a characteristic property is that the most electronegative element is found in the terminal positions; all akin to the II4− form of the pentaiodide ion. The analogies to the halides of the chalcogenides also include the closely related, square-planar halide complexes of tellurium, viz. TeI42−, that were synthesized and structurally characterized by Krebs and co-workers.10 In this context, also the unique and to this work relevant gold coordination compound with noble-gas ligands, AuXe42+ isolated by Seppelt and Seidel, should be mentioned.11
A search of the CSD for square-planar I5 structural fragments offers no relevant hits. A handful of structures can be described in terms of catenated pentaiodide fragments with a coordination resembling that of a distorted tetrahedron, but none can be regarded as an isolated square-planar II4− complex. As will be shown below, such a pentaiodide coordination complex resides in a well-defined minimum on the pentaiodide potential energy surface (PES), albeit energetically less stable than the V-shaped form shown in Fig. 1. This work will attempt to answer the question why such a coordination species has not been found among the many pentaiodide structures known; at least not yet.
Computational
Most quantum chemical computations of static models were performed using Gaussian 16 (Rev. B.01 and C.01).12 ECP-based basis sets were used for I and Au employing the Stuttgart–Dresden–Cologne group, relativistic, effective-core potentials (ECPs; MDF60 for Au and MDF28 for I) and valence spaces of (14s11p10d3f2g1h)/[6s6p5d3f2g1h] quality for Au,13,14 and (14s11p12d2f1g)[6s6p4d2f1g] for I.15 Calculations were performed at density-functional theory (DFT) level using the hybrid functional cam-B3LYP.16 2nd order relativistic effects including spin–orbit coupling were considered using and the def2-QZVP/def2-QZVP-2c basis sets and associated ECPs for iodine including spin–orbit coupling coefficients and based on the Gaussian formulation of the hybrid functional B3LYP as implemented in Turbomole 7.2.17–20 Molecular dynamics simulations were performed at DFT level using TeraChem v1.9-2021.05 employing the hybrid functional B3LYP and ECP-based basis sets of single-valence quality.21,22 A Langevin thermostat was employed at a fixed nominal temperature and a time step of 1 fs extending to in total 100 ps of simulation starting with an optimized structure of the system under study. Semi-empirical molecular dynamics simulations were performed using the xTB v.6.4.0 at GFN2 level in an NVT ensemble and a Berendsen thermostat.23,24 The acetone droplets consisting of 100 acetone molecules with the density of pure acetone were constructed using Packmol, and the solutes were confined to the droplet centre in the xTB simulations.25 Also the semi-empirical simulations were extended to 100 ps with 1 fs time steps, and data were collected at every 50 fs. Both TeraChem and xTB were operated on graphical processor units.Analyses of the computed molecular orbitals were made using the Natural Bond Order (NBO) and Natural Energy Decomposition Analysis (NEDA) functionalities in NBO 7.0 as interfaced with Gaussian 16.26 Results from the simulations were visualized using VMD 1.9 and GaussView 6.27,28 Details on the evolution of atom-atom distances with simulation time in the different systems studied are provided in the ESI.†
Results and discussion
Energies of the conformers
The initial investigation was performed to identify if a potential, square-planar II4− complex at all is energetically feasible with respect to both the most stable conformer, the V-shaped I5− ion, as well as with respect to the fragments I3− and I2 and the fundamental fragments consisting of one I− ion and two I2 molecules. The relative total energies compared to the V-shaped I5− ion are collected in Table 1.| No spin–orbit coupling | Spin–orbit coupling | ||
|---|---|---|---|
| I5− system | I–I distance/Å | ΔE/eV | ΔE/eV |
| (I−)·2I2, V-shaped | 2.815, 3.093 | 0 | 0 |
| II4−, square-planar | 2.923 | +0.94 | +0.91 |
| I− & two I2 | 2.663 | +2.09 | +2.08 |
| I3− & I2 | 2.663, 2.952 | +0.67 | +0.67 |
We can extract two pieces of qualitative information from the results in Table 1; first, both molecular ions are stable relative the components of an iodide ion and two iodine molecules, albeit the square-planar II4− is slightly less stable than the combination of a triiodide ion and an iodine molecule; second, the square-planar II4− complex is energetically stable, though less so than the commonly found V-shaped structure and should therefore be regarded as a meta-stable form of the pentaiodide ion. Although inclusion of spin–orbit coupling significantly shifts the total energies of the systems, the relative energies remain essentially unchanged and therefore do not alter these conclusions. In this context, it can be noted that the square-planar II4− complex displays no negative eigenvalues in the Hessian upon a vibrational spectroscopic analysis, and the totally symmetric vibration mode can be found at 118 cm−1, which because of significant overlap with typical polyiodide Raman bands would be difficult to single out in a normal Raman spectrum of a polyiodide system. Nevertheless, II4− represents a true (local) minimum on the PES of the pentaiodide ion system.
Bonding analyses of the V-shaped and square-planar pentaiodide forms using NBO7 and NEDA show a significant degree of covalency in both ions with Wiberg bond indices of 0.33 and 0.45, between the top/central iodine atom and the closest neighbours, respectively, and a corresponding NLMO bond order of 0.20 and 0.34. As a reference, from electron counts based on simple molecular-orbital diagrams one would expect the corresponding I–I bond order in I2 to be 1 and in a centrosymmetric and linear I3− to be 1/2. The Wiberg and NLMO bond indices, as anticipated, emerge as 1.03/1.01 and 0.53/0.52, respectively, for the two molecular systems. NEDA analyses of the energy contributions to the interaction energy for the V-shaped I5− ion and I3− (the latter, although centrosymmetric, treated as an I−–I2 unit) show that the charge-transfer contributions to the overall bonding energy are quite similar in the two systems, indicating that covalent interaction contributes significantly to the interaction between the I− ion and the two iodine molecules in the V-shaped I5− polyiodide ion. In the square-planar conformer, the charge-transfer contribution is significantly higher and dominates completely with a factor of about 8 with respect to polarization contributions.
It can be noted that although different types of frontier orbitals will be predominant bonding orbitals in the two analogous, square-planar AuI4− and II4− complexes, from a qualitative point of view the bonding scheme will be similar. Bonding in polyhalides is typically dominated by the halogen p-orbitals, as shown by Pimentel, Rundle and others.29,30 Thus, visualizing the ligand iodine atoms as iodide ions, they will predominantly dominate electron density from their filled p-orbitals into available empty orbitals on the central atom, viz. Au(+3) or I(+3). The available orbitals on gold(III) will be d-orbitals (i.e. the d(x2−y2)-orbital) and on iodine(III) p-orbitals (i.e. the p(x)- and p(y)-orbitals), if assigning bonding to the (x,y)-plane. In terms of electron density, the resulting systems will be highly similar, although symmetry will of course differ considering that d-orbitals are centrosymmetric and p-orbitals are not, as highlighted in Fig. 3.
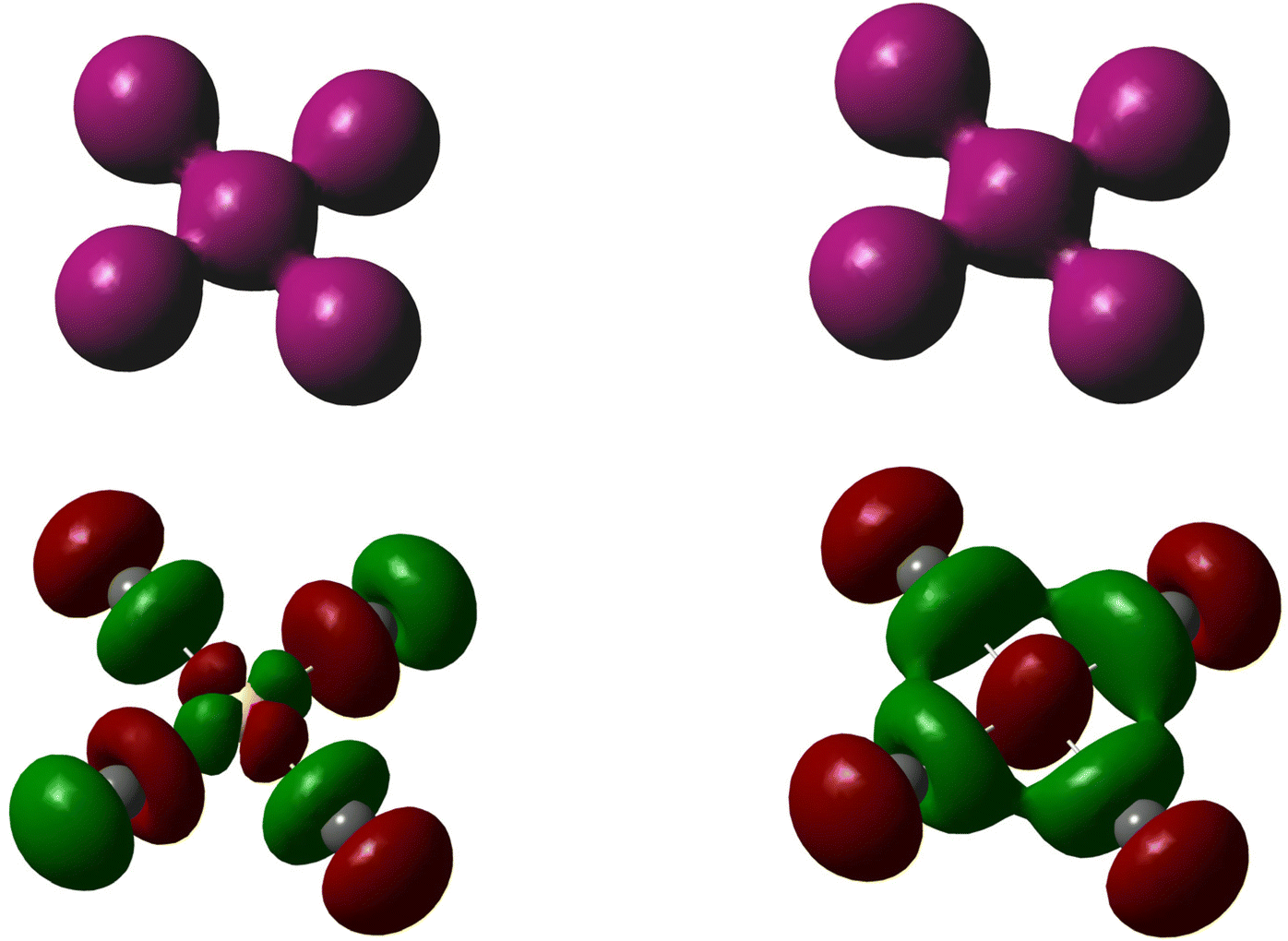 | ||
| Fig. 3 The total electron density surfaces of AuI4− (top left) and II4− (top right), and the corresponding surfaces of the highest occupied molecular orbitals (HOMOs, bottom); iso-surface values used are 0.04 e/(au)3 for the total electron density and 0.025 [e/(au)3]1/2 for the molecular orbitals. | ||
Transitions
Having established that the square-planar conformer of the pentaiodide ion is energetically feasible representing a local energy minimum on the pentaiodide PES, a logical next step will be to investigate if there is a straightforward route of transition between the square-planar and the commonly observed V-shaped conformers. Two different approaches were utilized.The first approach described in this section involved the extraction or addition of I2 units to an I3− core placing the iodine molecule either with all atoms in the same plane or the I2 unit perpendicularly to the triiodide ion. As is obvious from Fig. 4, in none of the investigated systems a straightforward pathway from or to the square-planar II4− by addition of or extraction of a neutral I2 molecule can be identified. In addition, none of the nine normal vibrational modes of the square-planar II4− ion offer any insights as for potential routes of transformation into a more commonly encountered form of the pentaiodide ion.
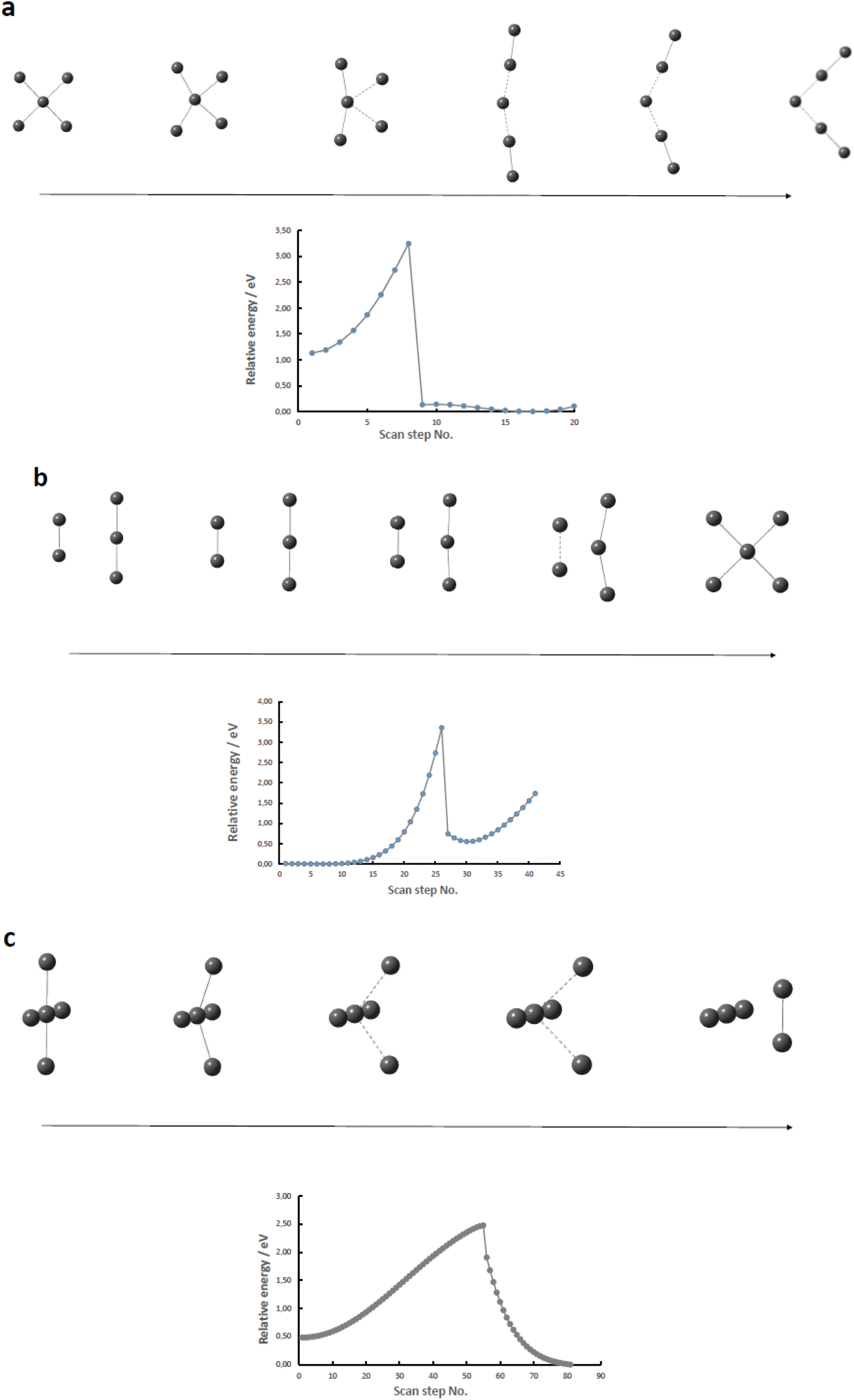 | ||
| Fig. 4 Relative energy scans of (a) in-plane I2 extraction from II4−; (b) in-plane I2 addition to I3−; (c) perpendicular extraction of I2 from II4− (with a linearly restricted I3− unit); iodine atoms (grey). | ||
The second type of analysis described in the subsequent section will involve molecular dynamics at ab initio and semi-empirical level in order to allow the investigation of the effects of dynamics, as well as potentially allow the identification of reaction routes of inter-conversion not formulated a priori.
Dynamics
In the above analysis, the isolated polyiodide fragments were investigated. However, in real-life syntheses both solvent and counter ions are involved, and in this section such effects will be taken into account.The first molecular dynamics simulations at ab initio level focussed on the isolated V-shaped and square-planar pentaiodide units extending to 100 ps. A primitive methodology to provoke reactive events is to increase the nominal simulation temperature in order to increase the probability of reaction events to take place. Therefore, the simulations at ambient temperature were complemented by further ones at 300 and 600 °C. The square-planar II4− ion remained intact during all simulations emphasizing that it should be regarded as a locally highly stable entity. However, upon increasing the simulation temperature for the V-shaped and commonly encountered I5− ion, that ion showed an increasing tendency to split into its expected and smaller building blocks, I3− and I2; at 600 °C, that separation was observed (towards the end of the simulation time) and persisted throughout the simulation time (see Fig. S1† for more details). This may reflect that the square-planar II4− ion does not have well-defined reaction pathways with relatively low reaction barriers to smaller fragments, whereas the V-shaped I5− does.
When synthesizing polyiodides, useful solvents are polar organic molecular systems, such as ethanol or acetone. It is therefore a natural first extension of the study to involve a solvent in the molecular dynamics studies. Acetone was selected as a suitable system, since it is commonly employed in polyiodide synthesis and will not complicate the polyiodide system by strong hydrogen bonding (known to have effects on the speciation and the structure of polyiodide compounds synthesized). A droplet consisting of 100 acetone molecules at a typical density for the solvent was constructed and the two conformers of the pentaiodide ion were placed in the system centres. The systems are quite big, and therefore semi-empirical simulations were employed. The simulations were extended to 100 ps at ambient temperature, see Fig. 5 and Fig. S2.† Again, the square-planar II4− complex remained intact over the entire time of simulation, whereas the V-shaped I5− almost immediately split into the expected I3− and I2 fragments with I2 diffusing to the outer rim of the acetone droplet.
 | ||
| Fig. 5 The two initial models of square-planar II4− (top left) and V-shaped I5− (top right), and the end results after 100 ps simulation under ambient conditions (bottom left and right, respectively); iodine atoms (bronze; different atom colour from the other figures for sake of clarity). | ||
As a side-comment, the diffusion of hydrophobic entities to the droplet rim during simulations in polar solvent models is a quite common phenomenon, in particular when studying biomolecules in water. Sometimes this is referred to as vacuum being hydrophobic. However, that is not a fully accurate description of the driving forces for the observed effect, since it is essentially the molecule-molecule interactions between the polar solvent components that are disturbed or reduced, and as a consequence the less polar solutes are pushed to the position where they interfere with the intrinsic solvent interactions the least, i.e. to the outskirts of the droplet. This is also the phenomenon that we observe for the I2 unit split off from the V-shaped I5− ion. In summary, we observe the same trends as noted for the non-solvated pentaiodide ions, although the polar solvent accentuates the tendency of the V-shaped I5− ion to decompose into I3− and I2 also at low simulation temperatures.
In polyiodide synthesis of course electroneutrality must be fulfilled, and thus the anionic reactants are by necessity accompanied by cationic counter ions. It is also in this aspect that the use of polar organic solvents makes sense, since they are good solvents for both the iodide salts and for the more non-polar iodine molecules. Depending on what type, structure and size of polyiodide is your target product, different cations and solvents can be selected. In the subsequent study, two quite different types of simple cations have been selected for aiMD simulations, Li+ and Au+. The lithium cation was selected because it is small and polarizing, and therefore will enhance any disrupting effects of a cation on the pentaiodide structures. It is well known from literature that cations of this type interact strongly with the polyiodide fragments and often in a bridging position between the ionic and neutral building blocks (like between the I− and I2 fragments). In this section, only the square-planar pentaiodide system was studied, since the interactions of the V-shaped pentaiodide with cations is well investigated and described in the literature. Thus, the main target is to provoke any type of reaction of the square-planar II4− complex in the presence of cations in order to extract insights into the reasons why such a species has not been observed albeit that it obviously, if formed, should be considerably stable.
The aiMD simulations for the anionic II4− system was repeated at ambient temperature and at 300 °C, but no reactions could be observed. However, at 600 °C, the square-planar II4− eventually split into the notorious building blocks I3− and I2, where the I3− ion is coordinated by the Li+ ion in the expected asymmetric coordinating position found in most solid polyiodide structures, see Fig. 6 and Fig. S3.† The dynamics of decomposition is not straightforward; after approximately 40 ps of simulation LiI splits off from the system leaving the remaining four iodine atoms in a less ordered heap, whereafter LiI again approaches the iodine heap to extract away an I2 entity and as a result the Li-I3 and I2 units diffuse away from each other. Further details are given in Fig. S3 in the ESI.† The main conclusion is that cation interaction very well may disfavour the formation of the square-planar II4− complex to the benefit of the regularly known, chain-like polyiodide building blocks.
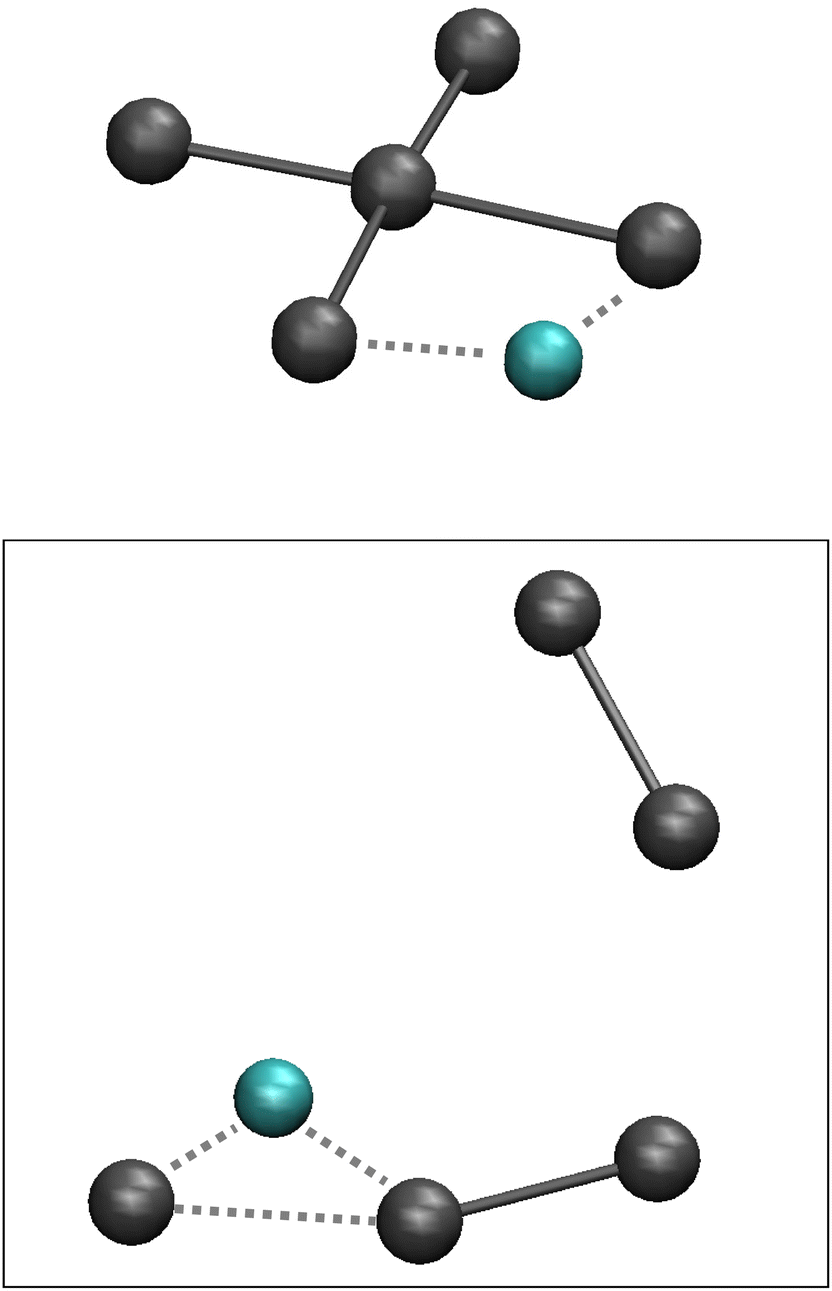 | ||
| Fig. 6 The initial Li+-II4− model as obtained from static geometry optimization (top), and a snapshot of the end products after approximately 60 ps simulation; iodine atoms (grey), lithium atoms (cyan). | ||
The Au+ ion was included mainly for two reasons; first, it represents a completely different type of cation than Li+; 2, the resemblance of the gold and iodine coordination chemistry may provide some new insights into the interaction between gold and iodine in the combined systems. Indeed, already at ambient conditions complex reactions can be observed; not always with easily interpreted results in terms of expected coordination compounds, though. At 25 °C, Au+ almost immediately becomes integrated into the II4− structure forming a linear AuI2-type of entity at the edge, but with unclear interaction with the remaining three iodine atoms.
After about 50 ps of simulation I2 is separated from the other atoms leaving behind, from a coordination chemistry point of view, a rather obscure entity; an odd-looking AuI3−, see Fig. 7. Comparisons to regular AuI2− and AuI4− complexes, as well as to a more non-intuitive AuI(I2) unit consisting of Au+ coordinated by an iodide ion on the one side and an iodine molecule on the other, the latter constellation comes out as the most likely end product in the aiMD simulation. A separately constructed and geometrically optimized AuI(I2) complex at DFT level offers highly analogous results. Both NBO charges and atom-atom distances match the end product of the MD simulation quite well (Au, I and I2: +0.29, −0.44, and close to zero in the simulation, +0.34, −0.40 and close to zero in the optimized structure; Au–I, Au–I2 and I–I: 2.589, 2.615 and 2.897 Å in the simulation, 2.537, 2.623 and 2.712 Å in the optimized structure). The simulations at higher temperatures display the same outcome, although arriving at the end components after much shorter time of simulation (see Fig. S4 with associated discussion in the ESI†). The rather odd end product of AuI(I2) can most likely be regarded as a product of how the scene of chemistry/composition was set in the simulations; inclusion of further electron donors or Lewis acids, viz. more iodide ions, will produce more common gold-iodide complexes together with more common polyiodide building blocks. The addition of one further iodide ion to the molecular dynamics system results in what can be described a more regular and linear gold(I) complex with the two ligands comprising one iodide ion and the II4− complex. At 600 °C the additional iodide ion and the thermally induced dynamics splits off one I2 unit leaving an AuI(I3) complex behind showing occasional further extrusion of I2. One further iodide ion (two extra in total) in the starting configuration results in two separate entities, one linear AuI2− complex and the quite stable square-planar II4− pentaiodide complex. In this context, it can be mentioned that the only compound we have found in the literature that structurally offers a fragment close to an II4− unit displays a complex one-dimensional structure of the composition [Ag5I6−]n containing rather flat, pentagonal pyramidal hexaiodide units bridged by the Ag+ cations.31 The I–I distances from the central iodine atom in this pentagonal pyramid spans 2.92 to 3.01 Å, but the typically encountered Ag–I distances indicate that the interesting hexaiodide configuration is coincidental, emerging as result of the Ag–I coordination in the complex cage structure.
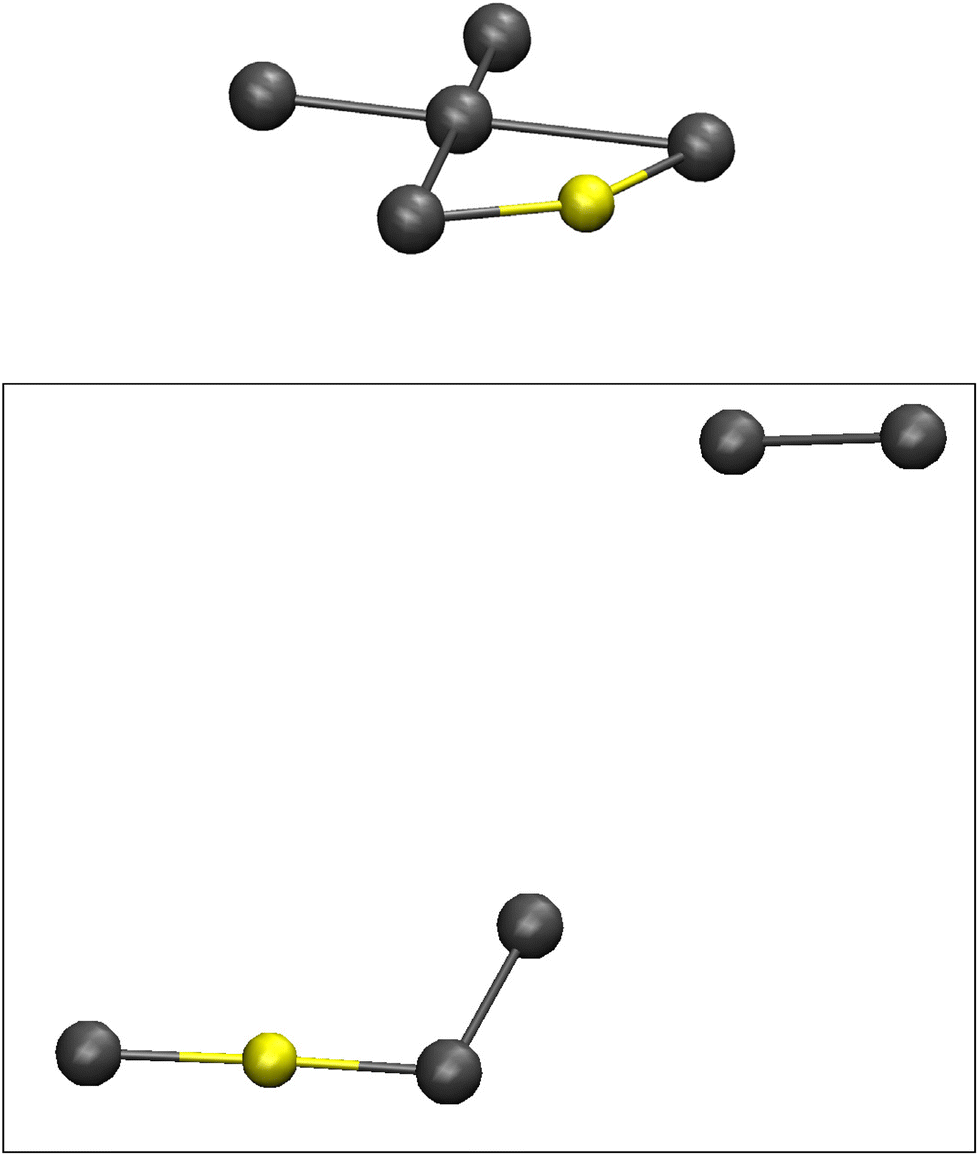 | ||
| Fig. 7 The initial Au-II4− model as obtained from static geometry optimization (top), and a snapshot of the end products after approximately 60 ps simulation; iodine (grey), gold (yellow). | ||
The main conclusion is that gold and iodine forms a platform for much interesting chemistry involving a large flexibility regarding both oxidation states and coordination geometries.
Conclusions
The current study shows that the AuI4−-like coordination complex of II4− indeed represents a local minimum on the pentaiodide PES. It is less stable than the V-shaped form typically found in solid pentaiodide compounds and should therefore be regarded as meta-stable. No straightforward pathway of transformation between the square-planar and V-shaped forms could be identified. However, the inclusion of attached cations to the square-planar II4− complex triggers decomposition into smaller polyiodide building blocks. This may hint to why the coordination compound version of the pentaiodide to our knowledge so far has not been identified in solid polyiodide compounds. The intrinsic stability of the square-planar II4− version suggests that it should be possible to isolate also this form of the pentaiodide system providing that proper synthetic conditions are applied. Two synthetic strategies appear most straightforward: either using rather large and weakly coordinating organic cations or alternatively (Fig. 7) smaller polarizing cations in combination with a strongly solvating, polar solvent.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The current work was funded by a grant from the Swedish Research Council (2020-06701).References
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS PubMed.
- L. Kloo, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, ch. 1.08, vol. 1, pp. 233–249 Search PubMed.
- J. H. Stenlid and T. Brinck, J. Am. Chem. Soc., 2017, 139, 11012–11015 CrossRef CAS PubMed.
- L. Kloo, J. Rosdahl and P. H. Svensson, Eur. J. Inorg. Chem., 2002, 1203–1209 CrossRef CAS.
- T. A. Shestimerova, A. V. Bykov, A. N. Kuznetsov, A. Y. Grishko, Z. Wei, E. V. Dikarev and A. V. Shevelkov, Z. Anorg. Allg. Chem., 2022, e202200039, 1–7 Search PubMed.
- Cambridge Structural Database (CSD, https://www.ccdc.cam.ac.uk/).
- S. Yu and J. S. Ward, Dalton Trans., 2022, 51, 4668–4674 RSC.
- J. Linnera, S. I. Ivlev, F. Kraus and A. J. Karttunen, Z. Anorg. Allg. Chem., 2019, 645, 284–291 CrossRef CAS.
- W.-L. Li, H.-T. Liu, T. Jian, G. V. Lopez, Z. A. Piazza, D.-L. Huang, T.-T. Chen, J. Su, P. Yang, X. Chen, L.-S. Wang and J. Li, Chem. Sci., 2016, 7, 475–481 RSC.
- S. Pohl, W. Saak and B. Krebs, Z. Naturforsch., 1985, 40B, 251–257 CrossRef CAS.
- S. Seidel and K. Seppelt, Science, 2000, 290, 117–118 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 Search PubMed.
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877–13883 CrossRef CAS PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, available from https://www.turbomole.com, 2017, TURBOMOLE V.7.2.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- I. S. Ufimtsev and T. J. Martinez, J. Chem. Theory Comput., 2009, 5, 2619–2628 CrossRef CAS PubMed.
- A. V. Titov, I. S. Ufimtsev, N. Luehr and T. J. Martinez, J. Chem. Theory Comput., 2013, 9, 213–221 CrossRef CAS PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2020, 11, 1–49 Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- L. Martinez, R. Andrade, E. G. Birgin and J. M. Martinez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- T. A. Keith and J. M. Millam, GaussView 6.1, 2016 Search PubMed.
- C. G. Pimentel, J. Chem. Phys., 1951, 19, 446–448 CrossRef.
- R. J. Hach and R. E. Rundle, J. Am. Chem. Soc., 1951, 73, 4321–4324 CrossRef CAS.
- X. Jin, K. Tang, W. Liu and Y. Tang, Heteroat. Chem., 1995, 6, 41–43 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt02501a |
| This journal is © The Royal Society of Chemistry 2022 |