
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterocycle-containing Noyori–Ikariya catalysts for asymmetric transfer hydrogenation of ketones†
Noha
Khamis
 ab,
Guy J.
Clarkson
a and
Martin
Wills
*a
ab,
Guy J.
Clarkson
a and
Martin
Wills
*a
aDepartment of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: m.wills@warwick.ac.uk
bDepartment of Chemistry, Faculty of science, University of Alexandria, Alexandria, Egypt
First published on 19th August 2022
Abstract
The synthesis of a range of N-(heterocyclesulfonyl)-functionalised Noyori–Ikariya catalysts is described. The complexes were prepared through a short sequence from C2-symmetric 1,2-diphenylethylene-1,2-diamine (DPEN) and were characterised by a range of methods including X-ray crystallography. The complexes were active catalysts for the asymmetric transfer hydrogenation (ATH) of a range of acetophenone derivatives, giving products of high ee in most cases, with notably good results for ortho-substituted acetophenones.
Introduction
Asymmetric transfer hydrogenation (ATH) of ketones and imines can be achieved in high enantiomeric excess (ee) using [(arene)Ru(L)Cl] complexes (Noyori–Ikariya catalysts).1–4 The bidentate ligand L is a homochiral, C2-symmetric 1,2-diamine in which one of the amines has been sulfonylated and in most cases the ligand employed is N-tosyl-1,2-diphenylethylene-1,2-diamine (TsDPEN), as illustrated in (R,R,SRu)-1, the favoured diastereoisomer.2 Under the reaction conditions, where typically either iPrOH or formic acid is used as the hydrogen source, the complex is converted via an unsaturated intermediate to hydride 2, in the same favoured diastereoisomeric form.3 Transfer of hydrogen from 2 to the substrate in a well-defined diastereoselective manner results in asymmetric reduction of the substrate with a predictable stereochemical outcome (Fig. 1).4 | ||
| Fig. 1 (a) Noyori–Ikariya catalyst (R,R,SRu)-1 (containing (R,R)-TsDPEN ligand and p-cymene arene), (b) hydride (R,R,SRu)-2 is formed in a diastereoselective manner from (R,R,SRu)-1, (c) asymmetric transfer hydrogenation (ATH) of ketones using Noyori–Ikariya catalysts. (d) stereochemical mode of hydride transfer from (R,R,SRu)-2 to an acetophenone derivative. | ||
The reactivity of catalysts of this type can be moderated through a number of modifications (Fig. 2, example complexes 3–10), including; to the diamine ligand,5 η6-arene6 and through an intramolecular link from the η6-arene to the diamine.7 Substitution of the non-tosylated amine is also tolerated.8 The ligand can also be modified at the sulfonamide with functional groups that can, for example, improve the solubility of the catalysts in water.9 Although there are many other reported modifications to the sulfonamide component,10 we were aware of very few examples of the replacement of the tosyl group with a heterocyclic sulfonamide, a modification which could potentially alter the reactivity and selectivity of the catalysts. There have been multiple reports of the application of piperidine and pyrrolidine-containing catalysts such as 8 to ATH,11 including tethered derivatives such as 9.7 An imidazolium derivative 10, has been applied to ketone ATH in ionic liquids,12 and the use of a DPEN derivative containing a quinoline ring (also described below) has been reported in the ATH of acetophenone and propiophenone, using [(p-cymene)RuCl2]2 as the metal source.13 Apart from moderating the selectivity and activity of the catalysts, heterocyclic groups may also help to facilitate the recovery of the catalysts after use, and improve their solubility in specific solvents, including water, as has been the case with other modifications.5–10
 | ||
| Fig. 2 (a) Examples of modifications to Noyori–Ikariya catalysts; modification to the DPEN component; modification of the η6-arene, substitution of the sulfonamide component; introduction of a link from the diamine to the η6-arene; additions to the non-sulfonylated nitrogen; changes to the sulfonamide component. (b) Catalysts containing heterocyclic rings in the sulfonamide unit. In all cases, the relative configuration has either been determined by X-ray crystallography or assigned by analogy as (R,R,SRu). | ||
Given their potential value, we have designed and prepared a range of heterocycle-containing Noyori–Ikariya complexes and tested them in the ATH of ketones.
Results and discussion
A series of heterocycle-containing complexes were prepared from 1,2-diphenylethene-1-2-diamine (DPEN). With aim of preparing a diverse series, the planned heterocycles included thiophene, benzothiophene, thioazole, quinoline, pyridine and furan. The first stage was formation of the ligands through direct substitution with the appropriate sulfonyl chloride to generate 11a–11d, 11f and 11g, where 11d is the previously reported quinoline derivative.13 This was then followed by complexation with [(p-cymene)RuCl2]2 dimer following the reported procedure3a for Noyori–Ikariya complex 1 (Fig. 3) to form the required complexes 12a–c, 12e–g. Some complexes were prepared from (R,R)-DPEN and others from its enantiomer, as indicated. Although the series included the reported quinoline 11d, the other ligands and complexes are novel. | ||
| Fig. 3 Preparation of [(p-cymene)Ru(HetSO2DPEN)Cl] complexes. (i) DPEN, HetSO2Cl, Et3N, THF, 0 °C. (ii) [(cymene)RuCl2]2, Et3N, iPrOH, 80 °C, 1 h. | ||
The novel complexes were formed in good yields and could be purified by column chromatography, reflecting their stability. The complexes were characterised by NMR spectroscopy, IR and MS and the X-ray crystal structures of five of the complexes were also obtained (Fig. 4, ESI†). In four cases (12a–c, 12g), the complexes possessed a structure analogous to the TsDPEN-derived examples, where the relative configuration at ruthenium to that of the diamine ligand is (R,R,SRu) or (S,S,RRu). In the case of the quinoline complex, however, the reduction of the heterocyclic ring of the ligand 11d was observed under our reaction conditions to give the tetrahydroquinoline complex 12e as the isolated product, quinoline reductions under ATH conditions have been reported,14 therefore this appears to be an example of self-catalysis of reduction by the catalyst as it is formed in the reaction, the hydrogen presumably coming from the isopropanol solvent. Additionally, complex 12e was found to have with the opposite relative stereochemistry at Ru relative to the ligand to what would be expected (i.e. (S,S,SRu). Complex 12e also exhibits an unusual H-bond from the NH of the isoquinoline ring to the N atom adjacent to the sulfonyl group (the total N–H–N bond length, is 3.096(8) Å). This may be the reason for the change in relative configuration for this complex, i.e. through stabilisation of the observed configuration. It should be noted that, in each case, the heterocycles are stacked against a DPEN phenyl group to maximize dispersion stabilization.
 | ||
| Fig. 4 X-ray crystal structures of five of the novel complexes prepared in this work. | ||
A comparison of key bond lengths and angles around the metal centre (Table 1, ESI†) otherwise showed only small differences between each structure. For the complexes of ‘conventional’ relative stereochemistry, the bond angles and lengths were very similar. In contrast, for the ‘inverted’ diastereoisomer (S,S,SRu)-12e, the dimensions were slightly different, presumably due to the alternative relative positioning of groups in the complex.
| (S,S,RRu)-12a | (R,R,SRu)-12b | (R,R,SRu)-12c | (S,S,SRu)-12e | (S,S,RRu)-12g | |
|---|---|---|---|---|---|
| Ru–Cl/Å | 2.4292 (10) | 2.4493 (13) | 2.4171 (15) | 2.4464 (16) | 2.4312 (7) |
| Ru-NH2/Å | 2.117 (3) | 2.127 (4) | 2.113 (4) | 2.134 (5) | 2.117 (2) |
| Ru-NTs/Å | 2.152 (3) | 2.130 (4) | 2.151 (4) | 2.163 (5) | 2.157 (2) |
| Cl–Ru-NTs/° | 87.68 (10) | 86.98 (13) | 88.01 (11) | 85.36 (14) | 88.01 (7) |
| Cl–Ru-NH2/° | 82.20 (11) | 82.37 (13) | 83.40 (13) | 87.09 (15) | 82.30 (8) |
| NH2-Ru-NTs/° | 79.01 (13) | 77.73 (16) | 78.36 (16) | 77.68 (19) | 78.89 (9) |
| NTs-S-CHet/° | 108.1 (2) | 107.5 (2) | 107.4 (2) | 106.7 (3) | 108.37 (14) |
Complex 12a was first tested in the ATH of acetophenone under a variety of conditions (Table 2). In previous studies, we have typically used a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 azeotrope of formic acid/triethylamine (FA/TEA) either neat or with a cosolvent, at rt. Under these conditions, the cosolvent-free reduction gave a product of ca. 97% ee, comparable to the result with catalyst 1,2 although the use of DCM cosolvent resulted in a slightly faster reaction and marginally higher product ee. The reduction product was also formed in high ee using MeCN and MeOH as cosolvents. With DCM cosolvent, the reaction was completed more rapidly at 40 °C and after a much shorter time at 60 °C although with decreased ees of 96.0% and 91.2% respectively. The complex was also capable of catalysing ATH in water, although the activity was slightly reduced. Variation of the FA/TEA ratio (known to influence catalyst activity in some cases)14d,15 did not significantly alter the reaction in this case, however.
:2 azeotrope of formic acid/triethylamine (FA/TEA) either neat or with a cosolvent, at rt. Under these conditions, the cosolvent-free reduction gave a product of ca. 97% ee, comparable to the result with catalyst 1,2 although the use of DCM cosolvent resulted in a slightly faster reaction and marginally higher product ee. The reduction product was also formed in high ee using MeCN and MeOH as cosolvents. With DCM cosolvent, the reaction was completed more rapidly at 40 °C and after a much shorter time at 60 °C although with decreased ees of 96.0% and 91.2% respectively. The complex was also capable of catalysing ATH in water, although the activity was slightly reduced. Variation of the FA/TEA ratio (known to influence catalyst activity in some cases)14d,15 did not significantly alter the reaction in this case, however.
a
| Cat. | Co-solvent | t/h | FA:TEA |
Conv./% | Ee/% | R/S |
|---|---|---|---|---|---|---|
|
a Conditions; Fig. 1c summarises the transformation, 1 mmol ketone, 1% catalyst, 0.5 mL FA:TEA, 0.5 mL cosolvent where indicated, rt (ca. 20 °C) unless otherwise indicated, S = [1.0], followed by chiral GC.
b Run at 40 °C.
c Run at 60 °C, without DCM.
|
||||||
| (S,S,RRu)-12a | DCM | 22 | 5:2 |
99 | 97.8 | S |
| (S,S,RRu)-12a | None | 56 | 5:2 |
97 | 96.6 | S |
| (S,S,RRu)-12a | MeCN | 61 | 5:2 |
98 | 96.2 | S |
| (S,S,RRu)-12a | MeOH | 35 | 5:2 |
99 | 96.6 | S |
| (S,S,RRu)-12a | DCMb | 16 | 5:2 |
99 | 96.0 | S |
| (S,S,RRu)-12a | —c | 6 | 5:2 |
97 | 91.2 | S |
| (S,S,RRu)-12a | H2O | 56 | 5:2 |
66 | 95.4 | S |
| (S,S,RRu)-12a | DCM | 26 | 1.2:1 |
99 | 96.6 | S |
| (S,S,RRu)-12a | DCM | 26 | 0.2:1 |
97 | 96.4 | S |
| (S,S,RRu)-1 | DCM | 40 | 5:2 |
97 | 96.8 | S |
The ATH of acetophenone using each of the catalysts was followed both by 1H NMR and by sampling from an ongoing reaction followed by analysis by chiral GC (ESI,†Table 3). The NMR experiment allowed a comparison of the relative activities of each catalyst to be obtained, whilst the chiral GC reaction provided information about ee variation over time. In the NMR experiments, a short induction period was observed, which may reflect the formation of a ruthenium hydride at the start of the reaction. The most active catalysts were the furan 12g and thiophene 12a, whilst the tetrahydroisoquinoline complex 12e was the least active. The reaction rate of the furan complex 12a was faster than the Noyori–Ikariya catalyst 1 by a factor of 1.77, while the tetrahydroquinoline complex 12e is slower by a factor of 0.5 than 1 under the conditions that we tested. Whilst it is difficult to make accurate comparisons, the furan and thiophene heterocycles may be slightly more active due to the smaller size of these groups. Alternatively, subtle changes to the electronic structure or conformations of the catalysts may be responsible. The tetrahydroisoquinoline complex 12e may be less active because a proportion of the catalyst is unavailable due to stabilisation of the (presumably inactive) intramolecularly H-bond form observed in the crystal structure. However, further studies are required in order to identify the reasons for observed the rate variations.
a
| Substrate | Catalyst | t/h | Conv./% | Yield/% | ee/% | R/S |
|---|---|---|---|---|---|---|
|
a Conditions; 1 mmol ketone, 1% catalyst, 0.5 mL FA:TEA, 0.5 mL DCM, S = [1], rt. Product configurations assigned by comparison to the literature data. Conv. = conversion by GC, yield = isolated product yield. ee determined by chiral GC. Where >99% ee is shown, a minor HPLC peak was not observed.
|
||||||
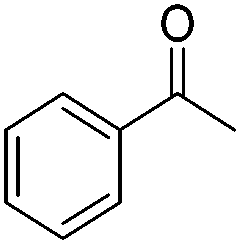
|
(S,S,RRu)-12a | 22 | 100 | 66 | 97.8 | S |
| (R,R,SRu)-12b | 22 | 98 | 91 | >99 | R | |
| (R,R,SRu)-12c | 27 | 99 | 73 | 98.6 | R | |
| (S,S,SRu)-12e | 49 | 100 | 77 | 96.8 | S | |
| (R,R,SRu)-12f | 46 | 98 | 88 | >99 | R | |
| (S,S,RRu)-12g | 46 | 99 | 91 | 96.6 | S | |

|
(S,S,RRu)-12a | 40 | 100 | 72 | 94.0 | S |
| (R,R,SRu)-12b | 26 | 88 | 70 | >99 | R | |
| (R,R,SRu)-12c | 26 | 89 | 79 | >99 | R | |
| (S,S,SRu)-12e | 91 | 99 | 79 | 97.4 | S | |
| (R,R,SRu)-12f | 47 | 99 | 92 | >99 | R | |
| (S,S,RRu)-12g | 48 | 99 | 93 | 91.2 | S | |

|
(S,S,RRu)-12a | 48 | 100 | 76 | 95.8 | S |
| (R,R,SRu)-12b | 21 | 100 | 72 | 96.2 | R | |
| (R,R,SRu)-12c | 27 | 100 | 72 | 96.8 | R | |
| (S,S,SRu)-12e | 161 | 100 | 80 | 87.4 | S | |
| (R,R,SRu)-12f | 165 | 100 | 93 | 97.2 | R | |
| (S,S,RRu)-12g | 23 | 100 | 88 | 96.2 | S | |
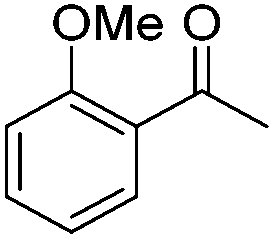
|
(S,S,RRu)-12a | 97 | 99 | 64 | 91.6 | S |
| (R,R,SRu)-12b | 111 | 99 | 71 | 92.0 | R | |
| (R,R,SRu)-12c | 96 | 100 | 73 | 96.4 | R | |
| (S,S,SRu)-12e | 183 | 30 | - | 89.4 | S | |
| (R,R,SRu)-12f | 165 | 99 | 81 | 95.0 | R | |
| (S,S,RRu)-12g | 96 | 98 | 71 | 91.2 | S | |

|
(S,S,RRu)-12a | 167 | 97 | 89 | 94.2 | S |
| (R,R,SRu)-12b | 142 | 98 | 91 | 98.3 | R | |
| (R,R,SRu)-12c | 142 | 99 | 81 | 96.7 | R | |
| (S,S,SRu)-12e | 167 | 51 | 44 | 98.9 | S | |
| (R,R,SRu)-12f | 165 | 98 | 95 | 97.2 | R | |
| (S,S,RRu)-12g | 187 | 96 | 82 | 93.6 | S | |

|
(S,S,RRu)-12a | 120 | 99 | 46 | 97.6 | S |
| (R,R,SRu)-12b | 120 | 99 | 41 | >99 | R | |
| (R,R,SRu)-12c | 120 | 99 | 59 | >99 | R | |
| (S,S,SRu)-12e | 145 | 97 | 65 | 97.6 | S | |
| (R,R,SRu)-12f | 165 | 100 | 97 | >99 | R | |
| (S,S,RRu)-12g | 96 | 98 | 42 | 98.4 | S | |

|
(S,S,RRu)-12a | 26 | 93 | 70 | 99.4 | S |
| (R,R,SRu)-12b | 26 | 99 | 72 | >99 | R | |
| (R,R,SRu)-12c | 26 | 99 | 72 | >99 | R | |
| (S,S,SRu)-12e | 26 | 99 | 85 | 99.0 | S | |
| (R,R,SRu)-12f | 22 | 93 | 70 | >99 | R | |
| (S,S,RRu)-12g | 23 | 98 | 81 | 96.4 | S | |

|
(S,S,RRu)-12a | 27 | 98 | 97 | 99.1 | S |
| (R,R,SRu)-12b | 27 | 99 | 91 | >99 | R | |
| (R,R,SRu)-12c | 27 | 99 | 93 | >99 | R | |
| (S,S,SRu)-12e | 27 | 100 | 90 | 99.3 | S | |
| (R,R,SRu)-12f | 22 | 99 | 82 | >99 | R | |
| (S,S,RRu)-12g | 23 | 99 | 98 | >99 | S | |

|
(S,S,RRu)-12a | 111 | 99 | 71 | >99 | S |
| (R,R,SRu)-12b | 111 | 99 | 82 | 92.8 | R | |
| (R,R,SRu)-12c | 160 | 99 | 82 | >99 | R | |
| (S,S,SRu)-12e | 160 | 99 | 82 | 99.0 | S | |
| (R,R,SRu)-12f | 22 | 99 | 88 | >99 | R | |
| (S,S,RRu)-12g | 48 | 99 | 87 | 99.6 | S | |

|
(S,S,RRu)-12a | 15 | 100 | 82 | 99.2 | R |
| (R,R,SRu)-12b | 15 | 100 | 74.6 | 98.9 | S | |
| (R,R,SRu)-12c | 15 | 100 | 73 | >99 | S | |
| (S,S,SRu)-12e | 15 | 100 | 86 | >99 | R | |
| (R,R,SRu)-12f | 22 | 100 | 93 | 98.4 | S | |
| (S,S,RRu)-12g | 15 | 100 | 88 | 95.8 | R | |

|
(S,S,RRu)-12a | 23 | 99 | 91 | 96.8 | R |
| (R,R,SRu)-12b | 23 | 99 | 84 | 94.6 | S | |
| (R,R,SRu)-12c | 23 | 99 | 84 | 98.1 | S | |
| (S,S,SRu)-12e | 23 | 98 | 87 | 97.2 | R | |
| (R,R,SRu)-12f | 22 | 99 | 93 | 96.2 | S | |
| (S,S,RRu)-12g | 23 | 99 | 87 | 94.6 | R | |

|
(S,S,RRu)-12a | 15 | 100 | 68 | 8.0 | R |
| (R,R,SRu)-12b | 15 | 100 | 69 | 20.2 | S | |
| (R,R,SRu)-12c | 15 | 100 | 74 | 33.8 | S | |
| (S,S,SRu)-12e | 15 | 100 | 78 | 9.8 | R | |
| (R,R,SRu)-12f | 22 | 100 | 82 | 35.2 | S | |
| (S,S,RRu)-12g | 15 | 100 | 76 | 1.2 | R | |
The enantiomeric excesses remained relative constant throughout the reductions for each catalyst. In the reported application of ligand 11d,13a through in situ formation of the catalyst, a combination of iPrOH and KOH was used as the reaction medium and reducing agent, and a product of 92.1% ee was obtained, which is similar to our result for complex 12e. Under the iPrOH/KOH conditions, the quinoline ring may have remained unreduced, or 12e may have been formed in situ.
The new complexes proved to be effective in the ATH of a range of ketone substrates (Table 3). In many cases, the results were excellent, with products of high ee formed. For relatively unhindered ketones such as acetophenone, para-substituted acetophenones, and heterocyclic analogues containing furan and thiophene rings, several of the catalysts gave alcohols in high yields and very high ee. Across this class, catalysts 12b and 12c gave the most consistently high product ee, although other catalysts also performed well. In one case, catalyst 12e gave significantly lower than 100% conversion for electron-rich para-methoxyacetophone. Notably, complex (S,S,SRu)-12a gave the same configuration of alcohol as other complexes derived from (R,R)-DPEN, despite the alternative relative configuration at Ru, indicating a similar mode of hydride transfer (Fig. 1c). Examples of fused-ring ketones, i.e. 4-chromanone and tetralone, were also reduced in very high ee; these compounds are known to be very compatible with ATH using Noyori–Ikariya catalysts.2,5–10,16 In addition, substrates containing substituents at the α-position to the ketone, including chloro and phenoxy, were also compatible substrates. The ATH of the pentafluorophenyl analogue of acetophenone is known to be difficult to achieve in high ee due to the electron-poor nature of the aromatic ring,4d but could be reduced in up to ca. 34% ee by catalyst 12c, which was the best of those tested. It is was also demonstrated that the ATH of ketones can also be achieved through in situ formation of the catalysts; using (R,R)-12f with [(p-cymene)RuCl2]2 in FA/TEA/DCM as before resulted in reduction of acetophenone in equivalent ee to that achieved using the preformed catalyst.
In the case of challenging ortho-substituted acetophenones, specifically ortho-chloro (up to 97.2% ee) and ortho-methoxy acetophenone (up to 96.4% ee), the product ees and rates were high, and better than for several established Noyori–Ikariya ATH catalysts which have commonly been employed, including tethered derivatives.7 Under the conditions in Table 3, ortho-methoxyacetophenone was reduced in 91.4% ee but at only 80.4% conversion after 170 h using catalyst (R,R)-1. Tethered catalyst (R,R)-6 is reported to give a product of 70% ee for the same substrate,17 although a derivative containing a OMe group on the η6-arene ring gave a product of 96% ee.18 Since ortho-phenyl substituted ketones were found to be particularly good substrates, other ketones with ortho-substituents were investigated with two of the most effective catalysts and these also gave products of high ee, in high conversions and good yields (Fig. 5), underlining the versatility of the catalysts, and opening possibilities for the asymmetric synthesis of otherwise challenging products of this type. The ATH reaction of (ortho-OiPr)acetophenone was successful but the enantiomers could not be resolved by chiral GC or HPLC.
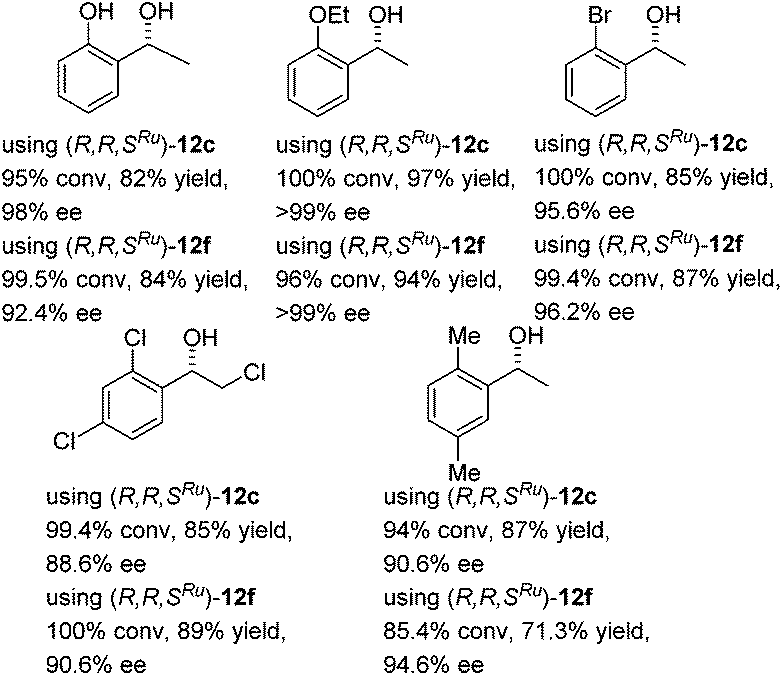 | ||
| Fig. 5 Further ortho-substituted products of ATH using the thioazole (12c) and 3-pyridyl (12f) catalysts. Configurations assigned by analogy with 2-methoxyacetophenone reduction. | ||
Conclusions
In conclusion, we report the results of the first comprehensive study of the preparation and applications to ATH of ketones of a series of DPEN-derived catalysts containing heterocyclic sulfonamide groups. The catalysts proved to be robust, readily characterised and highly active in the ATH of a series of ketones, giving alcohols in high conversion and enantiopurity. In some cases, the observed product ees exceeded those for established catalysts of this class. Although each catalyst generated an ATH product of similar ee in most cases, several variations were noted, suggesting the involvement of secondary directing effects. Although the precise nature of these effects cannot be fully identified at this point, these and further applications remain the subject of ongoing studies.Data availability
The research data (and/or materials) supporting this publication can be accessed at https://wrap.warwick.ac.uk/.Author contributions
The manuscript was written through contributions of all authors. MW planned the investigation, provided supervision, analyzed the data and wrote the manuscript. NK planned the investigation, carried out the practical work, analyzed the data and wrote the manuscript. GJC carried out the X-ray crystal structure analyses and provided the data for the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministry of Higher Education of the Arab Republic of Egypt for support of NK through a full funded Newton-Mosharafa scholarship [NMM4/20]. The X-ray diffraction instrument was obtained through the Science City Project with support from the AWM and part funded by the ERDF.References
- Reviews: (a) D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed; (b) A. E. Cotman, Escaping from Flatland: Stereoconvergent Synthesis of Three-Dimensional Scaffolds via Ruthenium(II)-Catalyzed Noyori–Ikariya Transfer Hydrogenation, Chem. – Eur. J., 2021, 27, 39–53 CrossRef CAS PubMed.
- Early publications: (a) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid-triethylamine mixture, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS; (b) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS; (c) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, Asymmetric transfer hydrogenation of alpha,beta-acetylenic ketones, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS.
- Structural studies: (a) K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, The Catalyst Precursor, Catalyst, and Intermediate in the RuII-Promoted Asymmetric Hydrogen Transfer between Alcohols and Ketones, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS; (b) A. M. R. Hall, P. Dong, A. Codina, J. P. Lowe and U. Hintermair, Kinetics of Asymmetric Transfer Hydrogenation, Catalyst Deactivation, and Inhibition with Noyori Complexes as Revealed by Real-Time High-Resolution FlowNMR. Spectroscopy, ACS Catal., 2019, 9, 2079–2090 CrossRef CAS.
- Mechanistic studies: (a) M. Yamakawa, I. Yamada and R. Noyori, CH/π Attraction: The Origin of Enantioselectivity in Transfer Hydrogenation of Aromatic Carbonyl Compounds Catalyzed by Chiral η6-Arene-Ruthenium(II) Complexes, Angew. Chem., Int. Ed., 2001, 40, 2818–2821 CrossRef CAS; (b) P. A. Dub and J. C. Gordon, The mechanism of enantioselective ketone reduction with Noyori and Noyori–Ikariya bifunctional catalysts, Dalton Trans., 2016, 45, 6756–6781 RSC; (c) P. A. Dub and J. C. Gordon, Metal–Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS; (d) P. A. Dub, N. V. Tkachenko, V. K. Vyas, M. Wills, J. S. Smith and S. Tretiak, Enantioselectivity in the Noyori-Ikariya Asymmetric Transfer Hydrogenation of Ketones, Organometallics, 2021, 40, 1402–1410 CrossRef CAS; (e) A. M. R. Hall, D. B. G. Berry, J. N. Crossley, A. Codina, I. Clegg, J. P. Lowe, A. Buchard and U. Hintermair, Does the Configuration at the Metal Matter in Noyori–Ikariya Type Asymmetric Transfer Hydrogenation Catalysts?, ACS Catal., 2021, 11, 13649–13659 CrossRef CAS PubMed.
- Modifications of diamine phenyl rings: (a) B. Zhang, M.-H. Xu and G.-Q. Lin, Catalytic Enantioselective Synthesis of Chiral Phthalides by Efficient Reductive Cyclization of 2-Acylarylcarboxylates under Aqueous Transfer Hydrogenation Conditions, Org. Lett., 2009, 11, 4712–4715 CrossRef CAS PubMed; (b) A. Amano, D. Igarashi and N. Sayo, Preparation of optically active transition metal/diamine complex and process for producing optically active alcohol with the complex, World Intellectual Property Organization, WO2005092830A1, 2005 Search PubMed; (c) D. Cartigny, K. Püntener, T. Ayad, M. Scalone and V. Ratovelomanana-Vidal, Highly Diastereo- and Enantioselective Synthesis of Monodifferentiated syn-1,2-Diol Derivatives through Asymmetric Transfer Hydrogenation via Dynamic Kinetic Resolution, Org. Lett., 2010, 12(17), 3788–3791 CrossRef CAS PubMed.
- Arene modifications: (a) J. Soleimannejad, A. Sisson and C. White, Functionalized-arene ruthenium half-sandwich compounds as enantioselective hydrogen transfer catalysts. Crystal structures of [RuCl{TsNCH(R)CH(R)NH2}(η6-C6H5OCH2CH2OH)] (R=H or Ph), Inorg. Chim. Acta, 2003, 352, 121–128 CrossRef CAS; (b) T. J. Geldbach and P. J. Dyson, A Versatile Ruthenium Precursor for Biphasic Catalysis and Its Application in Ionic Liquid Biphasic Transfer Hydrogenation: Conventional vs Task-Specific Catalysts, J. Am. Chem. Soc., 2004, 126, 8114–8115 CrossRef CAS PubMed; (c) S. Doherty, J. G. Knight, H. Alshaikh, J. Wilson, P. G. Waddell, C. Wills and C. M. Dixon, Arene-Immobilized Ru(II)/TsDPEN Complexes: Synthesis and Applications to the Asymmetric Transfer Hydrogenation of Ketones, Eur. J. Inorg. Chem., 2021, 226–235 CrossRef CAS; (d) M. Ito, Y. Endo and T. Ikariya, Well-Defined Triflylamide-Tethered Arene−Ru(Tsdpen) Complexes for Catalytic Asymmetric Hydrogenation of Ketones, Organometallics, 2008, 27, 6053–6055 CrossRef CAS.
- Tethered catalysts: (a) H. G. Nedden, A. Zanotti-Gerosa and M. Wills, The Development of Phosphine-Free “Tethered” Ruthenium(II) Catalysts for the Asymmetric Reduction of Ketones and Imines, Chem. Rec., 2016, 16, 2623–2643 CrossRef PubMed; (b) V. K. Vyas, G. J. Clarkson and M. Wills, Enantioselective Synthesis of Bicyclopentane-Containing Alcohols via Asymmetric Transfer Hydrogenation, Org. Lett., 2021, 23, 3179–3183 CrossRef CAS PubMed; (c) T. Touge, T. Hakamata, H. Nara, T. Kobayashi, N. Sayo, T. Saito, Y. Kayaki and T. Ikariya, Oxo-Tethered Ruthenium(II) Complex as a Bifunctional Catalyst for Asymmetric Transfer Hydrogenation and H2 Hydrogenation, J. Am. Chem. Soc., 2011, 133, 14960–14963 CrossRef CAS PubMed; (d) V. Parekh, J. A. Ramsden and M. Wills, Ether-tethered Ru(II)/TsDPEN complexes; synthesis and applications to asymmetric transfer hydrogenation, Catal. Sci. Technol., 2012, 2, 406–414 RSC; (e) A. Kišić, M. Stephan and B. Mohar, ansa–Ruthenium(II) Complexes of DPEN–SO2N(Me)(CH2)n(η6−aryl) Conjugate Ligands for Asymmetric Transfer Hydrogenation of Aryl Ketones, Adv. Synth. Catal., 2014, 356, 3193–3198 CrossRef; (f) R. Soni, K. E. Jolley, S. Gosiewska, G. J. Clarkson, Z. Fang, T. H. Hall, B. N. Treloar, R. C. Knighton and M. Wills, Synthesis of Enantiomerically Pure and Racemic Benzyl-Tethered Ru(II)/TsDPEN Complexes by Direct Arene Substitution: Further Complexes and Applications, Organometallics, 2018, 37, 48–64 CrossRef CAS; (g) A. Kisic, M. Stephan and B. Mohar, ansa-Ruthenium(II) Complexes of R2NSO2DPEN-(CH2)n(η2-Aryl) Conjugate Ligands for Asymmetric Transfer Hydrogenation of Aryl Ketones, Adv. Synth. Catal., 2015, 357, 2540–2546 CrossRef CAS.
- Alkylation of non-tosylated amine: (a) J. Barrios-Rivera, Y. Xu and M. Wills, Probing the effects of heterocyclic functionality in [(benzene)Ru(TsDPENR)Cl] catalysts for Asymmetric Transfer Hydrogenation, Org. Lett., 2019, 21, 7223–7227 CrossRef CAS PubMed; (b) J. E. D. Martins, G. J. Clarkson and M. Wills, Ru(II) Complexes of N-Alkylated TsDPEN Ligands in Asymmetric Transfer Hydrogenation of Ketones and Imines, Org. Lett., 2009, 11, 847–850 CrossRef CAS PubMed; (c) W. Shan, F. Meng, Y. Wu, F. Mao and X. Li, The synthesis of a new nitrogen joined N-PEG-TsDPEN ligand and its application in asymmetric transfer hydrogenation of ketones in neat water, J. Organomet. Chem., 2011, 696, 1687–1690 CrossRef CAS; (d) J. M. Zimbron, M. Dauphinais and A. B. Charette, Arylphosphonium Noyori–Ikariya catalyst supported on tetra-arylphosphonium salt for asymmetric transfer hydrogenation in water, Green Chem., 2015, 17, 3255–3259 RSC; (e) N. A. Cortez, C. Z. Flores-Lopez, R. Rodriguez-Apodaca, L. Z. Flores-Lopez, M. Parra-Hake and R. Somanathan, Transfer hydrogenation reduction of acetophenone catalyzed by Ru(II) and Rh(I) complexes with ligands derived from (1R,2R)-cyclohexane-1,2-diamine, ARKIVOC (Gainesville, FL, U. S.), 2005, 6, 162–171 CrossRef.
- Modifications of sulfonamide to improve water solubility: (a) T. Thorpe, J. Blacker, S. M. Brown, C. Bubert, J. Crosby, S. Fitzjohn, J. P. Muxworthy and J. M. J. Williams, Efficient rhodium and iridium-catalyzed asymmetric transfer hydrogenation using water-soluble amino sulfonamide ligands, Tetrahedron Lett., 2001, 42, 4041–4043 CrossRef CAS; (b) C. Bubert, J. Blacker, S. M. Brown, J. Crosby, S. Fitzjohn, J. P. Muxworthy, T. Thorpe and J. M. J. Williams, Synthesis of water-soluble aminosulfonamide ligands and their application in enantioselective transfer hydrogenation, Tetrahedron Lett., 2001, 42, 4037–4039 CrossRef CAS; (c) Z. Zhou, Q. Ma, Y. Sun, A. Zhang and L. Li, Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of aromatic ketones in water using novel water-soluble chiral monosulfonamide ligands, Heteroat. Chem., 2010, 21, 505–514 CrossRef CAS; (d) Z. Zhou and Y. Sun, Water-soluble chiral amino-sulfonamides as ligands for ruthenium(II)-catalyzed asymmetric transfer hydrogenation, Catal. Commun., 2009, 10, 1685–1688 CrossRef CAS.
- Further sulfonamide modifications: (a) D. Sterk, M. S. Stephan and B. Mohar, Transfer hydrogenation of activated ketones using novel chiral Ru(II)-N-arenesulfonyl-1,2-diphenylethylenediamine complexes, Tetrahedron Lett., 2004, 45, 535–537 CrossRef CAS; (b) X. Xu, R. Wang, J. Wan, X. Ma and J. Peng, Phosphonate-containing polystyrene copolymer-supported Ru catalyst for asymmetric transfer hydrogenation in water, RSC Adv., 2013, 3, 6747–6751 RSC; (c) K. M. Steward, M. T. Corbett, C. G. Goodman and J. S. Johnson, Asymmetric Synthesis of Diverse Glycolic Acid Scaffolds via Dynamic Kinetic Resolution of α-Keto Esters, J. Am. Chem. Soc., 2012, 134, 20197–20206 CrossRef CAS PubMed; (d) Z. Zhou and Y. Sun, Synthesis of polyethylene glycol supported chiral monosulfonamide and its application in asymmetric transfer hydrogenation of prochiral ketones, React. Kinet., Mech. Catal., 2010, 99, 391–396 CAS; (e) Y.-C. Chen, T.-F. Wu, L. Jiang, J.-G. Deng, H. Liu, J. Zhu and Y.-Z. Jiang, Synthesis of Dendritic Catalysts and Application in Asymmetric Transfer Hydrogenation, Org. Chem., 2005, 70, 1006–1010 CrossRef CAS PubMed; (f) Y.-C. Chen, T.-F. Wu, J.-G. Deng, H. Liu, X. Cui, J. Zhu, Y.-Z. Jiang, M. C. K. Choi and A. S. C. Chan, Multiple dendritic catalysts for asymmetric transfer hydrogenation, J. Org. Chem., 2002, 67, 5301–5306 CrossRef CAS PubMed; (g) G. Sun, W. Jian, Z. Luo, T. Sun, C. Li, J. Zhang and Z. Wang, Development of an Efficient and Scalable Asymmetric Synthesis of Eliglustat via Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation, Org. Process Res. Dev., 2019, 23, 1204–1212 CrossRef CAS; (h) Z. Luo, G. Sun, Z. Zhou, G. Liu, B. Luan, Y. Lin, L. Zhang and Z. Wang, Stereogenic cis-2-substituted-N-acetyl-3-hydroxy-indolines via ruthenium(II)-catalyzed dynamic kinetic resolution-asymmetric transfer hydrogenation, Chem. Commun., 2018, 54, 13503–13506 RSC; (i) O. Soltani, M. A. Ariger, H. Vazquez-Villa and E. M. Carreira, Transfer hydrogenation in water: enantioselective, catalytic reduction of α-cyano and α-nitro substituted acetophenones, Org. Lett., 2010, 12, 2893–2895 CrossRef CAS PubMed; (j) B. Mohar, A. Valleix, J.-R. Desmurs, M. Felemez, A. Wagner and C. Mioskowski, Highly enantioselective synthesis via dynamic kinetic resolution under transfer hydrogenation using Ru(η6-arene)-N-perfluorosulfonyl-1,2-diamine catalysts: a first insight into the relationship of the ligand's pKa and the catalyst activity, Chem. Commun., 2001, 24, 2572–2573 RSC.
- Dialkylaminosulfonyl, untethered: (a) D. Sterk, M. Stephan and B. Mohar, Highly Enantioselective Transfer Hydrogenation of Fluoroalkyl Ketones, Org. Lett., 2006, 8, 5935–5938 CrossRef CAS PubMed; (b) D. Sterk, M. S. Stephan and B. Mohar, New chiral N-(N,N-dialkylamino)sulfamoyl-1,2-diamine ligands for highly enantioselective transfer hydrogenation of ketones, Tetrahedron: Asymmetry, 2002, 13, 2605–2608 CrossRef CAS; (c) S. Zhang, F. Chen, Y.-M. He and Q.-H. Fan, Asymmetric Hydrogenation of Dibenzo[c,e]azepine Derivatives with Chiral Cationic Ruthenium Diamine Catalysts, Org. Lett., 2019, 21(14), 5538–5541 CrossRef CAS PubMed.
- H. Uchimoto, M. Ikeda, A. Matsushita, T. Shigeta, K. Arimitsu, H. Yasui, T. Tsuji, M. Ozeki, M. Yamashita, K. Nishide and I. Kawasaki, Development of new ligands for the recyclable catalytic asymmetric transfer hydrogenation in ionic liquid, Heterocycles, 2017, 94, 465–483 CrossRef CAS.
- (a) C. E. Dong, J. L. Zhang, W. Z. Zheng, Z. L. Yu and L. F. Zhang, Asymmetric transfer-hydrogenation of ketone catalyzed by novel Ru(II)-chiral sulfonamide complex, Chin. Chem. Lett., 2000, 11, 383–384 CAS; (b) J. Zhu, L. Zhu, Y. Wu, L. Cheng, H. Wang, X. Sun, J. Shen, Y. Zhou and Y. Ke, A novel C2 symmetric chiral stationary phase with N-[(4-Methylphenyl)sulfonyl]-L-leucine as chiral side chains, J. Sep. Sci., 2020, 43, 2338–2348 CrossRef CAS PubMed.
- (a) Y.-E. Luo, Y.-M. He and Q.-H. Fan, Asymmetric Hydrogenation of Quinoline Derivatives Catalyzed by Cationic Transition Metal Complexes of Chiral Diamine Ligands: Scope, Mechanism and Catalyst Recycling, Chem. Rec., 2016, 16, 2697–2711 CrossRef PubMed; (b) T. Wang, L.-G. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q.-H. Fan, J. Xiang, Z.-X. Yu and A. S. C. Chan, Highly Enantioselective Hydrogenation of Quinolines Using Phosphine-Free Chiral Cationic Ruthenium Catalysts: Scope, Mechanism, and Origin of Enantioselectivity, J. Am. Chem. Soc., 2011, 133(25), 9878–9891 CrossRef CAS PubMed; (c) V. Parekh, J. A. Ramsden and M. Wills, Asymmetric transfer hydrogenation of quinolines using tethered Ru(II) catalysts, Tetrahedron: Asymmetry, 2010, 21, 1549–1556 CrossRef CAS; (d) C. Wang, C. Li, X. Wu, A. Pettman and J. Xiao, pH-Regulated Asymmetric Transfer Hydrogenation of Quinolines in Water, Angew. Chem., Int. Ed., 2009, 48, 6524–6528 CrossRef CAS PubMed; (e) T. Wang, L.-G. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q.-H. Fan, J. Xiang, Z.-X. Yu and A. S. C. Chan, quinolines: Highly Enantioselective Hydrogenation of Quinolines Using Phosphine-Free Chiral Cationic Ruthenium Catalysts: Scope, Mechanism, and Origin of Enantioselectivity, J. Am. Chem. Soc., 2011, 133, 9878–9891 CrossRef CAS PubMed.
- (a) X. Wu, X. Li, F. King and J. Xiao, Insight into and Practical Application of pH-Controlled Asymmetric Transfer Hydrogenation of Aromatic Ketones in Water, Angew. Chem., Int. Ed., 2005, 44, 3407–3411 CrossRef CAS PubMed; (b) X. Zhou, X. Wu, B. Yang and J. Xiao, Varying the ratio of formic acid to triethylamine impacts on asymmetric transfer hydrogenation of ketones, J. Mol. Catal. A: Chem., 2012, 357, 133–140 CrossRef CAS.
-
(a) A. E. Cotman, M. Lozinšek, B. Wang, M. Stephan and B. Mohar,
trans-Diastereoselective Ru(II)-Catalyzed Asymmetric Transfer Hydrogenation of α-Acetamido Benzocyclic Ketones via Dynamic Kinetic Resolution, Org. Lett., 2019, 21(10), 3644–3648 CrossRef CAS PubMed;
(b) V. K. Vyas and B. M. Bhanage, Kinetic Resolution Driven Diastereo- and Enantioselective Synthesis of cis-β-Heteroaryl Amino Cycloalkanols by Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation, Org. Lett., 2016, 18, 6436–6439 CrossRef CAS PubMed;
(c) G. S. Caleffi, J. de O. C. Brum, A. T. Costa, J. L. O. Domingos and P. R. R. Costa, Asymmetric Transfer Hydrogenation of Arylidene-Substituted Chromanones and Tetralones Catalyzed by Noyori–Ikariya Ru(II) Complexes: One-Pot Reduction of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds, J. Org. Chem., 2021, 86, 4849–4858 CrossRef CAS PubMed;
(d) A. Keßberg, T. Lübken and P. Metz, Enantioselective Total Synthesis of Natural Isoflavans: Asymmetric Transfer Hydrogenation/Deoxygenation of Isoflavanones with Dynamic Kinetic Resolution, Org. Lett., 2018, 20, 3006–3009 CrossRef PubMed;
(e) S. Kwon, S. Lee, M. Heo, B. Lee, X. Fei, T. W. Corson and S.-Y. Seo, Total Synthesis of Naturally Occurring 5,7,8-Trioxygenated Homoisoflavonoids, ACS Omega, 2020, 5, 11043–11057 CrossRef CAS PubMed.
C and CO bonds, J. Org. Chem., 2021, 86, 4849–4858 CrossRef CAS PubMed;
(d) A. Keßberg, T. Lübken and P. Metz, Enantioselective Total Synthesis of Natural Isoflavans: Asymmetric Transfer Hydrogenation/Deoxygenation of Isoflavanones with Dynamic Kinetic Resolution, Org. Lett., 2018, 20, 3006–3009 CrossRef PubMed;
(e) S. Kwon, S. Lee, M. Heo, B. Lee, X. Fei, T. W. Corson and S.-Y. Seo, Total Synthesis of Naturally Occurring 5,7,8-Trioxygenated Homoisoflavonoids, ACS Omega, 2020, 5, 11043–11057 CrossRef CAS PubMed. - D. J. Morris, A. M. Hayes and M. Wills, The ‘reverse-tethered’ ruthenium(II) catalyst for asymmetric transfer hydrogenation: further applications, J. Org. Chem., 2006, 71, 7035–7044 CrossRef CAS PubMed.
- R. Soni, K. E. Jolley, G. J. Clarkson and M. Wills, Direct Formation of Tethered Ru(II) Catalysts Using Arene Exchange, Org. Lett., 2013, 15, 5110–5113 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, chiral HPLC spectra and X-ray crystallographic data for structures. CCDC 2182537–2182541. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02411j |
| This journal is © The Royal Society of Chemistry 2022 |