
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of the magnetic and photophysical properties in 3d–4f and 4f–4f′ heterobimetallic complexes involving a tetrathiafulvalene-based ligand†
Haiet
Douib
ab,
Jessica
Flores Gonzalez
a,
Saskia
Speed
cd,
Vincent
Montigaud
a,
Bertrand
Lefeuvre
a,
Vincent
Dorcet
a,
François
Riobé
 c,
Olivier
Maury
c,
Abdelkrim
Gouasmia
b,
Boris
Le Guennic
a,
Olivier
Cador
*a and
Fabrice
Pointillart
*a
c,
Olivier
Maury
c,
Abdelkrim
Gouasmia
b,
Boris
Le Guennic
a,
Olivier
Cador
*a and
Fabrice
Pointillart
*a
aInstitut des Sciences Chimiques de Rennes UMR 6226 CNRS-UR1, Université de Rennes 1, 35042 Rennes Cedex, France. E-mail: olivier.cador@univ-rennes1.fr; fabrice.pointillart@univ-rennes1.fr
bLaboratoire des Matériaux Organiques et Hétérochimie (LMOH), Département des sciences de la matière, Université Larbi Tébessi de Tébessa, Route de Constantine 12002, Tébessa, Algeria
cENSL, CNRS, Laboratoire de Chimie UMR 5182, 46 allée d'Italie, 69364 Lyon, France
dDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Martí i Franquès 1-11, 08028 Barcelona, Spain
First published on 11th October 2022
Abstract
The reaction between the 2-(1-(2,6-di(pyrazol-1-yl)-4-methylpyridyl)-4,5-(4,5-bis(propylthio)-tetrathiafulvalenyl)-1H-benzimidazol-2-yl)-pyridine ligand (L), 1 equivalent of Ln(hfac)3·2H2O/Dy(tta)3·2H2O (hfac− = 1,1,1,5,5,5-hexafluoroacetylacetonate, tta− = 2-thenoyltrifluoroacetonate) and M(hfac)2·2H2O leads to the formation of heteroleptic 3d–4f dinuclear complexes of formula [MLn(hfac)5(L)]n (M(II) = Cd, Zn, Co, Mn, Ni and Ln(III) = Dy, Yb, Nd) and [ZnDy(tta)2(hfac)3(L)]·(CH2Cl2). Their X-ray structures reveal that the two coordination sites are occupied by one Ln(III) ion and one M(II) transition metal respectively. The M(II) ions are coordinated to the benzoimidazolylpyridine (bzip) moiety in a N2O4 coordination sphere, while the Ln(III) ions are coordinated to the 2,6-di(pyrazol-1-yl)-4-pyridine (dpp) moiety in a N3O6 surrounding. When Dy(III) ion is used a field-induced Single-Molecule Magnet (SMM) behavior is detected with a magnetic relaxation time slightly dependent to the nature of the vicinal divalent transition metal. On the other hand, when the Yb(III) is used, intense, moderated or quenched 2F5/2 → 2F7/2 NIR luminescence is observed when the Yb(III) ion is respectively associated with the Zn(II), Mn(II) and Ni(II)/Co(II) ion. The emission intensity can be modulated in function of the metal-to-ligand charge transfer and d–d transition intensities. The replacement of the divalent transition metal by a trivalent lanthanide leads to the formation of heteroleptic 4f–4f′ dinuclear complexes of formula [Ln2−xLn′x(hfac)6(L)]·a(CH2Cl2)·b(C6H14) and [Dy1.11Nd0.89(tta)3(hfac)3(L)]. The coordination selectivity is based on the radius. Among the 4f–4f′ series, the Dy(III) derivatives displayed such ion in N2O6 eight-coordinated sphere allowing the observation of SMM behavior. The three compounds [Dy1.21Nd0.79(hfac)6(L)]·2(CH2Cl2)·(C6H14), [Yb1.04Nd0.96(hfac)6(L)] and [YbPr(hfac)6(L)] displayed respectively Nd(III), modarated Yb(III) and intense Yb(III) NIR emissions.
Introduction
The great success of the use of lanthanide ions in coordination chemistry and in the design of supramolecular edifices comes from their intrinsic multiple properties i.e. magnetic and optical properties.1,2 On the one hand, the high magnetic moment and strong magnetic anisotropy were exploited in the molecular magnetism field of research to design Single Molecule Magnets (SMMs) and targeted potential applications such as high-density data storage devices, quantum computing or spintronics.3–8 On the other hand, their specific luminescence ranging from the UV-visible to the Near Infrared (NIR) region, make lanthanide-based coordination complexes suitable for a plethora of other applications in the fields of OLEDs,9 time-resolved fluoro-immunoassays,10 biosensors11,12 and time-resolved imaging.13,14For both research fields, a challenge is to find efficient strategies to enhance the performance of these aforementioned properties. In a magnetic point of view, chemists combined 3d and 4f ions in order to combine the advantages of the two ions i.e. strong magnetic anisotropy of the 4f elements with significant magnetic interactions with the paramagnetic 3d elements15–18 and even quenching of the Quantum Tunneling of Magnetization (QTM).19 It is worth to notice that even the association with diamagnetic transition metals could lead to enhancement of the magnetic properties due to its electrostatic effect on the magnetic anisotropy of the 4f elements.20–22 Such electrostatic effect can be induced by trivalent lanthanide ions as demonstrated by M.-L. Tong in 2013.23–25 In other special cases, the 3d site can highlight exciting magnetic properties such as spin-crossover and influence the whole physical properties of the system.26 In an photophysic point of view, the emitting f excited states have very weak absorption coefficients with long lifetimes due to the Laporte rules.27 Thus direct sensitization is not efficient for 4f luminescence especially in diluted solution, and strategies based on the use of organic chromophores, which strongly absorb light in the UV-visible range, have been developed:28 (i) triplet excited-state sensitization, (ii) singlet excited-state with intra-ligand charge transfer (ILCT) which has been intensively used in the case of push–pull ligands such as tetrathiafulvalene (TTF) functionalized derivatives29 (iii) intramolecular f → f energy transfer process30,31 and (iv) induced triplet metal-to-ligand charge transfer (MLCT). As previously listed, one of the strategies to sensitize the luminescence is to inject transition metals in the architecture, for example a diamagnetic Zn(II)-Schiff base unit was used as an organometallic antenna for visible luminescence sensitization,32 while diamagnetic Pt(II) and Ru(II), and even paramagnetic Cr(III) were used to extend the lifetimes of NIR luminescence33 and optimization of molecular light-upconversion.34 On the contrary, paramagnetic transition metals are described as potential quenchers the 4f luminescence.35
Consequently, it is easy to be convinced that d–4f and 4f–4f′ heterobimetallic complexes could be a promising strategy to enhance both magnetic and optical properties but the challenge is to succeed to selectively coordinate the two different metal ions. To reach a perfect control of the step-by-step synthesis of the final targets, the chemists elaborated metallo-ligand approach36–38 and multi-coordination-site ligands. For the second strategy, chemists played with the size, charge, denticity, and hard/soft hetero-atoms of the coordinating units. The most famous examples of such ligands are the pseudo-helicenic ligands based on the tridentate chelating 6-(carbamoyl)pyridine-2-yl-1H-benzimidazole and bidentate chelating pyridine-2-yl-1alkylbenzimidazole moieties developed by the group of C. Piguet,26,39–41 the Schiff Base ligands such as Salicylideneimine derivatives42–47 and the macrocycle derived from 1,4,7,10-tetraazadodecane-1,4,7,10-tetraacetic acid (DOTA) derivated ligands.48 Using the latter family of ligands decorated with a TTF core, some of us succeeded to elaborate 3d–4f heterobimetallic complexes.49,50 Then we developed the synthesis of a TTF-based ligand involving one bis-chelating (bzip = benzimidazol-2-yl-pyridyl) and one tris-chelating (dpp = di-(pyrazol-1-yl)-4-pyridyl) coordination site for the design of homoleptic Dy(III) and Yb(III) complexes which displayed multi-SMM behavior51 and multi-emission52 in a single molecule.
In this article, we go one step forward using the previously mentioned 2-{1-[2,6-di(pyrazol-1-yl)-4-methylpyridyl]-4,5-[4,5-bis(propylthio)-tetrathiafulvalenyl]-1H-benzimidazol-2-yl}pyridine ligand (L) (Scheme 1) to design heteroleptic nd-4f (n = 3, 4) and 4f–4f′ complexes. These complexes have been characterized by X-ray diffraction, absorption spectroscopy and cyclic voltammetry.
 | ||
| Scheme 1 Molecular structure of ligand L highligting the two sites of coordination. | ||
The magnetic and emission properties of the complexes as well as the influence of the two associated metal ions were investigated.
Results and discussion
Synthesis
The L ligand was selected to design heterobimetallic complexes due to its two sites of coordination with different denticities.The first part of this article is focused on the design of 3d–4f heterobimetallic complexes. To do so, the first step consisted in the coordination of the d transition metal (M(hfac)2·2H2O) which must be coordinated to the bidentate chelating site (benzimidazole-2-yl-pyridine, bzip) of L. The second step consisted in the coordination of the 4f element (Ln(hfac)3·2H2O) to the remaining free tridentate chelating site of coordination (2,6-di(pyrazol-1-yl)-4-methylpyridine, dpp). The combination of the coordination number limitation of the d element and the non-lability of such element guarantees a optimal selectivity of the coordination for d and f elements on the bzip and dpp moieties, respectively. Finally, the Dy(hfac)3·2H2O was replaced by the Dy(tta)3·2H2O precursor keeping the same step-by-step protocol in order to evaluate the lability of the ancillary ligands in case of 3d ions. The step-by-step synthetic strategy was selected to be on line with the one used for the f–f′ complexes.
The second part of the article is focused on the design of 4f–4f′ heterobimetallic complexes. To reach this more challenging aim, the preference of the large lanthanide for the dpp site of coordination was demonstrated with the selective coordination of the Pr(III) ion in 14. The coordination selectivity is demonstrated at room temperature while heating the reaction mixture led to the coordination of the two bzip and dpp sites (see ESI† for X-ray structure of [Nd2(hfac)6(L)]·C6H14 (18)). After the selective coordination of the larger lanthanide (Nd(III)) on the dpp site, the smaller lanthanide (Dy(III)) was added for coordination to the bpiz site. The ratio between the two lanthanide ions are determined by Energy Dispersive Spectrometry (EDS) and given values are the average value between at least three measurements (see ESI† for details). This first step by step strategy lead to the formation of the [Dy1.21Nd0.79(hfac)6(L)]·2(CH2Cl2)·(C6H14) (15) compound. In order to increase the discrimination between the two sites, three strategies were used: (i) playing with the size of the ancillary ligand i.e. the Dy(hfac)3·2H2O was replaced by Dy(tta)3·2H2O, (ii) using a smaller lanthanide than Dy(III), i.e. the Dy(hfac)3·2H2O, was replaced by Yb(hfac)3·2H2O and (iii) using simultaneously a smaller lanthanide than Dy(III) (i.e. Yb(III)) and a bigger lanthanide than Nd(III) (i.e. Pr(III)). From isolated single crystals, one could notice that the first strategy led to the formation of the [Dy1.11Nd0.89(tta)3(hfac)3(L)] (16) compound while the second strategy led to the [Yb1.04Nd0.96(hfac)6(L)] (17) compound, and finally the third strategy lead to [YbPr(hfac)6(L)] (19). It is worth to notice that the latter compound can also be obtained starting from complex 14. The three strategies have been a success increasing the discrimination.
Crystal structure analysis
The whole series of compounds 1–10 possesses a similar molecular structure, the differences come from the crystallization space group which depends on the number and the nature of the interstitial solvent molecules. The latter ones depend of the size of the 3d and 4f metal ions. Consequently a general description of the structure is given for the whole series and only the significant structural changes are point out. | ||
| Fig. 1 Representative molecular structure of the series of compounds 1–10 (M = Cd, Zn, Mn, Co and Ni; Ln = Dy and Yb). Hydrogen atoms and solvent molecules of crystallization are omitted for clarity. | ||
The M1 ion is surrounded by four oxygen atoms that belong to two hfac− ligands and two nitrogen atoms from the bzip coordination site of L. The average Ln–O distances are shorter than the average Ln–N distances (Table S4†) due to the oxophilic character of the lanthanide, as usually observed. At the transition metal level, the difference of bond lengths between M–O and M–N are not as significant as for the lanthanide ions (Table S4†). The arrangement of the ligands around the lanthanide ions leads to a spherical tricapped trigonal prism (D3h symmetry) and spherical capped square antiprism (D2d symmetry) as coordination polyhedra (Table S5†). The distortion is visualized by continuous shape measures performed with SHAPE 2.1 (Table S5†).53 The central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths of the TTF core confirm the neutral form of L in all the compounds 1–10.
C bond lengths of the TTF core confirm the neutral form of L in all the compounds 1–10.
The crystal packing reveals the formation of head-to-tail dimers of L (called Donor–Acceptor dimers, D–A). Then each D–A dimer forms a column of L along the a axis thanks to Donor–Donor (D–D) interaction through S⋯S short contacts (Fig. 2 and S6–S9, Table S6†). Finally each column is separated by the Ln(hfac)3 and M(hfac)2 units along the b and c axis, respectively. The shortest intermolecular Ln–Ln distances as well as the M–Ln inter- and intra-molecular distances are listed in Table S6.† It is worth to notice that the most important structural differences in this series are observed in the case of 1. In fact one of the ligands is clearly more distorted with an α value lower than for the other 3d4f hetero-bimetallic compounds and a stronger overlap between the head-to-tail π systems of L allowing the formation of short S11⋯O2 contacts (Table S6†).
 | ||
| Fig. 2 Representative crystal packing of the series of compounds 1–10. The {4,5-[4,5-bis(propylthio)-tetrathiafulvalenyl]-1H-benzimidazol-2-yl}pyridine fragment is shown in spacefill representation while the dpp moiety and the organometallic precursors M(hfac)2 and Ln(hfac)3 are shown in ball and sticks representation. | ||
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) triclinic space group (Table S1†). The asymmetric unit is composed by one molecule of [ZnDy(tta)2(hfac)3(L)] and one crystallization molecule of dichloromethane. An Ortep view of 11 is depiced in Fig. S10.† The X-ray structures revealed that the Zn(II) and Dy(III) ions are respectively coordinated to the bzip and dpp fragments as expected. Nevertheless the starting nature of the precursors i.e. Zn(hfac)2 and Dy(tta)3 do not remain intact. In fact a reorganisation of the β-diketonate anions is observed so that the steric hindrance around the metal is optimal.
(no. 2) triclinic space group (Table S1†). The asymmetric unit is composed by one molecule of [ZnDy(tta)2(hfac)3(L)] and one crystallization molecule of dichloromethane. An Ortep view of 11 is depiced in Fig. S10.† The X-ray structures revealed that the Zn(II) and Dy(III) ions are respectively coordinated to the bzip and dpp fragments as expected. Nevertheless the starting nature of the precursors i.e. Zn(hfac)2 and Dy(tta)3 do not remain intact. In fact a reorganisation of the β-diketonate anions is observed so that the steric hindrance around the metal is optimal.
Consequently, the comproportionation reaction leads to the formation of Zn(hfac)(tta) and Dy(hfac)2(tta) units that are coordinated to the bzip and dpp sites (Fig. 3). Such reorganisation was already observed for the design of homo-metallic lanthanide complexes involving L starting from the Dy(hfac)3 and Dy(tta)3 precursors.51
 | ||
| Fig. 3 Molecular structure of 11. Hydrogen atoms and solvent molecules of crystallization are omitted for clarity. | ||
The Zn(II) and Ln(III) ions remain in N2O4 and N3O6 environment. The arrangement of the ligands around the lanthanide ion leads to a spherical tricapped trigonal prism (D3h symmetry) as coordination polyhedra (Table S5†).53 The reorganization of the ancillary ligands does not induce significant changes in the M–X and Ln–X (X = O and N) distances (Table S4†) neither in the crystal packing in which the formation of columns involving D–D and D–A dimers is observed (Table S6 and Fig. S11†).
 | ||
| Fig. 4 Molecular structure of 14. Hydrogen atoms and solvent molecules of crystallization are omitted for clarity. | ||
The X-ray structure reveals that only the tris-chelating coordination site (dpp moiety) of L is coordinated to one Pr(hfac)3 moiety. This selectivity can be explained by the larger ionic radius of the Pr(III) ion which is too large to fit in the bis-coordinating site. Complex 14 nicely illustrates the preference of lanthanide ions to one specific coordination sites. The Pr(III) ion is surrounded by six oxygen atoms that belong to three hfac− ligands and three nitrogen atoms coming from the tris-chelating coordination site (dpp moiety). The average Pr–O distances are shorter (2.479(10) Å) than the average Pr–N distances (2.649(11) Å) (Table S7†). The arrangement of the ligands leads to distorted spherical tricapped trigonal prism (D3h) and Muffin (Cs) as coordination polyhedra for the Pr1 and Pr2 ions, respectively (Table S5†). The central CC bonds of the TTF cores are equal to 1.324(2) Å which confirms the neutral form of L. The two crystallographically independent complexes form dimers by short S⋯S contacts (S4⋯S9 = 3.714 Å, S4⋯S10 = 3.719 Å, S5⋯S11 = 3.714 Å and S5⋯S12 = 3.732 Å) (Table S8†). The crystal packing reveals that the dimers further arrange as dimers of dimers isolated by Pr(hfac)3 units, with the shortest S⋯S contacts being S6⋯S7 = 3.645 Å and S6⋯S10 = 3.696 Å (Fig. S16†). The shortest intermolecular Pr–Pr distance is 9.592 Å (Pr1–Pr2). Compound 14 can be seen as an intermediate to obtain heterobimetallic complexes in which the PrIII ion or lightest lanthanide could be associated with another 4f metal ion.
 | ||
| Fig. 5 Representative molecular structure of compounds 15 and 17–19. Hydrogen atoms and solvent molecules of crystallization are omitted for clarity. | ||
The average Ln1–O and Ln1–N distances observed in the eight-coordinated sites respectively in 15 and 17/19 are in agreement with those observed in the isostructural [Dy2(hfac)6(L)]·2CH2Cl2·C6H14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 51 and [Yb2(hfac)6(L)]·2CH2Cl2·C6H1452 structures. It thus seems reasonable to consider these sites to be fully occupy by either the Dy(III) and Yb(III) ions. The fraction of Dy or Yb ions in the nine-coordinated site is determined by Scanning Electron Microscopy equal to 21% in 15, 4% in 17 and close to 0% in 19. This result shows that the higher the difference of ionic radius between the eight-coordinated lanthanide ion (Ln1) and the nine-coordinated lanthanide ion (Nd1/Ln2), the higher is the selectivity. The arrangement of the ligands leads to a distorted square anti-prism (D4d symmetry) and spherical tricapped trigonal prism (D3h symmetry) as coordination polyhedra for Ln1 and Nd1/Ln2 ions, respectively. The central CC bond of the TTF core confirms the neutral form of L.
51 and [Yb2(hfac)6(L)]·2CH2Cl2·C6H1452 structures. It thus seems reasonable to consider these sites to be fully occupy by either the Dy(III) and Yb(III) ions. The fraction of Dy or Yb ions in the nine-coordinated site is determined by Scanning Electron Microscopy equal to 21% in 15, 4% in 17 and close to 0% in 19. This result shows that the higher the difference of ionic radius between the eight-coordinated lanthanide ion (Ln1) and the nine-coordinated lanthanide ion (Nd1/Ln2), the higher is the selectivity. The arrangement of the ligands leads to a distorted square anti-prism (D4d symmetry) and spherical tricapped trigonal prism (D3h symmetry) as coordination polyhedra for Ln1 and Nd1/Ln2 ions, respectively. The central CC bond of the TTF core confirms the neutral form of L.
The crystal packing reveales the formation of head-to-tail dimers of L along the b axis (Fig. S21†). A one-dimensional organic network of L is formed along this axis thanks to S⋯S contacts between the head-to-tail dimers (Table S8†). Each column of L is separated by the Ln(hfac)3 moieties localized in the nine-coordination sites. The shortest intra- and intermolecular Ln–Ln distances are depicted in Table S7.†
 | ||
| Fig. 6 Molecular structure of 16. Hydrogen atoms are omitted for clarity. | ||
One could notice that even if ligand-scrambling took place, the two remaining coordinated tta-anions to Dy(III) centre led to an increase of the coordination selectivity. In other words, the use of tta− increases the discrimination between the coordination sites because it favors a perfect match between the size of the two lanthanide ions and the coordination sites for a minimum of steric hindrance. The crystal packing reveals the formation of isolated “head-to-tail” dimers (Fig. S23†).
Electrochemical properties
The redox properties of the complexes 1–19 are investigated by cyclic voltammetry (Fig. S24 and S25†), the values of the oxidation potentials are listed in Table S10.† The cyclic voltammograms for all the complexes show two mono-electronic oxidations at about 0.47–0.67 V for the first oxidation and about 0.90–0.99 V for the second oxidation, corresponding to the formation of a radical cation and a dication TTF fragment, respectively. The electrochemical properties attest that the reversibility of the oxidation potentials and the redox-activity of ligand (L) is conserved after complexation.Photophysical properties
500–27500 cm−1 energy range were attributed to Intra-Ligand Charge Transfer (ILCT) from the TTF to benzoimidazolpyridine fragment. This ILCT excitation is mainly due to the HOMO to LUMO transition and it is localized at lower energy after complexation of the electron-withdrawing metallic precursors as observed in the solid-state absorption spectra (Fig. S28 and S29†). The red-shift of the ILCT band after complexation attests the integrity of the complexes in CH2Cl2 solution. The following energy range of 27500–35000 cm−1 is constituted of intra-TTF and ILCT excitations as well as Intra-hfac− transitions (around 33000 cm−1). It is worth to notice that in this energy range, compound 16 is slightly different than all the other compounds because of the existence of lower-energy Intra-tta− transitions (around 29500 cm−1). Finally the highest-energy part of the absorption spectra (above 35000 cm−1) is devoted to the π–π* intra-dpp transitions.
Emission spectroscopy
091, and 10172 cm−1, but there are also shoulders corresponding either to the presence of two crystallographically independent Yb(III) centers with distinct coordination polyhedron symmetry (Table S5†) leading to two different crystal field splittings and/or additional transitions coming from MJ excited states of the 2F5/2 multiplet as frequently observed in previous studies.52,55,56
 | ||
| Fig. 7 Solid state NIR luminescence spectrum of 3 (a) and 5 (b) at 77 K, λex = 600 nm (16667 cm−1). | ||
The same light irradiation to complex 5 leads to a similar NIR Yb(III) centered luminescence. The resolution is lower but the four main transition are still observed centered at 9699 cm−1, 9766 cm−1, 9921 cm−1, and 10183 cm−1 (Fig. 7b).
The main difference between 3 and 5 is the stronger intensity of the emission for 3 than for 5. Finally the NIR luminescence for 8 and 10 is totally quenched. Among the series of 3d4f heterobimetallic compounds, three different optical behaviors can be identified: (1) an intense NIR emission when the Yb(III) ion is associated with the d10 closed shell electronic structure Zn(II), (2) a complete quenching of the Yb(III) NIR luminescence the presence Co(II) and Ni(II) ions and (3) a partial quenching in the presence of Mn(II).
The non-detrimental effect of close shell metal ion like Zn(II) is well established, it mainly acts as Lewis acid red-shifting ILCT transition and contributes to rigidify of the structure restricting nonradiative deactivation processes.57–59 In marked contrast, paramagnetic transition metals like Cu(II), Co(II), Ni(II) are generally good fluorescence quenchers due to the presence of low-lying d–d and/or Metal–Ligand Charge Transfer (LMCT) state favouring non radiative processes. These metal ions enhance the non radiative desexcitation pathways of the antenna and this later is no more able to sensitise the lanthanide ion resulting in a very strong emission quenching.60,61 Finally the partial emission quenching observed in the presence of paramagnetic Mn(II) may be due to a moderate spin–orbit coupling, it does not highlight strong MLCT and the weak d–d transitions are higher in energy than the π–π* singlet state. Thus it is does not favor nonradiative decays and compound 5 is one of the rare examples of NIR emitters containing a paramagnetic transition metal.
099 cm−1 (4F3/2 → 4I9/2) (Fig. 8a) while no emission of the Dy(III) ion is detected because of the non-energy matching between the emissive level of the Dy(III) ion and the ILCT. Compound 17 is composed of two potentially emissive lanthanides with the eight coordinated Yb(III) and the nine coordinated Nd(III). Irradiation of 17 at 600 nm at 77 K, leads to a single NIR emission at 9690 cm−1, 9930 cm−1 and 10193 cm−1 which is characteristic of the 2F5/2 → 2F7/2 transition of the Yb(III) ion (Fig. 8b). Based on the previous works62–66 and on the much more resolved Yb(III)-centered luminescence (Fig. 8c) once Nd(III) has been changed by a Pr(III) ion in 19, the absence of Nd(III)-centered emission in 17 might be an indication of an efficient Nd-to-Yb Energy Transfer (ET).
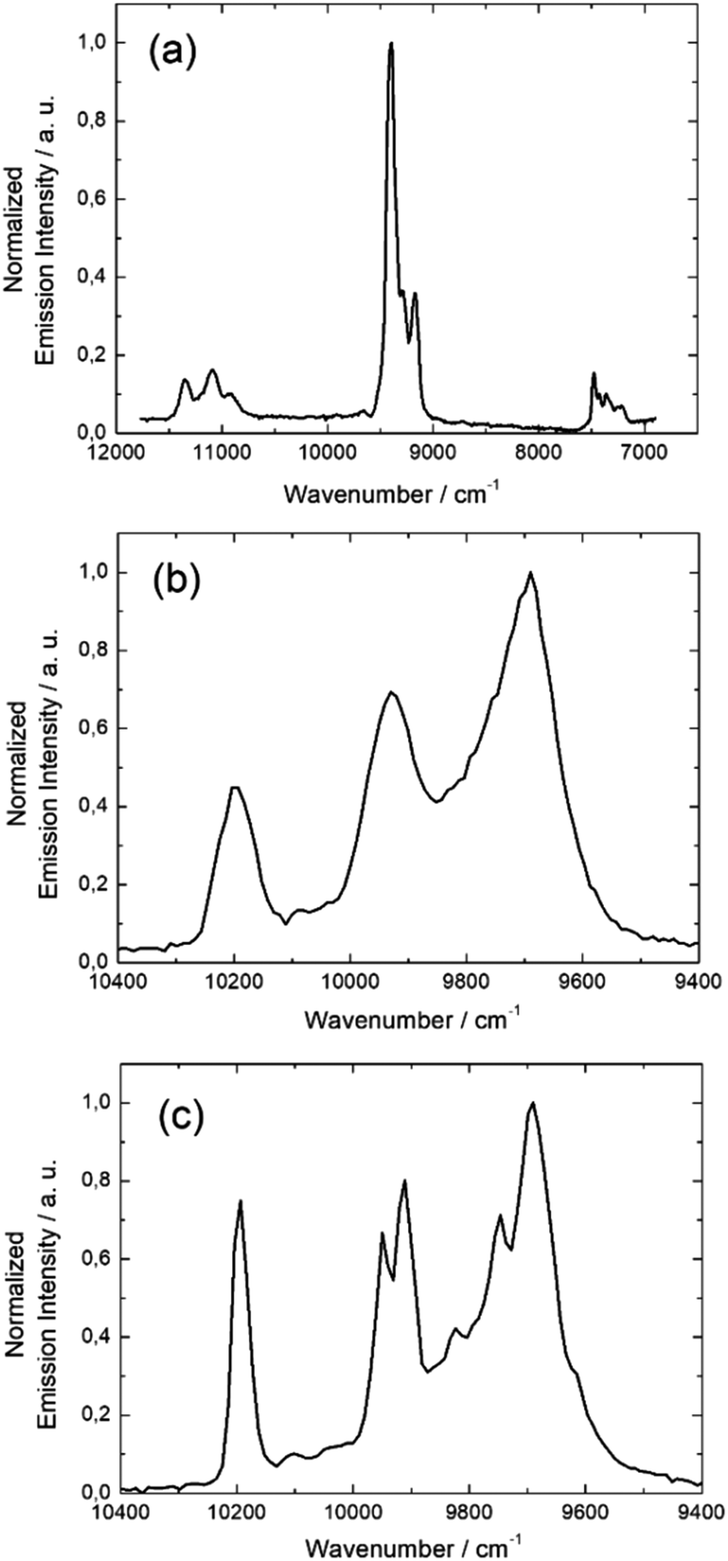 | ||
| Fig. 8 Solid state NIR luminescence spectra of 15 (a), -17 (b) and 19 (c) at 77 K, λex = 600 nm (16667 cm−1). | ||
Magnetic properties
 the Stevens operators which can expressed as polynomials of the total
angular momentum matrices (J2, Jz, J+ and J−).68,69
the Stevens operators which can expressed as polynomials of the total
angular momentum matrices (J2, Jz, J+ and J−).68,69
The parameters to adjust are Bqk's. The best fit is obtained with B02 = 5.5 cm−1, B04 = 2.4 × 10−3 cm−1, B06 = 6.1 × 10−5 cm−1 and B66 = 0.1 cm−1, and a small amount of paramagnetic impurities (χimp = 0.022 cm3 mol−1) to account for the monotonic increase of χMvs. T curve at lowest temperatures (Fig. S54†). Analysis reveal that the ground state singlet is non magnetic and is a mixture of the two MJ = ±3 components of the ground state multiplet 3H4 of Pr(III) as a result of the strong transverse component  which mixes states with |ΔMJ| = 6. Field variation of the magnetization for 14 at 2 K is quasi linear and well reproduced by the simulated curve obtained with previously fitted parameters (Fig. S54†). χMT vs. T plot for compound 15 is represented on Fig. S55.† At room temperature χMT is equal to 20.44 cm3 K mol−1 which is much higher than the Curie limit expected for one Dy(III) ion in the octa-coordinated site (6H15/2: 14.17 cm3 K mol−1) plus one Nd(III) ion in the nona-coordinated site (4I9/2: 1.64 cm3 K mol−1). This suggests that Dy(III) ions are also found in the N3O6 environment. To get a rough estimation of the distribution we have performed a Curie–Weiss analysis (χM = C/(T − θ)) between 100 and 300 K (Fig. S55†). The best fit is obtained with C = 20.99 cm3 K mol−1 (Fig. S55†), from which we estimate the chemical formula [Dy1.41Nd0.59(hfac)6(L)] with a nona-coordinated site occupation Dy0.41/Nd0.59. This proportion of Dy(III) in nine-coordination is slightly higher than the one determined from Scanning Electron Microscopy analysis. Same considerations apply to 16. With C = 17.72 cm3 K mol−1 (Fig. S56†), one finds the chemical formula [Dy1.15Nd0.85(hfac)3(tta)3(L)] with nona-coordinated site occupation Dy0.15/Nd0.85 in excellent agreement with the Scanning Electron Microscopy analysis. For compound 17, with Yb(III) (2F7/2: 2.57 cm3 K mol−1) the chemical formula writes [Yb1.15Nd0.85(hfac)6(L)] with C = 4.35 cm3 K mol−1 (Fig. S57†) also in very good agreement with the Scanning Electron Microscopy analysis. Finally, for 19, one can write [Yb0.79Pr1.21(hfac)6(L)] with C = 4.52 cm3 K mol−1 and the 3H4 multiplet ground state of Pr(III) (Fig. S58†).
which mixes states with |ΔMJ| = 6. Field variation of the magnetization for 14 at 2 K is quasi linear and well reproduced by the simulated curve obtained with previously fitted parameters (Fig. S54†). χMT vs. T plot for compound 15 is represented on Fig. S55.† At room temperature χMT is equal to 20.44 cm3 K mol−1 which is much higher than the Curie limit expected for one Dy(III) ion in the octa-coordinated site (6H15/2: 14.17 cm3 K mol−1) plus one Nd(III) ion in the nona-coordinated site (4I9/2: 1.64 cm3 K mol−1). This suggests that Dy(III) ions are also found in the N3O6 environment. To get a rough estimation of the distribution we have performed a Curie–Weiss analysis (χM = C/(T − θ)) between 100 and 300 K (Fig. S55†). The best fit is obtained with C = 20.99 cm3 K mol−1 (Fig. S55†), from which we estimate the chemical formula [Dy1.41Nd0.59(hfac)6(L)] with a nona-coordinated site occupation Dy0.41/Nd0.59. This proportion of Dy(III) in nine-coordination is slightly higher than the one determined from Scanning Electron Microscopy analysis. Same considerations apply to 16. With C = 17.72 cm3 K mol−1 (Fig. S56†), one finds the chemical formula [Dy1.15Nd0.85(hfac)3(tta)3(L)] with nona-coordinated site occupation Dy0.15/Nd0.85 in excellent agreement with the Scanning Electron Microscopy analysis. For compound 17, with Yb(III) (2F7/2: 2.57 cm3 K mol−1) the chemical formula writes [Yb1.15Nd0.85(hfac)6(L)] with C = 4.35 cm3 K mol−1 (Fig. S57†) also in very good agreement with the Scanning Electron Microscopy analysis. Finally, for 19, one can write [Yb0.79Pr1.21(hfac)6(L)] with C = 4.52 cm3 K mol−1 and the 3H4 multiplet ground state of Pr(III) (Fig. S58†).
One could qualitatively analyse the frequency maxima values of the four compounds 2, 4, 7 and 9 concluding that the slowest magnetic relaxation (40 Hz) is observed for the Dy(III) associated with the transition metal of highest spin state (Mn(II) S = 5/2) while more the value of the M(II) spin state is weak more the corresponding system relax faster (Fig. 9 and S60–S65†). Such observation might signify that the dipolar interactions between the Dy(III) and associated M(II) ions could play a role on the dynamics of the 3d4f complexes even if effect of weak modification of the crystal field around the lanthanide cannot be rule out. The relaxation time τ was extracted at each temperature using an extended Debye model (details in ESI, Table S11–S15†) in fitting simultaneously the frequency dependence of χ′′M and χ′M for 1, 2, 4, 7 and 11 or manually selecting the frequencies maxima of χ′′M for 9. Normalized Cole–Cole plots of the series 1, 2, 4, 7 and 11 are given in Fig. S66–S70.† In-field Arrhenius plots were fitted considering the two thermally dependent Orbach and Raman processes (eqn (1)).
 | (1) |
 | ||
| Fig. 9 Arrhenius plots of the relaxation times in a 1500 Oe applied magnetic field for compounds 1 (full circles), 2 (down triangles), 4 (up triangles), 7 (squares), 9 (pentagons) and 11 (stars). Full red lines are the best-fit curves (see text). | ||
The best fits for each of the compounds were obtained with the parameters given in Table S17.† For compounds 1, 2, 4, 7, 9 and 11, separated contributions from Orbach and Raman processes are provided in Fig. S70–S76.†
Compound 15 behaves also in a standard way. Indeed, in zero external field it shows slowing down of the relaxation of the magnetic moment which can be safely due to the Dy(III) ions in the N2O6 coordination polyhedron made of three hfac− anions (Fig. S77†).71 On contrary, the small fraction of Dy(III) ions in the N3O6 environment does not contribute in zero field. Once an external field is applied, both octa- and nona-coordinated sites move to lower frequencies with two clear massifs (Fig. S77†) with altitudes that qualitatively fit the 0.41/1 ratio between two sites and the fastest relaxation for the nona-coordinated site.
Furthermore, the high frequency massif appears at the slowest frequency for an external dc field which is close to 1.5 kOe at 2 K as expected from previous studies.70 The thermal dependence of the relaxation time for each of the Dy centers were extracted from a combination of two extended debye model and the resulting plots fitted with a Raman process while the orbach one became negligible (Fig. S78, Fig. 10, Tables S16 and S17†). Such relaxation of the magnetization for a Dy(III) in D4d N2O6 environment is in agreement with what some of us experimentally and computationally determined.72 One could remark that the n values obtained from our best fits for compounds (1, 2, 4, 7, 9, 11 and 15) are much lower than the expected n value for Kramers ions (n = 9) (Table S17†). Such value can decrease down to 4, depending on the energies of the ground state doublets.73,74 Recently, such exponent reduced close to 2 because of the presence of both acoustic and optic phonons in specific ligand environments.75
 | ||
| Fig. 10 Arrhenius plots of the relaxation times in a 1500 Oe applied magnetic field for Dy1 (full circles) and Dy2 (open circles) metallic sites in 15. | ||
In compound 16, the nona-coordinated site is only occupied by 15% of Dy(III) which is a too low value to clearly be identified from ac magnetometry and therefore only the octa-coordinated site might be observed. According to ref. 4, Dy(III) in N2O6 environment made of two tta− and one hfac− ligands should relax at 1 Hz at 2 K and 3 kOe with an amplitude at the maximum of χ′′M slightly below 0.8 cm3 mol−1. This is close to what we observe for 16 (Fig. S79†) with the maximum falling at 10 Hz in the same sample environment and a magnetic relaxation for Dy1 (Fig. 11) which occurs through an Orbach (Table S17†) and remaining QTM (τTI = 0.0146(6) s) processes as previously observed for Dy(III) in N2O6 environment made of two tta− and one hfac− ligands.70
 | ||
| Fig. 11 Arrhenius plots of the relaxation times in a 3000 Oe applied magnetic field for the Dy1 metallic site in 16. | ||
Conclusions
In the previous lines, it was demonstrated that the ligand 2-(1-(2,6-di(pyrazol-1-yl)-4-methylpyridyl)-4,5-(4,5-bis(propylthio)-tetrathiafulvalenyl)-1H-benzimidazol-2-yl)-pyridine is suitable for the design of d4f and 4f4f′ heteroleptic complexes through selective coordination on both benzoimidazolylpyridine (bzip) and 2,6-di(pyrazol-1-yl)-4-pyridine (dpp) moieties. The key parameter of the coordination selection for the d–4f complexes is the difference of coordination number between transition metals (coordination number = 6) and lanthanide ions (coordination number = 8, 9) while for the 4f–4f′ complexes the key parameter is the radius size. All the heterobimetallic complexes involving eight or nine-coordinated Dy(III) ion displayed field-induced SMM with a slight influence of the spin state value of the associated transition metal through dipolar interactions and/or weak change of electronic distribution around the 4f ion. In a photo-physical point of view, the use of the Yb(III) in the design of the 3d–4f complexes leads to an intense, moderated or quenched 2F5/2 → 2F7/2 NIR luminescence depending if the Yb(III) ion is respectively associated with the Zn(II), Mn(II) or Ni(II)/Co(II) ion. Light irradiation at 600 nm provokes the observation of the 4F3/2 → 4I13/2/4I11/2/4I9/2 Nd(III) centred emission for the [Dy1.21Nd0.79(hfac)6(L)]·2(CH2Cl2)·(C6H14) complex and the 2F5/2 → 2F7/2 Yb(III) centred emission for the [YbPr(hfac)6(L)] complex. When the Pr(III) ion is replaced by the Nd(III) ion ([Yb1.04Nd0.96(hfac)6(L)]) no dual NIR emission is detected.Finally the compound 15 displayed both slow magnetic relaxation and NIR luminescence and can be described as a field-induced luminescent single-molecule magnet.
Author contributions
H. D. and B. L. performed the organic syntheses, the coordination chemistry and crystallisations; V. D. realised the single crystal X-ray diffraction experiments and refined the X-ray structures; J. F. G. and O. C. performed and analysed the magnetic measurements; S. S., F. R., O. M. performed and analysed the photophysical measurements; A. G., B. L. G. and V. M. discussed the idea and the results and commented on the manuscript; F. P. and O. C. contributed to the writing of the article. F. P. conceived and designed the experiments and was granted of the ANR and ERC funded projects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Région Bretagne, Rennes Métropole, CNRS, Université de Rennes 1, FEDER, the ANR (ANR-13-BS07-0022-01) and the European Commission through the ERC-CoG 725184 MULTIPROSMM (project no. 725184). S. S. would like to thank Obra Social de la Fundació “La Caixa” and Fundació Universitària Agustí Pedro i Pons for financial support.References
- C. Piguet and J.-C. G. Bünzli, Chem. Soc. Rev., 1999, 28, 347–358 RSC.
- S. Comby and J.-C. G. Bünzli, Handbook on the Physics and Chemistry of Rare Earths, Elsevier BV, Amsterdam, The Netherland, 2007, ch. 235, vol. 37 Search PubMed.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, New York, 2006 Search PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- J. Lehmann, A. Gaita-Arino, E. Coronado and D. Loss, Nat. Nanotechnol., 2007, 2, 312–317 CrossRef CAS PubMed.
- M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Nanotechnol., 2013, 8, 165–169 CrossRef CAS PubMed.
- K. Kuriki, Y. Koike and Y. Okamoto, Chem. Rev., 2002, 102, 2347–2356 CrossRef CAS PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed and references therein.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- A. Beeby, S. W. Botchway, I. M. Clarkson, S. Faulkner, A. M. Parker, D. Parker and J. A. G. Williams, J. Photochem. Photobiol., B, 2000, 57, 83–89 CrossRef CAS.
- A. Grichine, A. Haefele, S. Pascal, A. Duperray, R. Michel, C. Andraud and O. Maury, Chem. Sci., 2014, 5, 3475–3485 RSC.
- R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS.
- T. N. Hooper, J. Schnack, S. Piligkos, M. Evangelesti and E. K. Brechin, Angew. Chem., Int. Ed., 2012, 51, 4633–4636 CrossRef CAS PubMed.
- J. W. Sharples and D. Collison, Coord. Chem. Rev., 2014, 260, 1–20 CrossRef CAS PubMed.
- K. R. Vignesh, S. K. Langley, K. S. Murray and G. Rajaraman, Chem. – Eur. J., 2017, 23, 1654–1166 CrossRef CAS PubMed.
- H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2014, 276, 1–33 CrossRef CAS.
- I. Oyarzabal, J. Ruiz, E. Ruiz, D. Aravena, J. Seco and E. Colacio, Chem. Commun., 2015, 51, 12353–12356 RSC.
- W.-B. Sun, P.-F. Yan, S.-D. Jiang, B.-W. Wang, Y.-Q. Zhang, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 684–691 RSC.
- M. Shamugam, A. Upadhyay, C. Das, S. Vaidya, S. K. Singh, T. Gupta, R. Mondal, S. Langley, K. Murray and G. Rajaraman, Chem. – Eur. J., 2017, 23, 4903–4916 CrossRef PubMed.
- J.-D. Leng, J.-L. Liu, Y.-Z. Zheng, L. Ungur, L. F. Chibotaru, F.-S. Guo and M.-L. Tong, Chem. Commun., 2013, 49, 158–160 RSC.
- Q.-W. Li, J.-L. Liu, J.-H. Jia, J.-D. Leng, W.-Q. Lin, Y.-C. Chen and M.-L. Tong, Dalton Trans., 2013, 42, 11262 RSC.
- F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150–175 CrossRef CAS.
- C. Edder, C. Piguet, G. Bernardinelli, J. Mareda, C. Bochet, J.-C. G. Bünzli and G. Hopfgartner, Inorg. Chem., 2000, 39, 5059–5073 CrossRef CAS PubMed.
- J. H. van Vleck, J. Phys. Chem., 1937, 41, 67–80 CrossRef CAS.
- A. D'Aléo, F. Pointillart, L. Ouahab, C. Andraud and O. Maury, Coord. Chem. Rev., 2012, 256, 1604–1620 CrossRef.
- F. Pointillart, B. Le Guennic, O. Cador, O. Maury and L. Ouahab, Acc. Chem. Res., 2015, 48, 2834–2842 CrossRef CAS PubMed.
- S. Faulkner and S. J. Pope, J. Am. Chem. Soc., 2003, 125, 10526–10527 CrossRef CAS PubMed.
- T. J. Sørensen, M. Tropiano, O. A. Blackburn, J. A. Tilney, A. M. Kenwright and S. Faulkner, Chem. Commun., 2013, 49, 783–785 RSC.
- J. Long, R. Vallat, R. A. S. Ferreira, L. D. Carlos, F. A. Almeida Paz, Y. Guari and J. Larionova, Chem. Commun., 2012, 48, 9974–9976 RSC.
- D. Imbert, M. Cantuel, J.-C. G. Bünzli, G. Bernardinelli and C. Piguet, J. Am. Chem. Soc., 2003, 125, 15698–15699 CrossRef CAS PubMed.
- D. Zare, Y. Suffren, H. Nozary, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2017, 56, 14612–14617 CrossRef CAS PubMed.
- H.-R. Wen, P.-P. Dong, S.-J. Liu, J.-S. Liao, F.-Y. Liang and C.-M. Liu, Dalton Trans., 2017, 46, 1153–1162 RSC.
- Y. Liu, Y.-C. Chen, J. Liu, W.-B. Chen, G.-Z. Huang, S.-G. Wu, J. Wang, J.-L. Liu and M.-L. Tong, Inorg. Chem., 2020, 59, 687–694 CrossRef CAS PubMed.
- K. Fan, S.-S. Bao, R. Huo, X.-D. Huang, Y.-J. Liu, Z.-W. Yu, M. Kurmoo and L.-M. Zheng, Inorg. Chem. Front., 2020, 7, 4580–4592 RSC.
- J. Wang, J. J. Zakrzewski, M. Zychowicz, V. Vieru, L. F. Chibotaru, K. Nakabayashi, S. Chorazy and S. Ohkoshi, Chem. Sci., 2021, 12, 730–741 RSC.
- N. André, T. B. Jensen, R. Scopelliti, D. Imbert, M. Elhabiri, G. Hopfgartner, C. Piguet and J.-C. G. Bünzli, Inorg. Chem., 2004, 43, 515–529 CrossRef PubMed.
- N. Dalla-Favera, J. Hamacek, M. Borkovec, D. Jeannerat, G. Ercolani and C. Piguet, Inorg. Chem., 2007, 46, 9312–9322 CrossRef CAS PubMed.
- P. E. Ryan, G. Canard, S. Koeller, B. Bocquet and C. Piguet, Inorg. Chem., 2012, 51, 10012–10024 CrossRef CAS PubMed.
- R. E. P. Winpenny, Chem. Soc. Rev., 1998, 27, 447–452 RSC.
- M. Sakamoto, K. Manseki and H. Okawa, Coord. Chem. Rev., 2001, 219, 379–414 CrossRef.
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369–2388 CrossRef CAS PubMed.
- M. Andruh, J. P. Costes, C. Diaz and S. Gao, Inorg. Chem., 2009, 48, 3342–3359 CrossRef CAS PubMed (Forum Article).
- M. Andruh, Chem. Commun., 2011, 47, 3025–3042 RSC.
- L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC.
- T. J. Sorensen and S. Faulkner, Acc. Chem. Res., 2018, 51, 2493–2501 CrossRef PubMed.
- G. Cosquer, F. Pointillart, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2011, 17, 12502–12511 CrossRef CAS PubMed.
- G. Cosquer, F. Pointillart, B. Le Guennic, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2012, 51, 8488–8501 CrossRef CAS PubMed.
- M. Feng, F. Pointillart, B. Lefeuvre, V. Dorcet, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2015, 54, 4021–4028 CrossRef CAS PubMed.
- S. Speed, M. Feng, G. Fernandez-Garcia, F. Pointillart, B. Lefeuvre, F. Riobé, S. Golhen, B. Le Guennic, F. Totti, Y. Guyot, O. Cador, O. Maury and L. Ouahab, Inorg. Chem. Front., 2017, 4, 604–617 RSC.
- M. Llunell, D. Casanova, J. Cirera, J. M. Bofill, P. Alemany and S. Alvarez, S. SHAPE (version 2.1), Barcelona, 2013 Search PubMed.
- G. Cosquer, F. Pointillart, J. Jung, B. Le Guennic, S. Golhen, O. Cador, Y. Guyot, A. Brenier, O. Maury and L. Ouahab, Eur. J. Inorg. Chem., 2014, 69–82 CrossRef CAS.
- P. Goldner, F. Pell, D. Meichenin and F. Auzel, J. Lumin., 1997, 71, 137–150 CrossRef CAS.
- X. Yi, K. Bernot, V. Le Corre, G. Calvez, F. Pointillart, O. Cador, B. Le Guennic, J. Jung, O. Maury, V. Placide, Y. Guyot, T. Roisnel, C. Daiguebonne and O. Guillou, Chem. – Eur. J., 2014, 20, 1569–1576 CrossRef CAS PubMed.
- L. Tian, M. Zhang, P. Lu, W. Zhang, B. Yang and Y. Ma, Chin. Sci. Bull., 2004, 49, 246–248 CAS.
- T. D. Pasatoiu, C. Tiseanu, A. M. Madalan, B. Jurca, C. Duhayon, J.-P. Sutter and M. Andruh, Inorg. Chem., 2011, 50, 5879–5889 CrossRef CAS PubMed.
- O. Maury, J.-P. Guégan, T. Renouard, A. Hilton, P. Dupau, N. Sandon, L. Toupet and H. Le Bozec, New J. Chem., 2001, 25, 1553–1566 RSC.
- J. Song, C.-R. Li, Q. Xu, X.-T. Xu, L.-X. Sun and Y.-H. Xing, Spectrochim. Acta, Part A, 2015, 150, 308–315 CrossRef CAS PubMed.
- M. Maity, M. C. Majee, S. Kundu, S. K. Samanta, E. C. Sanudo, S. Ghosh and M. Chaudhury, Inorg. Chem., 2015, 54, 9715–9726 CrossRef CAS PubMed.
- T. Förster, Faraday Soc., 1959, 27, 7–17 RSC.
- F. Artizzu, A. Serpe, L. Marchio, M. Saba, A. Mura, M. L. Mercuri, G. Bongiovanni, P. Deplano and F. Quochi, J. Mater. Chem. C, 2015, 3, 11524–11530 RSC.
- A. D. Sontakke, K. Biswas, R. Sen and K. Annapurna, Opt. Mater., 2010, 32, 1333–1336 CrossRef.
- R.-X. Chen, T. Gao, W.-B. Sun, H.-F. Li, Y.-H. Wu, M.-M. Xu, X.-Y. Zou and P.-F. Yan, Inorg. Chem. Commun., 2015, 56, 79–82 CrossRef CAS.
- L. Abad Galan, D. Aguila, Y. Guyot, V. Velasco, O. Roubeau, S. T. Teat, M. Massi and G. Aromi, Chem. – Eur. J., 2021, 27, 7288–7299 CrossRef CAS PubMed.
- O. Kahn, Molecular Magnetism, VCH, 1993 Search PubMed.
- R. Orbach, Proc. R. Soc. London, Ser. A, 1961, 264, 458–484 CAS.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Oxford University Press, Oxford, Reprint edn, 2012 Search PubMed.
- M. Feng, F. Pointillart, B. Lefeuvre, V. Dorcet, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2015, 54, 4021–4028 CrossRef CAS PubMed.
- G. Cosquer, F. Pointillart, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2013, 19, 7895–7903 CrossRef CAS PubMed.
- F. Pointillart, J.-K. Ou-Yang, G. Fernandez-Garcia, V. Montigaud, J. Flores Gonzalez, R. Marchal, L. Favereau, F. Totti, J. Crassous, O. Cador, L. Ouahab and B. Le Guennic, Inorg. Chem., 2019, 58, 52–56 CrossRef CAS PubMed.
- C. Dekker, A. F. M. Arts, H. W. de Wijn, A. J. van Duyneveldt and J. A. Mydosh, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 11243–11251 CrossRef CAS PubMed.
- J. Tang and P. Zhang, Lanthanide single molecule magnets, Springer, 2015 Search PubMed.
- C. A. P. Goodwin, D. Reta, F. Ortu, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2017, 139, 18714–18724 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for synthesis and physical characterisation. Crystallographic information in CIF format, Ortep view of 1, 3, 4, 6, 8, 11, 13, 15, 17–19 (Fig. S1–S5, S10, S12, S14, S17–S20 and S22), packing views of 3, 4, 6, 8, 11, 17–19 (Fig. S6–S9, S11, S13, S15, S16, S21 and S23), cyclic voltammetry for compounds 1–13 (Fig. S24) and 14–19 (Fig. S25), visible absorption in CH2Cl2 solution (Fig. S26 and S27), in KBr solid-state (Fig. S28 and S29), visible emission at room temperature and 77 K (Fig. S30–S43), dc magnetic data (Fig. S44–S58) and ac magnetic data (Fig. S59–S79). X-ray crystallographic data (Tables S1–S4 and S6–S8), SHAPE analysis (Tables S5 and S9), oxidation potentials (Table S10) and additional ac parameters (Tables S11–S17). CCDC 1865127–1865130 and 1865135–1865140. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02375j |
| This journal is © The Royal Society of Chemistry 2022 |