
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-redox reactivity of V(II) and Fe(II) formamidinates towards CO2 resulting in the formation of novel M(II) carbamates†
Krzesimir
Korona
 a,
Arkadiusz
Kornowicz
b,
Iwona
Justyniak
b,
Michał
Terlecki
a,
Artur
Błachowski
c and
Janusz
Lewiński
*ab
a,
Arkadiusz
Kornowicz
b,
Iwona
Justyniak
b,
Michał
Terlecki
a,
Artur
Błachowski
c and
Janusz
Lewiński
*ab
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warszawa, Poland. E-mail: lewin@ch.pw.edu.pl
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warszawa, Poland
cAGH University of Science and Technology, Faculty of Geology, Geophysics and Environmental Protection, al. Mickiewicza 30, 30-059 Kraków, Poland
First published on 13th October 2022
Abstract
Chemical fixation of CO2 is a powerful tool for the preparation of novel multinuclear metal complexes and functional materials. Particularly, the insertion of CO2 into a metal–X bond (X = H, C, N, O) often is a key elementary step in the various processes transforming this greenhouse gas into valuable products. Herein, we report on the reactivity between CO2 and V(II) and Fe(II) complexes supported by N,N′-bis(2,6-diisopropylphenyl)formamidinate ligands (DippF). The reactions proceeded with multiple insertions of CO2 into the M–N bonds leading to the isolation of three novel complexes: [(κ2-DippFCO2)(THF)V(μ-DippFCO2)3V(THF)], [(κ2-DippFCO2)Fe(μ-DippFCO2)2(μ-DippF)Fe(THF)] and [(κ2-DippFCO2)Fe(μ-DippFCO2)3Fe(κ1-DippFH)], which were characterised using single-crystal X-ray diffraction, FTIR and 57Fe Mössbauer spectroscopy (for the diiron compounds). We provide the first well-documented studies of the CO2 reactivity towards the V–N bond and broaden the state-of-the-art of the undeveloped area of the reactivity of low-valent V(II) complexes. Moreover, we showed that the effectivity of the examined CO2 insertion processes strongly depends on the used solvent's characteristics (for the Fe(II) system) and the metal centre's coordination sphere geometry (for the V(II) system).
Introduction
The insertion of CO2 into the metal–N bond is essential in transforming this most widespread greenhouse gas into value-added products, like ureas, polyurethanes, isocyanates and carbamates.1–6 Metal carbamates are easily formed for non-redox active and mild reducing metal centres,1,5,7–9 but their formation with the participation of low-valent metal centres is still poorly developed. In turn, redox-active transition metal centres in a low oxidation state are known for CO2 reduction and C–O bond cleavage after direct electron transfer from the metal centre to a coordinated CO2 molecule.7,10–16 In particular, the reduction of CO2 at iron centres seems the most complex and challenging due to the variety of reaction pathways and possible products. Fe(0) and Fe(I) centres can reduce CO2 to carbonyl or oxalate,17–20 but the definitely more common Fe(II) complexes are usually able to activate CO2 only via the ligand-based reduction21,22 or the insertion into a Fe–X bond.23,24 In turn, there are also known examples of the reductive activation of CO2 by Fe(II) centres, as in a tetraisocyanide complex25 or a multinuclear [NiFe4S4] cluster,26 so the activation of CO2 on iron-based systems still requires further fundamental-level research. In contrast to late first-row transition metals, reports concerning CO2 activation by early transition metal complexes are relatively rare.10 For example, it was only in 2017 that Gambarotta et al. reported the first well-documented processes of CO2 reduction by V(II) and V(III) systems.27–29 As part of our continuous effort in designing various new reaction systems for small molecules activation30,31 and using of CO2 as a substrate in the preparation of functional materials,32,33 herein we examine the reactivity of model mononuclear amidinate complexes of V(II) and Fe(II) towards CO2 and present their ability to capture multiple equivalents of CO2.Results and discussion
Synthesis and structural characterisation of mononuclear V(II) and Fe(II) bis(formamidinates)
Mononuclear complexes [V(DTolF)2(THF)2] (1) [V(DippF)2(THF)] (2), and [Fe(DippF)2] (3) were synthesised in THF via the salt metathesis reaction between MCl2 (M = V, Fe) and a potassium salt of formamidinate ligand: N,N′-bis(2,6-diisopropylphenyl)-formamidinate (DippF) and N,N′-di(p-tolyl)formamidinate (DTolF), respectively (Scheme 1). Interestingly, in a similar reaction between LiDTolF and in situ generated VCl2 (with NaHBEt3 as a reductant), Cotton et al. obtained a divanadium paddlewheel-type complex [V2(DTolF)4].34 In turn, recently, we revealed that the reaction of FeCl2 with DTolF in THF resulted in the isolation of the non-centrosymmetric binuclear complex [Fe(μ-DTolF)3Fe(κ2-DTolF)].35 These results clearly demonstrate that subtle modification of the formamidinate ligand backbone profoundly affects the character of the resulting molecular complexes. The resulting complexes 1–3 were characterised spectroscopically (for details see Experimental section and ESI†), and their molecular structures were confirmed using single-crystal X-ray diffraction. | ||
| Scheme 1 Synthesis of V(II) and Fe(II) formamidinates. | ||
The molecular structure of 1 (Fig. 1a) can be described as a THF-solvated mononuclear V(II) amidinate bischelate complex with a distorted octahedral metal centre. The octahedral geometry of the metal centre's coordination sphere is distorted by a narrow bite angle of amidinate ligands (61.2(1)). The amidinate ligands lie in the equatorial plane, and two solvated THF molecules are positioned in the axial positions. The V–N bond lengths are equal 2.193(1) and 2.195(1) Å, and the V–O bonds are 2.135(1) Å. The molecular structure of 2 (Fig. 1b) revealed a five-coordinate mononuclear V(II) bis(amidinate) complex, solvated by one THF molecule. Three V–N bonds (to N1, N3, N4) are coplanar with a similar length of 2.160–2.164 Å, while the fourth V–N bond is slightly shorter (2.133(1) Å) and deflected from this plane by about 40°. The resulting biplanar angle between two amidinate NCN groups is 39.1(1)°. The THF molecule is coordinated perpendicular to the V–N1–N3–N4 plane, on the opposite side to the out-of-plane V–N2 bond (the V–O bond is 2.124(1) Å), which results in the coordination sphere geometry close to a distorted trigonal bipyramidal (for detailed CShM analysis see ESI, Table S14†). In contrast to 1 and 2, complex 3 does not comprise any coordinated THF molecules, and its molecular structure can be described as a tetracoordinate mononuclear amidinate bischelate complex (Fig. 1c). All Fe–N bond distances fall into the range of 2.034–2.054 Å, and the amidinate ligands are twisted by ca. 40° (the biplanar angle between two NCN groups is 38.7(6)°). It results in the coordination sphere geometry close to a distorted square planar (Table S15†). We note that compounds 1–3 were synthesised in similar conditions, which suggests a stronger affinity of THF molecules to the V(II) centre than to Fe(II) one.
 | ||
| Fig. 1 The molecular structures of 1 (a), 2 (b), 3 (c). Hydrogen atoms were omitted for clarity. | ||
Reactions of mononuclear M(II) formamidinates 1–3 with CO2
Carbon dioxide molecules are susceptible to insert into M–N bonds.3,4 However, to the best of our knowledge, such reactions involving V(II) and Fe(II) centres have not been reported yet. Bearing in mind the potential impact of donor solvents on the fixation of CO2 molecules on metal centres, we decided to carry out reactions for the mononuclear M(II) formamidinates 1–3 using both THF and toluene as a donor and non-coordinating solvent, respectively (Scheme 2). When compound 1 was exposed to CO2, no colour change was observed, and followed crystallisation resulted in the isolation of the parent complex in high yield. As we mentioned, V(II) centres show a strong affinity for THF, and the presence of a coordinatively saturated six-coordinated V(II) centre likely hampers the fixation of CO2 molecules. Treatment of a THF solution of 2 with an excess of CO2 at ambient temperature resulted in a quick colour change from green to light purple. Thus, the presence of the unsaturated five-coordinated V(II) centre is essential for CO2 activation proceeding. Crystallisation from a THF–pentane solution resulted in rectangular purple crystals of a novel binuclear carbamate [(κ2-DippFCO2)(THF)V(μ-DippFCO2)3V(THF)] (4), which is formed due to the complete N-carboxylation at the V–N amidinate-binding in conjunction with an aggregation of the anticipated (bis)carbamate mononuclear species. The valence bond calculations indicate that both V(II) centres remained at the +II oxidation state, so the reaction with CO2 did not trigger any redox processes on the low-valent metal centres. As mentioned by Gambarotta et al., the insertion of CO2 is unavoidable for strongly nucleophilic ligands despite the low oxidation state of the metal centre,28 which has been proven herein. We note that the analogous reaction carried out in toluene as a non-coordinating solvent also afforded 4. | ||
| Scheme 2 Reactions of mononuclear M(II) formamidinates 1–3 with CO2. | ||
In the case of Fe(II) complex 3, the reaction with an excess of CO2 in THF solution is evidenced by a rapid colour change from pale green to beige. Followed crystallisation from a THF–hexane solution led to the isolation of beige crystals of a heteroleptic Fe(II) complex [(κ2-DippFCO2)Fe(μ-DippFCO2)2(μ-DippF)Fe(THF)] (5). The molecular structure of 5 contains three carbamate ligands and one intact amidinate.
Remarkably, the reaction of 3 with CO2 in toluene led to the formation of compound [(κ2-DippFCO2)Fe(μ-DippFCO2)3Fe(κ1-DippFH)] (6), which was isolated after crystallisation from toluene–hexane solution. The binuclear complex 6 contains three bridging carbamate ligands, one chelating carbamate at the Fe1 centre, and the DippFH molecule coordinated as a neutral ligand to the Fe2 centre. Thus, the results demonstrate that two major factors control the effectivity of CO2 capturing: (i) the coordination state of the metal centre (for the V(II) complexes); (ii) the character of a solvent, i.e. the presence of a donor solvent decreases the effectivity of CO2 capture (for the Fe(II) system). It seems reasonable to assume that in both cases, an initial CO2 molecule insertion into the Fe–N amidinate-binding is similar and leads to an intermediate Fe(amidinate)(carbamate) species. The resulting intermediate species feature smaller steric hindrances at the metal centre and likely form aggregates of higher nuclearities, which then can participate in the following CO2 insertion processes. However, in the case of 5, a significant competition between THF and CO2 for coordination at Fe(II) centres probably hampers an insertion process, leading to the formation of the aggregate of mononuclear bis(carbamate) and mono(carbamate) intermediates. In the absence of THF, the complete N-carboxylation at the Fe–N amidinate-binding occurs, and the resulting product 6 incorporates four carbamate ligands. Remarkably, complex 6 is additionally stabilised by a DippFH molecule, and the neutral formamidine may be likely generated via a side reaction. Finally, the isolated compounds 4–6 were characterized spectroscopically and using single-crystal X-ray diffraction. Their 1H NMR spectra were rather complex for these M(II) carbamates and difficult for unequivocal interpretation (Fig. S10–12†). We also note that the insertion reactions of CO2 are sometimes reversible;8,36 however, during the experimental work, we have not noticed any signs of insertion reversibility.
Structural characterisation of 4–6
The molecular structure of 4 can be described as an asymmetric binuclear carbamate V(II) complex, which arose due to the insertion of two CO2 molecules into V–N bonds of 2 (Fig. 2a); detailed geometric parameters for 4 are listed in ESI, Table S8.† Additionally, each V(II) centre coordinates one THF molecule. The distance between vanadium atoms (3.140(1) Å) excludes the presence of significant metal–metal interactions.37 Both vanadium centres adopt a similar distorted octahedral geometry of the coordination sphere (Table S16†) but differ in their composition; the V1 centre comprises six oxygen atoms, whereas the V2 centre involves five oxygens and one nitrogen atom. It is a result of the unique coordination diversity of the structurally identical carbamate anions, which act as O,O′-chelating (κ2(O,O′)), O,O′-bridging (μ2–κ2(O,O′):κ1(O) and μ2–κ1(O):κ1(O′)), or O,N-bridging (μ2–κ2(O,N):κ1(O)) ligands as shown in Fig. 2d. In the κ2(O,O′) and μ2κ1(O):κ1(O′)-coordinated ligands, both oxygen donor centres are equivalent with similar C–O bond lengths in the range of 1.255–1.265 Å. Contrary, in the μ2–κ2(O,O′):κ1(O) mode, the oxygen atoms are variously coordinated to the V(II) centres, which significantly differentiates the two C–O bonds ((1.247(4) and 1.291(4) Å, respectively). In the O,N-bridging μ2–κ2(O,N):κ1(O) ligand, only one oxygen atom from the CO2− moiety coordinates to the metal centres, which causes the high differentiation between C–Obridging (1.287(4) Å) and C–Oterminal (1.216(4) Å) bonds; the observed contracted C–Oterminal bond length suggests essentially localised C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. The V–O bond lengths are in a range of 2.064–2.214 Å; the lowest values correspond to μ2–κ2(O,N):κ1(O) and κ2(O,O′) coordinated ligands, and the highest ones involve μ2-bridging O4 atom from μ2–κ2(O,O′):κ1(O) mode. The solid-state FTIR spectrum of 4 contains one strong band centred at 1717 cm−1 (Fig. S4†) corresponding to the νC–O asymmetric stretching vibration of the monodentate carboxylate group.38 Other νC–O asymmetric stretching vibration bands, derived from variously coordinated carbamate ligands, lie between 1664–1559 cm−1 and likely overlap with C–N bands of amidinate groups.1 In the 1H NMR spectrum of 4, the signals from the four variously coordinated ligands are overlapping in the region 6.5–7.7 ppm and are hard to unequivocal interpretation (Fig. S10†).
O bond. The V–O bond lengths are in a range of 2.064–2.214 Å; the lowest values correspond to μ2–κ2(O,N):κ1(O) and κ2(O,O′) coordinated ligands, and the highest ones involve μ2-bridging O4 atom from μ2–κ2(O,O′):κ1(O) mode. The solid-state FTIR spectrum of 4 contains one strong band centred at 1717 cm−1 (Fig. S4†) corresponding to the νC–O asymmetric stretching vibration of the monodentate carboxylate group.38 Other νC–O asymmetric stretching vibration bands, derived from variously coordinated carbamate ligands, lie between 1664–1559 cm−1 and likely overlap with C–N bands of amidinate groups.1 In the 1H NMR spectrum of 4, the signals from the four variously coordinated ligands are overlapping in the region 6.5–7.7 ppm and are hard to unequivocal interpretation (Fig. S10†).
 | ||
| Fig. 2 The molecular structures of 4 (a), 5 (b), 6 (c) and coordination modes of the carbamate ligand (d). Hydrogen atoms were omitted for clarity. Ar = 2,6-di(i-propyl)phenyl. | ||
The binuclear compound 5 was formed through the reaction of two molecules of 3 and three equivalents of CO2, thus its molecular structure contains three carbamate and one amidinate ligands (Fig. 2b). Both metal centres are in the +II oxidation state (see 57Fe Mössbauer spectroscopy analysis), and the distance between them is long (over 3.3 Å), which clearly excludes any effective Fe–Fe interactions.37 Two carbamate and one amidinate anions bridge Fe centres, and the additional O,O′-chelating carbamate anion and THF molecule are coordinated in the axial positions, differentiating both metal centres. Thus, the Fe1 centre adopts the coordination sphere geometry between trigonal bipyramidal and tetragonal pyramidal involving four O- and one N-donor atoms, while Fe2 has a tetrahedral coordination environment with three O and one N atoms (Table S14†). The metal-donor atom bonds at the four-coordinated Fe2 centre are significantly shorter than those at the five-coordinated Fe1 centre (for the Fe–O bonds 1.986–2.090 Å vs. 2.046–2.273 Å, for the Fe–N bonds 1.993(2) vs. 2.065(2) Å); detailed geometric parameters for 5 are listed in ESI, Table S10.†
The molecular structure of 6 (Fig. 2c) resembles that observed for compound 5 and contains one terminally chelating carbamate ligand to the Fe1 centre, three bridging carbamates, and the DippFH molecule coordinated as a neutral ligand to the Fe2 centre; such terminal coordination mode was commonly observed for neutral formamidine ligands in transition and main group metals’ complexes.39–41 Similarly to 4 and 5, the significant metal–metal interactions are precluded as the Fe–Fe distance is 3.686(1);37 for detailed geometric parameters for 6 see Table S12.† Both Fe(II) centres have different coordination sphere geometries, close to trigonal bipyramidal and tetrahedral for Fe1 and Fe2, respectively (Table S18†). The Fe–O bonds at bridging ligands are only slightly shorter at the four-coordinate Fe2 centre than those at the five-coordinate Fe1 centre (1.992–2.001 Å vs. 2.007–2.056 Å), and the longest Fe–O distances (2.072(2) and 2.171(2) Å) correspond with the chelating ligand at Fe1 centre. All the lengths of Fe–O and Fe–N bonds in 5 and 6 are consistent with other multinuclear iron(II) complexes containing carbamate or amidinate ligands.1,35,42 A thorough analysis of the FTIR spectra of 5–6 (Fig. S5 and 6†) is featureless because the observed bands derived from carboxylate and amidinate groups overlap each other in the range of 1665–1544 cm−1.
For additional insight into the electronic environment of diiron compounds 5 and 6, the 57Fe Mössbauer spectroscopy measurements were performed; spectra were collected at 80 K (Fig. 3). Complex 5 has two distinct iron centres, so we attempted to fit the spectrum with two quadrupole doublets, but in this way, the contributions of two spectral components were far from equal. The spectrum is best fitted with three components with isomer shifts δ in a range of 1.08–1.40 mm s−1 and quadrupole splitting parameters ΔEQ of 2.3–2.9 mm s−1. These hyperfine parameters are typical for high-spin Fe(II) centres. The occurrence of three spectral components may be explained by the existence of two forms of compound 5 in the measured sample – THF ligand could partially dissociate while drying the sample in the vacuum. To support this hypothesis, we performed TGA for compound 5 (Fig. S19†). The weight loss in the range of 140–155 °C is 4.8%, similar to one THF molecule share (ca. 4.1%) in 5. The subsequent weight loss (5.2%) in the range of 155–190 °C may be correlated with a partial CO2 release. Following the assumption of the THF molecule dissociation, the quadrupole doublet at δ = 1.19 mm s−1 (ΔEQ = 2.90 mm s−1) can be assigned to fifth-coordinate Fe1 centre (46% contribution), and the two doublets at δ = 1.08 mm s−1 (ΔEQ = 2.30 mm s−1) and δ = 1.40 mm s−1 (ΔEQ = 2.77 mm s−1) correspond with Fe2 centre, with and without THF ligand (together 54% contribution). The spectrum of 6 is best fitted by two quadrupole doublets at δ = 1.21 mm s−1 (ΔEQ = 3.00 mm s−1) and δ = 1.24 mm s−1 (ΔEQ = 2.61 mm s−1); these parameters clearly suggest the presence of two high-spin Fe(II) centres, and proving the occurrence of neutral formamidinate ligand at Fe2 centre. Both fifth-coordinate Fe centres in complexes 5 and 6 have similar coordination environments, so, by analogy, the doublet at δ = 1.21 mm s−1 for complex 6 can be assigned to the Fe1 centre – the replacing one oxygen atom with nitrogen increased the δ by 0.02 mm s−1 and the ΔEQ by 0.1 mm s−1.
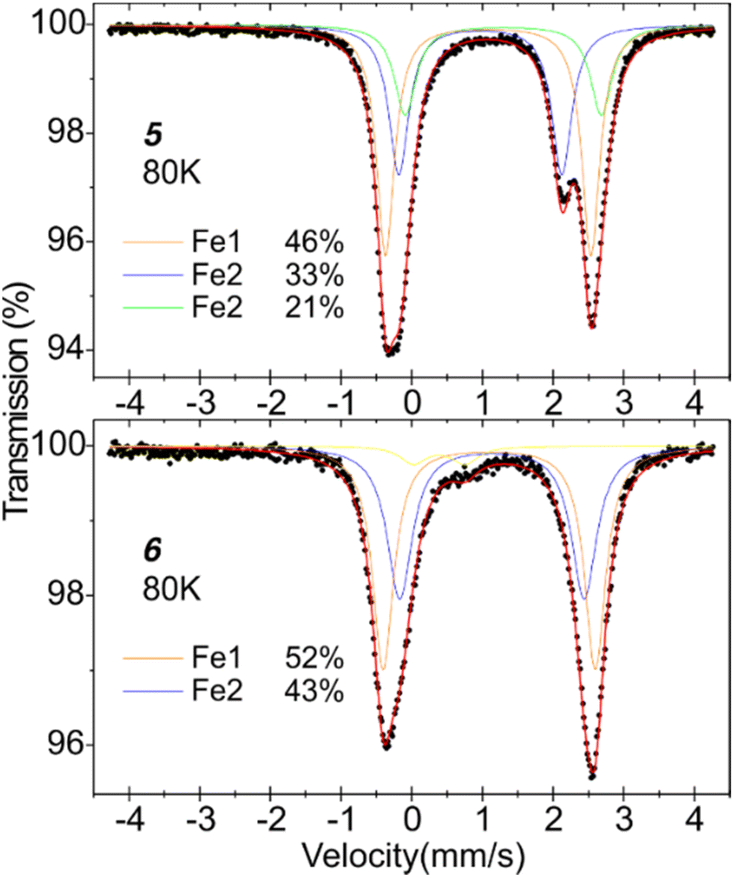 | ||
| Fig. 3 57Fe Mössbauer spectra for 5 and 6 (at 80 K). The Mössbauer parameters δ(ΔEQ) in mm s−1 of the individual spectral components are as follows: [for 5] 1.19(2.90) (orange line), 1.08(2.30) (blue), 1.40(2.77) (green); [for 6] 1.21(3.00) (orange), 1.24(2.61) (blue). The yellow line at the chart below corresponds to Fe(III) impurity with 0.50(0.73). | ||
Conclusions
We performed a case study on the reactivity of monomeric V(II) and Fe(II) bis(amidinates) towards CO2. Our investigations demonstrated that these type complexes are easily N-carboxylated at the M–N amidinate-binding in conjunction with an aggregation of the resulting intermediate species. Remarkably, the effectivity of the examined CO2 insertion processes strongly depends on the used solvent's character and the metal centre's coordination sphere geometry. The CO2 insertion proceeded only for the coordinatively unsaturated metal centres. The observed CO2 insertion into the V–N bonds of 2, which resulted in the formation of divanadium tetra(carbamate) compound 4, contrasts with documented by Gambarotta multielectron reductions of CO2 by V(II) and V(III) systems.27–29 The lack of oxidation of the low-valent V(II) centre and the following C–O bond cleavage could seem surprising. This phenomenon was probably caused by the strong nucleophilicity of the N-donor amidinate ligand. Comparative studies with Fe(II) bis(amidinate) 3 also resulted in the formation of binuclear carbamates, and the identity of the isolated products 5 and 6 was determined by the character of the solvent used. The N-carboxylation at the Fe–N amidinate-binding was complete for the reaction in the non-coordinating solvent (toluene) and less effective in THF as a donor solvent. Thus in all cases, the N-carboxylation led to dinuclear complexes, and it seems reasonable that the initial trapping of a CO2 molecule affords intermediate M(amidinate)(carbamate) species featuring smaller steric hindrances at the metal centre, and then they likely form binuclear aggregates vulnerable to the subsequent CO2 insertion processes.Experimental section
General experimental methods
Unless otherwise stated, all reactions involving air- and moisture-sensitive organometallic compounds were conducted under argon atmosphere using standard Schlenk techniques and glovebox techniques (MBraun UniLab Plus; <0.1 ppm O2, <0.1 ppm H2O). All glassware was stored in a 150 °C oven overnight before use. All solvents were purified by passage through activated aluminium oxide (MBraun SPS) and stored over 3 Å molecular sieves. N,N′-Bis(2,6-diisopropylphenyl)formamidine (DippFH) was synthesised according to the literature.43 The potassium salt of a formamidinate (DippFK) was prepared by deprotonating neutral formamidines by KHMDS (Sigma-Aldrich). VCl3 and FeCl2 were purchased from Alfa Aesar; Na(Hg) was purchased from abcr GmbH; CO2 (5.5 purity) was purchased from Air Products.Synthesis of compounds 1–6
Instrumentation
Elemental analysis was performed using an UNICUBE (Elementar Analysensysteme GmbH). Fourier-transform infrared attenuated total reflectance (FTIR-ATR). FT-IR spectra were acquired on Bruker TENSOR II FTIR spectrometer; the samples were transferred under Paratone-N oil. NMR spectra were acquired on Varian Mercury 400 MHz spectrometer at 298 K. 57Fe Mössbauer spectroscopy measurements were performed at 80 K in transmission geometry applying the RENON MsAa-4 spectrometer44 equipped with the LND Kr-filled proportional detector. The He–Ne laser-based interferometer was used to calibrate a velocity scale. A commercial 57Co(Rh) source made by RITVERC GmbH was used. A transmission integral approximation has been applied to fit Mössbauer spectra using the MOSGRAF data processing software suite. The SVT-400 cryostat by Janis Research Inc. was used to maintain the temperature of absorbers. The spectral isomer (centre) shifts δ are reported with respect to the isomer (centre) shift of room temperature α-Fe. The absorbers for Mössbauer measurements were prepared using 9 mg cm−2 of investigated materials.Single crystal X-ray diffraction
The crystals of all complexes were selected under Paratone-N oil, mounted on the nylon loops and positioned in the cold stream on the diffractometer. The X-ray data for complexes 1–6 were collected at 100(2) K on a SuperNova Agilent diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). The data were processed with CrysAlisPro.45 The structures 1 and 3–5 were solved by direct methods using the SHELXT program and were refined by full matrix least-squares on F2 using the program SHELXL.46 The structures 2 and 6 were refined with the OLEX2.refine refinement package using Gauss–Newton minimisation.47 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were added to the structure model at geometrically idealised coordinates and refined as riding atoms. Crystallographic data (excluding structure factors) for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as a supplementary publication. CCDC: 2175762 (1), 2161489 (2), 2161490 (3), 2161491 (4), 2161492 (5), 2161493 (6).†Crystal data for 1–6
Crystal data for 1 (CCDC-2175762†), C38H46N4O2V: M = 641.73, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.479(5) Å, b = 12.620(5) Å, c = 15.960(5) Å, α = 108.573(5)°, β = 92.537(5)°, γ = 107.338(5)°, U = 1707.0(12) Å3, Z = 2, F(000) = 682, Dc = 1.249 g cm−3, μ(Mo-Kα) = 0.329 mm−1, θmax = 26.994°, 7355 unique reflections. Refinement converged at R1 = 0.0462, wR2 = 0.0926 for all data (R1 = 0.0376, wR2 = 0.0867 for 6276 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.034.
(no. 2), a = 9.479(5) Å, b = 12.620(5) Å, c = 15.960(5) Å, α = 108.573(5)°, β = 92.537(5)°, γ = 107.338(5)°, U = 1707.0(12) Å3, Z = 2, F(000) = 682, Dc = 1.249 g cm−3, μ(Mo-Kα) = 0.329 mm−1, θmax = 26.994°, 7355 unique reflections. Refinement converged at R1 = 0.0462, wR2 = 0.0926 for all data (R1 = 0.0376, wR2 = 0.0867 for 6276 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.034.
Crystal data for 2 (CCDC-2161489†), C54H78N4OV: M = 850.19, monoclinic, space group P21/n (no. 14), a = 15.2867(3) Å, b = 14.7719(2) Å, c = 25.4469(4) Å, β = 107.502(2)°, U = 5480.24(17) Å3, Z = 4, F(000) = 1846, Dc = 1.0304 g cm−3, μ(Mo-Kα) = 0.218 mm−1, θmax = 26.50°, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 308 unique reflections. Refinement converged at R1 = 0.0461, wR2 = 0.1218 for all data (R1 = 0.0388, wR2 = 0.1132 for 9702 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 0.8625.
308 unique reflections. Refinement converged at R1 = 0.0461, wR2 = 0.1218 for all data (R1 = 0.0388, wR2 = 0.1132 for 9702 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 0.8625.
Crystal data for 3 (CCDC-2161490†), C50H70FeN4: M = 782.95, monoclinic, space group P21 (no. 4), a = 12.2223(8) Å, b = 16.0549(9) Å, c = 12.3894(9) Å, β = 107.628(7)°, U = 2317.0(3) Å3, Z = 2, F(000) = 848, Dc = 1.122 g cm−3, μ(Mo-Kα) = 0.361 mm−1, θmax = 26.495°, 7617 unique reflections. Refinement converged at R1 = 0.0730, wR2 = 0.1759 for all data (R1 = 0.0674, wR2 = 0.1686 for 7069 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.034.
Crystal data for 4 (CCDC-2161491†), C229H324N16O20V4: M = 3824.78, monoclinic, space group P21/n (no. 14), a = 15.3254(4) Å, b = 48.4643(15) Å, c = 15.4889(2) Å, β = 90.021(2)°, U = 11504.1(5) Å3, Z = 2, F(000) = 4124, Dc = 1.104 g cm−3, μ(Mo-Kα) = 0.219 mm−1, θmax = 26.499°, 18423 unique reflections. Refinement converged at R1 = 0.0930, wR2 = 0.1759 for all data (R1 = 0.0723, wR2 = 0.1644 for 14625 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.068.
Crystal data for 5 (CCDC-2161492†), C221H304Fe4N16O14: M = 3632.18, monoclinic, space group P21/n (no. 14), a = 14.00932(15) Å, b = 64.6462(8) Å, c = 25.1469(3) Å, β = 92.0150(10)°, U = 22760.2(5) Å3, Z = 4, F(000) = 7832, Dc = 1.060 g cm−3, μ(Mo-Kα) = 0.307 mm−1, θmax = 28.249°, 50059 unique reflections. Refinement converged at R1 = 0.0965, wR2 = 0.1490 for all data (R1 = 0.0755, wR2 = 0.1411 for 38802 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.072.
Crystal data for 6 (CCDC-2161493†), C141H204Fe2N10O8: M = 2278.94, monoclinic, space group P21/n (no. 14), a = 21.550(5) Å, b = 29.745(5) Å, c = 22.858(5) Å, β = 92.430(5)°, U = 14639(5) Å3, Z = 4, F(000) = 5144, Dc = 1.0340 g cm−3, μ(Mo-Kα) = 0.251 mm−1, θmax = 26.500°, 30281 unique reflections. Refinement converged at R1 = 0.0736, wR2 = 0.1356 for all data (R1 = 0.0516, wR2 = 0.11987 for 22994 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal 1.0676.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Science Centre, Poland for financial support – grant OPUS 2017/25/B/ST5/02484. 57Fe Mössbauer spectroscopy measurements were performed by AB using equipment of the Mössbauer Spectroscopy Laboratory, Pedagogical University, Kraków, Poland.References
- D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857–3898 CrossRef PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- N. Hazari and J. E. Heimann, Inorg. Chem., 2017, 56, 13655–13678 CrossRef CAS PubMed.
- M. Aresta, Coord. Chem. Rev., 2017, 334, 150–183 CrossRef CAS.
- G. Bresciani, L. Biancalana, G. Pampaloni and F. Marchetti, Molecules, 2020, 25, 3603 CrossRef CAS PubMed.
- U. Bayer and R. Anwander, Dalton Trans., 2020, 49, 17472–17493 RSC.
- E. A. Hamilton, A. F. R. Kilpatrick, Z. R. Turner, D. A. X. Fraser, J.-C. Buffet and D. O'Hare, Dalton Trans., 2021, 50, 4494–4498 RSC.
- U. Bayer, D. Werner, C. Maichle-Mössmer and R. Anwander, Angew. Chem., Int. Ed., 2020, 59, 5830–5836 CrossRef CAS PubMed.
- R. M. Gauld, R. McLellan, A. R. Kennedy, J. Barker, J. Reid and R. E. Mulvey, Chem. – Eur. J., 2019, 25, 14728–14734 CrossRef CAS PubMed.
- K. A. Grice, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS.
- A. Paparo and J. Okuda, Coord. Chem. Rev., 2017, 334, 136–149 CrossRef CAS.
- L. Roy, M. H. Al-Afyouni, D. E. DeRosha, B. Mondal, I. M. DiMucci, K. M. Lancaster, J. Shearer, E. Bill, W. W. Brennessel, F. Neese, S. Ye and P. L. Holland, Chem. Sci., 2019, 10, 918–929 RSC.
- D. Sahoo, C. Yoo and Y. Lee, J. Am. Chem. Soc., 2018, 140, 2179–2185 CrossRef CAS PubMed.
- C. Yoo and Y. Lee, Inorg. Chem. Front., 2016, 3, 849–855 RSC.
- N. S. Labrum, C. Chen and K. G. Caulton, Chem. – Eur. J., 2019, 25, 7935–7940 CrossRef CAS PubMed.
- J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582–14585 CrossRef CAS PubMed.
- P. M. Jurd, H. L. Li, M. Bhadbhade and L. D. Field, Organometallics, 2020, 39, 2011–2018 CrossRef CAS.
- D. Z. Zee, M. Nippe, A. E. King, C. J. Chang and J. R. Long, Inorg. Chem., 2020, 59, 5206–5217 CrossRef CAS PubMed.
- L. Fohlmeister and C. Jones, Aust. J. Chem., 2014, 67, 1011–1016 CrossRef CAS.
- C. T. Saouma, C. C. Lu, M. W. Day and J. C. Peters, Chem. Sci., 2013, 4, 4042–4051 RSC.
- D. L. J. Broere, B. Q. Mercado and P. L. Holland, Angew. Chem., Int. Ed., 2018, 57, 6507–6511 CrossRef CAS PubMed.
- A. R. Sadique, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2008, 47, 784–786 CrossRef CAS PubMed.
- H. Gehring, R. Metzinger, B. Braun, C. Herwig, S. Harder, K. Ray and C. Limberg, Dalton Trans., 2016, 45, 2989–2996 RSC.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- C. C. Mokhtarzadeh, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2017, 56, 10894–10899 CrossRef CAS PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- C. J. Viasus, N. P. Alderman, S. Licciulli, I. Korobkov and S. Gambarotta, Chem. – Eur. J., 2017, 23, 17269–17278 CrossRef CAS PubMed.
- C. J. Viasus, N. P. Alderman, B. Gabidullin and S. Gambarotta, Angew. Chem., Int. Ed., 2018, 57, 10928–10932 CrossRef CAS PubMed.
- C. J. Viasus, B. Gabidullin and S. Gambarotta, Angew. Chem., Int. Ed., 2019, 58, 14887–14890 CrossRef CAS PubMed.
- T. Pietrzak, I. Justyniak, M. Kubisiak, E. Bojarski and J. Lewiński, Angew. Chem., Int. Ed., 2019, 58, 8526–8530 CrossRef CAS PubMed.
- A. Tulewicz, M. Wolska-Pietkiewicz, M. Jędrzejewska, T. Ratajczyk, I. Justyniak and J. Lewiński, Chem. – Eur. J., 2019, 25, 14072–14080 CrossRef CAS PubMed.
- M. K. Leszczyński, D. Kornacki, M. Terlecki, I. Justyniak, G. M. Miletić, I. Halasz, P. Bernatowicz, V. Szejko and J. Lewiński, ACS Sustainable Chem. Eng., 2022, 10, 4374–4380 CrossRef PubMed.
- K. Sokołowski, W. Bury, I. Justyniak, D. Fairen-Jimenez, K. Sołtys, D. Prochowicz, S. Yang, M. Schröder and J. Lewiński, Angew. Chem., Int. Ed., 2013, 52, 13414–13418 CrossRef PubMed.
- F. A. Cotton, L. M. Daniels and C. A. Murillo, Angew. Chem., Int. Ed. Engl., 1992, 31, 737–738 CrossRef.
- K. Korona, M. Terlecki, I. Justyniak, M. Magott, J. Żukrowski, A. Kornowicz, D. Pinkowicz, A. Kubas and J. Lewinski, Chem. – Eur. J., 2022, 28 DOI:10.1002/chem.202200620.
- L. J. Murphy, K. N. Robertson, R. A. Kemp, H. M. Tuononen and J. A. C. Clyburne, Chem. Commun., 2015, 51, 3942–3956 RSC.
- R. H. Duncan Lyngdoh, H. F. Schaefer and R. B. King, Chem. Rev., 2018, 118, 11626–11706 CrossRef CAS PubMed.
- W. H. Bernskoetter, E. Lobkovsky and P. J. Chirik, Angew. Chem., Int. Ed., 2007, 46, 2858–2861 CrossRef CAS PubMed.
- F. A. Cotton, L. M. Daniels, D. J. Maloney, J. H. Matonic and C. A. Murillo, Polyhedron, 1994, 13, 815–823 CrossRef CAS.
- S. H. Oakley, D. B. Soria, M. P. Coles and P. B. Hitchcock, Dalton Trans., 2004, 537 RSC.
- Y. Yamaguchi, K. Ogata, K. Kobayashi and T. Ito, Bull. Chem. Soc. Jpn., 2004, 77, 303–309 CrossRef CAS.
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple bonds between metal atoms, Springer, New York, 2005 Search PubMed.
- C. Loh, S. Seupel, H. Görls, S. Krieck and M. Westerhausen, Eur. J. Inorg. Chem., 2014, 2014, 1312–1321 CrossRef CAS.
- A. Błachowski, K. Ruebenbauer, J. Żukrowski and R. Górnicki, Acta Phys. Pol., A, 2008, 114, 1707–1713 CrossRef.
- Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, IR and 1H NMR data. CCDC 2175762 and 2161489–2161493. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02274e |
| This journal is © The Royal Society of Chemistry 2022 |