
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Curcumin-based ionic Pt(II) complexes: antioxidant and antimicrobial activity†
Rossella
Caligiuri
a,
Giuseppe
Di Maio
a,
Nicolas
Godbert
 a,
Francesca
Scarpelli
a,
Angela
Candreva
ab,
Isabella
Rimoldi
c,
Giorgio
Facchetti
c,
Maria Giovanna
Lupo
d,
Emilia
Sicilia
e,
Gloria
Mazzone
e,
Fortuna
Ponte
e,
Isabella
Romeo
fg,
Massimo
La Deda
ab,
Alessandra
Crispini
a,
Renata
De Rose
h and
Iolinda
Aiello
*ab
a,
Francesca
Scarpelli
a,
Angela
Candreva
ab,
Isabella
Rimoldi
c,
Giorgio
Facchetti
c,
Maria Giovanna
Lupo
d,
Emilia
Sicilia
e,
Gloria
Mazzone
e,
Fortuna
Ponte
e,
Isabella
Romeo
fg,
Massimo
La Deda
ab,
Alessandra
Crispini
a,
Renata
De Rose
h and
Iolinda
Aiello
*ab
aMAT-INLAB, LASCAMM CR-INSTM, Unità INSTM della Calabria, Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, 87036 Arcavacata di Rende, CS, Italy. E-mail: iolinda.aiello@unical.it
bCNR NANOTEC-Istituto di Nanotecnologia UOS Cosenza, 87036 Arcavacata di Rende, CS, Italy
cDipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, 20133 Milan, Italy
dDipartimento di Medicina, Università degli Studi di Padova, 35128 Padova, Italy
eDipartimento di Chimica e Tecnologie Chimiche, 87036 Arcavacata di Rende, CS, Italy
fDipartimento di Scienze della Salute, Università degli Studi “Magna Græcia” di Catanzaro, Campus “S. Venuta”, Viale Europa, 88100 Catanzaro, Italy
gNet4Science Academic Spin-Off, Università degli Studi “Magna Græcia” di Catanzaro, Campus “S. Venuta”, Viale Europa, 88100 Catanzaro, Italy
hLAB CF-INABEC, Dipartimento di Chimica e Tecnologie Chimiche, 87036 Arcavacata di Rende, CS, Italy
First published on 11th October 2022
Abstract
A series of novel cationic curcumin-based Pt(II) complexes with neutral (N^N) ligands and triflate anions as counterions, [(N^N)Pt(curc)]CF3SO3, 1–4, were synthesised and fully characterised. The antioxidant radical scavenging activity of complexes 1–4 was measured spectrophotometrically using DPPH as the internal probe. Computational strategies have been exploited to ascertain the mechanism of antioxidant action of curcumin (H(curc)) and its Pt(II) complexes. Finally, compounds 1–4 were tested in vitro for their growth inhibitory activity against two bacteria (Staphylococcus aureus and Escherichia coli) by the disk diffusion technique at different Pt(II) complex solution concentrations. The effect of the complexation of H(curc) was investigated.
1. Introduction
Metal complexes based on naturally occurring bioactive molecules are recently spreading as pharmaceuticals and many research works are dedicated to the investigation of their biological activities. The interaction of metal ions with coordinating ligands is found to be responsible for the relevant changes in their chemical behaviour and biological properties, resulting in improved chemical stability, increased pharmacological activity, and reduction of side effects.1–5 Moreover, the presence of a metal centre that is able to react with different nucleophiles at the cellular level makes these compounds active as therapeutic tools. Indeed, they are able to modulate different biological processes at once, an aspect otherwise unrealizable by carbon-based molecules or simple encapsulation processes.6Within bioactive metal chelating ligands of natural extraction, particular interest has been devoted to curcumin, [1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptane-3,5-dione] (H(curc)), a lipophilic polyphenol extracted from the powdered rhizome of Curcuma longa L. as the principal colouring agent. Curcumin has been widely used as a spice and cosmetic component in traditional Ayurvedic and Chinese herbal medicine for a very long time.7,8 Consumption of H(curc) has been associated with a plethora of beneficial effects on human health, spanning from anti-inflammatory, antithrombotic, and anti-arthritic to immunomodulatory and neuroprotective abilities.9,10 H(curc) has also been extensively investigated for its antiproliferative, antimetastatic, and antiangiogenic properties.11,12 Furthermore, H(curc) acts as a free radical scavenger and antioxidant.13,14 Its potent activity is correlated to its peculiar structural characteristics, in particular, the position of the hydroxyl groups, the enol form of the diketone moiety, and the extended conjugated structure.15–17 In addition, H(curc) is well known as an antimicrobial agent with a broad spectrum of activity, including against Gram-positive and Gram-negative bacteria, as well as several viruses.18
Thus in the last few decades, the study of the properties and mechanism of action of H(curc) has attracted ever-increasing attention. However, poor solubility, hydrolytic instability, very low bioavailability, and photodegradation are responsible for its still limited use in the pharmaceutical field, despite its promising biological preclinical and clinical effects.19,20 Different strategies have been attempted to overcome these limitations, for example, its encapsulation in nanoparticles or its linkage to liposomes or micelles.21–26 However, H(curc) complexation with a proper metal ion still provides the most fruitful approach to circumvent the challenges related to its low pharmacokinetic profile.27,28 Taking advantage of the presence of the central β-diketo unit, H(curc) can form a large variety of metal chelates of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 metal-to-ligand ratio with various divalent and trivalent metal ions, stabilizing its more resistant monoanionic enolic form and reducing its photodegradation.27,28 Curc-based metal complexes have been widely probed for their anticancer properties, showing improved stability, better bioavailability and greater cytotoxicity in their effectiveness as anticancer agents with respect to pure H(curc).29–42 Despite the well-known antioxidant and antibacterial activity shown by the H(curc) free ligand, the derived curc-based metal complexes have been much less investigated with regard to these potential applications. While only a few curc-based metal complexes have shown better antioxidant properties than pure H(curc), in most cases, the measured antioxidant activity has been found to be comparable to that of the free ligand.29–32 Moreover, to the best of our knowledge, all the investigated metal derivatives are homoleptic complexes with the only exception of one of Cu(II).31 Despite the fact that metal complexes containing antimicrobial agents could display synergistic activity as reported by Song et al. in the case of curc-based rare earth metal complexes, in most cases reported so far, curc-based metal complexes display comparable antibacterial activity with respect to pure H(curc).29,33,34
:1 and 1:2 metal-to-ligand ratio with various divalent and trivalent metal ions, stabilizing its more resistant monoanionic enolic form and reducing its photodegradation.27,28 Curc-based metal complexes have been widely probed for their anticancer properties, showing improved stability, better bioavailability and greater cytotoxicity in their effectiveness as anticancer agents with respect to pure H(curc).29–42 Despite the well-known antioxidant and antibacterial activity shown by the H(curc) free ligand, the derived curc-based metal complexes have been much less investigated with regard to these potential applications. While only a few curc-based metal complexes have shown better antioxidant properties than pure H(curc), in most cases, the measured antioxidant activity has been found to be comparable to that of the free ligand.29–32 Moreover, to the best of our knowledge, all the investigated metal derivatives are homoleptic complexes with the only exception of one of Cu(II).31 Despite the fact that metal complexes containing antimicrobial agents could display synergistic activity as reported by Song et al. in the case of curc-based rare earth metal complexes, in most cases reported so far, curc-based metal complexes display comparable antibacterial activity with respect to pure H(curc).29,33,34
Herein, based on our experience in the study of ionic bioactive Pt(II) complexes,35–38 we designed and studied novel heteroleptic cationic Pt(II) complexes of the general formula [(N^N)Pt(curc)]CF3SO3 with the intention to extend the studies on the potential application of transition metal complexes as antioxidant and antimicrobial curc carrier agents. To achieve ionic derivates that are able to introduce favourable electrostatic interactions and thus improve the bioactive properties of the resulting complexes, (N^N) ligands with different electronic and steric effects [(N^N) = 1,3-diaminopropane (pn), 2-picolylamine (pic), 2,2′-bipyridine (bipy) and 4,4′-dinonyl-2,2′-bipyridine (bipy-C9)] have been selected. In particular, the structural differences of the (N^N) ligands in terms of the flexibility of the six and five-membered chelated rings formed upon the coordination to the Pt(II) metal ion as well as the extension of the aromatic character can be a suitable tool to appropriately tune the lipophilicity of the resulting synthesized complexes. Moreover, the choice to introduce triflate as the counterion, instead of the nitrate anion, which is usually used for the preparation of analogous cationic Pt(II) complexes, relies on the recognition of nitrate as a toxic agent, whereas for the triflate anion, non-clinical toxicological data are still available.39–42 In addition, according to a recent study, the triflate anion could improve the solubility of the cationic species with respect to the nitrate counterpart.43
2. Experimental
2.1. Materials and methods
Curcumin was purchased from Carbosynth and used without further purification as we already reported the highest grade of purity of this supplier with respect to other commercially available sources.44 All other reagents and solvents were purchased from Aldrich Chemical Co. or Alfa Aesar and were used without further purification. Cycloplatinated precursors [(pn)PtCl2], I,45 [(pic)PtCl2], II,46 [(bipy)PtCl2], III,46 and [(bipy-C9)PtCl2], IV47 were synthesised following methods already reported. The Pt(II) complexes 1–4 were synthesised through microwave assisted syntheses that were carried out using a CEM Discover Synthesis Unit (CEM Corp., Matthews, NC) following our previously reported procedure for the preparation of ionic Pt(II) or Pt(IV) complexes.36,48 Melting points were determined using a Leica DMLP polarising microscope equipped with a Leica DFC280 camera and a CalCTec (Italy) heating stage. Elemental analyses were performed using a PerkinElmer 2400 analyser CHNS/O. Conductivity measurements were performed in DMF or MeOH solutions using an InoLab Cond Level 1-720 conductometer equipped with an LR 325/001 immersion cell at ca. 1 mM. The IR spectra (KBr pellets) were recorded using a Spectrum One PerkinElmer FT-IR spectrometer. The 1H NMR spectra were recorded using a Bruker Avance 300 MHz spectrometer in DMSO-d6 with TMS as the internal standard. The 13C NMR spectra were recorded in DMSO-d6 using a Bruker DRX Avance 400 MHz equipped with a non-reverse probe. MS analyses were performed by using a Thermo Finnigan (MA, USA) LCQ Advantage system MS spectrometer with an electrospray ionization source and an ‘Ion Trap’ mass analyser. The MS spectra were obtained by direct infusion of a sample solution in MeOH. Eagle's minimum essential medium (MEM) was purchased from Sigma Aldrich, while trypsin–EDTA, penicillin, streptomycin, sodium pyruvate, non-essential amino acid solution, fetal calf serum (FCS), plates and Petri dishes were purchased from EuroClone. The Pt(II) complexes were dissolved in dimethyl sulfoxide (DMSO) at the maximal concentration of 200 μM. The amount of DMSO did not exceed 0.25% of the culture media volume. The UV/vis absorption spectra were recorded using a PerkinElmer Lambda 900 spectrophotometer using 1 cm path length quartz cuvettes. Stability measurements were carried out in MeOH, DMSO and 10% DMSO/PBS at a concentration of 10 μM registering the absorption spectra of the samples in a 48 h time interval. The samples in 10% DMSO/PBS (phosphate buffered saline in H2O, pH = 7.4) were prepared starting from a DMSO mother solution and then directly diluting in a quartz cuvette to reach the desired concentration. Between measurements, the samples were stored at 25 °C in the dark.2.2. Syntheses
2.3. Cell viability assay
A sulphorhodamine B (SRB) assay was performed to assess cell viability after treatments. 5 × 103 cells per well were seeded in a 96-well tray in triplicate. After 24 h of incubation, the cells were treated with different concentrations of compounds. An SRB assay was performed after 48 h as previously described.492.4. DPPH radical scavenging activity
The antioxidant activity was measured using 2,2-diphenyl-1-picrylhydrazyl (DPPH) as a scavenger adapting a spectrophotometric method already reported.50,51 DPPH is a stable radical chemical species that presents an intense band in the visible region (515 nm) of the absorption spectrum (Fig. S1, ESI†), due to the presence of an unpaired electron, that produces an intense violet colour. Antioxidant molecules are able to acquire the unpaired electron, resulting in a reduction of the 515 nm band absorbance of the probe and a consequent decolouration of the initial solution.50,51 Thus, the antioxidant activity can be spectrophotometrically quantified by measuring the decrease in this band intensity by mixing a known amount of the analyte with the DPPH solution. Various concentrations of each sample solution were investigated (i.e. 10, 20, 30, 40, 50 μM) by mixing them with a fixed concentration of the probe (100 μM) in 3 mL of spectroscopic grade MeOH. Absorbance was measured 3 h after sample preparation to ensure that stationary conditions were reached: during this time, the samples were incubated in the dark at room temperature. Each measure was conducted in triplicate, on three independent preparations. The antioxidant activity for each sample was calculated by determining the decrease in the absorbance (A) of DPPH at 515 nm according to the following equation:where At=0sample is the absorbance at 515 nm of the sample at time t = 0 (obtained by the mathematical sum of the absorption spectra of DPPH and the absorption spectra of the analyte); At=xsample is the absorbance at 515 nm of the sample at the investigated time (3 h). Finally, α is a correction factor, which takes into account a slight decrease in the optical density of the DPPH solution in MeOH (Fig. S1, ESI†), that overestimates the antioxidant activity of each sample:

| α = At=0DPPH − At=xDPPH |
2.5. Computational details
All the calculations were performed using the Gaussian16 software package.53 Geometry optimizations and frequency calculations were carried out by employing the hybrid B3LYP as the exchange and correlation functional54,55 and the standard triple-ζ basis set 6-311++G(d,p) for all the atoms except for platinum, for which the SDD effective core potential and the corresponding split valence basis set were used. The continuum solvation model based on density (SMD)56 was included in all the calculations to simulate the MeOH environment experimentally used, with a dielectric constant of 32.6. The local minima and transition states (TSs) were identified by the number of imaginary frequencies (0 and 1, respectively). The intrinsic reaction coordinate (IRC) calculations were used to verify whether the located transition states are properly connected with the relative minima.57,58 Unrestricted calculations were used for open shell systems, where radicals are involved. No spin contamination was found, the 〈S2〉 value being about 0.750 in all cases. Thermodynamic corrections at 298.15 K were included in the calculation of the relative energies.The absorption spectra were simulated in the MeOH environment within the time-dependent framework of density functional theory. For this purpose, fifty excitation states were computed by using the MN15 exchange and correlation functional59 coupled with the basis sets employed in the optimization calculations.
The O–H bond dissociation enthalpy (BDE), the ionization potential (IP), the O–H proton dissociation enthalpy (PDE), the anion proton affinity (PA) and the electron transfer enthalpy (ETE), accounting for the different oxidation mechanisms described below, were calculated under a MeOH implicit environment at 298.15 K, according to the following expressions:
| BDE = H(Hn−1Antiox˙) + H(H˙) − H(HnAntiox) |
| IP = H(HnAntiox˙+) + H(e−) − H(HnAntiox) |
| PDE = H(Hn−1Antiox˙) + H(H+) − H(HnAntiox˙+) |
| PA = H(Hn−1Antiox−) + H(H+) − H(HnAntiox) |
| ETE = H(Hn−1Antiox˙) + H(e−) − H(Hn−1Antiox−) |
Proton and electron solvation enthalpies computed at the same level of theory were used.60
To predict the pharmacokinetic properties of all the complexes, the SwissADME (https://www.swissadme.ch/) web tool was used. It calculates the adsorption, distribution, metabolism, and excretion (ADME) parameters, pharmacokinetics, bioavailability, and drug-like behaviour. The Brain Or IntestinaL EstimateD permeation method (BOILED-Egg) was used to estimate the solubility, polarity, and lipophilicity of the investigated complexes.61
2.6. Experimental measurement of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) Pow
Pow
RP-HPLC analysis was performed to correlate the hydrophobicity of the complexes with their retention time. The chromatograms were analyzed using a reversed-phase HPLC column (Partisil C18-ODS), at 25 °C, using KI as the internal standard and water/methanol in the ratio of 80/20 as mobile phase in the presence of HCOOH 15 mM (flow rate: 1 mL min−1, λ = 210 nm). The calibration curve was realized in comparison with the reference compounds (cisplatin, carboplatin and oxaliplatin).35,79
2.7. Antibacterial activity
The in vitro antibacterial activity of H(curc) and complexes 1–4 against Gram-positive (Staphylococcus aureus) and Gram-negative (Escherichia coli) bacteria were tested and evaluated using the modified Kirby–Bauer disk diffusion method.62,63S. aureus (DSM 346) and E. coli (DSM 1576) were provided by DSMZ (German Collection of Microorganisms and Cell Cultures, Braunschweig) and grown respectively in 50 mL of DSM medium 92 and DSM medium 1 for 24 h at 37 °C. When the bacterial cultures of each microorganism were in their exponential phase of growth, dilution with sterile 0.8% saline solution was performed to reach a suspension of 108 cells per mL, and then the concentrations were spectrophotometrically determined. Then, 100 μL of bacterial culture suspensions were spread onto the surface of Nutrient Agar (NA) (Beef extract, peptone, sodium chloride and agar) plates. H(curc) and complexes 1–4 were dissolved in DMSO at concentrations of 0.5 μg μL−1 (w/v). The other diluted concentrations used (0.4–0.05 μg μL−1) were prepared by separately diluting them in the culture medium, so that the effective DMSO concentration did not exceed 0.1%. At this concentration, DMSO had no visually observable growth inhibiting effect. Filter paper disks of 6 mm diameter, previously sterilized in an autoclave, were placed on the surface of NA inoculated plates, pressed gently, and directly wet with 10 μL of each concentration of H(curc) and the tested complexes. Wet disks with fresh DMSO were used as the negative control. The inhibition zones were measured after overnight incubation in the dark at 37 °C. Each inhibition zone was measured three times with a calibre to obtain an average value. The antibacterial activity was calculated as a mean of three replicated experiments and all data are presented as mean ±standard deviation (SD). The absence of bacterial growth in the area around the disks was regarded as positive antibacterial activity. The minimal inhibitory concentration (MIC) was approximately deduced considering the lowest concentration at which the compound was still able to produce a transparent halo of inhibition larger than 6 mm (diameter of the disk paper).3. Results and discussion
3.1. Synthesis
The novel series of cationic Pt(II) complexes were synthesized starting from their corresponding precursors [(N^N)PtCl2], I–IV,45–47 through reaction with an equimolar amount of AgCF3SO3 in DMF solution. To the resulting solution containing the in situ formed solvate intermediate, [(N^N)Pt(solv)2][CF3SO3]2, was directly added a solution of the triethylammonium curc salt. The resulting mixture was placed in a microwave oven following the procedure already used in previous works for the synthesis of metal complexes48,64,65 to afford the derivatives [(N^N)Pt(curc)][CF3SO3] 1–4 in a very short reaction time (Scheme 1). | ||
| Scheme 1 Synthetic pathway of the [(N^N)Pt(curc)]CF3SO3 complexes, 1–4. Reagents and conditions: (i) AgCF3SO3, DMF, N2, r.t., dark, overnight; (ii) DMF, H(curc), NEt3, 60 °C, 150 W, 10 min. | ||
Complexes 1–4 were obtained as reddish-brown or orange-yellow powders in moderate yields, similar to already known curc–Pt(II) complexes.39–41 They are stable in the air and soluble in MeOH, DMF and DMSO. All complexes were characterized by the determination of their melting point, elemental analyses, mass spectrometry, molar conductivity, and FT-IR, 1H and 13C NMR and UV/vis spectroscopy. The molar conductivity values in either MeOH (30–50 S cm3 mol−1) or DMF (40–45 S cm3 mol−1), according to complexes 1–4 solubility, confirm the 1:1 electrolytic nature of the synthesized compounds.66 Considering the IR spectra (Fig. S2, S6, S10, and S14, ESI†), the curc coordination is confirmed by the shift in bands predominantly related to the overlapping stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO. Indeed, in the free ligand, these bands are found at 1628 cm−1 and 1603 cm−1 while in complexes 1–4 they are shifted to lower wavenumbers, in particular within the frequency ranges of 1619–1623 cm−1 and 1594–1596 cm−1, respectively.67 The shift of these absorption bands always occurs upon the coordination of curc to a metal centre, due to the redistribution of the electron density in the curc complexes.34,68 Moreover, the characteristic stretching bands associated with the presence of the counterion are an additional confirmation of the complex's formation and its ionic nature. In particular, the intense absorption stretching band at ca. 1270 cm−1 is related to the asymmetric stretching vibration of the SO3− ionic group.69 In the 1H NMR spectra of the Pt(II) complexes (Fig. S3, S7, S11, and S15, ESI†), the characteristic signals of the γ-proton of the methylene group and the methoxy groups of the coordinated curc are shifted to high fields in comparison with those of H(curc), confirming the metal–ligand coordination.67 In particular, a singlet of γ-proton is present in the ppm range of 5.86–6.13 and those of methoxy groups at the ppm value of ca. 3.90. Furthermore, the effective coordination of curc to the Pt(II) fragment is also supported by the shift to the lower field of signals related to the (N^N) ligands. In addition, regarding 13C-NMR, data are in agreement with the Pt(II) complexes analogues already reported.39–41
C and CO. Indeed, in the free ligand, these bands are found at 1628 cm−1 and 1603 cm−1 while in complexes 1–4 they are shifted to lower wavenumbers, in particular within the frequency ranges of 1619–1623 cm−1 and 1594–1596 cm−1, respectively.67 The shift of these absorption bands always occurs upon the coordination of curc to a metal centre, due to the redistribution of the electron density in the curc complexes.34,68 Moreover, the characteristic stretching bands associated with the presence of the counterion are an additional confirmation of the complex's formation and its ionic nature. In particular, the intense absorption stretching band at ca. 1270 cm−1 is related to the asymmetric stretching vibration of the SO3− ionic group.69 In the 1H NMR spectra of the Pt(II) complexes (Fig. S3, S7, S11, and S15, ESI†), the characteristic signals of the γ-proton of the methylene group and the methoxy groups of the coordinated curc are shifted to high fields in comparison with those of H(curc), confirming the metal–ligand coordination.67 In particular, a singlet of γ-proton is present in the ppm range of 5.86–6.13 and those of methoxy groups at the ppm value of ca. 3.90. Furthermore, the effective coordination of curc to the Pt(II) fragment is also supported by the shift to the lower field of signals related to the (N^N) ligands. In addition, regarding 13C-NMR, data are in agreement with the Pt(II) complexes analogues already reported.39–41
3.2. UV/vis spectroscopy
The absorption spectra of H(curc) and 1–4 were recorded for all samples in MeOH diluted solutions (Fig. 1). The H(curc) absorption spectrum shows a low intensity band in the UV region with the maximum peak at 258 nm and a broad and intense absorption band in the visible portion of the electromagnetic spectrum with the maximum peak centred at 420 nm and a shoulder peak at 432 nm, both attributed to the ππ* transitions of its enolic form.70,71 | ||
| Fig. 1 Absorption spectra of H(curc) (violet) and complexes 1 (blue), 2 (red), 3 (orange) and 4 (red) in MeOH dilute solution [10 μM]. | ||
For a better description of the spectral features of 1–4, two distinct regions of the absorption spectra, i.e., the visible and the UV regions have been separately considered. In the visible region, the shape of the spectra is rather similar for all complexes. This is due to the presence of the curc–Pt electronic transitions that generate a large double-peaked band with a shoulder centred around 400 nm. The red shifted peak and the shoulder peak around 400 nm are attributed to the ligand-to-metal charge transfer (LMCT) transition from curc to the metallic centre, while the blue shifted peak is due to a ππ* ligand centred (LC) transition on curc. The latter is red shifted with respect to the analogous transition in H(curc) due to the coordination to Pt(II) ion. The only difference observed in the visible region is regarding the exact position of this band. For complexes 3 and 4, a red shift of the band, with respect to the analogous transitions of 1 and 2, is observed, which is attributed to the rigidity imparted by the presence of the bipy and bipy-C9 ligands, respectively. The simulated spectra (Fig. S18 and Table S1, ESI†) and the corresponding natural transition orbitals (NTOs, Fig. S19, ESI†) confirm the assignment of the lowest energy band for free ligand H(curc) and Pt(II) cationic complexes 1–4. The broad and intense bands observed for all the complexes around 450 nm are LMCT in character and involve the HOMO and LUMO; they are red-shifted because of the evident participation of the metal in the NTOs (Fig. S11, ESI†). Whilst the transition around 400 nm (computed to be around 360 nm) starts from the HOMO−1 orbitals thus becoming of the LC type, though a little participation of MLCT can be observed for all the complexes.
In the UV region, the spectra show differences due to the LC electronic transitions localized on the different N^N ligands. For complexes 1 and 2 only the ππ* LC transition of curc, centred respectively at 260 nm and 265 nm, is observed. The slight red-shift of this transition in 2 is caused by the greater rigidity imparted by the pic ligand with respect to the pn one. As expected, the non-aromatic N^N ligand in 1 does not take part in the electronic transitions and the band corresponds to the excitation from the primarily metal d-orbital (HOMO−1) with the population of the ligand-localized orbital (LUMO+1) generating an absorption band of MLCT character, though a considerable part of the CT is centred on the curc ligand in this case as in the other. On the other hand, the LUTO associated with such a transition in 2 shows the evident participation of the aromatic portion of the pic ligand into the orbital (Fig. S19, ESI†). Finally, for complexes 3 and 4, the broad and destructured band observed at low wavelengths is attributed to a combination of LC transition on curc and bipy aromatic rings. The typical ππ* electronic transitions on bipy aromatic rings are also observed at 305 and 320 nm.31 Indeed, the computed band originate from the electronic transitions centred on the bipy ligand as well as from the CT from orbitals primary localized on curc with the population of orbitals centred on the bipy ligand.
3.3. Stability
Complexes 1–4 and H(curc) show a pale-yellow colour both in MeOH and DMSO dilute solutions. In MeOH, they are stable in the time laps of 48 h. The absorption spectra retained the same shape with no significant variations in the intensity band ratio (Fig. S20–S24, ESI†). The slight variations in absorption intensity observed for complexes 1, 2, and 3 are probably due to their negligible crystallization taking place over time rather than effective degradation. These variations are minimal at the wavelength of 515 nm eventually proving that the acquired data for antioxidant activity (see section 3.4) are only dependent on the scavenging activity of the analytes. To study the behaviour of complexes 1–4 with respect to H(curc) under biological pH conditions, their stability in DMSO and an aqueous solution of 10% DMSO/PBS was also evaluated. The absorption spectra of all samples over time show only slight variations in intensity, with no significant variations in the interband ratio, again attributed to negligible crystallization event (Fig. S25–S29, ESI†).72 When diluted with PBS aqueous solution, a colour variation from yellow to pink was observed for all the samples. This variation is more visible for complexes 1–4 than H(curc). Comparing the spectra registered in DMSO and in PBS solution (Fig. S30–S34, ESI†) at t = 0 h two aspects can be highlighted. The lower intensity of the spectra and the greater destructuration with respect to their analogues in DMSO solution are due to the worst solubility of the samples in an aqueous environment. The presence of a new absorption band (centred above 540 nm for complexes 1–4 and 530 nm for H(curc) and responsible for the pink coloration of the buffer) is attributed to the weak deprotonation of the H(curc) hydroxyl groups, due to the slight alkalinity of the buffer solution.73 Considering the absorption spectra over time in DMSO/PBS solution, different behaviour for complexes 1–3 than 4 and H(curc) were observed. H(curc), as known, experienced strong degradation under biological pH conditions. This is proved by the strong intensity reduction of the spectra with increasing time and by the dramatic variation of the ratio between the absorption bands centred at 365 and 430 nm, already observed after 1 h in the DMSO/PBS mixture. This ultimately results in the complete discoloration of its solution after 48 h (Fig. S30, ESI†). It is believed that the degradation of H(curc) goes through an autoxidation mechanism. Reacting with O2 the main degradation product is a bicyclopentadione formed by the double cyclization of the heptadienedione chain between the two methoxyphenol rings.74 This cyclization causes the cleavage of the strongly conjugated heptadienedione chain, responsible for the strong reduction of the absorption maximum intensity (80%). Interestingly, this strong reduction of the absorption band and the consequent variation of the interband ratio already after 1 h were only partially observed in complex 4; They were not observed at all for complexes 1–3. For complex 4 after t = 3 h a strong reduction in absorption intensity was observed (of about 50% on the absorption maximum), accompanied by a variation in the ratio between the three absorption peaks. From this point on, only a weak reduction in the intensity of the entire spectrum was observed, suggesting the stabilization of the sample with the only crystallization event (confirmed by the observation of the observed presence of a powder at the bottom of the cuvette). Conversely, complexes 1–3 did not undergo any degradation. Only a reduction in intensity over time was observed with no variations in the interband ratio, attributable to the worst solubility in the aqueous solution that induces partial crystallization of the powders over time. Thus, we can conclude that the complexation of curc with Pt(II) and the various (N^N) ligands improves its stability in biological pH conditions. This is true in particular for complexes 1–3 that did not show any degradation, and from this point of view are more suitable for their application in biological fields.3.4. Antioxidant activity
It is well known that Pt(II) metal complexes are toxic for humans and, thanks to their capability in particular to reduce the vitality of the cancer cell lines, are often used as antitumor agents.75 Considering the possibility to utilize Pt(II) complexes for treating alternative diseases than cancer, their potential antiproliferative effects must be evaluated. Complexes 1–4 are not cytotoxic with IC50 values up to 200 μM against one breast cancer cell line (MDA-MB-231),49,76 thus opening the possibility of using them as antioxidant and antibacterial agents.The antioxidant activity of complexes 1–4 was evaluated spectrophotometrically (see the Experimental section), and compared with that of H(curc). The % antioxidant activity plotted against the concentration is reported in the histogram presented in Fig. 2. The data follow a sigmoidal curve, i.e., the antioxidant activity increases monotonically with concentration, with slow growth when the data approach 0 and 100%. The corresponding IC50 value and the correlation coefficient (i.e., R2) extrapolated from the plot are reported in Table 1. The R2 coefficient of determination approaches unity for all the curves, confirming the goodness of the model adopted. The value of IC50 for H(curc) of 24 ± 2 μM is found similar to that reported by Sökmen et al. (8.4 μg mL−1 that corresponds to 22.8 μM), while Halevas et al. obtained an IC50 of 16.6 ± 2 μM.31,77 This discrepancy could be due to the choice of fitting the data with a linear function rather than a non-linear equation. The IC50 values for complexes 1–4 are all within the limit of the experimental errors and comparable with that of H(curc) (Table 1). A similar behaviour was already observed for a Cu(II) complex with the same ligands as complex 3.31 Thus, both the complexation with Pt(II) and the presence of different (N^N) ligands left the antioxidant properties of H(curc) unchanged. However, the best bioavailability of 1–3 evidenced by stability measurement in 10% DMSO/PBS (see section 3.3) makes these new curc-based metal complexes more suitable as antioxidant agents.
 | ||
| Fig. 2 Antioxidant activity of H(curc) and complexes 1–4 as a function of concentration. | ||
| Compound | H(curc) | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| IC50 (μM) [correlation coefficient, R2] | 24 ± 2 [0.9941] | 23 ± 1 [0.9992] | 27 ± 1 [0.9995] | 26 ± 3 [0.9891] | 26 ± 1 [0.9987] |
To get a deep insight into the mechanism of action of 1–4 as antioxidants, computational tools were exploited. A primary antioxidant like H(curc), generally indicated as HnAntiox, can prevent oxidation by acting as a free radical scavenger. The main mechanisms by which the antioxidant activity can be exerted are essentially three:
(i) hydrogen atom transfer (HAT)
| HnAntiox + ˙R → Hn−1Antiox˙ + RH |
| HnAntiox + ˙R → HnAntiox+˙ + R− |
| HnAntiox+˙ → Hn−1Antiox˙ + H+ |
| HnAntiox → Hn−1Antiox− + H+ |
| Hn−1Antiox− + ˙R → Hn−1Antiox˙ + R− |
Hydrogen abstraction (HAT) by the free radical ˙R to form the HnAntiox˙ radical occurs in one single step. Instead in the SET-PT mechanism, first the radical cation HnAntiox+˙ is formed via electron transfer from the antioxidant to the free radical, which in turn deprotonates yielding the HnAntiox˙ radical. In addition, in the SPLET mechanism, two consecutive steps have to take place: the deprotonation of the phenolic compound and the subsequent electron transfer from the phenoxide anion to ˙R to form the HnAntiox˙ radical. In both the stepwise mechanisms, SET-PT and SPLET, the RH is formed in the last step of the whole reaction mechanism (R− + H+ → RH).
The ability of a compound to act as an antioxidant can be predicted by computing some of its intrinsic properties, accounting for the different mechanistic hypotheses described above: (i) bond dissociation enthalpy (BDE), (ii) adiabatic ionization potential (IP) and O–H dissociation enthalpy (PDE), (iii) proton affinity (PA) and electron transfer enthalpy (ETE) for the three reaction mechanisms, respectively.
On the basis of calculations previously reported on H(curc), the main mechanism considered viable for H(curc) and its derivatives is the HAT one.78,79 It involves the direct transfer of an H atom from the hydroxyl group of the antioxidant to the free radical. Accordingly, among the descriptors cited above, BDE represents the best reliable thermochemical parameter to describe such a mechanism. However, in order to ascertain the mechanism through which even the curc-based Pt(II) complexes 1–4 can exhibit the antioxidant activity, all the descriptors were estimated. For the sake of comparison, the same parameters were computed even for neutral H(curc), the most abundant species at physiological pH,80 simulated in its enol form, as it is reported to be more stable than the keto one.78 The outcomes of these calculations are shown in Fig. S27.† These data show that homolytic breaking of the H(curc) O–H bond requires the lowest amount of energy to take place, as both IP and ETE values, accounting for the rate determining steps of SET-PT and SPLET, respectively, entail a greater amount of energy for such reactions to occur. Accordingly, it can be concluded that the HAT mechanism remains the preferred pathway through which the curc bound to Pt(II) centre in 1–4 exerts its antioxidant activity. In particular, looking at the BDE values computed for the complexes (reported in Table 2), it appears evident that the radicalization of the hydroxyl group of the curc ligand requires essentially the same amount of energy expense in all cases, similar to that of the free ligand. Moreover, as the lowest BDE (weakest O–H bond) is expected to lead to the greatest antioxidant activity, comparing the computed BDE values with that of the other antioxidant molecules, the hydrogen transfer should occur rather fast. Even looking at the band gap (Fig. 3) no significant differences were found between H(curc) and its ionic Pt(II) complexes, though both the HOMO and LUMO are stabilized by the complexation. The HOMO plots of the complexes show the typical π-like molecular orbital characteristics of the curcumin ligand, whilst the LUMO plots show the implications of the metal centre in all the cases even if outspread on the whole curc π system.
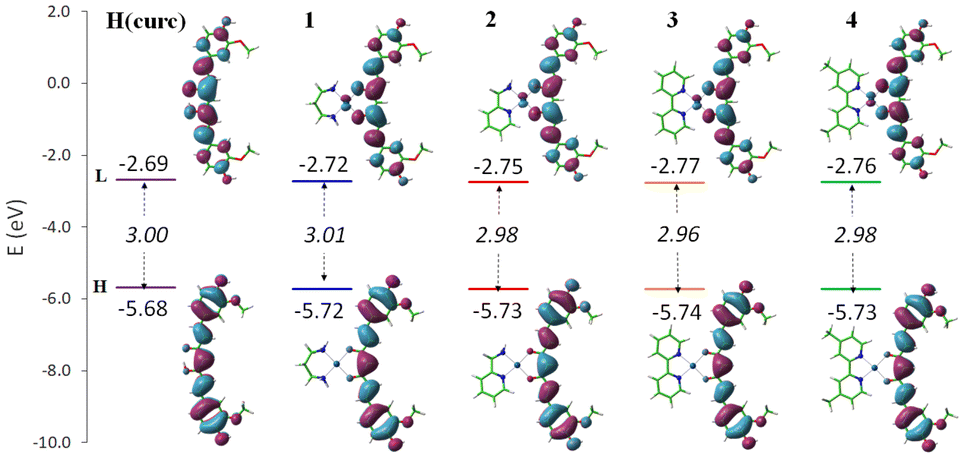 | ||
| Fig. 3 Band gap (eV) in italic and the plots of frontier molecular orbitals, HOMO (H) and LUMO(L). | ||
| H(curc) | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| BDE | 76.3 | 76.6 | 76.7 | 76.7 | 76.7 |
| ΔGf | −9.8 | −10.0 | −9.7 | −9.6 | −9.7 |
| ΔG‡ | 13.5 | 12.8 | 13.6 | 12.9 | 13.4 |
Therefore, the computation of the thermochemical parameters allows to ascertain that the H(curc) HAT mechanism remains the preferred one even upon Pt(II) complexation. The free radical scavenging activity was further explored by analysing the HAT mechanism even from a kinetic point of view. This was performed by taking into consideration the hydroperoxyl radical (HOO˙) as the reactive oxygen species to be quenched. Peroxyl radicals (ROO˙) are, indeed, the major compounds causing oxidative stress in biological systems.81 Moreover, since they display low to moderate reactivity, they represent a proper choice for exploring the antioxidant activity of the ionic Pt(II) complexes. The HAT mechanism was, thus, simulated by the quenching of the smallest member of the peroxyl family, HOO˙, as it plays a key role in the toxic side effects accompanying aerobic respiration.82 The relative activation and formation of free energies are included in Table 2.
From these data it emerges that the exergonicity of the HAT reaction obtained for the radical quenching by curcumin or its complexes remains in a range of less than 1 kcal mol−1. Similarly, the energy barrier that has to be overcome for H abstraction from H(curc) is essentially the same whether or not curcumin is coordinated to the metal centre. The energy expense of no more than 10 kcal mol−1 makes the antioxidant particularly active in quenching the free radical. The obtained results for H(curc) are in agreement with a very recent study on H(curc) and its analogue behaviour in the gas phase and H2O environment, for which the activation energy was computed to be in the range of 9.9–13.3 kcal mol−1.79
Furthermore, in order to predict the pharmacokinetics of the curc–metal based compounds, their ADME physiochemical descriptors were estimated. As it is known, the path of each administered compound in the body relies on the physiological factors that regulate its absorption, distribution, metabolism and excretion (ADME). We considered lipophilicity, size, polarity, solubility, and the ability to satisfy the drug-likeness criteria of these Pt(II) complexes. The obtained results are collected in Table S2 in the ESI.† All the complexes have only one violation of Lipinski's rule of five associated with a high MW, the value ranging from 636.75 to 971.11, with the exception of complex 4 for which an additional violation related to logPow was found. The investigated complexes show the following properties: H-bond acceptors = 6, 2 < H-bond donors < 4, −0.32 < logPow < 8.01. The logPow value, used to denote the permeation, of the compounds to have a reasonable probability of being well absorbed, must not be greater than 5.0. Therefore, complexes 1–3 could be able to better cross the cell lipid bilayer membrane to reach the site of action, characterized by increased lipophilicity as a function of the (N^N) ligand size increase. This is indeed asserted by the experimental determination of the logPow which follows the same trend than the computed one (Table S3, ESI†). A BOILED-Egg diagram, that represents an intuitive model to predict the passive human gastrointestinal absorption (HIA) and the blood–brain barrier (BBB) permeation, is built-up using the additional data extracted from the ADME analysis. Such a diagram (Fig. S36 in ESI†) indicates that 1–3 (in the white region) can be passively absorbed by the gastrointestinal tract and are predicted to be a substrate for the efflux transporter P-glycoprotein from the central nervous system.
3.5. Antimicrobial activity
In the investigation of the antimicrobial activity of complexes 1–4, the nature of the N-donor ligands and their chelation effects should be evaluated. Moreover, the molecular structure, the charge and the presence and nature of the counterions all have their influence on this activity.83The antibacterial activity of 1–4 against E. coli and S. aureus are tested using H(curc) and DMSO as the positive and negative controls, respectively. The complexes are solubilised in DMSO at different concentrations in the range of 0.05–0.5 μg μL−1. The antibacterial activities expressed as the diameter of growth inhibition area in mm with the subtraction of paper disk diameter (6 mm), are listed in Table S3, ESI.† The inhibition halos of complexes 1–4 and H(curc) at maximum concentration value for both microorganisms are reported in Fig. S37.†
The presented data clearly show that, in the concentration range considered, each Pt(II) complex and H(curc) exhibit varying degrees of inhibitory effect on the growth of the tested bacterial species. DMSO did not display any bacterial asset, confirming that the observed inhibitory effects are attributable only to the Pt(II) complexes or H(curc). Complexes 1–4 exhibit antibacterial activity against both Gram-positive and Gram-negative microorganisms with a trend depending on concentration, but with differences related to the diversity of the different (N^N) ligands coordinated to the metal centre ion. As regards E. coli, the bacterial growth inhibition of complexes 1–4 increases with the increase in the aromaticity of the (N^N) ligands and their overall rigidity as highlighted by the increase in the bacterial inhibition area diameter measured at various concentrations (Fig. 4). Noteworthy, in E. coli all the Pt(II) complexes perform better than H(curc). This increased activity with respect to H(curc) could be explained on the basis of Tweedy's chelation theory for which the polarity of the ligand and the central metal ion are reduced through charge equilibration over the whole chelated ring.84 The sharing of the positive charge of the metal centre with the donor groups of coordinated ligands and the π-electron delocalization on the aromatic rings increases the lipophilic character of the metal complex and hence its liposolubility. These features favour the permeation of metal chelates through the lipidic layer of the bacterial membranes, causing the death of the organism.85 It is worth noting that the lipid membrane surrounding the cells favours the passage of lipid-soluble agents, an important factor controlling the antimicrobial activity (Overton's concept). Moreover, the data obtained from the antimicrobial activity with respect to E. coli are in accordance with the theoretically calculated lipophilicity of complexes 1–4 (Table S2, ESI†). Indeed, the induced increase in lipophilicity should enhance the penetration of the complexes into the lipid membranes, and eventually blocks the metal binding sites in the enzymes of microorganisms. Remarkably, this trend is common for all diluted concentrations considered even for 0.05 μg μL−1 while at this concentration H(curc) does not show any activity (Fig. 4).
 | ||
| Fig. 4 The trend of bacterial inhibition area diameter against E. coli at various concentrations. | ||
As regards S. aureus, data suggest that all complexes 1–4 and H(curc) are less active than in E. coli as is evident from Fig. 5, Table S4,† and Table 3. Accordingly, the estimation of MIC (minimum inhibitory concentration) shows that lower amounts of the complexes are needed to inhibit E. coli than S. aureus, demonstrating a greater sensitivity of E. coli to these complexes (Table 3).
 | ||
| Fig. 5 The trend of bacterial inhibition area diameter against S. aureus at various concentrations. | ||
Although the antibacterial activity of all complexes against this microorganism is lower than that measured against E. coli, the inhibitory effect is once again higher than the free ligand which reduces its activity until 0.3 μg μL−1. In terms of halos of inhibition, the trend 4 > 3 > 2 > 1 observed for E. coli is almost confirmed also for S. aureus, except for complex 2 which shows a higher resistance with respect to 1. Thus, probably, in the case of S. aureus, other mechanisms, that go beyond the lipophilic parameter, have to be taken into consideration. In any case, the non-compliant behaviour of complex 2 is still better than H(curc) at low concentrations (0.2 μg μL−1) because of the loss of the activity of the free ligand at that value.
4. Conclusions
With the aim to develop new curc–metal based compounds with multi-functional biological properties, nominally antioxidant and antimicrobial, new cationic curcumin–Pt(II) complexes, of the general formula [(N^N)Pt(curc)]CF3SO3, 1–4, were synthesized and fully characterised. Different (N^N) bidentate ligands (pn, pic, bipy and phen) were used to evaluate the influence on both the physical and biological properties of the derived Pt(II) complexes. From the photophysical characterization data, under physiological pH conditions, the coordination of H(curc) to the Pt(II) ion in complexes 1–4 appears evident, implying consistent structural modifications of free H(curc) and inducing its stability, albeit with some differences related to the (N^N) chelated unit. Indeed, complexes 1–3 did not show any degradation under these conditions, overcoming the major drawbacks of H(curc) related to poor bioavailability and easy oxidability and photodegradability. Otherwise, the presence of substituents on the 2,2′-bipyridine ligand results in the destabilization of the derived complex 4 which, in the biological environment, undergoes a certain level of degradation. Once the absence of cytotoxic activity against one breast cancer cell line (MDA-MB-231) is tested, the antioxidant properties of complexes 1–4 were evaluated spectrophotometrically and compared with that of H(curc). As a result, the antioxidant activity of H(curc) remains unchanged upon coordination to the Pt(II) ion, with no differences related to the different (N^N) chelating ligands. Moreover, the same mechanism of antioxidant action is confirmed by both experimental and theoretical studies. For better stability under biological conditions (at least in the case of complexes 1–3) and their antioxidant activity, these new curc-based metal complexes are therefore more suitable antioxidant agents with respect to H(curc).Finally, complexes 1–4, showed enhanced antimicrobial activity with respect to pure H(curc), as revealed by the growth inhibition of E. coli and S. aureus microorganisms even at low concentrations. The antibacterial activity is found to be concentration dependent and related to the nature of the (N^N) chelating ligands. Indeed, the bacterial growth inhibition of complexes 1–4 increases with the increase in the aromaticity of the (N^N) ligands, which implies an increase of lipophilicity, as confirmed through theoretical calculations.
Concluding the complexation of H(curc) to Pt(II) metal ions for the formation of the new ionic [(N^N)Pt(curc)]CF3SO3 complexes appears to be a successful way to obtain photochemical metal-based curc carrier antioxidant agents and more efficient antimicrobial species. Moreover, the careful choice of the ancillary (N^N) ligands is able to modulate the lipophilicity properties of the derived Pt(II) complexes, necessary for the enhancement of the antibacterial activity.
Author contributions
R. C. and G. D. M. contributed equally to this work as first authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the project PON “Ricerca e Innovazione” 2014-2020–Progetto STAR 2–PIR01_00008 funded by MIUR (Ministero dell'Università e della Ricerca) and the project PON ARS01_00401, “Sviluppo di tecnologie di materiali e di tracciabilità per la sicurezza e la qualità dei cibi—Demetra”. I. A. is grateful to Alessio Giovinazzo for his contribution to the preparation of the selected complexes and to Prof. Nicola Ferri for the cytotoxic activity measurements, respectively.References
- A. K. Renfrew, Metallomics, 2014, 6, 1324–1335 CrossRef CAS PubMed.
- K. Karami, Z. Mehri Lighvan, H. Farrokhpour, M. Dehdashti Jahromi and A. A. Momtazi-borojeni, J. Biomol. Struct. Dyn., 2018, 36, 3324–3340 CrossRef CAS PubMed.
- P. Štarha and Z. Trávníček, Coord. Chem. Rev., 2019, 395, 130–145 CrossRef.
- B. Sanz Mendiguchia, I. Aiello and A. Crispini, Dalton Trans., 2015, 44, 9321–9334 RSC.
- M. G. A. El-Wahed, M. S. Refat and S. M. El-Megharbel, J. Mol. Struct., 2008, 892, 402–413 CrossRef CAS.
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS PubMed.
- S. C. Gupta, B. Sung, J. H. Kim, S. Prasad, S. Li and B. B. Aggarwal, Mol. Nutr. Food Res., 2013, 57, 1510–1528 CrossRef CAS PubMed.
- Z. Stanić, Plant Foods Hum. Nutr., 2017, 72, 1–12 CrossRef PubMed.
- S. Hewlings and D. Kalman, Foods, 2017, 6, 92 CrossRef PubMed.
- B. Salehi, Z. Stojanović-Radić, J. Matejić, M. Sharifi-Rad, N. V. Anil Kumar, N. Martins and J. Sharifi-Rad, Eur. J. Med. Chem., 2019, 163, 527–545 CrossRef CAS PubMed.
- M. Urošević, L. Nikolić, I. Gajić, V. Nikolić, A. Dinić and V. Miljković, Antibiotics, 2022, 11, 135 CrossRef PubMed.
- K. Bairwa, J. Grover, M. Kania and S. M. Jachak, RSC Adv., 2014, 4, 13946 RSC.
- T. Ak and I. Gülçin, Chem.-Biol. Interact., 2008, 174, 27–37 CrossRef CAS PubMed.
- G. K. Jayaprakasha, L. Jaganmohan Rao and K. K. Sakariah, Food Chem., 2006, 98, 720–724 CrossRef CAS.
- K. I. Priyadarsini, D. K. Maity, G. H. Naik, M. S. Kumar, M. K. Unnikrishnan, J. G. Satav and H. Mohan, Free Radicals Biol. Med., 2003, 35, 475–484 CrossRef CAS PubMed.
- Y. M. Sun, H. Y. Zhang, D. Z. Chen and C. B. Liu, Org. Lett., 2002, 4, 2909–2911 CrossRef CAS PubMed.
- L. Shen and H. F. Ji, Spectrochim. Acta, Part A, 2007, 67, 619–623 CrossRef PubMed.
- S. Zorofchian Moghadamtousi, H. Abdul Kadir, P. Hassandarvish, H. Tajik, S. Abubakar and K. Zandi, BioMed Res. Int., 2014, 2014, 1–12 CrossRef PubMed.
- C. D. Lao, M. T. Ruffin IV, D. Normolle, D. D. Heath, S. I. Murray, J. M. Bailey, M. E. Boggs, J. Crowell, C. L. Rock and D. E. Brenner, BMC Complementary Altern. Med., 2006, 6, 4–7 CrossRef PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- Z. Hussain, H. E. Thu, S. F. Ng, S. Khan and H. Katas, Colloids Surf., B, 2017, 150, 223–241 CrossRef CAS PubMed.
- A. C. da Silva, P. D. de F. Santos, J. T. do P. Silva, F. V. Leimann, L. Bracht and O. H. Gonçalves, Trends Food Sci. Technol., 2018, 72, 74–82 CrossRef.
- M. F. S. Jadid, B. Shademan, R. Chavoshi, N. Seyyedsani, E. Aghaei, E. Taheri, P. Goleij, S. Hajazimian, V. Karamad, J. Behroozi, M. N. Sabet, A. Isazadeh and B. Baradaran, Environ. Toxicol., 2021, 36, 1043–1051 CrossRef CAS PubMed.
- A. Zarrabi, A. Zarepour, A. Khosravi, Z. Alimohammadi and V. K. Thakur, Fibers, 2021, 9, 1–17 CrossRef.
- S. Sun, X. Du, M. Fu, A. R. Khan, J. Ji, W. Liu and G. Zhai, Int. J. Pharm., 2021, 595, 120227 CrossRef CAS PubMed.
- M. Bagheri, M. H. Fens, T. G. Kleijn, R. B. Capomaccio, D. Mehn, P. M. Krawczyk, E. M. Scutigliani, A. Gurinov, M. Baldus, N. C. H. Van Kronenburg, R. J. Kok, M. Heger, C. F. Van Nostrum and W. E. Hennink, Mol. Pharm., 2021, 18, 1247–1263 CrossRef CAS PubMed.
- A. Shakeri, Y. Panahi, T. P. Johnston and A. Sahebkar, BioFactors, 2019, 45, 304–317 CrossRef CAS PubMed.
- S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC.
- T. Q. Hieu and D. T. T. Thao, J. Chem., 2019, 2019, 1–8 CrossRef.
- Q. H. Tran and T. T. Doan, New J. Chem., 2020, 44, 13036–13045 RSC.
- E. Halevas, A. Pekou, R. Papi, B. Mavroidi, A. G. Hatzidimitriou, G. Zahariou, G. Litsardakis, M. Sagnou, M. Pelecanou and A. A. Pantazaki, J. Inorg. Biochem., 2020, 208, 111083 CrossRef CAS PubMed.
- L. Gorgannezhad, G. Dehghan, S. Y. Ebrahimipour, A. Naseri and J. E. Nazhad Dolatabadi, J. Mol. Struct., 2016, 1109, 139–145 CrossRef CAS.
- E. H. Ismail, D. Y. Sabry, H. Mahdy and M. M. H. Khalil, J. Sci. Res., 2014, 6, 509–519 CrossRef.
- Y. M. Song, J. P. Xu, L. Ding, Q. Hou, J. W. Liu and Z. L. Zhu, J. Inorg. Biochem., 2009, 103, 396–400 CrossRef CAS PubMed.
- G. Mazzone, S. Scoditti, R. Caligiuri, L. Ricciardi, E. Sicilia, M. G. Lupo, I. Rimoldi, N. Godbert, M. La Deda, A. Ionescu, M. Ghedini, I. Aiello and G. Facchetti, Inorg. Chem., 2022, 61, 7188–7200 CrossRef CAS PubMed.
- L. Ricciardi, M. La Deda, A. Ionescu, N. Godbert, I. Aiello and M. Ghedini, Dalton Trans., 2017, 46, 12625–12635 RSC.
- A. Ionescu, R. Caligiuri, N. Godbert, L. Ricciardi, M. La Deda, M. Ghedini, N. Ferri, M. G. Lupo, G. Facchetti, I. Rimoldi and I. Aiello, Appl. Organomet. Chem., 2020, 34(3), 1–11 CrossRef.
- A. Ionescu, N. Godbert, L. Ricciardi, M. La Deda, I. Aiello, M. Ghedini, I. Rimoldi, E. Cesarotti, G. Facchetti, G. Mazzeo, G. Longhi, S. Abbate and M. Fusè, Inorg. Chim. Acta, 2017, 461, 267–274 CrossRef CAS.
- K. Mitra, S. Gautam, P. Kondaiah and A. R. Chakravarty, Angew. Chem., Int. Ed., 2015, 54, 13989–13993 CrossRef CAS PubMed.
- K. Mitra, S. Gautam, P. Kondaiah and A. R. Chakravarty, Eur. J. Inorg. Chem., 2017, 2017, 1753–1763 CrossRef CAS.
- A. Upadhyay, S. Gautam, V. Ramu, P. Kondaiah and A. R. Chakravarty, Dalton Trans., 2019, 48, 17556–17565 RSC.
- E. A. Thackaberry, Expert Opin. Drug Metab. Toxicol., 2012, 8, 1419–1433 CrossRef CAS PubMed.
- P. Sacchi, L. Loconte, G. Macetti, S. Rizzato and L. Lo Presti, Cryst. Growth Des., 2019, 19, 1399–1410 CrossRef CAS.
- A. A. Abe, C. Oliviero Rossi, P. Caputo, M. P. De Santo, N. Godbert and I. Aiello, Materials, 2021, 14, 1622 CrossRef CAS PubMed.
- J. Josephsen, Inorg. Chim. Acta, 2018, 478, 54–58 CrossRef CAS.
- K. A. Mitchell and C. M. Jensen, Inorg. Chem., 1995, 34, 4441–4446 CrossRef CAS.
- M. Kato and M. Ikemori, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m25–m26 CrossRef PubMed.
- A. Ionescu, N. Godbert, I. Aiello, L. Ricciardi, M. La Deda, A. Crispini, E. Sicilia and M. Ghedini, Dalton Trans., 2018, 47, 11645–11657 RSC.
- I. Rimoldi, G. Facchetti, G. Lucchini, E. Castiglioni, S. Marchianò and N. Ferri, Bioorg. Med. Chem., 2017, 25, 1907–1913 CrossRef CAS PubMed.
- M. S. Blois, Nature, 1958, 181, 1199–1200 CrossRef CAS.
- S. B. Kedare and R. P. Singh, J. Food Sci. Technol., 2011, 48, 412–422 CrossRef CAS PubMed.
- Z. Chen, R. Bertin and G. Froldi, Food Chem., 2013, 138, 414–420 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallin, 2019 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- P. J. Stephen, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, in Theory and Applications of Computational Chemistry: The First 40 Years, 2005, pp. 195–249 Search PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- Z. Marković, J. Tošović, D. Milenković and S. Marković, Comput. Theor. Chem., 2016, 1077, 11–17 CrossRef.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- J. H. Ortez, in Manual of Antimicrobial Susceptibility Testing, ed. M. B. Coyle, 2005, pp. 39–52 Search PubMed.
- A. W. Bauer, M. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 493–496 CrossRef CAS PubMed.
- N. Godbert, T. Pugliese, I. Aiello, A. Bellusci, A. Crispini and M. Ghedini, Dalton Trans., 2007, 32, 5105–5111 Search PubMed.
- F. Scarpelli, A. Ionescu, L. Ricciardi, P. Plastina, I. Aiello, M. La Deda, A. Crispini, M. Ghedini and N. Godbert, Dalton Trans., 2016, 45, 17264–17273 RSC.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- R. Benassi, E. Ferrari, S. Lazzari, F. Spagnolo and M. Saladini, J. Mol. Struct., 2008, 892, 168–176 CrossRef CAS.
- M. A. Subhan, K. Alam, M. S. Rahaman, M. A. Rahman and R. Awal, J. Sci. Res., 2013, 6, 97–109 CrossRef.
- D. H. Johnston and D. F. Shriver, Inorg. Chem., 1993, 32, 1045–1047 CrossRef CAS.
- C. Párkányi, M. R. Stem-Beren, O. R. Martínez, J. J. Aaron, M. Bulaceanu-MacNair and A. F. Arrieta, Spectrochim. Acta, Part A, 2004, 60, 1805–1810 CrossRef PubMed.
- K. I. Priyadarsini, J. Photochem. Photobiol., C, 2009, 10, 81–95 CrossRef CAS.
- All the differences observed between the absorption spectra in MeOH and DMSO are attributable to both the different polarity of the solvents and to the solubility of each species in these media.
- Y. B. Tsaplev, V. A. Lapina and A. V. Trofimov, Dyes Pigm., 2020, 177, 108327 CrossRef CAS.
- M. Griesser, V. Pistis, T. Suzuki, N. Tejera, D. A. Pratt and C. Schneider, J. Biol. Chem., 2011, 286, 1114–1124 CrossRef CAS PubMed.
- G. Facchetti and I. Rimoldi, Bioorg. Med. Chem. Lett., 2019, 29, 1257–1263 CrossRef CAS PubMed.
- G. Facchetti, N. Ferri, M. G. Lupo, L. Giorgio and I. Rimoldi, Eur. J. Inorg. Chem., 2019, 2019, 3389–3395 CrossRef CAS.
- M. Sökmen and M. Akram Khan, Inflammopharmacology, 2016, 24, 81–86 CrossRef PubMed.
- A. Galano, R. Álvarez-Diduk, M. T. Ramírez-Silva, G. Alarcón-Ángeles and A. Rojas-Hernández, Chem. Phys., 2009, 363, 13–23 CrossRef CAS.
- A. Purushothaman, K. S. Teena Rose, J. M. Jacob, R. Varatharaj, K. Shashikala and D. Janardanan, Food Chem., 2022, 373, 131499 CrossRef CAS PubMed.
- M. Bernabé-Pineda, M. T. Ramírez-Silva, M. Romero-Romo, E. González-Vergara and A. Rojas-Hernández, Spectrochim. Acta, Part A, 2004, 60, 1091–1097 CrossRef.
- P. Terpinc and H. Abramovič, Food Chem., 2010, 121, 366–371 CrossRef CAS.
- A. D. N. J. De Grey, DNA Cell Biol., 2002, 21, 251–257 CrossRef CAS PubMed.
- A. Regiel-Futyra, J. M. Dąbrowski, O. Mazuryk, K. Śpiewak, A. Kyzioł, B. Pucelik, M. Brindell and G. Stochel, Coord. Chem. Rev., 2017, 351, 76–117 CrossRef CAS.
- G. B. Tweedy, Phytopathology, 1964, 55, 910–914 Search PubMed.
- S. J. Sabounchei, P. Shahriary, S. Salehzadeh, Y. Gholiee and A. Chehregani, C. R. Chim., 2015, 18, 564–572 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H- and 13C-NMR, ESI-MS spectra, absorption and emission spectra in MeOH, DMSO and 10%DMSO/PBS varying time, theoretical UV-Vis spectra and data, thermodynamic data, theoretical calculated lipophilicity, antimicrobial data. See DOI: https://doi.org/10.1039/d2dt01653b |
| This journal is © The Royal Society of Chemistry 2022 |