
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 capture from ambient air via crystallization with tetraalkylammonium hydroxides†
Manish Kumar
Mishra‡
 a,
Volodymyr
Smetana
b,
Ethan A.
Hiti
a,
Hannah B.
Wineinger
a,
Fengrui
Qu
a,
Anja-Verena
Mudring
*bc and
Robin D.
Rogers
*a
a,
Volodymyr
Smetana
b,
Ethan A.
Hiti
a,
Hannah B.
Wineinger
a,
Fengrui
Qu
a,
Anja-Verena
Mudring
*bc and
Robin D.
Rogers
*a
aDepartment of Chemistry & Biochemistry, The University of Alabama, Tuscaloosa, AL 35487, USA
bDepartment of Materials and Environmental Chemistry, Stockholm University, 10691 Stockholm, Sweden
cDepartment of Chemistry and iNANO, 253 Aarhus University, 8000 Aarhus C, Denmark
First published on 8th November 2022
Abstract
Aqueous solutions of a series of short carbon chain tetra(n-alkyl)ammonium hydroxides, [Nnnnn][OH] with n = 2: n-ethyl, 3: n-propyl, 4: n-butyl, have been serendipitously found to be potential candidates for direct air carbon capture (DAC) when being used as reagents in more complicated reactions. Aqueous solutions of [N3333][OH], [N2222][OH], or [N3333][OH] with UO2SO4·3H2O and 1,4-diamidoximylbenzene, and [N4444][OH] with cytosine (HCyt) directly absorb CO2 from the atmosphere upon mild heating in the open atmosphere crystallizing in complexes reaching up to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO2/[Nnnnn]OH ratio. [N2222][HCO3]·3H2O (1), [N2222]2[H(HCO3)3]·5H2O (2), [N3333][HCO3]·0.5H2O (3), [N3333][H(HCO3)2] (4), [N3333]2[(tpa)(H2CO3)2] (5; tpa = terephthalate), [N4444][H(Cyt)(HCO3)]·H2O (6) and [N4444][H2(Cyt)2(HCO3)]·H2O (7) have been isolated in crystalline form and structurally characterized by single crystal X-ray diffraction. The compounds are characterized by complex polyanionic formations from bicarbonate dimers ([(HCO3)2·(H2O)]24−) or chains ([H(HCO3)2]nn− or [H2(tpa)(HCO3)2]n2n−) to water-bicarbonate associates ([(HCO3)2·6H2O]2− and [(H2CO3·(HCO3)2)2·6H2O·2H2O]2−) and three-component anionic layers ([H(Cyt)(HCO3)·H2O]nn− and [H2(Cyt)2(HCO3)·H2O]nn−) frequently showing proton sharing. While some hydroxides themselves can maintain a high CO2/[Nnnnn][OH] ratio, particularly 2 and 4, the presence of secondary hydrogen bond donors/acceptors may increase the sorption efficiency through decreased solubility and enhanced crystallization.
:1 CO2/[Nnnnn]OH ratio. [N2222][HCO3]·3H2O (1), [N2222]2[H(HCO3)3]·5H2O (2), [N3333][HCO3]·0.5H2O (3), [N3333][H(HCO3)2] (4), [N3333]2[(tpa)(H2CO3)2] (5; tpa = terephthalate), [N4444][H(Cyt)(HCO3)]·H2O (6) and [N4444][H2(Cyt)2(HCO3)]·H2O (7) have been isolated in crystalline form and structurally characterized by single crystal X-ray diffraction. The compounds are characterized by complex polyanionic formations from bicarbonate dimers ([(HCO3)2·(H2O)]24−) or chains ([H(HCO3)2]nn− or [H2(tpa)(HCO3)2]n2n−) to water-bicarbonate associates ([(HCO3)2·6H2O]2− and [(H2CO3·(HCO3)2)2·6H2O·2H2O]2−) and three-component anionic layers ([H(Cyt)(HCO3)·H2O]nn− and [H2(Cyt)2(HCO3)·H2O]nn−) frequently showing proton sharing. While some hydroxides themselves can maintain a high CO2/[Nnnnn][OH] ratio, particularly 2 and 4, the presence of secondary hydrogen bond donors/acceptors may increase the sorption efficiency through decreased solubility and enhanced crystallization.
1. Introduction
Carbon dioxide emissions to the atmosphere significantly contribute to negative environmental impacts causing the greenhouse effect and, consequently, leading to accelerating global warming1 and related regional and global climate changes.2 In this light, efficient CO2 uptake from the flue gasses and removal from the atmosphere is a critical challenge to be solved in the near future.3 Direct air capture (DAC) methods4 are currently based on absorption by liquids solvents, either hydroxide-carbonate systems5 or amine-functionalized sorbents.6 However, high material cost and/or energy consumption for their recovery are serious obstacles preventing these technologies from implementation on an industrial scale affecting equally capture, desorption, and further conversion of CO2.7–11 Moreover, thermal degradation of the more promising amine-functionalized sorbents during recycling due to the formation/breaking of covalent bonds still represents a technical problem and may further contribute to the method's cost,12 and additional environmental problems.13 Therefore, new highly efficient materials and methods need to be developed considering both technological and economic aspects.During the last decades, ionic liquids (ILs)14–17 and deep eutectic solvents (DESs)18 have been proposed as “greener” alternatives. Despite many analogous properties and functionalities, these two classes offer different advantages and have different drawbacks. Although DESs as CO2 sorbents have yet not been intensively investigated and need further developments, they may offer low production costs and are generally biodegradable.19 Despite a few drawbacks such as high price, high viscosity and potentially toxicity, ILs have already attracted significant attention as a reliable replacement to the amine-functionalized sorbents.20 Their main advantages are low melting points combined with negligible vapor pressure,21 possibilities of designed functionalization towards desired chemical and physical properties,22,23 possibility of separation,24 and variety of binding modes they can offer for CO2 sorption.25 Preferred physical absorption in ILs may also offer economic advantages through reduced energy penalty and significantly declined degradation rate during recycling.
Along with the classical imidazolium,26,27 pyridinium,28 and pyrrolidinium29 ILs, a variety of different cation/anion pairs30 have been tested recently for the CO2 capture performance including blends with conventional sorbents.31 Among them, quaternary ammonium and phosphonium salts have not been intensively investigated but already have revealed promising performance and possible industrial applications.30,32–34 For example, 10% aqueous solutions of tetrabutylammonium hydroxide N4444OH showed high CO2 solubility at elevated pressures.35 Bulky tetraalkylammonium cations do not participate in strong intermolecular bonding, but do provide a framework supporting such interactions between other components of the reaction.36
Here we suggest the power of crystal engineering allows the design of improved materials for DAC, however, such approaches need real structural data. While exploring the use of tetraalkylammonium hydroxides to deprotonate complexing ligands and thus facilitate f-element binding, we have detected evidence of the ready capture of CO2 in the form of crystalline tetraalkylammonium salts. We directly observed that aqueous solutions of small carbon chain tetraalkylammonium hydroxides have hidden potential for direct carbon capture through crystallization in addition to previously observed dissolution at higher pressures. This potential may further be enhanced through the introduction of additional hydrogen bond acceptors expanding bonding patterns and affecting the solubility of the products.
2. Experimental section
2.1. Chemicals and materials
Uranyl sulfate trihydrate (UO2SO4·3H2O) was obtained from International Bio-Analytical Industries, Inc. (Boca Raton, FL). Hydroxylamine hydrochloride, tetraethylammonium hydroxide ([N2222][OH]; 25 wt% in water), tetrabutylammonium hydroxide ([N4444][OH]; 55 wt% in water), and tetrapropylammonium hydroxide ([N3333][OH]; 40 wt% in water) were purchased from Fisher Scientific (Hampton, NH). Cytosine (99%), 1,4-dicyanobenzene, and potassium carbonate were obtained from Sigma Aldrich (St Louis, MO). Ethanol with purity ≥95% (The British Drug Houses, BDH chemicals, Middle East LLC, Dubai, UAE) was obtained from VWR international (Radnor, PA). Deionized (DI) water with a specific resistivity of 17.38 MΩ cm at 25 °C was obtained from an in-house system (Culligan Water Systems, Rosemont, IL). All commercially available chemicals were used as received without further purification.2.2. Synthesis
2.3. NMR
1H and 13C NMR were measured using a Bruker Avance 300 MHz NMR spectrometer (Bruker Biospin Corporation, Billerica, MA) in DMSO-d6 (Cambridge Isotope Laboratories, Inc., Tewksbury, MA). The spectrum for both 1H and 13C NMR were referenced to the DMSO solvent peak at 2.50 ppm and confirmed the structure.371H NMR: δH (300 MHz; DMSO-d6) 5.83 (2H, s), 7.66 (2H, s), 9.68 (1H, s); 13C NMR: δC (300 MHz; DMSO-d6) 125.47, 134.07, 150.90.2.4. Crystallization
The above reaction was repeated but left heating in the sand bath longer. After 8.5 h a white slurry was produced in the reaction mixture. The reaction was taken off heat and stored under an atmosphere of CO2. After 1 week, the reaction had formed a semisolid paste with crystalline solid dispersed throughout, and a sample of the white semisolid crystalline product was scraped out, stored in a small vial under an atmosphere of CO2.
2.5. Single crystal X-ray diffraction (SXCRD)
Crystals of 1–7 decompose readily when heated and are relatively unstable, liquefying when left out at ambient temperatures. This made SCXRD studies somewhat problematic. Nonetheless, SCXRD data for single crystal structural determinations of complexes 1–2, and 4–5 were collected on a Bruker diffractometer equipped with a PLATFORM 3-circle goniometer and an Apex II CCD area detector (Bruker AXS, Madison, WI) using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Suitable irregular-shaped crystals were mounted on a nylon loop with paratone-N (Hampton Research, Aliso Viejo, USA) oil and cooled to −50 °C by a cold stream of N2(g) using an N-Helix cryostat (Oxford Cryosystems, Oxford, UK). Data were collected in 0.5° frames using a strategy of scans about the omega and phi axes. Data collection, unit cell determination, data reduction and integration, absorption correction, and scaling were performed using the Apex-2 software suite (Bruker, Madison, WI).All full occupancy non-hydrogen atoms were refined anisotropically. Hydrogen atoms bonded to oxygen atoms were located from the difference map. Their coordinates were allowed to refine while their thermal parameters were constrained to ride on the carrier atoms. Hydrogen atoms bonded to carbon atoms were placed in calculated positions, and their coordinates and thermal parameters were constrained to ride on the carrier atoms. All the crystal structures were solved by intrinsic phasing with SHELXT41 and refined by full-matrix least squares methods on F2 using SHELXL-201442 within the APEX3 software suite (Bruker AXS WI, 2017).
The data for 3 were collected on an XtaLAB Synergy R, DW system, HyPix diffractometer, at −100 °C. Data were measured using ω scans with Mo-Kα radiation. The diffraction pattern was indexed and the total number of runs and images was based on the strategy calculation from the program CrysAlisPro 1.171.41.118a (Rigaku OD, Yarnton, England 2021). The maximum resolution that was achieved was θ = 33.390° (0.65 Å). Data reduction, scaling and absorption corrections were performed using CrysAlisPro 1.171.41.118a. A Gaussian absorption correction was performed using CrysAlisPro 1.171.41.118a. The numerical absorption correction was based on Gaussian integration over a multifaceted crystal model Empirical absorption correction using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm.
Data for 6 and 7 were collected on a Bruker D8 Advance diffractometer with a Photon 100 CMOS area detector and an IμS microfocus X-ray source using Mo-Kα radiation. Crystals were coated with Paratone oil and cooled to −100 °C under a cold stream of nitrogen using an Oxford cryostat (Oxford Cryosystems, Oxford, UK). Hemispheres of data out to a resolution of at least 0.80 Å were collected by a strategy of φ and ω scans. Unit cell determination, data collection, data reduction, correction for absorption, structural solution, and refinement were all conducted using the Apex3 software suite.
The structures of 3, 6, and 7 were solved by direct methods and refined on F2 by full-matrix least-squares procedures using SHELXTL v.2017. Non-hydrogen atoms were readily located and refined anisotropically. Hydrogen atoms bonded to carbon were placed in calculated positions, and the coordinates were constrained to ride on the carrier atoms. Methyl group hydrogen atoms were refined using a riding-rotating model. Hydrogen atoms bonded to nitrogen were located from the difference Fourier maps in all cases, except for those in 3 and 7 where hydrogen atoms on protonated nitrogen atoms were placed in calculated positions and constrained to ride on the nitrogen atoms. Hydrogen atoms bonded to oxygen were located from the difference Fourier maps in all cases, except for those in 7 and those on lattice water molecules in 2 which were omitted from the refinement but included in the formula. All hydrogen atom thermal parameters were constrained to ride on the carrier atoms.
3. Results and discussion
3.1. Synthesis
As part of our investigation into the selective complexation of uranyl ions by amidoxime ligands suitable for extraction of uranium from seawater,37,38 we began using tetraalkylammonium hydroxides to deprotonate the ligand and promote uranyl binding. Instead of isolating the complexes we sought, we observed the formation of colorless crystals of bicarbonate salts. For example, when UO2SO4·3H2O and 1,4-diamidoximylbenzene were mixed with aqueous solutions of tetraethyl- or tetrapropylammonium hydroxides ([N2222][OH] and [N3333][OH], respectively) and heated unsealed to 80 °C overnight, single crystals were obtained of [N2222][HCO3]·3H2O (1), [N2222]2[H(HCO3)3]·5H2O (2), [N3333][H(HCO3)2] (4), and [N3333]2[(tpa)(H2CO3)2] (5). In the reaction leading to 5, 1,4-diamidoximylbenzene was converted to terephthalate ([tpa]2−).These results led us to try and prepare the bicarbonate salts directly by bubbling CO2 at ambient pressure through aqueous solutions of tetralkylammonium hydroxides. The only related experiment we could find was a report noted in the Introduction where 10 wt% aqueous [N4444][OH] showed high CO2 solubility at elevated pressures,35 however, no solids were reported.
In our experiment, CO2 was bubbled at ambient pressure through a 40 wt% aqueous solution of [N3333][OH] at 90 °C for 2.5 h after which time a white slurry was produced. Cooling to RT and storage under CO2 led to a semisolid paste with crystalline solid dispersed throughout. SCXRD identified the crystalline product as [N3333][HCO3]·0.5H2O (3). Repeating this experiment with 8.5 h did not lead to identification of additional crystalline phases.
Finally, pulling from our recent work with complexes of nucleobases,36 and CO2 capture attempts using guainidine,43 we hypothesized that the addition of a nucleobase to aqueous tetraalkylammonium hydroxide could serve as a perfect reaction medium through formation of an extensive hydrogen bonding network and decrease of solubility. Directly mixing cytosine (HCyt) and [N4444][OH] (55 wt% in water) in a 1:1 ratio and heating unsealed at 90 °C overnight led to colorless block-shaped crystals of both [N4444][H(Cyt)(HCO3)]·H2O (6) and [N4444][H2(Cyt)2(HCO3)]·H2O (7) in the same reaction vessel.
In the reactions leading to 1, 2, and 4–7, CO2 gas was trapped directly from the atmosphere and precipitated at elevated (80–90 °C) temperatures. The preparation of 3 demonstrated that CO2 bubbled through the solution at ambient temperatures could also lead to crystallization of bicarbonate salts, effectively trapping CO2. The very low solubility of these salts in water (most crystallized even at elevated temperatures) suggest the potential for DAC technologies. While all of the investigated compounds easily trap 1 CO2 molecule per [Nnnnn]+ cation, some representatives showed enhanced performance of up to 1.5 (2) or even 2 (4) CO2 molecules per cation, which immediately leaves the solution through crystallization. To better understand the mechanism of CO2 capture we analyzed the obtained products to provide some insights on the binding mechanism and efficiency of different factors, e.g., cation size, water content, or secondary proton donors/acceptors.
3.2. Structural characterization: anionic chains and layers
Having easily isolated seven tetraalkylammonium bicarbonate salts (Table 1), we further investigated the structural details of these to ascertain any structural clues that would allow us to improve the amount of CO2 we could capture. Each structure contains CO2 trapped in the form of bicarbonate, or possibly carbonic acid in anionic dimers, oligomers, infinite chains, or layers with or without additional anions (i.e., [cyt]−, [tpa]2−).| Compound | SG | V, Å3 | Z | ρ, g cm−3 | V/Z, Å3 | V/NNnnnn, Å3 | NCO2/NNnnnn | |
|---|---|---|---|---|---|---|---|---|
| 1 | [N2222][HCO3]·3H2O | P21/c | 1372.0(2) | 4 | 1.188 | 343.0 | 343.0 | 1 |
| 2 | [N2222]2[H(HCO3)3]·5H2O | P21/c | 3023(4) | 4 | 1.175 | 755.8 | 378.0 | 1.5 |
| 3 | [N3333][HCO3]·0.5H2O | P21/c | 3080.2(1) | 4 | 1.106 | 770.1 | 385.0 | 1 |
| 4 | [N3333][H(HCO3)2] | P21/c | 5285.4(6) | 4 | 1.166 | 1321.4 | 440.5 | 2 |
| 5 | [N3333]2[(tpa)(H2CO3)2] | Pccn | 3994.9(5) | 4 | 1.099 | 998.7 | 499.4 | 1 |
| 6 | [N4444][H(Cyt)(HCO3)]·H2O | Pbca | 4868.6(9) | 8 | 1.180 | 608.6 | 608.6 | 1 |
| 7 | [N4444][H2(Cyt)2(HCO3)]·H2O |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
1511.5(3) | 2 | 1.195 | 755.8 | 755.8 | 1 |
We will first focus on the important hydrogen bonded anionic motifs (Fig. 1). The high water proportion in 1 and 2, serves as an independent structure-forming factor. The anionic layers in both compounds contain (H2O)6 hexagons. While the layer in 1 is formed of checkered (H2O)6 hexagons and (HCO3−)2 dimers, the one in 2 contains carbonic-acid-bicarbonate chains with the (H2O)6 hexagons and additional (H2O)2 dimers serving as connectors.
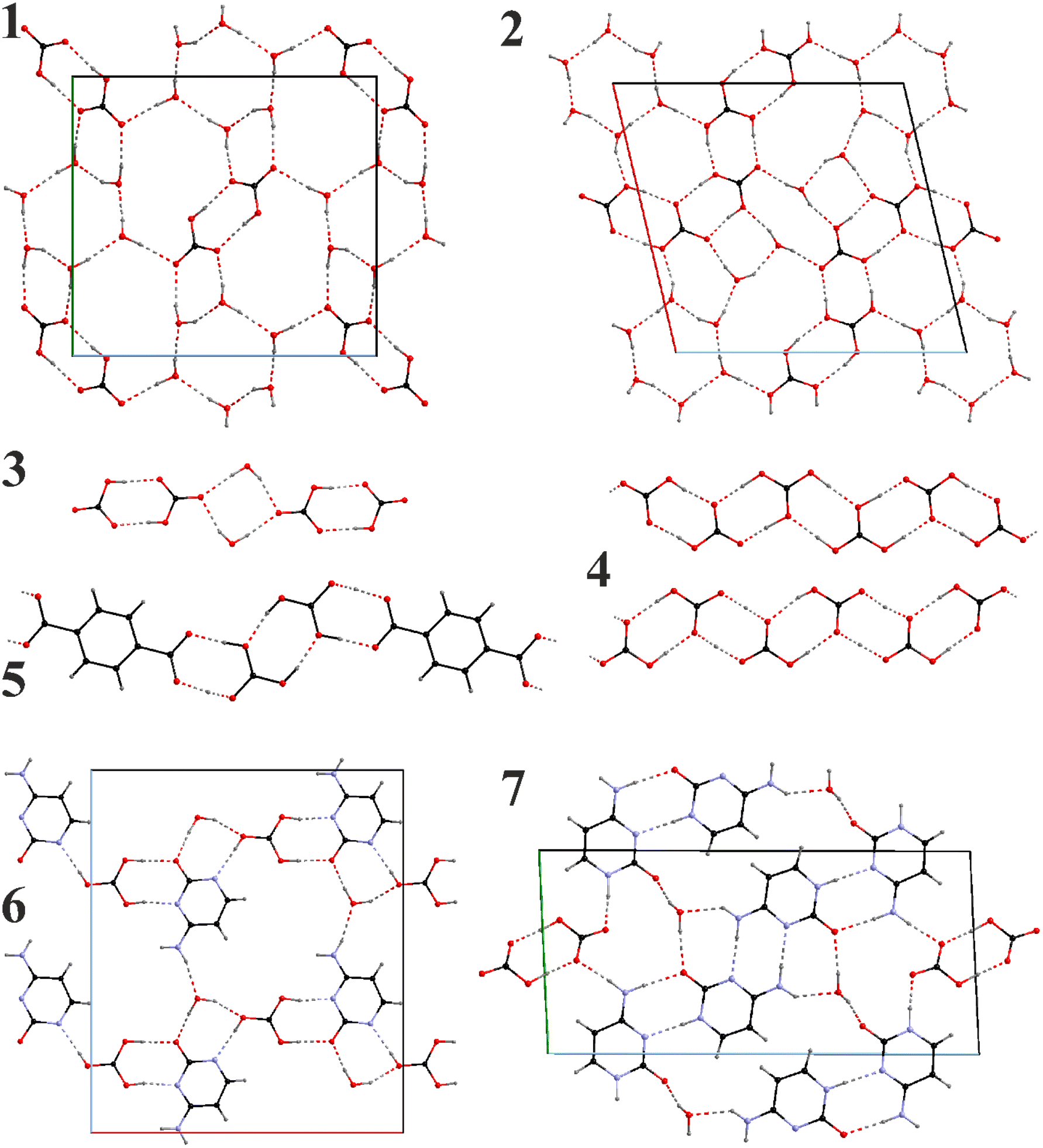 | ||
| Fig. 1 Water-bicarbonate layers in the crystal structures of [N2222][HCO3]·3H2O (1) and [N2222]2[H(HCO3)3]·5H2O (2); isolated short chain in the crystal structure of [N3333][HCO3]·0.5H2O (3); polymeric (infinite) bicarbonate chains in the crystal structures of [N3333][H(HCO3)2] (4), and [N3333]2[(tpa)(H2CO3)2] (5); water-cytosine-bicarbonate layers in the crystal structures of [N4444][H(Cyt)(HCO3)]·H2O (6) and [N4444][H2(Cyt)2(HCO3)]·H2O (7). | ||
In contrast to solely bicarbonate anions present in 1, the proportion of protons in 2 suggests the presence of carbonic acid. Taking into account comparable distances between the oxygen pairs of the neighboring carbonate groups and the charge balance requirement, we found that some H positions must be occupationally disordered. It is clear that partially occupied H positions could not be refined precisely, and the actual position can be slightly off; however, the selected model supplies two H-bond donors for each C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and one for each C–O(H).
O group and one for each C–O(H).
A more accurate criterion would be the C–O distances within the groups and here we observe a characteristic picture. In the fully ordered 1 (Fig. 1), CO and C–O(H) distances of the bicarbonate groups are fully separated (dCO = 1.246–1.259(2) Å and dC–O = 1.347(2) Å) and can serve as a standard. In 2 (Fig. 1), we observe a rather uniform distribution of the distances within each group – dC–O = 1.27–1.30(2); 1.35–1.38(2), and 1.35–1.42(2) Å. According to these distances one of the groups must be CO32−, while the other two – H2CO3; however, equal distribution of the distances within a group and positional disorder suggests a proton exchange within and between them which the crystallographic data cannot distinguish.
Anionic water-bicarbonate dimers are a unique feature of the crystal structure of 3 (Fig. 1). Two hydrogen bonded bicarbonate dimers anions are bridged by two water molecules via hydrogen bonding, forming discreet, [(HCO3)2·(H2O)]24− anions. Within the bicarbonate dimers, there is a clear differentiation between the CO and C–O(H) distances similarly to 1 (dC–O = 1.239–1.270(1) Å and dC–O = 1.350–1.361(1) Å) and all OH groups participate in the bicarbonate dimer formation. It should also be noted that dimeric bicarbonate motifs are among the most common associates observed in such systems in the literature.44
In the anhydrate 4 with the highest ratio of CO2 to cation (2), there are two inequivalent polycarbonate chains (Fig. 1), one of which is a uniform sinusoid-like strip, while the other is flatter but significantly twisted (Fig. 2). In the sinusoidal chain, there are two symmetry-independent carbonate species. In one of them, we observe two distinct C–O distances (dC–O = 1.250–1.282(5) Å and dC–O = 1.338(5) Å) corresponding to an ordered bicarbonate ion in good agreement with the refined proton position. The other one with dC–O = 1.348–1.370(5) Å must be a disordered carbonic acid with proton exchange within one molecule.
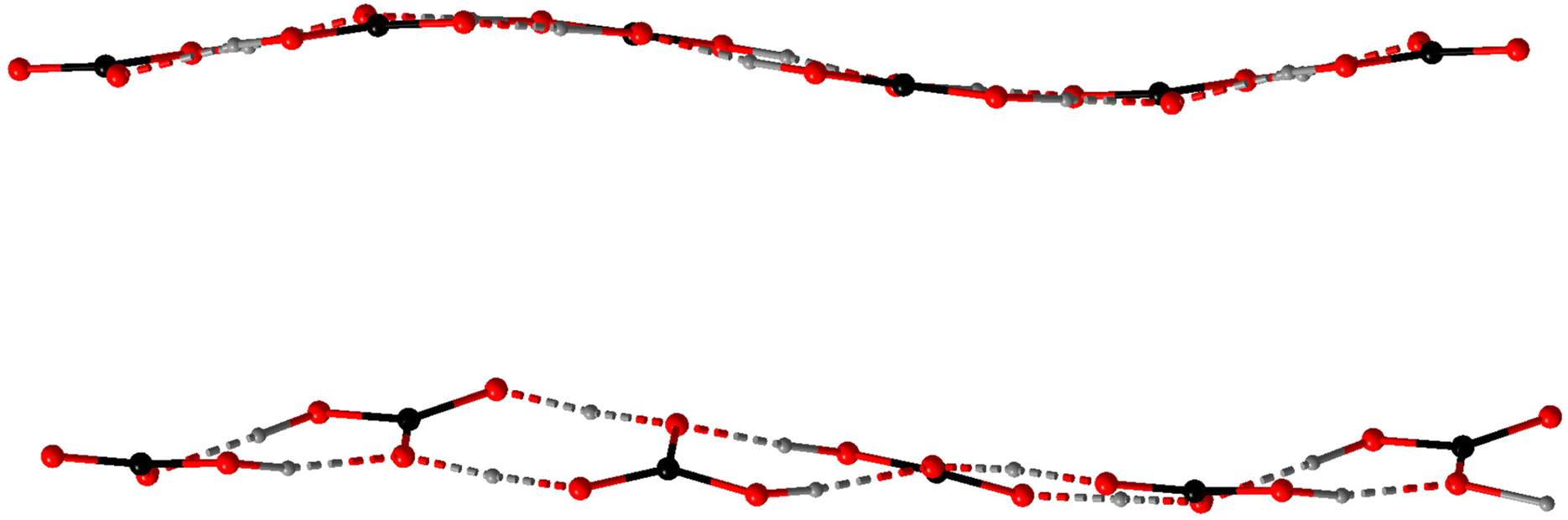 | ||
| Fig. 2 Bicarbonate chains in the crystal structure of 4, sinusoidal (top) and twisted (bottom). | ||
In the twisted chain, there are four symmetry-independent carbonate groups which involve significant proton disorder/exchange. Two unique features have been observed in this chain – all C–O distances are rather long starting a 1.293(5) Å, and a minor differentiation between them within the same carbonate group, e.g., 1.352(5) vs. 1.320(5) Å. Additionally, some proton positions could be refined exactly between the neighboring bicarbonate anions suggesting all CO32− oxygen positions must be at least partially protonated.
Analogous bicarbonate chains are also observed for the anhydrous 5, however with the caveat that the terephthalate anions serve as linkers via the carboxylate groups. It is interesting that the chain is practically planar. Another peculiar feature is the protonation of the carbonate groups. Two protons could be refined right next to the CO32− anions while one – closer to the center between the carbonate and carboxylate groups. All C–O distances in the carbonate groups are in the narrow range 1.366(6) Å supporting full protonation. On the other hand, the same distances within the carboxylate group are significantly shorter – 1.256(5) Å pointing towards tpa2−.
The introduction of cytosine in 6 and 7 (Fig. 1) brings new possibilities for hydrogen bonding. Depending on the cytosine/CO2 proportion, bicarbonate ions are either isolated or form dimers both participating in the formation of polymeric chains and again H2O binds them together. Similarly to 5, carbonate groups in 6 appear to be fully protonated with the C–O distances ranging between 1.364–1.374(2) Å though involving proton disorder within the carbonic acid. Carbonic acid and cytosine anions alternate along the a axis forming a chain connected via 1 hydrogen bond per each pair (taking into account the disordered proton position). A half of the pairs get reinforced through additional water bridges, which also serve as connectors between the chains to form layers.
In 7 only bicarbonate anions have been observed forming dimers. Two distinct C–O contact groups could be outlined, two with dC–O = 1.241–1.267(2) Å and one with dC–O = 1.348(2) Å. The bicarbonate dimers are completely isolated being surrounded by the cytosine-water motifs. Cytosine anions form homoleptic oligomeric chains with either double NH⋯N or NH⋯O connectivities. The chains themselves are connected via the water bridges (along the b axis) or the bicarbonate dimers (along the c axis).
3.3. Comparison of connectives in anionic dimers, chains, and layers
Despite different structural motifs and compositions, certain tendencies could be found in the series 1–7. The most common is the tendency of the carbonate groups to form oligomeric associates following the tendency already observed in the previous works.44 Isolated dimers have been observed in the crystal structures of 1, 3, 5, and 7. Additionally, the polymeric bicarbonate chains in 2 and 4 also show visible segregation of the short oligomeric constituents (from 2 to 4 carbonate units). The O⋯O distances within dimeric units in 1, 3, and 7 are all relatively short – 2.663(2), 2.576–2.676(1), and 2.577(2) Å, respectively, indicating strong associates. This though is in contrast to 5 where the connection between the carbonate and carboxylate groups are slightly shorter (dO–O = 2.601–2.631(3) Å) than between the carbonates themselves (dO–O = 2.708(3) Å). This may be due to the fact that in 5 neutral H2CO3 molecules and tpa2− anions are present.Homoleptic chains in 2 and 4 show that such formations cannot be perfectly uniform in the long range. In anhydrous 4 these chains even though compositionally identical, differentiate between themselves due to different protonation and therefore connectivity patterns. The twisted chains contain clearly segregated dimers of the composition [H·(HCO3)2]− lying practically in the same planes (the deviation from planarity is around 10°) and with O⋯O separations 2.597–2.664(4) Å. These dimer planes form an angle of 30.0(1)° leading to the total straight but helix-like appearance of the chain. The inter-dimer separations are also slightly longer – 2.695–2.738(4) Å. The sinusoidal chains are a little more complex and contain rather tetrameric building blocks due to the pairwise distribution of the carbonic acid and bicarbonate groups. The shortest contacts have naturally been observed between the bicarbonate dimers (dO⋯O = 2.616(4) Å). Slightly longer O⋯O contacts exist between the bicarbonate anion and the neighboring carbonic acid (dO⋯O = 2.627–2.689(4) Å), while H2CO3⋯H2CO3 separations are the longest (dO⋯O = 2.766(4) Å). Nevertheless, both bicarbonate and carbonic acid dimers are planar forming only a ∼10° angle between themselves which is responsible for the sinusoidal appearance.
The presence of water in the crystal packing of 2 does not have significant influence on the bicarbonate chains. Particularly, the higher water ratio in 2 compared to 1 does not prevent formation of the polybicarbonate chains, but helps to stabilize different connectivity patterns, i.e., trimeric associates [H2CO3·(HCO3)2]2−. The intergroup separations range between 2.50–2.56(1) Å, while those between the associates are significantly longer – 2.72–2.80(1) Å. Hexameric water associates in 2 are practically planar and show minor deviations from an ideal hexagon (dO⋯O = 2.75–2.77(1), ∠O–O–O = 112–130(1)°). Such water hexamers in 1 are in general very similar though exhibit weaker connectivities (dO⋯O = 2.798–2.846(2) Å) in line with stronger bicarbonate associates in this compound.
Finally, in the presence of cytosine (6–7) having multiple possibilities to act as both H-bond donor and acceptor, inter-carbonate interactions are no longer dominant in the structure. Both structures exhibit cytosine-bicarbonate chains though with different sequences, either 1:1 or 4:2. 7 with the higher cytosine ratio exhibits certain structural analogies with 1 having bicarbonate dimers isolated by homoleptic oligomeric (tetrameric) cytosine chains. Nevertheless, both 6 and 7 are three-component systems where each part plays an important role in the packing efficiency. Particularly, water molecules serve as bridges between the heteroleptic cytosine-bicarbonate chains in 6 or enforce their connectivity in 7.
3.4. Packing
A common feature for 1–7 is the clathrate nature of the packing where the [Nnnnn]+ cations form either separate layers or networks where van der Waals interactions dominate. The cations provide space and serve as templates (Fig. 3) for the anion substructures. Nonetheless, the structures of the cationic formations are dependent on the ability of the anionic parts to form hydrogen-bonded networks, and therefore correlate with the presence of water in the crystal structure. Completely anhydrous 4 and 5 form polymeric chains and the resulting cationic framework can be characterized as tunnel-like. In these compounds we observe the highest packing efficiency filling all possible voids/channels formed by alkyl chains. 3 is unique in this series as it forms short dimers and the packing is dominated by the cationic part encapsulating the anionic associates, nonetheless, we can outline mixed cationic-anionic layers separated by pure cationic ones. The remaining structures – 1, 2, 6, and 7 are layered with ideal alternation of the anionic and the cationic components. | ||
| Fig. 3 Packing of the cations and the anionic fragments in the crystal structures of [N2222][HCO3]·3H2O (1), [N2222]2[H(HCO3)3]·5H2O (2), [N3333][HCO3]·0.5H2O (3), [N3333][H(HCO3)2] (4), [N3333]2[(tpa)(H2CO3)2] (5), [N4444][H(Cyt)(HCO3)]·H2O (6) and [N4444][H2(Cyt)2(HCO3)]·H2O (7). | ||
3.5. Discussion
The sorption of CO2 by quaternary ammonium hydroxides has already been suggested as a promising way for post-combustion capture. For instance, 10 wt% aqueous [N4444][OH] showed high CO2 solubility (mol (CO2) per mol (absorbent)) of up to 3.5 at elevated pressures of up to 1 MPa but decreasing with temperature.35 The mechanism of this enhanced sorption was never investigated but was assumed to be physical. In this work, we have shown that higher CO2 capacity can be achieved through the formation of extended hydrogen-bonded chains or networks avoiding the formation of strong covalent bonds, but certainly going behind weak van der Waals (vdW) interactions. Both [N2222][OH] and [N3333][OH] showed enhanced CO2 capacity (up to 2) at ambient pressure and slightly elevated temperatures. The elevated temperature in this case is an advantage as potential decrease of solubility stimulates crystallization removing the trapped product from the solution.CO2 captured by aqueous solutions tends to form hydrated CO32−/HCO3−/H2CO3 guest species with a variety of predesigned receptors. The higher negative charge of the CO32− anion and the variety of coordination modes it can exhibit45 make it an easy target for multiple hydrogen bond donors, from mono- to multipodal and cyclic.44 We observed that the total CO2 capacity of the system depends on the number of secondary components in the compound (particularly water). Easy substitution of H2O by HCO3− between 1 and 2 suggests additional potential for the CO2 capacity.
Various amino-based ILs mixtures have been tested for the direct air capture of CO2 including aqueous [Nnnnn][OH] with amino acids.46 The main drawbacks of these solutions are the high concentration of CO2 required and the formation of covalent bonds, which may bring higher energy penalties and losses during recycle and release of the CO2.
A completely different approach has been reported recently with guanidine aqueous solutions.43,47 They may slowly trap CO2 directly from ambient air avoiding the formation of covalent C–N bonds. The main disadvantage of this system is still low CO2 capacity and potentially high production cost of the N-heterocycles. Application of the quaternary ammonium hydroxides or their aqueous solutions directly not only eliminates the need for additional hydrogen bond donors/acceptors but suggests potential for significant enhancement of the sorption efficiency through the chemical design.
4. Conclusions
In this work, several aqueous quaternary ammonium hydroxide solutions have been observed to directly capture CO2 from air by crystallization of bicarbonate salts. Upon mild heating (∼80–90 °C), crystals of seven different compounds could be observed overnight. Practically all compounds exhibit bicarbonate associates that with or without help of secondary hydrogen bond donors/acceptors, particularly H2O, extend in polymeric motifs. Although H2O was found to play a significant role in the initialization of the reaction, we cannot directly correlate its presence with the CO2 sorption efficiency. For instance anhydrous 4 shows the highest CO2/[Nnnnn]+ ratio (2), while 2 contains a significant proportion of water molecules per formula unit, but still allows a relatively high CO2/[Nnnnn]+ ratio (1.5) in contrast to 3 (1). The presence of other bulky hydrogen donors reduces the CO2/[Nnnnn]+ ratio to 1. In general, the highest proportion of CO2 per Nnnnn cation could be achieved for pure binary systems.It is important to note that although we did not quantify CO2 recovery, the crystals obtained in this study are not stable and CO2 can be easily released. The crystals redissolve when heated and slowly liquefy at over a period of a few days. A logical next step to this work would be to quantify CO2 recovery with mass balance, however, we would suggest optimizing the crystalline forms first.
Hydrogen bonding was found to be the primary and sufficient mechanism of the CO2 capture leading to the formation of strong carbonic acid-bicarbonate associates reducing the solubility. Formation of the anhydrous 4 is extraordinary as it not only allows the highest observed CO2/[Nnnnn]+ ratio but exhibits pure polymeric bicarbonate 1D chains. Though this compound could be obtained only in a mixture with generally lower CO2 sorption efficiency, the [Nnnnn][OH]/H2O (n = 2 and 3) show good potential for the direct air capture and further optimization of the sorption capacity. On the other hand, one or all alkyl chains can easily be modified suggesting broad possibilities for further development of the area and potential for further increase of the sorption efficiency.
Author contributions
M. K. M., E. A. H. and H. B. W.: Methodology, experiments. V. S.: Analysis, visualization, writing. F. Q.: analysis. A. V. M., R. D. R.: conceptualization, resources, supervision, writing, review and editing. All the authors participated in reviewing the entire manuscript as well.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported, in part, by the Royal Swedish Academy of Science through the Göran Gustafsson prize to A.-V. M., by the Swedish Research Council (Vetenskapsrådet, VR) through Grant 2020-05405 (A.-V. M.), and by a Tage Erlander Guest Professorship to R. D. R. (VR Grant 2018-00233). This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Elements program under Award DE-SC0019220. The SCXRD instrumentation at The University of Alabama was supported by U.S. NSF MRI 1828078.References
- T. R. Anderson, E. Hawkins and P. D. Jones, Endeavour, 2016, 40, 178–187 CrossRef PubMed.
- N. W. Arnell, J. A. Lowe, A. J. Challinor and T. J. Osborn, Clim. Change, 2019, 155, 377–391 CrossRef.
- D. W. Keith, Science, 2009, 325, 1654 CrossRef CAS PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- G. Holmes and D. W. Keith, Philos. Trans. R. Soc., A, 2012, 370, 4380–4403 CrossRef CAS PubMed.
- A. Goeppert, H. Zhang, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, ChemSusChem, 2014, 7, 1386–1397 CrossRef CAS PubMed.
- G. Realmonte, L. Drouet, A. Gambhir, J. Glynn, A. Hawkes, A. C. Köberle and M. Tavoni, Nat. Commun., 2019, 10, 3277 CrossRef CAS PubMed.
- T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- J. M. Hanusch, I. P. Kerschgens, F. Huber, M. Neuburger and K. Gademann, Chem. Commun., 2019, 55, 949–952 RSC.
- B. Shao, Y. Zhang, Z. Sun, J. Li, Z. Gao, Z. Xie, J. Hu and H. Liu, Green Chem. Eng., 2022, 3, 189–198 CrossRef.
- J. Davis and G. Rochelle, Energy Procedia, 2009, 1, 327–333 CrossRef CAS.
- M. Karl, R. F. Wright, T. F. Berglen and B. Denby, Int. J. Greenhouse Gas Control, 2011, 5, 439–447 CrossRef CAS.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS PubMed.
- M. Aghaie, N. Rezaei and S. Zendehboudi, Renewable Sustainable Energy Rev., 2018, 96, 502–525 CrossRef CAS.
- J. Huang and T. Rüther, Aust. J. Chem., 2009, 62, 298–308 CrossRef CAS.
- S. K. Shukla, S. G. Khokarale, T. Q. Bui and J.-P. T. Mikkola, Front. Mater., 2019, 6, 42 CrossRef.
- C. Ma, S. Sarmad, J.-P. Mikkola and X. Ji, Energy Procedia, 2017, 142, 3320–3325 CrossRef CAS.
- S. Sarmad, J.-P. Mikkola and X. Ji, ChemSusChem, 2017, 10, 324–352 CrossRef CAS PubMed.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834–2839 CrossRef CAS PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- E. Hayashi, M. L. Thomas, K. Hashimoto, S. Tsuzuki, A. Ito and M. Watanabe, ACS Appl. Polym. Mater., 2019, 1, 1579–1589 CrossRef CAS.
- D. Zhang, R. Qu, H. Zhang and F. Zhang, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2020, 35, 750–757 CrossRef CAS.
- S. Evjen, A. Fiksdahl, D. D. D. Pinto and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2018, 76, 167–174 CrossRef CAS.
- M. C. Corvo, J. Sardinha, T. Casimiro, G. Marin, M. Seferin, S. Einloft, S. C. Menezes, J. Dupont and E. J. Cabrita, ChemSusChem, 2015, 8, 1935–1946 CrossRef CAS PubMed.
- N. M. Yunus, M. I. A. Mutalib, Z. Man, M. A. Bustam and T. Murugesan, Chem. Eng. J., 2012, 189–190, 94–100 CrossRef CAS.
- L. C. Tomé, M. Isik, C. S. R. Freire, D. Mecerreyes and I. M. Marrucho, J. Membr. Sci., 2015, 483, 155–165 CrossRef.
- S. Supasitmongkol and P. Styring, Energy Environ. Sci., 2010, 3, 1961–1972 RSC.
- S. Evjen, A. Fiksdahl and H. K. Knuutila, Ind. Eng. Chem. Res., 2019, 58, 10533–10539 CrossRef CAS.
- A. S. Hamouda, M. S. Eldien and M. F. Abadir, Ain Shams Eng. J., 2020, 11, 1061–1067 CrossRef.
- T. Ema, K. Fukuhara, T. Sakai, M. Ohbo, F.-Q. Bai and J.-y. Hasegawa, Catal. Sci. Technol., 2015, 5, 2314–2321 RSC.
- A. Mohammadi, A. Kamran-Pirzaman and N. Rahmati, Pet. Sci. Technol., 2021, 39, 647–665 CAS.
- R. Safdar, A. A. Omar and B. Lal, J. Mol. Liq., 2018, 266, 522–528 CrossRef CAS.
- M. K. Mishra, S. P. Kelley, V. Smetana, D. A. Dixon, A. S. McNeill, A.-V. Mudring and R. D. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18224 CrossRef CAS PubMed.
- S. P. Kelley, P. S. Barber, P. H. K. Mullins and R. D. Rogers, Chem. Commun., 2014, 50, 12504–12507 RSC.
- N. Tang, J. Liang, C. Niu, H. Wang, Y. Luo, W. Xing, S. Ye, C. Liang, H. Guo, J. Guo, Y. Zhang and G. Zeng, J. Mater. Chem. A, 2020, 8, 7588–7625 RSC.
- H.-B. Pan, L.-J. Kuo, J. Wood, J. Strivens, G. A. Gill, C. J. Janke and C. M. Wai, RSC Adv., 2015, 5, 100715–100721 RSC.
- S. O. Kang, S. Vukovic, R. Custelcean and B. P. Hay, Ind. Eng. Chem. Res., 2012, 51, 6619–6624 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. A. Seipp, N. J. Williams, M. K. Kidder and R. Custelcean, Angew. Chem., Int. Ed., 2017, 56, 1042–1045 CrossRef CAS PubMed.
- U. Manna and G. Das, CrystEngComm, 2021, 23, 512–527 RSC.
- X.-Y. Zheng, J. Xie, X.-J. Kong, L.-S. Long and L.-S. Zheng, Coord. Chem. Rev., 2019, 378, 222–236 CrossRef CAS.
- H. Yu, Y.-T. Wu, Y.-Y. Jiang, Z. Zhou and Z.-B. Zhang, New J. Chem., 2009, 33, 2385–2390 RSC.
- R. Custelcean, K. A. Garrabrant, P. Agullo and N. J. Williams, Cell Rep. Phys. Sci., 2021, 2, 100385 CrossRef CAS.
Footnotes |
| † CCDC 2169751–2169757. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02262a |
| ‡ Present address: Department of Chemistry (SAS), Vellore Institute of Technology (VIT), Vellore 632014, Tamil Nadu, India. |
| This journal is © The Royal Society of Chemistry 2022 |