
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative NMR metabolomics of the responses of A2780 human ovarian cancer cells to clinically established Pt-based drugs†
Veronica
Ghini
ab,
Francesca
Magherini
c,
Lara
Massai
b,
Luigi
Messori
 *b and
Paola
Turano
*ab
*b and
Paola
Turano
*ab
aMagnetic Resonance Center (CERM), University of Florence, 50019 Sesto Fiorentino, Florence, Italy. E-mail: turano@cerm.unifi.it
bDICUS, Department of Chemistry, University of Florence, Via della Lastruccia 3-13, 50019 Sesto Fiorentino, Italy. E-mail: luigi.messori@unifi.it
cDepartment of Experimental and Clinical Biomedical Sciences “M. Serio”, University of Florence, 50134 Florence, Italy
First published on 28th July 2022
Abstract
Pt-Based drugs play a very important role in current cancer treatments; yet, their cellular and mechanistic aspects are not fully understood. NMR metabolomics provides a powerful tool to investigate the metabolic perturbations induced by Pt drugs in cancer cells and decipher their meaning in relation to the presumed molecular mechanisms. We have carried out a systematic and comparative 1H NMR metabolomics study to analyze the responses of A2780 human ovarian cancer cells to the main clinically established Pt drugs, i.e., cisplatin, carboplatin and oxaliplatin. Notably, NMR analysis revealed some moderate and consistent changes in the metabolomic profiles of A2780 cells treated with the 3 Pt drugs with respect to controls, but only very small differences among them. Beyond alterations at the level of nucleic acid precursors, the observed changes highlight in all cases the induction of a significant endoplasmic reticulum stress. Owing to the clinical relevance of platinum resistance, the behavior of a cisplatin resistant A2780 cancer cell line upon cisplatin treatment was also evaluated.
Introduction
Cisplatin (CIS), carboplatin (CARBO) and oxaliplatin (OXA) are the three most important platinum-based drugs approved worldwide for cancer treatment in clinics (Fig. 1A).1,2 All these Pt drugs are characterized by the presence of a square planar platinum(II) center that is primarily responsible for biological and pharmacological effects. In order to perform its biological actions, the platinum(II) center requires chemical activation that is typically achieved through the slow release of the more labile ligand(s) (i.e., the two chlorides in CIS, the cyclobutandicarboxylate ligand in CARBO and the oxalate ligand in OXA).3 The nature of the leaving group, beyond controlling the activation of the platinum(II) center, also modulates the overall pharmaceutical profile of these metallodrugs by affecting the lipophilicity, the aqueous solubility, the ability of crossing cell membranes, the intracellular biodistribution, and so on. The activated platinum(II) center shows a relevant and broad reactivity with a variety of biomolecules, such as nucleic acids and proteins; platinum(II) coordination to the exposed groups of these biomolecules and the resulting structural and functional perturbations are the main determinants of the cytotoxic and anticancer effects of this class of metallodrugs.1,4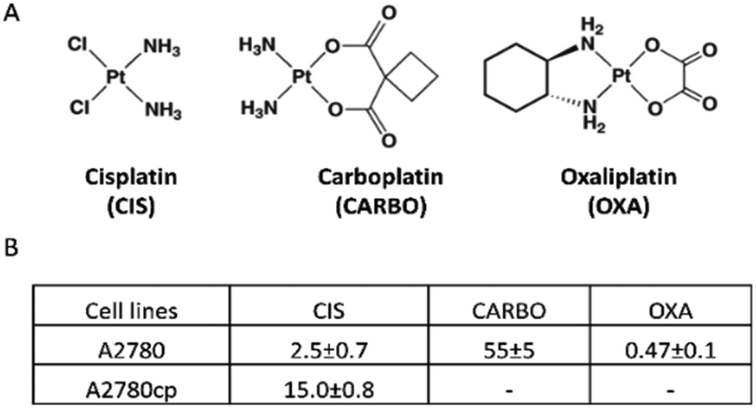 | ||
| Fig. 1 (A) The structures of cisplatin, carboplatin and oxaliplatin and (B) a table reporting IC50 values (μM) as obtained by the MTT assay using an exposure time of 72 h. | ||
Cisplatin was first approved for cancer treatment in 1978 following the pioneering studies conducted by Barnett Rosenberg. Carboplatin was later modelled on the basis of cisplatin's structure and reactivity and entered clinical use in 1988. Though basically both share the same mode of action, carboplatin manifests slower activation kinetics and, accordingly, a lower biological potency and a lower systemic toxicity than cisplatin. Owing to its lower toxicity, carboplatin replaces conveniently cisplatin in a number of therapeutic cancer protocols.5 Oxaliplatin obtained clinical approval in 2002. Oxaliplatin is characterized by the presence of oxalate as a bidentate ligand playing the role of a leaving group. Notably, oxaliplatin manifests a spectrum of anticancer actions that are significantly different from cisplatin and carboplatin, though the reasons for the observed pharmacological differences are not clear yet.
At variance with CIS and CARBO that are often used in the treatment of ovarian, lung and testicular cancers, OXA is almost exclusively used in the treatment of colon cancer.6
For a long time it was believed that the three main Pt drugs roughly shared the same mechanism of action mostly relying on the direct damage to genomic DNA as a result of platinum(II) coordination to DNA nucleobases and subsequent activation of cancer cell apoptosis (the so called “DNA paradigm” for the mode of action of Pt drugs).7,8 However, just a few years ago, Lippard and his group proposed – through an advanced and sophisticated experimental approach – that oxaliplatin possesses a mode of action largely distinct from cisplatin that is based on the induction of ribosome biogenesis stress.9
Untargeted nuclear magnetic resonance (NMR) metabolomics is a powerful tool to characterize the metabolome of cultured cancer cells, to monitor the cellular responses to treatment and to relate the observed metabolic changes to the mode of action of the tested drugs.10–14 Despite being far less sensitive, NMR metabolomics shows some important advantages over MS metabolomics such as greater reproducibility, much less sample handling, and an easier and more straightforward interpretation of the obtained results.15,16
Several studies in the literature already exploited NMR metabolomics to evaluate the metabolic responses of cancer cells to metal based drugs, in particular Pt drugs. The effects of Cisplatin were studied in a variety of cancer cells including lung,17 brain,18–20 breast,21,22 and ovarian23,24 cancer cells, as well as osteosarcoma,25,26 mostly by analyzing whole or lysed cells or focusing on cell extracts and cell media. These studies revealed some common trends in the induced metabolic changes such as a direct effect on lipid metabolism and membrane integrity, a decrease in the overall metabolic activities of the treated cells, and the alteration of some other diagnostic metabolites, e.g., UDP sugars. However, the picture arising from the NMR metabolomic studies conducted so far on cisplatin is still ill-defined and fragmentary owing to the large variability of the observed cellular responses, to the several investigated cancer cell lines and to the often largely different conditions of the various experiments. On the other hand, only very few studies have analyzed so far the behavior of carboplatin and oxaliplatin. The metabolic impact of OXA on the polar extracts of hepatocellular carcinoma cells was studied by NMR metabolomics,27 and that of carboplatin (in combination with other drugs) on the exo-metabolome of oral squamous carcinoma cells.28 A very recent paper by Ann Gil described comparatively the effects of cisplatin and oxaliplatin on human osteosarcoma cells; no major differences in the resulting patterns of metabolomic alterations were highlighted.29
Upon considering the available literature, we realized that no systematic investigation yet existed aimed at comparing the metabolic effects of cisplatin, carboplatin, and oxaliplatin in the same cancer cell line as a function of the time of treatment. Thus, we decided to characterize the alterations in the NMR metabolomic profiles of A2780 ovarian cancer cells elicited by Pt drug treatment, considering the changes produced both in the endo- and exo-metabolomes.
One of the main goals of our study was to reveal the possible metabolic changes between cisplatin and carboplatin on one hand and oxaliplatin on the other hand, in relation to recently described mechanistic differences.9 Moreover, to enlarge the description of the metabolomics effects induced by Pt drugs, we have extended our attention to an A2780 cancer cell line made resistant to cisplatin (A2780cp, hereafter) with the aim to explore the molecular basis of Pt resistance at the metabolic level and identify the possible biomarkers of Pt resistance.
Results
Preliminary characterization of the investigated cancer cell lines
The in vitro cytotoxic activity of CIS was preliminarily determined for the A2780 and A2780cp cell lines by the MTT assay using an exposure time of 72 h. The IC50 values are reported in Fig. 1B. The A2780cp cell line has an IC50 value for CIS of 15 μM compared with the value of 2.5 μM measured for its sensitive counterpart, leading to a resistance index of 6 (Fig. 1B). The cytotoxic activity of CARBO and OXA on the A2780 line was also measured; their IC50 values are about 22 times larger and 5 times lower than that of CIS, respectively. For the NMR analysis of the effects induced in A2780 cells we used a concentration of the Pt drugs equal to the respective IC50 value at 72 h (in such a way to work under equitoxic conditions), but for shorter times (24 and 48 h) in order to detect only or predominantly the metabolic derangements taking place before the occurrence of significant apoptosis. In the case of A2780cp, experiments were performed at two different concentrations of CIS, namely that corresponding to the IC50 of this cell line (to work at equal toxicity) and that of the IC50 of the A2780 cell line (to work with the same concentration of CIS).After this initial screening, 1H NMR spectra were acquired for A2780 and A2780cp untreated cells to evaluate their characteristic metabolomic fingerprints, both in terms of endo- and exo-metabolomes (Fig. 2 and 3). The standard RPMI 1640 medium supplemented with 2 mM glutamine, 10% of FCS (same batch for all the experiments) and antibiotics was used for all the experiments.
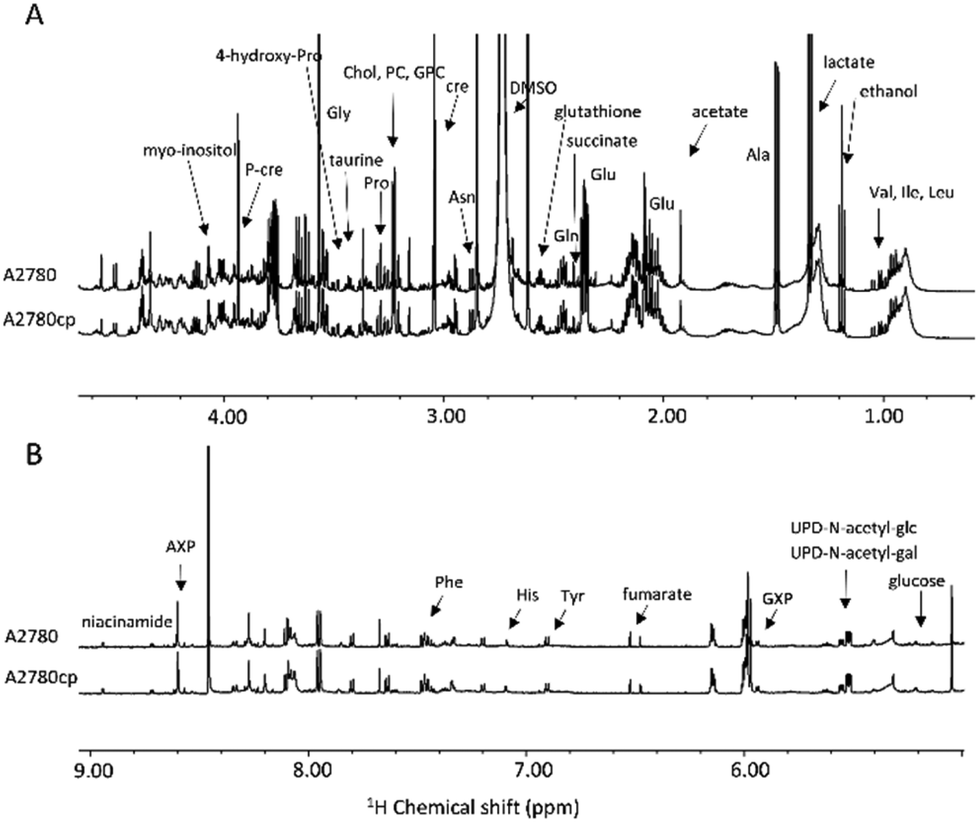 | ||
| Fig. 2 (A) Upfield (1.00–4.50 ppm) and (B) downfield (5.00–9.00 ppm) regions of the representative 1H NMR CPMG spectra of cell lysates (endo-metabolome of A2780 and A2780cp cell lines). | ||
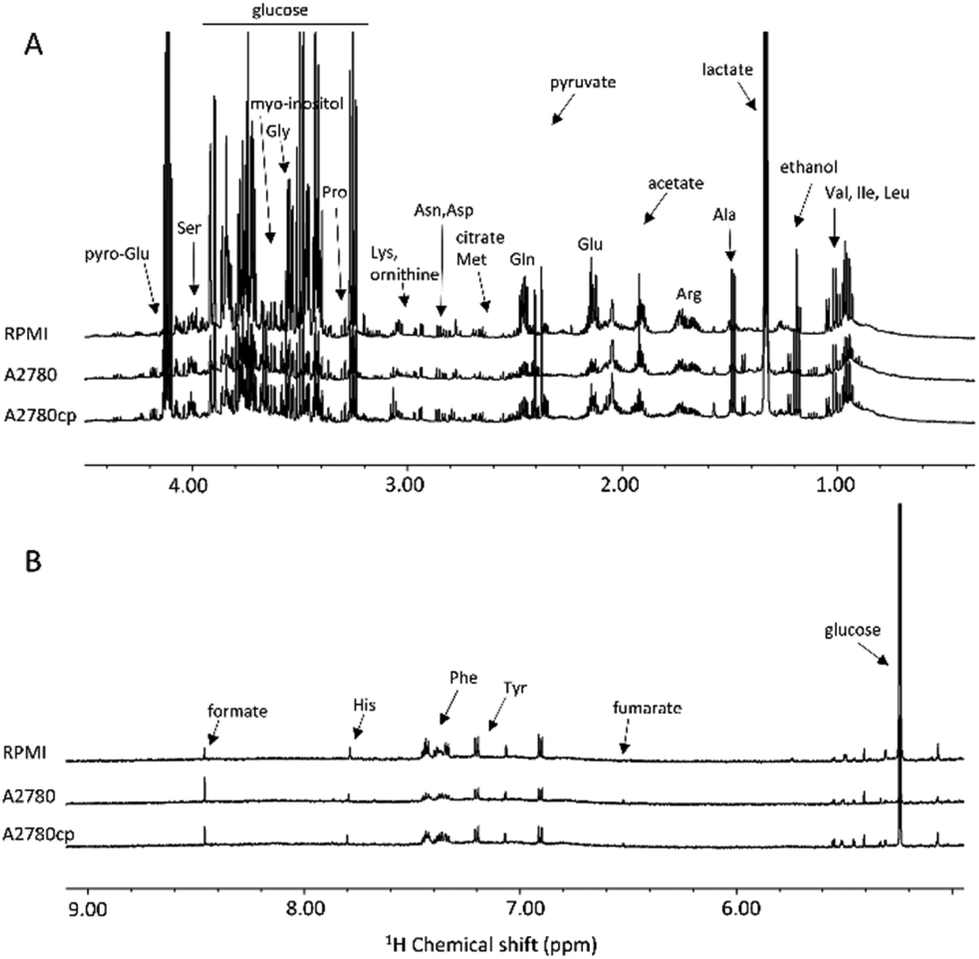 | ||
| Fig. 3 (A) Upfield (1.00–4.50 ppm) and (B) downfield (5.00–9.00 ppm) regions of the representative 1H NMR NOESY spectra of growth media (exo-metabolome of A2780 and A2780cp cell lines and fresh supplemented medium). | ||
The metabolites that could be identified in the spectra of the cell lysates and growth media are listed in Table S1;† they mainly comprise amino acids, intermediates of glycolysis and of the tricarboxylic acid cycle, choline-derivatives, carbohydrates and nucleotides, providing an overview of the metabolic state of the cell. Notably, in the two cell lines, we observed important differences in the relative concentrations of these molecules both in the lysates and in the growth media, but not in the chemical nature of the metabolites.
The effects of CIS, CARBO and OXA on A2780 cells
Fig. 4 shows the main changes in the endo- and exo-metabolomes of A2780 cells observed upon treatment with CIS, CARBO and OXA (2.5 μM, 55 μM, and 0.47 μM, respectively) after 24 or 48 h.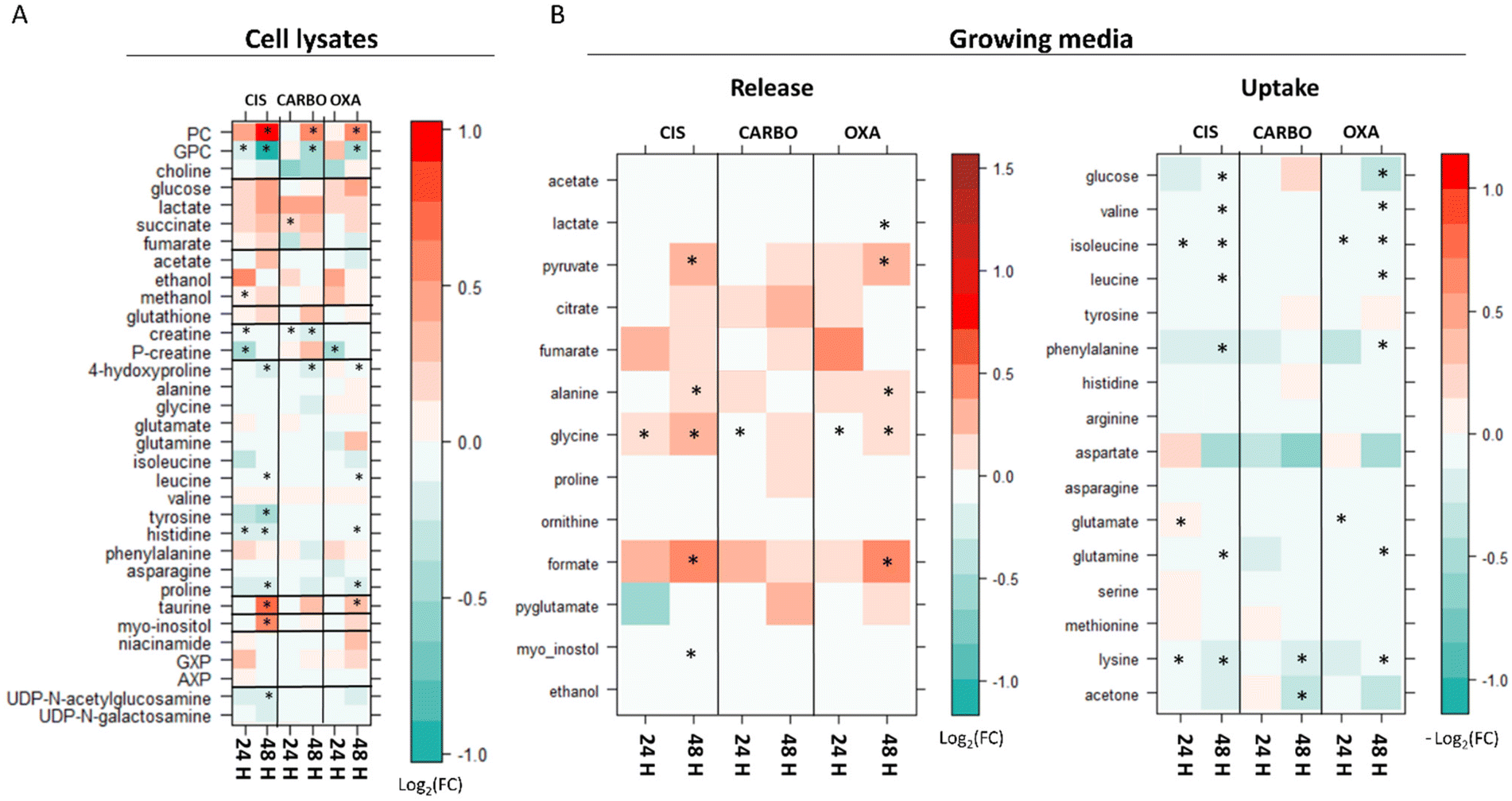 | ||
| Fig. 4 Level plots of fold change displaying the metabolites whose levels vary upon 24 h and 48 h of treatment of A2780 cells with CIS, CARBO and OXA. (A) Cell lysates: red/green parameters indicate the higher/lower concentration of Pt drug-treated cells with respect to control samples (log2(FC)). (B) Growth media: red/green parameters indicate the higher/lower process of uptake (−log2(FC)) or release (log2(FC)) in Pt drug-treated cells with respect to control samples. The brightness of the colours corresponds to the magnitude of FC. Asterisks indicate statistical significance (p-value <0.05). | ||
It is evident that the alterations induced by the treatment are on average relatively modest both in number and intensity. The observed changes occur quite slowly being typically less intense at 24 h compared to 48 h. Among the three Pt drugs, CIS is the one that produces the largest and earliest changes, both at the level of the endo- and exo-metabolomes. For this reason, the effects of CIS were also evaluated at 6 hours, Fig. S1.† The overall pattern of the observed changes is generally conserved for the three Pt drugs although the effects of CARBO are, on average, less evident.
In the cell lysates, the most prominent effects are associated with the fact that all three Pt-drugs induce a net change in the intracellular concentrations of the choline derivatives: in particular, a significant increase of phosphocholine (PC) along with a decrease in glycerol phosphocholine (GPC) is invariably observed; choline, instead, is not altered by the treatment. While for carboplatin and oxaliplatin these changes appear only after 48 h of treatment, cisplatin induces a significant increase in PC already at 6 h and 24 h (Fig. 4 and Fig. S1 and S2†). Accordingly, at 48 h, the ratio between GPC and PC (GPC/PC) significantly decreases for all three treatments going from 0.7 in controls to 0.2, 0.3 and 0.3 for CIS, OXA and CARBO, respectively.
At 24 h, the GPC/PC ratio significantly changes only for CIS, going from 0.3 in controls cells to 0.2 (Fig. S2†).
Additional Pt drug-induced general changes in the cell lysates are the following (see Fig. 4A): (i) an increase of taurine at 48 h; this effect is significant for cisplatin and oxaliplatin; (ii) an increase in myo-inositol at 48 h (significant only for cisplatin); (iii) a significant decrease of creatine and/or phosphocreatine at 24 h; (iv) a general but modest decrease in the intracellular levels of a few amino acids over time (4-hydroxy-Pro, Leu, Tyr, His and Pro).
In the growth media, there is a gradual down-regulation of the uptake of several amino acids (such as Val, Ile, Leu, Phe and Lys) (Fig. 4B). Moreover, an increase in the release of a few important metabolites like formate, Gly, Ala, and pyruvate is observed as a consequence of the three treatments over time. The release of formate, pyruvate and Ala is significantly larger after 48 h of treatment in CIS- and OXA-treated cells; the concentration of Gly in the media starts to be significantly higher in all the Pt drug-treated cells with respect to control cells at 24 hours, while at 48 h it was found to be significantly different in CIS- and OXA-treated cells. Finally, at 48 h, for CIS and OXA, we can detect a very slight but significant decrease in glucose uptake along with a slightly greater release of lactate detected only in OXA treated cells at 48 h. These changes can be evocative of a small decrease in the glycolytic flux, which appears to be more prominent for OXA.
A2780 cisplatin-resistant cells
In paragraph 1 (preliminary characterization of the investigated cancer cell lines) we have described the general fingerprints of A2780 and A2780cp cells highlighting that the differences do not reside in the chemical nature of the detected metabolites but, rather, in their relative concentrations, both in the lysates and in the growth media. In line with this observation, principal components analysis (PCA) score plots shown in Fig. 5A and B revealed that A2780 and A2780cp segregate into different clusters; the bar plots, representing the log2 fold change (FC) of the metabolites in Fig. 5C and D, clearly indicate the differences in the relative amount of each metabolite.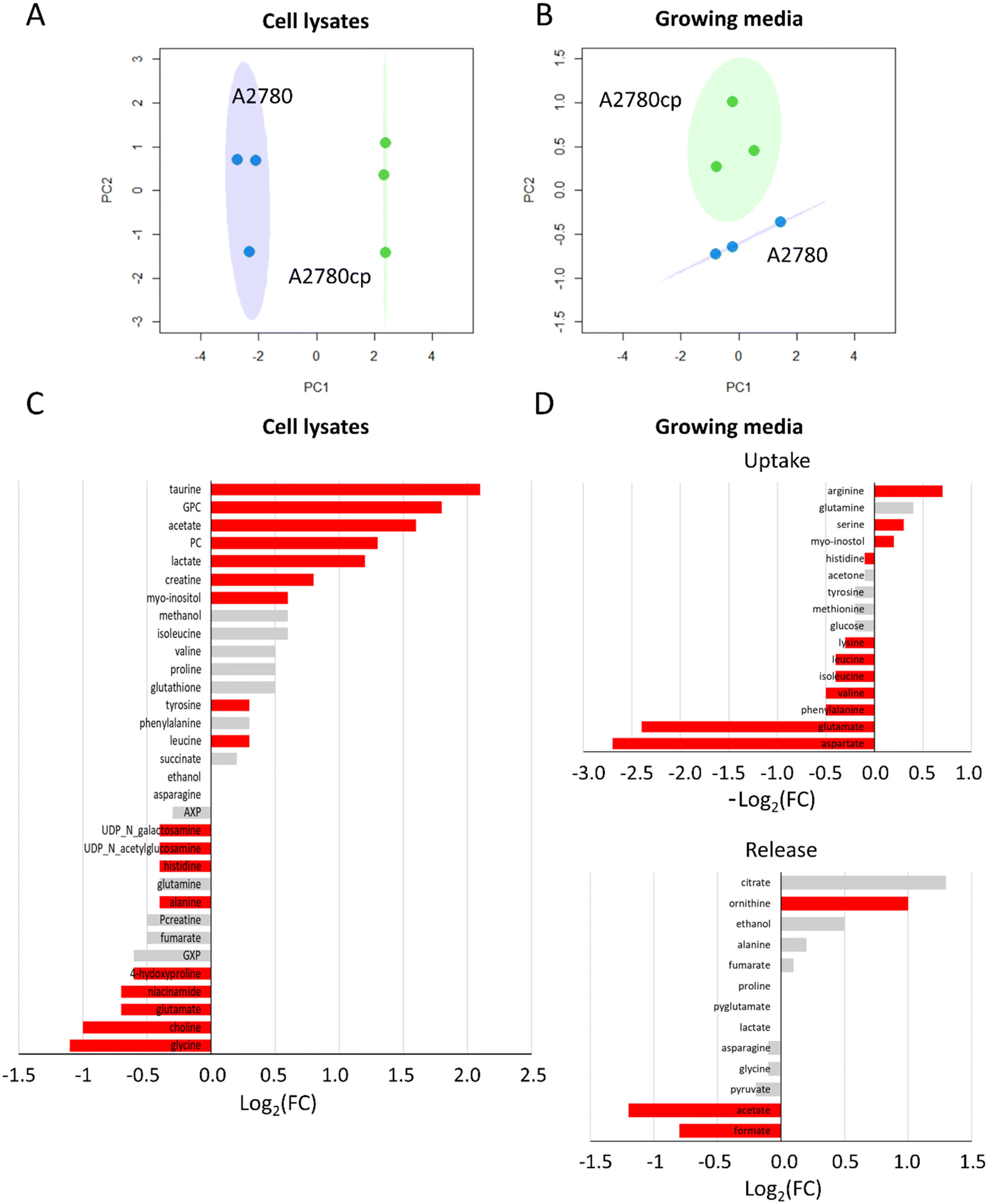 | ||
| Fig. 5 Comparison between untreated A2780 and A2780cp cells. PCA score plots with 95% covariance ellipses of (A) cell lysates (PC1: 78.22% and PC2: 14.57%) and (B) growth media (PC1: 56.71% and PC2: 34.43%); blue dots (A2780) and green dots (A2780cp). (C and D) Bar plots of fold change for the comparison of A2780 cp cells and A2780 cells. (C) Cell lysates: positive/negative bars indicate the higher/lower concentration of A2780cp cells with respect to A2780 cells (log2(FC)). (D) Growth media: positive/negative bars indicate the higher/lower process of uptake (−log2(FC)) or release (log2(FC)) in A2780cp cells with respect to A2780 cells. Red bars indicate statistical significance (p-value <0.05). | ||
In A2780cp cell lysates, we observed much greater concentrations (log2(FC) > 1) with respect to A2780 cells for the two choline derivatives (PC and GPC), and for taurine, acetate, lactate, creatine and myo-inositol, and to a lesser extent for Tyr and Leu (Fig. 5C). In A2780cp cells, the GPC/PC ratio has a value of 1.0 in comparison to 0.7 in A2780 cells (Fig. S1†). Lower concentrations of UDP-N-acetylglucosamine (UDP-GlcNAc), UDP-N-galactosamine, His, Ala, 4-hydroxyproline, niacinamide, Glu, choline and Gly (log2(FC) < −1) were detected (Fig. 5C). Looking at the growth media (Fig. 5D), A2780cp cells show a lower uptake of amino acids, such as the branched chain amino acids (Val, Ile, Leu) and the aromatic amino acids (His, Phe and Tyr). Interestingly, A2780cp cells do not use the Asp and Glu present in the medium. In A2780cp cells an increase in ornithine release along with an increase in Arg uptake is detected; these changes are evocative of a dysregulation of the urea cycle. Resistant cells also show a significant decrease in the release of formate and acetate. At variance, glucose uptake and lactate and pyruvate release do not exhibit significant differences between the two cell lines.
Then, we analysed the effects of the treatment of A2780cp cells on CIS. In particular, we tested the condition of equal toxicity (i.e. 48 h treatment with 2.5 μM and 15 μM of CIS for A2780 cells and A2780cp cells, respectively) and the condition of equal concentration (i.e. 48 h treatment with 2.5 μM, corresponding to the 72 h IC50 value for A2780) (Fig. 6).
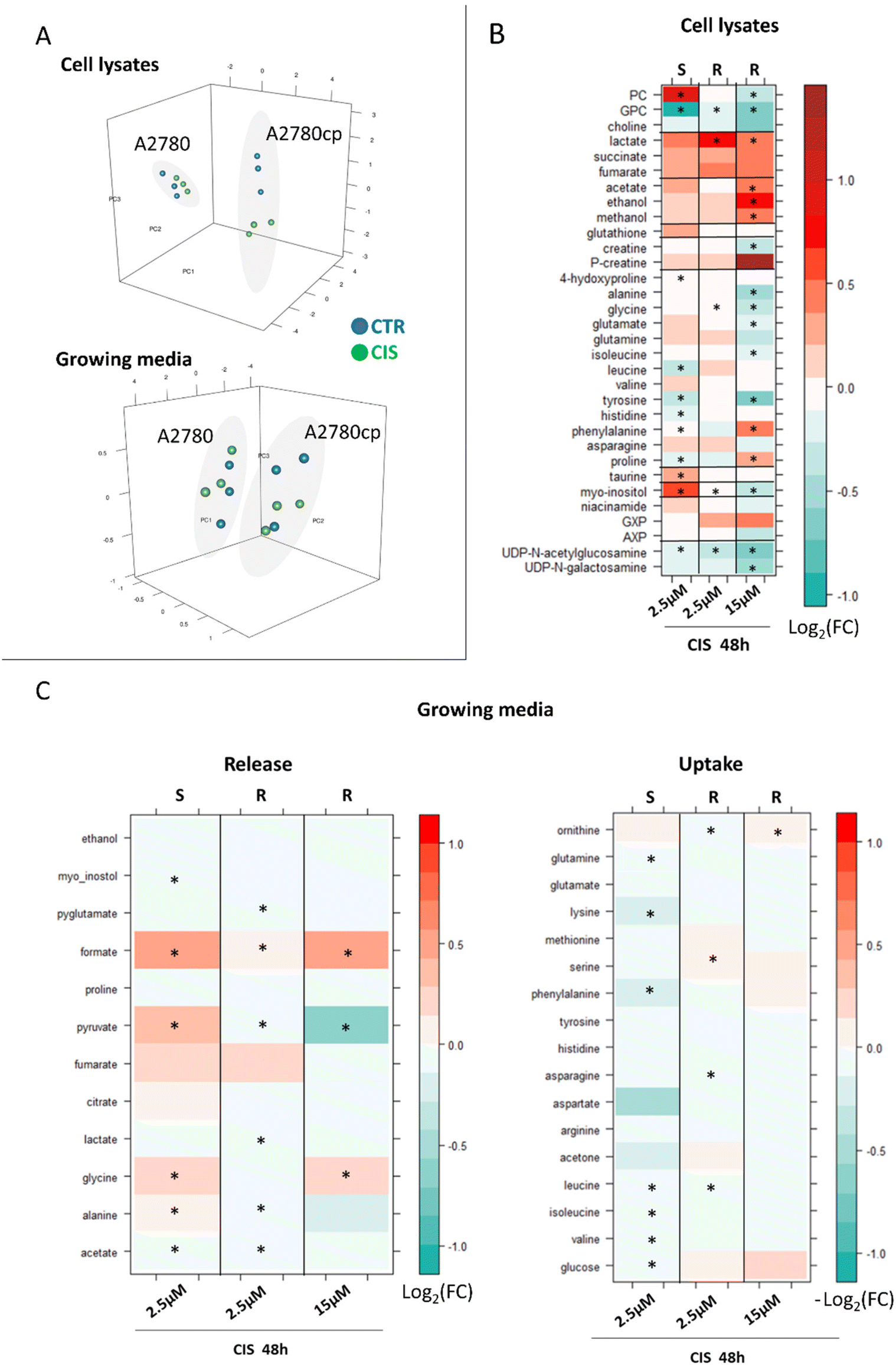 | ||
| Fig. 6 Metabolomic changes induced by CIS treatment: comparison between A2780 and A2780cp cells. (A) PCA score plots of cell lysates (PC1: 64.34%, PC2: 19.46%, and PC3: 0.75%) and growth media (PC1: 83.28%, PC2: 10.12%, and PC3: 0.32%) for the comparison of equal toxicity between A2780 and A2780cp cells; blue dots (control cells) and green dots (CIS-treated cells) and (B) level plots of fold change for cell lysates: red/green parameters indicate the higher/lower concentration of CIS-treated cells with respect to control samples (log2(FC)). (C) Growth media: red/green parameters indicate the higher/lower process of uptake (−log2(FC)) or release (log2(FC)) in CIS-treated cells with respect to control samples. The brightness of the colors corresponds to the magnitude of FC. Asterisks indicate statistical significance (p-value <0.05). S: sensitive A2780 cells and R: cisplatin-resistant A2780cp cells. | ||
When a CIS concentration corresponding to equal cytotoxicity was used, the changes detected in A2780cp lysates were found to be more evident than in A2780 lysates (PCA score plots in Fig. 6A), due to the greater number of affected metabolites and to the larger intensity of their changes, with 17 out of 34 metabolites significantly altered upon treatment, in comparison to 11 significantly altered metabolites in A2780 cells (Fig. 6B, 3rd column).
Most of the CIS-induced changes are coherent (in terms of direction) between the two cell lines, but 4 metabolites move in the opposite direction. Indeed, PC, glutamate and myo-inositol decrease upon treatment with A2780cp cells while they increase in the sensitive cells; proline shows the opposite trend. Upon cisplatin treatment with A2780cp, the GPC/PC ratio goes from 1.0 to 0.8 (Fig. S2†), a change to be compared with the decrease from 0.7 to 0.2 observed in A2780 cells.
Fewer differences are instead observed in A2780cp cells with respect to A2780 cells at the level of the exo-metabolites (Fig. 6C, 3rd column). In both cases the CIS treatment induces a significant increase in the release of formate and glycine. The opposite behaviour for the release of pyruvate and Ala (even if not significant) is instead observed.
As expected, when cells were treated with the equal concentration of CIS used for the 48 h treatment of A2780 cells (i.e. 2.5 μM), the observed changes were milder (in terms of log2(FC)) but all coherent (in terms of direction) with respect to the changes observed under the equal toxicity conditions (Fig. 6B, 2nd column). Four metabolites are significantly decreased upon treatment, i.e., GPC, glycine, myo-inositol and UDP-GlcNAc, while lactate levels are significantly increased.
The same trend is observed in the exo-metabolome; milder alterations in terms of log2(FC) and all coherent in terms of the direction of the changes with respect to the equal toxicity conditions, Fig. 6C, 2nd column.
Discussion
NMR metabolomics is an excellent tool for the detailed characterisation of the cellular effects of drugs. Though intrinsically not very sensitive, this NMR method has the potential to measure – in real time and in a very reproducible manner – the concentrations of the most abundant metabolites in untreated cells and their changes upon drug treatment at different time intervals; in turn, the concentrations of a few selected metabolites and their time dependent changes may reflect very closely the actual metabolic state of the cell and its possible dysregulations.As a matter of fact, numerous NMR studies during the last ten years have analysed and interpreted the metabolomic alterations induced by anticancer metallodrugs, in primis cisplatin, in cultured cancer cells;23,24 yet, the resulting picture appears not to be homogeneous nor conclusive. As stated above, this may be the consequence of the intrinsic large variability occurring among the several tested cancer cell lines and also of the highly different concentrations of Pt drugs used in those studies. These arguments led us to investigate in detail and comparatively, through 1H NMR metabolomics, the effects of the three main clinically established Pt drugs, i.e., cisplatin, carboplatin and oxaliplatin, in A2780 ovarian cancer cells, one of the most widely used cancer cell models.
The 1H NMR analysis of A2780 cancer cells before treatment allowed us to identify unambiguously and quantify 34 metabolites in the endo-metabolome and 30 metabolites in the exo-metabolome. The direct observation of these metabolites offers a chance to monitor directly the main metabolic pathways of the cell. Afterwards, A2780 cancer cells were treated with equitoxic amounts of the three mentioned platinum drugs (using drug concentrations corresponding to the respective IC50 values at 72 h), and the induced alterations in the endo-metabolome and exo-metabolome analysed through NMR at 24 and 48 h in order to detect the early cellular effects of these Pt drugs before significant cell death occurs. On the whole, the three Pt drugs turned out to affect a rather limited number of metabolites and to cause relatively moderate quantitative changes even after 48 h treatment; also, we noticed that the patterns of the induced changes were quite similar among the three Pt drugs, at least from the qualitative point of view.
Then, the clinical importance of acquired resistance to Pt drugs prompted us to analyse comparatively an additional A2780 ovarian cancer cell line made resistant to cisplatin (the A2780cp line). Notably, the A2780 and A2780cp cell lines show starting metabolomic profiles that are significantly different from each other in terms of the concentrations of many metabolites, suggesting that the acquired CIS resistance is associated with multiple metabolic alterations. Indeed, a number of studies have shown that CIS resistance is a multifaceted phenomenon that is mediated by a variety of cellular processes such as increased drug inactivation/efflux, increased DNA repair, alterations in drug toxicity and changes in the apoptotic processes.30 It was previously reported that platinum drugs interact with membranes and cause a relevant change in their biophysical properties; such alterations may activate apoptotic pathways but are also involved in Pt uptake and Pt resistance.31 Accordingly, the increased amount of PC and GPC and the decreased level of choline that we found in A2780cp cells in comparison to A2780 cells can be explained in terms of biophysical changes in the membranes of resistant cells, which in turn might influence drug transport. Consistently, we observe here the intracellular accumulation of taurine in A2780cp cells, which has been reported to be a marker of the inhibition of cisplatin uptake.32 In fact, taurine release is controlled by the same proteins that were suggested to be directly involved in the cellular uptake of cisplatin and that are found to be down-regulated in resistant cells.32 The observed increase in choline derivatives and decrease in UDP-GlcNAc and UDP-N-galactosamine are considered markers for cancer aggressiveness.33,34 Among the possible effects related to resistance, the proposed pro-survival increased production of glutathione23,35,36 was not visible in our experiments, whereas this phenomenon has been clearly observed by NMR for other metallodrugs.14 Even with these different starting situations, the responses to the CIS treatment of A2780cp and A2780 cells show a number of common traits. Two different conditions were tested to compare the response to CIS of A2780cp cells with that of A2780 cells, i.e. equal toxicity and equal concentration of platinum. On the whole, the detected changes were coherent with those observed in the A2780 cells with a few notable exceptions, as it will be discussed below.
The main metabolic changes elicited by Pt drugs in A2780 and A2780cp cells may be recapitulated and interpreted as follows.
Formate and Gly are precursors for the de novo synthesis of nucleotides and their increased release in the media for both A2780 and A2780cp cells could be the consequence of the reduced biosynthetic demand. This observation is broadly consistent with the primary mechanism of Pt drugs that are believed to exert their action mainly through the formation of Pt adducts with genomic DNA, thus causing inhibition of DNA replication.3 Beyond hitting their primary target (DNA), Pt-drugs may give rise to multiple off-target interactions, affecting cell signalling and other pathways. Details of these effects are given below. In this context, it is worth reminding that apoptosis mediated by endoplasmic reticulum (ER) stress has been proposed by several authors as an alternative cancer cell death mechanism characteristic of Pt drugs.37,38 Additionally, the group of Steve Lippard even suggested that the mode of action of OXA mostly relies on the induction of ribosome biogenesis stress rather than direct DNA damage.9
In our experiments, the change in PC and GPC concentrations is the most relevant event occurring in A2780 cells upon treatment with the 3 Pt drugs; a large and early decrease of GPC is accompanied by a later increase of PC. In A2780 cells, at 48 h, this trend translates into a GPC/PC ratio significantly decreased for all three treatments, going from 0.7 in controls to 0.2, 0.3 and 0.3 for CIS, OXA and CARBO, respectively. Instead, the CIS-treated A2780cp cells are characterized by a higher GPC/PC starting ratio (1.0 vs. 0.7), which slightly decreases down to 0.8 upon CIS treatment. Consistently with our findings, a favourable response to treatments is believed to be associated with a reduction in GPC concentration during the course of the treatment.39 Multiple mechanisms can contribute to the modulation of GPC and PC levels from membrane remodelling as a function of Pt drug uptake31 to the de novo lipogenic synthesis and/or lipolytic pathway.40 The increased level of intracellular PC in cancer cells is mainly attributed to the upregulation of choline kinase, the first enzyme in the Kennedy pathway responsible for choline phosphorylation, but can also be partly produced by the activation of phosphatidylcholine-specific phospholipase C.40 Notably, depletion of phosphatidylcholine affects the endoplasmic reticulum morphology,41 whereas choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis.42 Therefore, a close, albeit complex, interplay exists between lipid metabolism and ER stress.
The relevant changes in the levels of taurine and myo-inositol observed upon Pt drug treatment may be tentatively interpreted as follows. ER stress is intimately linked to the unfolded protein response (UPR), a cell-signaling system that readjusts ER folding capacity to restore protein homeostasis. The key UPR signal activator, the inositol requiring enzyme 1α/β (IRE1), responds to ER stress by propagating the UPR signal from the ER to the cytosol. The UPR has a dual function, as it promotes homeostasis and cell survival under mild ER stress but can lead to apoptosis and cell death under intense, persistent stress.43,44 The intracellular levels of myo-inositol are expected to modulate UPR via IRE1.45 The different signs of the fold change in myo-inositol induced by CIS in A2780 and A2780cp cells might be ascribed to the above UPR-modulation of cell survival/apoptosis. In addition, myo-inositol together with taurine is an osmolyte involved in modulating the cell volume.46 The increase in the intracellular levels of these two molecules upon platinum treatment in A2780 cells could be related to a progression towards an apoptotic phenotype and to cell adaptation in an attempt to contrast it. In particular, taurine corrects cell shrinkage, one of the main characteristics of the apoptotic process.32 It was recently demonstrated that in the A2780 cell line, cisplatin resistance correlates with an increased intracellular accumulation of taurine that in turn protects cells from volume reduction.32 The here-detected high levels of taurine (FC > 2) in A2780cp cells with respect to the sensitive cells are in line with this observation.
Another consistently observed change in all our examples is the decrease of UDP-GlcNAc levels; this variation is significant following CIS treatment in both A2780 and A2780cp cells, while with OXA and CARBO the decrease is not statistically significant. UDP-GlcNAc is the precursor of N-acetylglucosamine, which is added to Ser and Thr residues for O-GlcNAcylation and to Asn for N-glycosylation. Interestingly, disruption of this latter glycosylation leads to the accumulation of unfolded proteins in the ER lumen and induces the UPR.47 This inducible post-translational modification is differently modulated in different tumor types and has been proposed to regulate cancer cell metabolic reprograming and survival stress signalling, which also occurs in the ER.18,48
Notably, upon platinum treatment, in both sensitive and resistant cells, we observed a clear increase of intracellular succinate, fumarate and lactate. The variations of the levels of these metabolites appear stronger in A2780cp cells where the increase of the lactate level is statistically significant. In contrast, the changes in the exo-metabolome upon treatment are not coherent between the two cell lines. In A2780 cells, Pt drug treatment leads to a small, but significant, reduction of the glucose uptake. This tendency is more marked for CIS and OXA treatments; in the latter case we also observed a significant decrease of lactate excretion. Interestingly, ER-induced stress has been reported to be associated with greatly reduced glucose uptake and lactate production.49 Moreover, a significant increase in the release into the medium of pyruvate and alanine is here observed for A2780 cells. In contrast, CIS treatment of A2780cp cells establishes a more anaerobic metabolism, where a significant increase in the intracellular concentration of lactate is accompanied by an increase in the glucose uptake (although not significant); accordingly, the amounts of pyruvate and alanine excreted in the medium decrease significantly. The levels of extracellular lactate were not significantly altered by the treatment.
Finally, the redox metabolism, at least for what concerns the NMR-detectable part (which does not include ROS nor NOx), is in all cases scarcely affected by Pt drug treatment. In agreement with our results, no change in the GSH level was detected in the A549 lung cancer cell line treated for 48 h with cisplatin.17
Experimental
Cell culture and Pt drug treatment
A2780 cells (RRID: CVCL 0134) were purchased from Sigma-Aldrich and derived from the European Collection of Authenticated Cell Culture (human ovarian carcinoma; catalogue no.: 93112519; lot no. 16L020, passage no.: P + 8); the A2780 human cells have been authenticated using STR (or SNP) profiling. A2780cp cells were obtained via prolonged exposition to sub-lethal doses of cisplatin. All experiments were performed with mycoplasma-free cells.Cells were maintained in RPMI1640 medium supplemented with 2 mM glutamine, 10% of fetal calf serum (FCS) and antibiotics at 37 °C in a 5% CO2 atmosphere and sub-cultured twice weekly. Oxaliplatin and carboplatin were purchased from Alfa Aesar, with a declared purity of 100% and 99.2%, respectively. Cisplatin was purchased from Merck, with a declared purity of 99.9%.
The inhibition of cell proliferation by CIS, OXA and CARBO on A2780/A2780cp cell lines was evaluated by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) test using an exposure time of 72 h. Viable cells can convert MTT into formazan, which is coloured and detectable at 595 nm. From the absorbance measurements, the half-maximal inhibitory concentration (IC50) value was calculated using GraphPad Prism software version 6.0 (Graphpad Holdings, LLC, USA).
For drug treatment, 2.7 × 106 A2780 cells were seeded in a p100 plate and after 24 hours they were exposed to a concentration of the three Pt drugs, i.e. CIS, OXA and CARBO equal to their 72 h-exposure IC50 value (Fig. 1B). The drug incubations were stopped at two different time points, 24 h and 48 h. For CIS, 6 h incubation was also tested.
The A2780cp cells were exposed to a concentration of cisplatin equal to their 72 h-exposure IC50 value (Fig. 1B). The A2780cp cells were also exposed to a concentration of cisplatin equal to the 72 h-exposure IC50 value for A2780 sensible cell lines. The former condition was used to study the A2780cp and A2780 cells under equal toxicity. The latter condition was used, instead, to study both cell lines using equal concentrations of the drug. In both cases the drug incubation was stopped after 48 h of treatment.
All the experiments were performed on biological triplicates. At the end of each experiment, in agreement with the procedures for NMR sample preparation,10,14,50 the cells were washed with phosphate buffered saline (PBS, pH 7.4) and then scraped in PBS supplemented with a protease inhibitor cocktail diluted in DMSO to quench the enzymatic reactions and stabilize the sample composition; the cells were lysed by sonication in ice and then centrifuged at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min at 4 °C. 1 ml of the growth medium was also collected for the metabolomic analysis. All the samples were stored at −80 °C until further analyses.
000g for 30 min at 4 °C. 1 ml of the growth medium was also collected for the metabolomic analysis. All the samples were stored at −80 °C until further analyses.
NMR sample preparation
Frozen samples were thawed at room temperature and shaken before use. For cell lysates, 50 μl of 2H2O were added to 450 μl of each lysate sample (PBS, pH 7.4). In the case of cell culture media, 300 μl of sodium phosphate buffer (75 mM Na2HPO4; 20% v/v 2H2O, pH 7.4) was added to 300 μl of each sample. The mixtures were homogenized by vortexing for 30 s and transferred into 5 mm NMR tubes for analysis.NMR analysis
Cell lysates and the respective growth media were analysed using untargeted 1H NMR based metabolomics.15,16 Samples for NMR analyses were prepared according to procedures developed to obtain highly quality samples for cell metabolomics.15,16 All the 1H NMR spectra were recorded with a Bruker 600 MHz spectrometer (Bruker BioSpin) optimized for metabolomic investigations, operating at 600.13 MHz proton Larmor frequency and equipped with a 5 mm PATXI 1H–13C–15N and 2H-decoupling probe including a z-axis gradient coil, an automatic tuning-matching (ATM) and an automatic refrigerated sample changer (SampleJet, Bruker BioSpin). A BTO 2000 thermocouple was used for temperature stabilization at the level of approximately 0.1 K of the sample. Before measurement, to equilibrate temperature at 300 K, samples were kept for at least 5 minutes inside the NMR probe head.The spectra of cell lysates were acquired with the Carr–Purcell–Meiboom–Gill (CPMG) sequence using a one-dimensional (1D) spin–echo sequence with water presaturation, 512 scans, 73728 data points, a spectral width of 12019 Hz and a relaxation delay of 4 s.15
The spectra of the growth media were acquired with a 1D nuclear Overhauser enhancement spectroscopy (NOESY)-presaturation pulse sequence, 64 scans, 98304 data points, a spectral width of 18028 Hz and a relaxation delay of 4 s.15
The raw data were multiplied by a 0.3 Hz exponential line broadening before applying Fourier transform. Transformed spectra were automatically corrected for phase and baseline distortions and calibrated using TopSpin 3.6 (Bruker Biospin srl).
Statistical analysis
All the statistical analyses were performed using the “R” software. Multivariate principal component analysis (PCA) was applied on binned spectra. To this aim, each spectrum in the region of 10.00–0.2 ppm was divided into 0.02 ppm chemical shift bins, and the corresponding spectral areas were integrated using the AMIX software. The area of each bin was normalized to the total spectral area calculated with exclusion of the water and DMSO regions (4.50–5.00 ppm and 2.90–2.60 ppm, respectively). No scaling method was applied on the data. PCA was used as unsupervised exploratory analysis to obtain a general overview of the data and to detect the presence of clusters.The metabolites, whose peaks were well resolved, were assigned and their levels were analysed using a R script developed in-house. We identified and quantified 34 metabolites in the spectra of cell lysates and 30 metabolites in the spectra of the growth medium (Table S1†). The assignment was performed using an internal 1H NMR spectral library of pure organic compounds (BBIOREFCODE, Bruker BioSpin), stored reference NMR spectra of metabolites and spiking experiments. Matching between new NMR data and databases was performed using the AssureNMR software (Bruker BioSpin). The nonparametric Wilcoxon–Mann–Whitney test was used for the determination of meaningful metabolites; a p-value <0.05 was considered statistically significant. Log2 fold change (FC) was calculated for each metabolite to show how the metabolite levels vary upon the different comparisons. FC was calculated as the ratio of median metabolite concentrations in the spectra of the two groups (treated over controls). In the growth media, the metabolites were divided into two different classes, i.e. those that are taken up from the medium and those that are released into the medium. While for the molecules that are released, lower concentration levels upon treatment mean a lower release, for the molecules that are taken up from the starting media, lower levels upon treatment mean a greater consumption of nutrients, i.e. increased uptake, and vice versa. Thus, for the taken-up metabolites the quantity that is plotted is −log2(FC).
Conclusions
In conclusion, this study has provided detailed and comparative insight into the cellular effects of the three main Pt drugs in A2780 cancer cells in terms of NMR detectable metabolomic alterations. We have shown that Pt treatment produces, on the whole, similar and relatively moderate effects in A2780 cancer cells. In any case, the patterns of the observed changes were carefully analyzed and their meaning interpreted in the frame of the available mechanistic knowledge. In particular, a number of metabolomic alterations triggered by Pt drugs could be related to the induction of endoplasmic reticulum stress, in good agreement with previous observations. This study further supports the use of NMR metabolomics as an excellent investigative tool to characterize drug induced metabolic alterations.Author contributions
Conceptualization, L. M. and P. T.; methodology, V. G.; formal analysis, V. G. investigation, V. G. and F. M.; resources, La. M.; writing – original draft, L. M., P. T. and V. G.; writing–review and editing, F. M. and La. M.; supervision, L. M. and P. T. The work reported in the paper has been performed by the authors, unless clearly specified in the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. T. and V. G. acknowledge the support and the use of resources of Instruct-ERIC, a Landmark ESFRI project, and specifically the CERM/CIRMMP Italy Centre.Notes and references
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs, Chem. Rev., 2016, 116(5), 3436–3486 CrossRef CAS PubMed.
- L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nat. Rev. Cancer, 2007, 7(8), 573–584 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4(4), 307–320 CrossRef CAS PubMed.
- S. Schoch, S. Gajewski, J. Rothfuß, A. Hartwig and B. Köberle, Comparative Study of the Mode of Action of Clinically Approved Platinum-Based Chemotherapeutics, Int. J. Mol. Sci., 2020, 21(18), 6928 CrossRef CAS PubMed.
- R. S. Go and A. A. Adjei, Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin, J. Clin. Oncol., 1999, 17(1), 409–422 CrossRef CAS PubMed.
- J. Lokich, What Is the “Best” Platinum: Cisplatin, Carboplatin, or Oxaliplatin?, Cancer Invest., 2001, 19(7), 756–760 CrossRef CAS PubMed.
- M. P. M. Marques, D. Gianolio and G. Cibin, et al., A molecular view of cisplatin's mode of action: interplay with DNA bases and acquired resistance, Phys. Chem. Chem. Phys., 2015, 17(7), 5155–5171 RSC.
- C. W. Fong, Platinum anti-cancer drugs: Free radical mechanism of Pt-DNA adduct formation and anti-neoplastic effect, Free Radicals Biol. Med., 2016, 95, 216–229 CrossRef CAS PubMed.
- P. M. Bruno, Y. Liu and G. Y. Park, et al., A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress, Nat. Med., 2017, 23(4), 461–471 CrossRef CAS PubMed.
- G. D'Alessandro, D. Quaglio and L. Monaco, et al., 1H-NMR metabolomics reveals the Glabrescione B exacerbation of glycolytic metabolism beside the cell growth inhibitory effect in glioma, Cell Commun. Signaling, 2019, 17(1), 108 CrossRef PubMed.
- J. L. Griffin and J. P. Shockcor, Metabolic profiles of cancer cells, Nat. Rev. Cancer, 2004, 4(7), 551–561 CrossRef CAS PubMed.
- F. De Castro, M. Benedetti, L. Del Coco and F. P. Fanizzi, NMR-Based Metabolomics in Metal-Based Drug Research, Molecules, 2019, 24(12), 2240 CrossRef CAS PubMed.
- J. Zhang, C. Hang and T. Jiang, et al., Nuclear Magnetic Resonance-Based Metabolomic Analysis of the Anticancer Effect of Metformin Treatment on Cholangiocarcinoma Cells, Front. Oncol., 2020, 10, 570516 CrossRef PubMed.
- V. Ghini, T. Senzacqua and L. Massai, et al., NMR reveals the metabolic changes induced by auranofin in A2780 cancer cells: evidence for glutathione dysregulation, Dalton Trans., 2021, 50(18), 6349–6355 RSC.
- A. Vignoli, V. Ghini and G. Meoni, et al., High-Throughput Metabolomics by 1D NMR, Angew. Chem., Int. Ed., 2019, 58(4), 968–994 CrossRef CAS PubMed.
- P. G. Takis, V. Ghini, L. Tenori, P. Turano and C. Luchinat, Uniqueness of the NMR approach to metabolomics, TrAC, Trends Anal. Chem., 2019, 120, 115300 CrossRef.
- I. F. Duarte, A. F. Ladeirinha and I. Lamego, et al., Potential Markers of Cisplatin Treatment Response Unveiled by NMR Metabolomics of Human Lung Cells, Mol. Pharmaceutics, 2013, 10(11), 4242–4251 CrossRef CAS PubMed.
- X. Pan, M. Wilson and L. Mirbahai, et al., In vitro metabonomic study detects increases in UDP-GlcNAc and UDP-GalNAc, as early phase markers of cisplatin treatment response in brain tumor cells, J. Proteome Res., 2011, 10(8), 3493–3500 CrossRef CAS PubMed.
- X. Pan, M. Wilson and C. McConville, et al., Increased unsaturation of lipids in cytoplasmic lipid droplets in DAOY cancer cells in response to cisplatin treatment, Metabolomics, 2013, 9(3), 722–729 CrossRef CAS PubMed.
- T. J. Carneiro, M. Vojtek and S. Gonçalves-Monteiro, et al., Metabolic Impact of Anticancer Drugs Pd2Spermine and Cisplatin on the Brain of Healthy Mice, Pharmaceutics, 2022, 14(2), 259 CrossRef CAS PubMed.
- K. Resendiz-Acevedo, M. E. García-Aguilera, N. Esturau-Escofet and L. Ruiz-Azuara, 1H -NMR Metabolomics Study of the Effect of Cisplatin and Casiopeina IIgly on MDA-MB-231 Breast Tumor Cells, Front. Mol. Biosci., 2021, 8, 1135 Search PubMed.
- L. Del Coco, M. Majellaro and A. Boccarelli, et al., Novel Antiproliferative Biphenyl Nicotinamide: NMR Metabolomic Study of its Effect on the MCF-7 Cell in Comparison with Cisplatin and Vinblastine, Molecules, 2020, 25(15), 3502 CrossRef CAS PubMed.
- S. Alonezi, J. Tusiimire and J. Wallace, et al., Metabolomic Profiling of the Synergistic Effects of Melittin in Combination with Cisplatin on Ovarian Cancer Cells, Metabolites, 2017, 7(2), E14 CrossRef PubMed.
- F. De Castro, M. Benedetti and G. Antonaci, et al., Response of Cisplatin Resistant Skov-3 Cells to [Pt(O,O′-Acac)(γ-Acac)(DMS)] Treatment Revealed by a Metabolomic 1H-NMR Study, Molecules, 2018, 23(9), 2301 CrossRef PubMed.
- I. F. Duarte, I. Lamego and J. Marques, et al., Nuclear Magnetic Resonance (NMR) Study of the Effect of Cisplatin on the Metabolic Profile of MG-63 Osteosarcoma Cells, J. Proteome Res., 2010, 9(11), 5877–5886 CrossRef CAS PubMed.
- I. Lamego, I. F. Duarte, M. P. M. Marques and A. M. Gil, Metabolic Markers of MG-63 Osteosarcoma Cell Line Response to Doxorubicin and Methotrexate Treatment: Comparison to Cisplatin, J. Proteome Res., 2014, 13(12), 6033–6045 CrossRef CAS PubMed.
- C. Lin, J. Dong and Z. Wei, et al., 1H NMR-Based Metabolic Profiles Delineate the Anticancer Effect of Vitamin C and Oxaliplatin on Hepatocellular Carcinoma Cells, J. Proteome Res., 2020, 19(2), 781–793 CrossRef CAS PubMed.
- H. Wang, J. Chen and Y. Feng, et al., 1H nuclear magnetic resonance-based extracellular metabolomic analysis of multidrug resistant Tca8113 oral squamous carcinoma cells, Oncol. Lett., 2015, 9(6), 2551–2559 CrossRef CAS PubMed.
- A. S. Martins, A. L. M. Batista de Carvalho, M. P. M. Marques and A. M. Gil, Response of Osteosarcoma Cell Metabolism to Platinum and Palladium Chelates as Potential New Drugs, Molecules, 2021, 26(16), 4805 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla and I. Vitale, et al., Molecular mechanisms of cisplatin resistance, Oncogene, 2012, 31(15), 1869–1883 CrossRef CAS PubMed.
- N. Martinho, T. C. B. Santos, H. F. Florindo and L. C. Silva, Cisplatin-Membrane Interactions and Their Influence on Platinum Complexes Activity and Toxicity, Front. Physiol., 2019, 9, 1898 CrossRef PubMed.
- B. H. Sørensen, U. A. Thorsteinsdottir and I. H. Lambert, Acquired cisplatin resistance in human ovarian A2780 cancer cells correlates with shift in taurine homeostasis and ability to volume regulate, Am. J. Physiol.: Cell Physiol., 2014, 307(12), C1071–C1080 CrossRef PubMed.
- N. M. Akella, L. Ciraku and M. J. Reginato, Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer, BMC Biol., 2019, 17(1), 52 CrossRef CAS PubMed.
- K. Glunde, Z. M. Bhujwalla and S. M. Ronen, Choline metabolism in malignant transformation, Nat. Rev. Cancer, 2011, 11(12), 835–848 CrossRef CAS PubMed.
- Z. H. Siddik, Cisplatin: mode of cytotoxic action and molecular basis of resistance, Oncogene, 2003, 22(47), 7265–7279 CrossRef CAS PubMed.
- L. Amable, Cisplatin resistance and opportunities for precision medicine, Pharmacol. Res., 2016, 106, 27–36 CrossRef CAS PubMed.
- A. Mandic, J. Hansson, S. Linder and M. C. Shoshan, Cisplatin Induces Endoplasmic Reticulum Stress and Nucleus-independent Apoptotic Signaling, J. Biol. Chem., 2003, 278(11), 9100–9106 CrossRef CAS PubMed.
- J. Tian, R. Liu and Q. Qu, Role of endoplasmic reticulum stress on cisplatin resistance in ovarian carcinoma, Oncol. Lett., 2017, 13(3), 1437–1443 CrossRef CAS PubMed.
- S. A. Moestue, G. F. Giskeødegård, M. D. Cao, T. F. Bathen and I. S. Gribbestad, Glycerophosphocholine (GPC) is a poorly understood biomarker in breast cancer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(38), E2506–E2506 CrossRef CAS PubMed.
- F. Podo, L. Paris and S. Cecchetti, et al., Activation of Phosphatidylcholine-Specific Phospholipase C in Breast and Ovarian Cancer: Impact on MRS-Detected Choline Metabolic Profile and Perspectives for Targeted Therapy, Front. Oncol., 2016, 6, 171 Search PubMed.
- N. Testerink, M. H. M. van der Sanden, M. Houweling, J. B. Helms and A. B. Vaandrager, Depletion of phosphatidylcholine affects endoplasmic reticulum morphology and protein traffic at the Golgi complex, J. Lipid Res., 2009, 50(11), 2182–2192 CrossRef CAS PubMed.
- E. Sanchez-Lopez, T. Zimmerman and T. Gomez del Pulgar, et al., Choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis via CHOP in cancer cells, Cell Death Dis., 2013, 4, e933 CrossRef CAS PubMed.
- J. Y. Chan, J. Luzuriaga and E. L. Maxwell, et al., The balance between adaptive and apoptotic unfolded protein responses regulates β-cell death under ER stress conditions through XBP1, CHOP and JNK, Mol. Cell. Endocrinol., 2015, 413, 189–201 CrossRef CAS PubMed.
- N. Hiramatsu, W.-C. Chiang, T. D. Kurt, C. J. Sigurdson and J. H. Lin, Multiple Mechanisms of Unfolded Protein Response–Induced Cell Death, Am. J. Pathol., 2015, 185(7), 1800–1808 CrossRef CAS PubMed.
- M. Suliman, M. W. Schmidtke and M. L. Greenberg, The Role of the UPR Pathway in the Pathophysiology and Treatment of Bipolar Disorder, Front. Cell. Neurosci., 2021, 15, 735622 CrossRef CAS PubMed.
- F. Klaus, M. Palmada and R. Lindner, et al., Up-regulation of hypertonicity-activated myo-inositol transporter SMIT1 by the cell volume-sensitive protein kinase SGK1, J Physiol, 2008, 586(6), 1539–1547 CrossRef CAS PubMed.
- S. Vasseur and S. N. Manié, ER stress and hexosamine pathway during tumourigenesis: A pas de deux?, Semin. Cancer Biol., 2015, 33, 34–39 CrossRef CAS PubMed.
- R. M. de Queiroz, R. Madan, J. Chien, W. B. Dias and C. Slawson, Changes in O-Linked N-Acetylglucosamine (O-GlcNAc) Homeostasis Activate the p53 Pathway in Ovarian Cancer Cells, J. Biol. Chem., 2016, 291(36), 18897–18914 CrossRef CAS PubMed.
- X. Wang, C. O. Eno and B. J. Altman, et al., ER stress modulates cellular metabolism, Biochem. J., 2011, 435(1), 285–296 CrossRef CAS PubMed.
- E. Perrin, V. Ghini and M. Giovannini, et al., Diauxie and co-utilization of carbon sources can coexist during bacterial growth in nutritionally complex environments, Nat. Commun., 2020, 11(1), 3135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt02068h |
| This journal is © The Royal Society of Chemistry 2022 |