DOI:
10.1039/D2DT02063G
(Paper)
Dalton Trans., 2022,
51, 12502-12511
A niobium pentafulvene ethylene complex: synthesis, properties and reaction pathways†
Received
28th June 2022
, Accepted 25th July 2022
First published on 26th July 2022
Abstract
The π–η5:σ–η1 coordination mode of early transition metal pentafulvene ligands yields a strongly nucleophilic exocyclic carbon atom (Cexo). The substitution of the chlorido ligand of bis(η5:η1-(di-p-tolyl)pentafulvene)niobium chloride (1) by reaction with ethyl magnesium bromide is subsequently followed by a β-C–H activation employing this Cexo, forming the pentafulvene niobium ethylene complex 2. The intermediately formed ethyl complex can be intercepted with water, protonating both pentafulvene moieties and thereby retaining the ethyl moiety to give the terminal oxo complex 3. Complex 2 shows cooperative reactions of the remaining pentafulvene and the ethylene ligand. While the pentafulvene functions as a proton acceptor, the ethylene can be liberated to provide a NbIII metal center, available for E–H bond addition. Thereby, the imido hydride complex 4 and niobaaziridine hydride 5 are obtained. These hydride complex formations are investigated by deuterium labeling, concluding the redox mechanism. An alternative β-hydride elimination pathway is further disproven by purposely synthesizing a proposed intermediate and thermally treating it, to show that it does not undergo the β-hydride elimination under reaction conditions.
Introduction
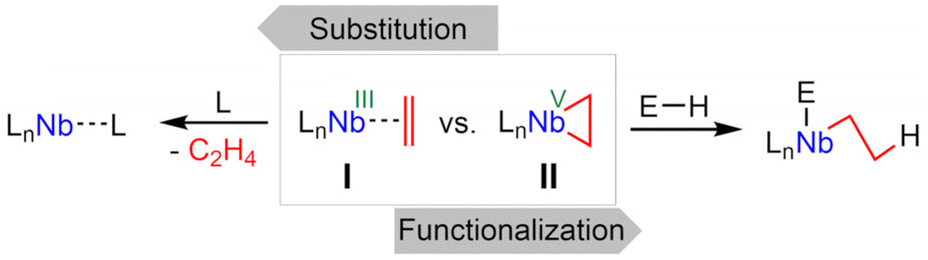
Low-valent early transition metals are attractive candidates for catalysis and synthesis.1 It is common to stabilize these typically highly reactive species, so that they can be released in situ if needed. Prominent examples are the Negishi (Cp2Zr(η2-butene)) and Rosenthal reagents (e.g. Cp2Ti(η2-Me3Si–C2–SiMe3)).2,3 Well-defined low-valent niobium complexes include classic CO, phosphine, and alkyne stabilized systems.4–6 A niobium(III) trisamido complex was found to be self-stabilizing by a reversible β-H activation forming niobaaziridine hydride complexes.7,8 Similarly, niobocene(III) alkyl complexes tautomerize to η2-alkene hydrides: extensive kinetic and mechanistic studies were done after synthesis by either treatment of niobocene trihydrides with alkenes,9–12 or by alkylation and subsequent rearrangements of niobocene dichlorides with suitable Grignard reagents.10–14 Other niobocene η2-alkene complexes have been found by reducing a suitable niobocene dichloride with an excess of alkylation agent, resulting in the coordination of an intramolecular alkene moiety, that can be exchanged with CO or isonitriles.15 Niobium η2-alkene complexes based on an (tBu3SiO)3Nb backbone have been prepared by ligand exchange,16 or reduction of the corresponding dichloride.17 A rearrangement of the alkene ligand giving alkylidene complexes was observed,17 and mechanistic studies yielded a silanolate ligand involved protonation and deprotonation step.18 Further, η2-cyclooctadiene15 and NMR evidence for unusual η2-alkene coordination to cationic d0 niobium complexes were reported.19 Reacting a niobium methylidyne complex with an excess of ethylene yielded the unexpected propenyl-ethylene complex, that can subsequently eliminate ethylene to yield a niobium(III) allyl complex.20 An unusual synthesis involves reduction of tris(propofolato) niobium dichloride and dehydrogenation of one isopropyl substituent, yielding an intramolecular niobium η2-alkene complex that works as a one-proton acceptor and allows insertion of multiple bond containing substrates into one Nb–C bond.21 On top of that, an in situ generated niobium η2-cyclopropene complex was shown to activate various C–H bonds.22 Generally, the reactivity of niobium alkene complexes can be separated into two groups (Scheme 1). Described as niobium η2-alkene complexes (I) stabilizing a d2 metal center, simple ligand exchange reactions become plain. Meanwhile the strong π-back-donation from the metal to the ligand is best described as a niobacyclopropan (II) and exhibits corresponding reactivity, for example the aforementioned insertion reactions and proton acceptor capabilities.
 |
| | Scheme 1 Reactivity of niobium η2-alkene complex(I) vs. reactivity of niobacyclopropane (II). | |
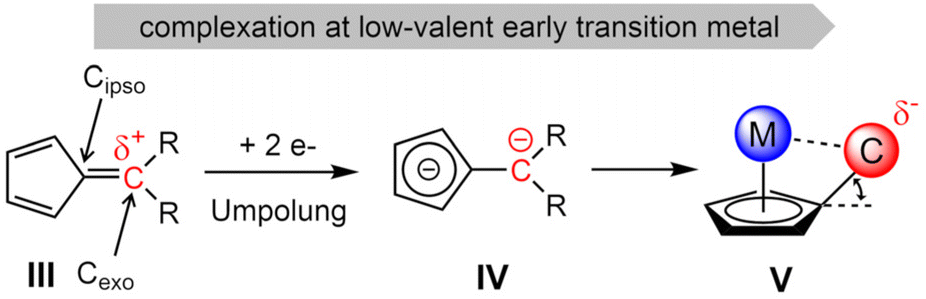
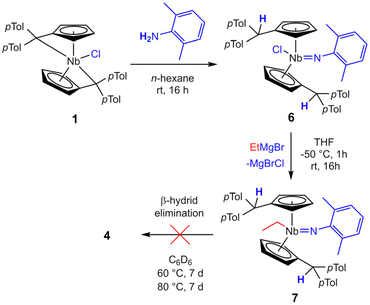
By the complexation of a pentafulvene (III) to a low-valent early transition metal center, a pentafulvene is reduced twice (IV), charge inverting the exocyclic carbon atom (Cexo, Scheme 2).23,24 The thereby achieved π–η5:σ–η1 coordination mode of the pentafulvene ligand (V) yields a highly reactive M–Cexo bond with a highly nucleophilic Cexo. Its bond to the sp2-hybridized Cipso pulls it back into the plane of the five-membered ring, limiting its desired interaction with the electrophilic metal center. This activated bond can be employed in numerous reactions, including E–H activations,25,26 and insertions of multiple bond containing substrates.27,28 Additionally, certain haptotropic flexibility of the pentafulvene ligand can be observed.29,30 Noteworthy, among the E–H activation reactions is the selective β-C–H activation of an ethyl ligand yielding a titanium ethylene complex.31 Recently reported, bis(η5:η1-(di-p-tolyl)pentafulvene)niobium chloride (1) is capable of selectively activating a σ-organyl α-C–H bond to generate an alkylidene complex.32 Here, we present the synthesis of a niobium ethylene complex, formed by β-C–H activation of an introduced ethyl ligand using a pentafulvene ligand as a proton acceptor. The formation, properties and reactivity of this niobium pentafulvene ethylene complex are discussed and possible mechanistic pathways are studied via deuterium labeling and synthesis of a postulated intermediate.
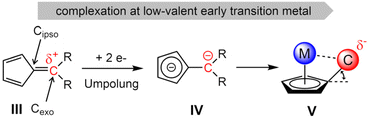 |
| | Scheme 2 Redox forms of a pentafulvene in the course of complexation to a low-valent early transition metal. | |
Results and discussion
Synthesis and characterization of niobium ethylene complex 2
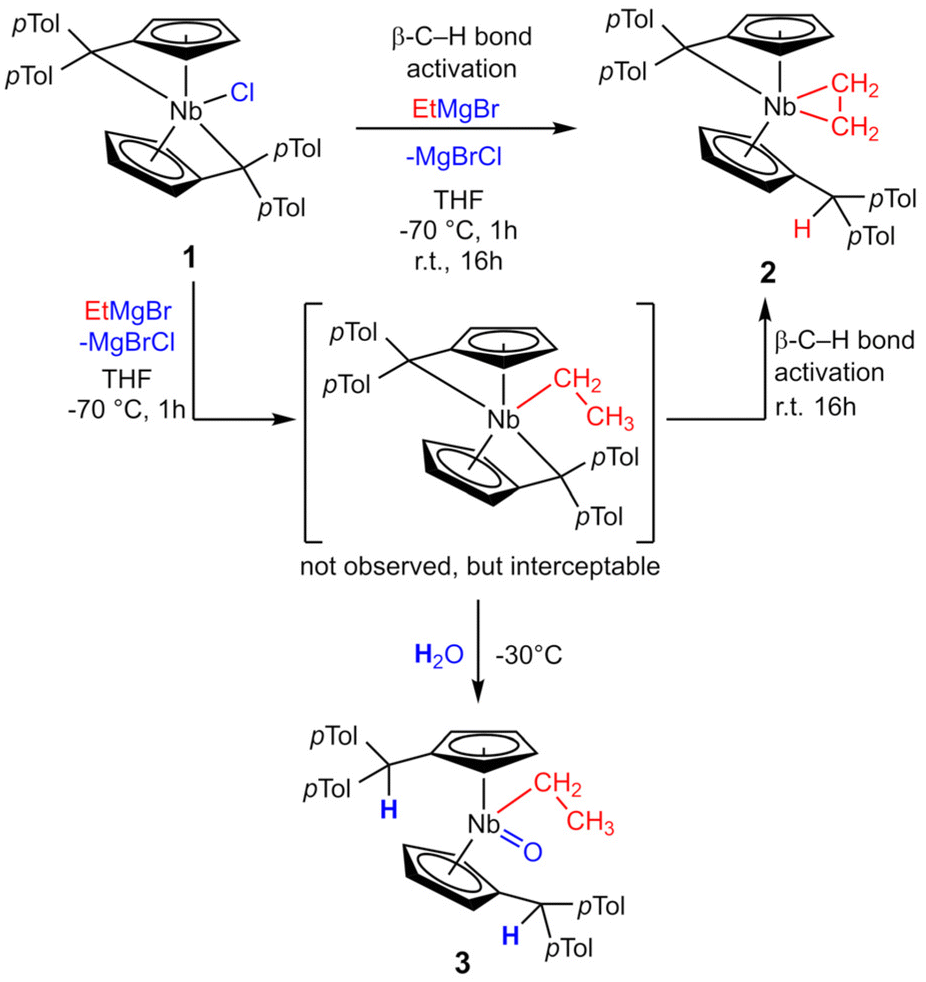
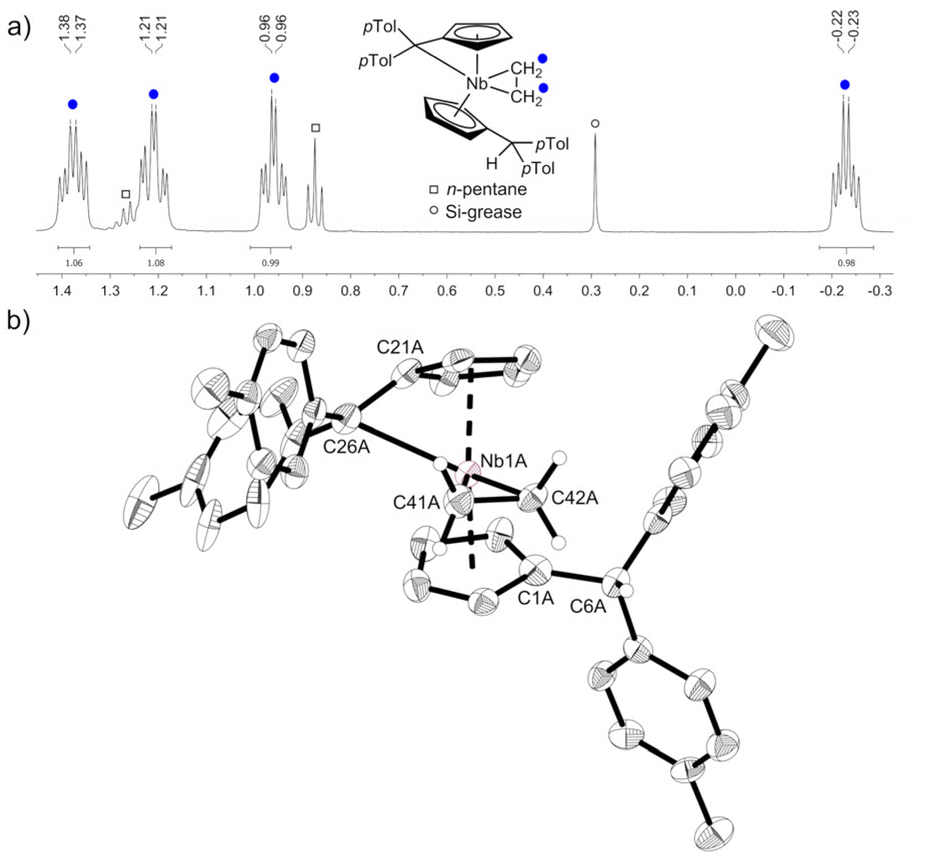
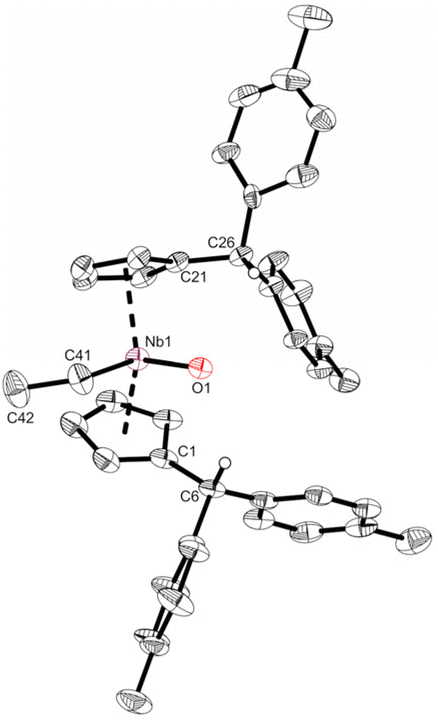
Reacting bis(η5:η1-(di-p-tolyl)pentafulvene)niobium chloride (1) with one equivalent of ethyl magnesium bromide at −70 °C in THF yields an immediate color change of the solution from dark red to dark orange. Bringing the solution to room temperature, the color changed to dark yellow. Niobium ethylene complex 2 is formed by β-C–H activation at the introduced ethyl substituent by the exocyclic carbon atom of one pentafulvene ligand (Scheme 3). After removal of the solvents and continuous extraction with pentane, subsequent decantation and drying in vacuum, 2 is obtained as a yellow microcrystalline powder in moderate isolated yield (62%). Complex 2 was fully characterized by NMR measurements. In the 1H NMR spectrum one signal for each ethylene proton is found (δ = −0.24; 0.95; 1.24; 1.38 ppm; compare Fig. 1a), indicating hindered rotation of the ethylene ligand, thus supporting strong back-donation of the metal center. Further, in the 13C{1H} NMR spectrum, the ethylene carbon atoms are found in the higher field (δ = 27.2; 29.4 ppm), and, determined by the non-decoupled spectrum, the 1JHC coupling constants (1JHC = 149; 150 Hz) are in the range of typical Csp3 coupling constants, Csp2 coupling constants are typically higher.33 Consequently, we display the ethylene complex 2 as a niobacyclopropane (Scheme 3). Characteristic of one remaining and one protonated pentafulvene ligand, in the 1H NMR spectrum, a sharp singlet shows this protonation (δ = 5.15 ppm), that is found between the eight signals for the five-membered ring protons (δ = 2.92–5.50 ppm). The structure of 2 was confirmed by X-ray diffraction (Fig. 1b). Single crystals were grown from a saturated n-hexane solution at room temperature.
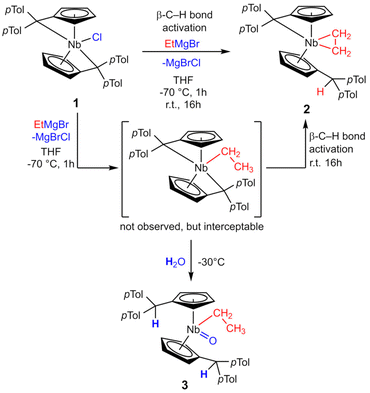 |
| | Scheme 3 Reaction of 1 with ethyl magnesium bromide to form ethylene complex 2. The bis(pentafulvene)niobium ethyl complex can be intercepted with water to form 3. | |
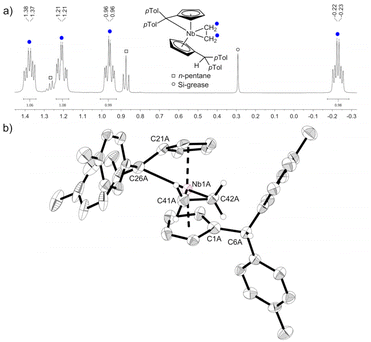 |
| | Fig. 1 (a) excerpt of the 1H NMR spectrum of 2 (500 MHz, C6D6, 305 K) showing the four signals of the ethylene ligand. (b) Molecular structure of compound 2 (one of the two non-equivalent, disordered molecules). Thermal ellipsoids are drawn at the 50% probability level. Most of the hydrogen atoms are omitted for clarity. | |
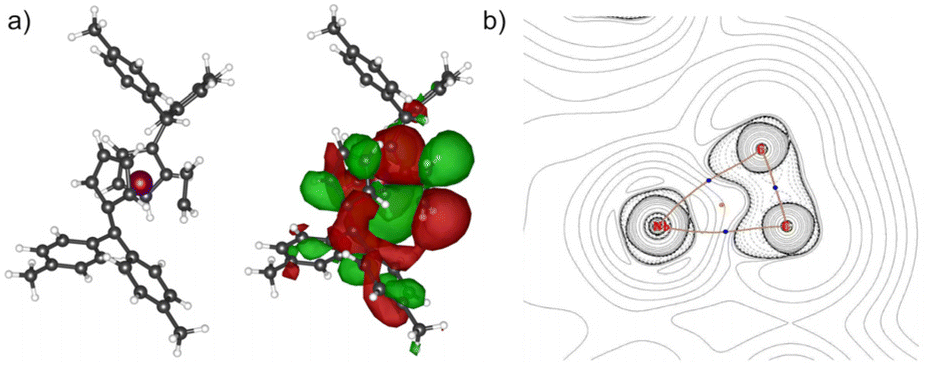
Complex 2 crystallizes in the orthorhombic space group Pna21 with two molecules in the unit cell, each position disordered by the two enantiomers (niobium pentafulvene fragments are overlapping while the Cp′ and ethylene ligands positions are inverted, compare Fig. S14†). The Nb–Cexo bonds of complex 2 (2.44–2.55 Å) are shorter than those in the reactant complex 1 (2.57–2.69 Å).34 Consistently, the Cipso–Cexo bonds of the fulvene ligands are longer in complex 2 than in complex 1 (1: 1.42–1.43 Å; 2: 1.45–1.46 Å) indicating a slightly stronger reduction of the ligand. The Cipso–Cexo bond of the formed Cp′ ligand is elongated to a Csp2–Csp3 single bond (1.51–1.54 Å) showing the protonation of the former pentafulvene ligand.35 The Nb–C bond length of the formed η2-ethylene ligand is located in the range of 2.19–2.29 Å and the average C–C bond length of 1.43 Å is, as expected, considerably longer than an ethylene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (1.33 Å)36 but not as long as a typical single bond in cyclopropanes (1.51 Å).35 Compared to recently reported niobium ethylene complexes, these parameters are in good agreement, overall showing a considerable niobacyclopropane character.14,20 To support the experimental molecular structure of 2, quantum chemical calculations at the density functional B3LYP/Def2-TZVP were used.37–40 Comparing the optimized molecular structure with the four structures obtained by X-ray diffraction, a respectable agreement is found despite the distorted system (Table S4†). The highest difference in the median bond lengths is found for the Nb–Cexo bond (4.4%). A comparably large part of the calculated HOMO of 2 is analogical to the ethylene π* orbital (Fig. 2a). Correspondingly, the 2D contour plot of the calculated Laplacian of the electron density in the ethylene niobium plane shows triangular bond paths (Fig. 2b), supporting the formulation of a niobacyclopropane.
C double bond (1.33 Å)36 but not as long as a typical single bond in cyclopropanes (1.51 Å).35 Compared to recently reported niobium ethylene complexes, these parameters are in good agreement, overall showing a considerable niobacyclopropane character.14,20 To support the experimental molecular structure of 2, quantum chemical calculations at the density functional B3LYP/Def2-TZVP were used.37–40 Comparing the optimized molecular structure with the four structures obtained by X-ray diffraction, a respectable agreement is found despite the distorted system (Table S4†). The highest difference in the median bond lengths is found for the Nb–Cexo bond (4.4%). A comparably large part of the calculated HOMO of 2 is analogical to the ethylene π* orbital (Fig. 2a). Correspondingly, the 2D contour plot of the calculated Laplacian of the electron density in the ethylene niobium plane shows triangular bond paths (Fig. 2b), supporting the formulation of a niobacyclopropane.
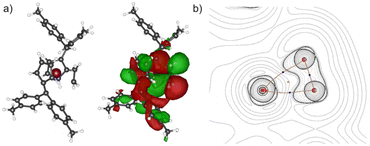 |
| | Fig. 2 (a) Calculated structure of 2 without and with the calculated surface diagram of the HOMO (isodensity value 0.008 at B3LYP/Def2-TZVP). (b) 2D contour plots of the calculated Laplacian of the electron density, ∇2ρ(r), in the niobium ethylene plane. Bond paths are shown in brown with the bond critical points in blue. The ring critical point is shown in orange. Dotted contours indicate regions of local charge accumulation (∇2ρ(r) < 0); solid contours indicate regions of local charge depletion ((∇2ρ(r) > 0). | |
In the formation of 2 the chloride of 1 is substituted by the ethyl ligand in a salt metathesis and subsequently the β-C–H bond is activated. For the related pentafulvene-induced α-C–H bond activation recently reported, the reaction is slow enough to monitor the intermediate by NMR spectroscopy.32 In the present case, this was unsuccessful. Instead, the postulated intermediate could be trapped with water, yielding the terminal niobocene oxo alkyl complex 3 (Scheme 3). Repeating the synthesis of 2, one equivalent of degassed water was added to the reaction solution at −30 °C. Warming to room temperature and stirring for 16 h, followed by extraction with n-hexane and drying in vacuum gave a brown solid, containing, among others, 2 and 3. Recrystallization of the obtained solid from n-hexane gives 3 in pure form as colorless crystals in low yield (5%). Compound 3 is air and moisture robust, well soluble in aromatic but poorly soluble in aliphatic solvents. 3 was fully characterized by NMR measurements. In the 1H NMR spectrum, the retained ethyl substituent gives a typical signal pattern (δ = 1.56 (t); 1.85 (q) ppm). Although both substituted Cp′ ligands are chemically identical, they are asymmetric in themselves giving e.g. four multiplets for the Cp′ protons (δ = 4.59; 5.29; 5.37; 5.69 ppm) due to the asymmetric substitution at the niobium center. The occurred protonation is shown by the sharp singlet of the two protonated Cexo positions (δ = 5.79 ppm). The structure of complex 3 was confirmed by single crystal X-ray diffraction (Fig. 3). Single crystals were obtained from a saturated n-hexane solution at room temperature. Oxo alkyl niobocene derivatives have been reported and characterized by XRD previously.41–43 Compound 2 shows similar bonding parameters as those differently substituted complexes, with a distorted tetrahedral coordination (bent niobocene 130.471(21)°), a strong Nb–O bond of 1.7559(19) Å and a Nb–C-bond of 2.249(3) Å.
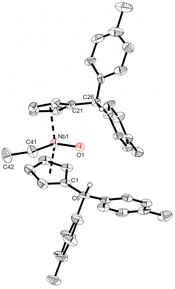 |
| | Fig. 3 Molecular structure of compound 3. Thermal ellipsoids are drawn at the 30% probability level. Selected bond lengths [Å] and angles [°]: Nb1–O1 1.756(2), Nb1–C41 2.248(4), Nb1–Ct1 2.1738(4), Nb1–Ct2 2.1699(4), C1–C6 1.515(4), C21–C26 1.507(4), O1–H6 2.313(2), O1–Nb1–C41 93.32(10), O1–Nb1–Ct1 109.11(6), O1–Nb1–Ct2 109.11(6), C41–Nb1–Ct1 101.96(9), C41–Nb1–Ct2 106.04(10), Ct1–Nb1–Ct2 130.47(2). | |
Reactivity of niobium ethylene complex 2
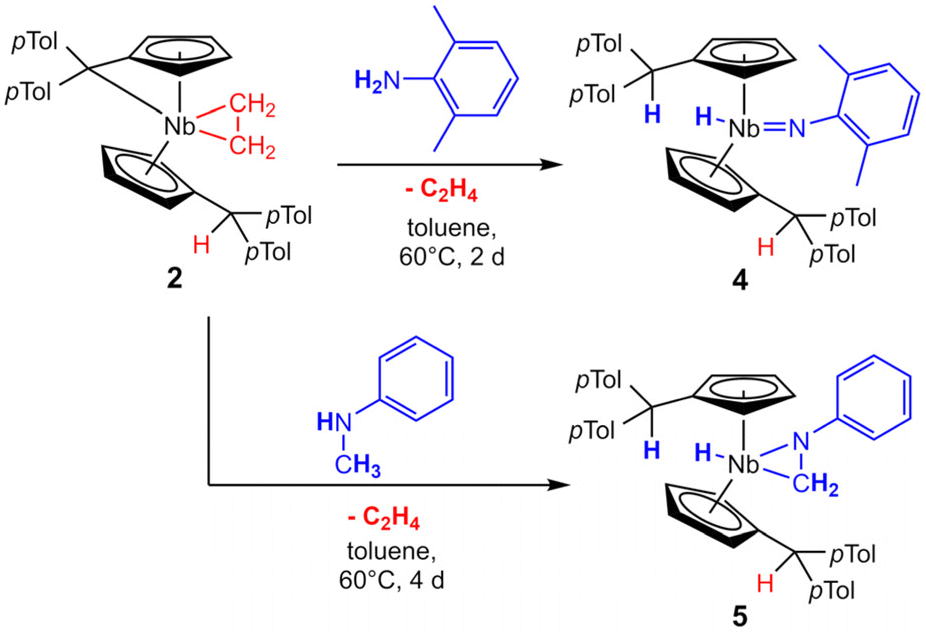
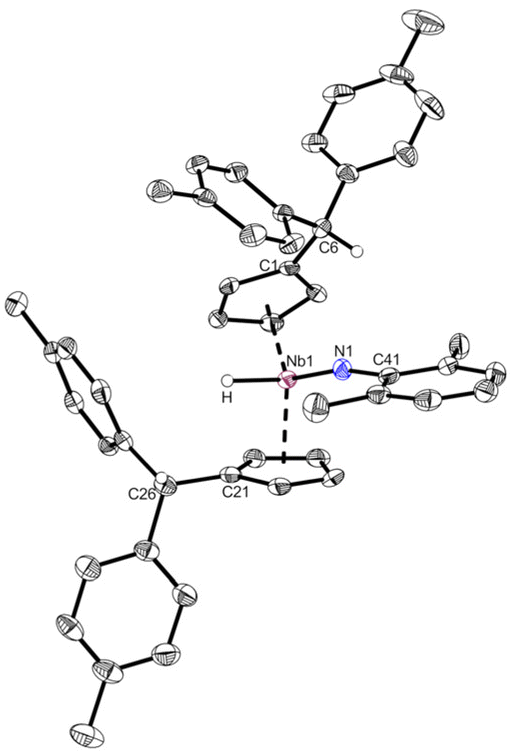
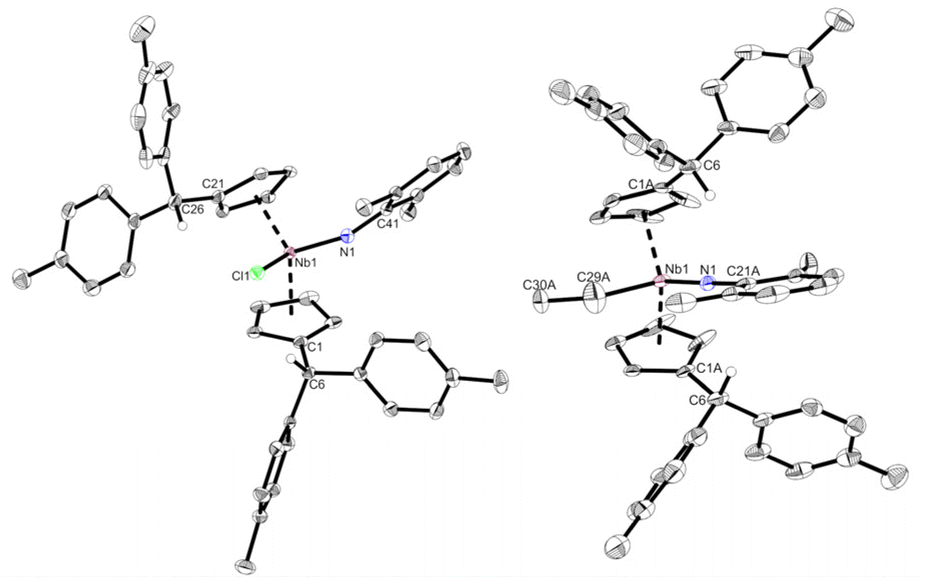
Complex 2 is highly air and moisture sensitive. Although tested at numerous conditions, no reaction of 2 with water to form 3 was achieved. The variable temperature NMR spectra of 2 showed no decomposition or ethylene liberation in toluene-d8 up to 100 °C. After repeating the experiment in bromobenzene-d5 to enable heating up to 150 °C, trace amounts of ethylene are detected already at room temperature. Matching the decomposition temperature of 2 in the solid state (110 °C), the breakdown of 2 in bromobenzene starts at 110 °C, showing ethylene liberation and indications for an intermediate (further details in ESI†). Testing multiple substrates (alkynes, NHCs, nitriles, isocyanides), yielded no selective ligand exchange of the ethylene, even at elevated temperatures. Relatively acidic E–H substrates (alkoholes, phenols, sec. amines) routinely show the formation of complex product mixtures. No selective reaction reforming the ethyl ligand was found. In spite of that, reacting 2 with 2,6-dimethylaniline for two days at 60 °C in toluene yields the imido complex 4 (Scheme 4, top). After removal of the solvent and recrystallization from n-hexane, 4 is isolated as a yellow powder in good yields (87%). It is air and moisture sensitive and well soluble in aromatic solvents. When the 1H NMR spectrum of 4 is compared to the one of 2, it lacks the distinct coupling pattern of the ethyl moiety. Instead, a broad singlet at 3.58 ppm is found, which shows no coupling in 1H–13C HMQC or 1H–13C HMBC experiments. It was therefore assigned to a niobium hydride moiety, also matching the 1H NMR chemical shift of a previously reported niobocene imido hydride (δ = 3.17 ppm).44 To investigate the fortune of the ethylene ligand, the reaction was repeated in C6D6 and indeed, the formation of ethylene was observed (5.25 ppm, Fig. S7†). The structure of complex 4 was confirmed by single crystal X-ray diffraction (Fig. 4). Single crystals were obtained from a saturated n-hexane solution at room temperature. To the best of our knowledge, 4 is the first niobocene imido hydride complex characterized by X-ray diffraction. Complex 4 is tetrahedrally coordinated with the niobocene bent at 129.64(1)°. The Nb1–N1 bond (1.818(2) Å) and the angle Nb1–N1–C41 (165.1(2) Å) are comparable with reported niobocene imido moieties.45,46 The Nb–H bond length (1.75(4) Å) lies in the range of reported niobium hydrides.8,47–49
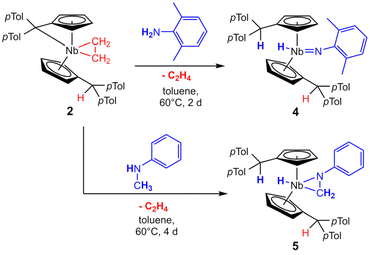 |
| | Scheme 4 Reaction of 2 with 2,6-dimethylaniline and N-methylaniline. | |
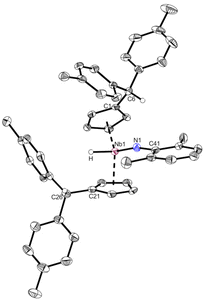 |
| | Fig. 4 Molecular structure of compound 4. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: Nb1–N1 1.818(2), Nb1–H 1.75(4), Nb1–Ct1 2.1708(3), Nb1–Ct2 2.1399(3), N1–C41 1.372(4), C1–C6 1.519(4), C21–C26 1.514(4), N1–H 2.59(4), N1–H6 2.653(2), N1–Nb1–H 93.0(13), Nb1–N1–C41 165.1(2), N1–Nb1–Ct1 114.15(7), N1–Nb1–Ct2 111.59(7), H–Nb1–Ct1 99.0(14), H–Nb1–Ct2 98.5(16), Ct1–Nb1–Ct2 129.64(1). | |
Similar to the formation of 4, reacting 2 with three equivalents of N-methylaniline in toluene at 60 °C for four days yields niobaaziridine hydride 5 (Scheme 4, bottom). Complex 5 tends to oil out of solution rather than crystallize, impeding full purification and structure elucidation by X-ray diffraction. Nevertheless, it is possible to fully characterize 5 by NMR measurements of the dried reaction mixture. The comparisons of key chemical shifts with similar known structures, and the comparison of the results of our mechanistic studies (see below) with the known formation of niobaaziridine hydrides by oxidative C–H bond addition to NbIII metal centers,7,8 strongly support the presented Lewis structure. In the 1H NMR spectrum the four typical signals for the four different protons of the Cp′ ligands (δ = 4.50, 4.54, 4.78, 4.83 ppm) are found, showcasing the CS symmetry of the niobocene. The singlet of the two protonated Cexo positions (δ = 4.90 ppm) displays the activation of the N-methylaniline. Further, the singlet of the niobaaziridine methylene group (δ = 1.47 ppm, 2H) is found, and this signal can be assigned to a methylene carbon in the 13C{1H} NMR spectrum (δ = 17.5 ppm). Consistent with the observed formation of ethylene (compare Fig. S10–12†), no signals for an ethyl ligand are found. Instead, the formed hydride is shown by the singlet at δ = 1.22 ppm that does not couple to a carbon atom in 1H–13C-HMQC NMR experiments. In the 1H–15N-HMBC NMR spectrum, the hydride does couple to the niobaaziridine nitrogen, but via a singlet instead of a doublet, which would be expected for an NH-group. This hydride signal is significantly shifted to higher fields compared to other niobaaziridine hydrides,7,8 but fits into the immense chemical shift range of reported niobocene hydride complexes.44,47,50 The assigned positions were elucidated by 2H NMR, repeating the reaction with deuterated amines. As expected, deuterium incorporation is found at the Cexo position, as well as at the niobaaziridine methylene group and the hydride ligand (compare mechanistic investigations). The determined 15N chemical shift of the niobaaziridine nitrogen atom (δ = 119 ppm), found by coupling to the hydride and the methylene protons, is close to the shift found for a related known niobaaziridine complex (hydride formally exchanged for neosilyl: δ = 129 ppm).32 The niobocene(III) alkyl and niobium(III) trisamido stabilization by oxidative β-C–H addition to the metal center was shown to be reversible by the addition of a suitable substrate.7,10,12 In the case of niobaaziridine hydride 5, an equilibrium would explain why crystallization or precipitation of 5 fails. Multiple attempts to reverse the β-C–H activation by common substrates yielded either no reaction (isocyanides, pyridines) or multi-component mixtures (NHCs, phosphine oxides). Further, in the case of other niobaaziridine hydrides, insertion reactions of multiple bond containing substrates were performed yielding stabilized niobaaziridines.7,8 Adapting this, no consecutive insertions of nitriles or ketones were observed.
Mechanistic studies
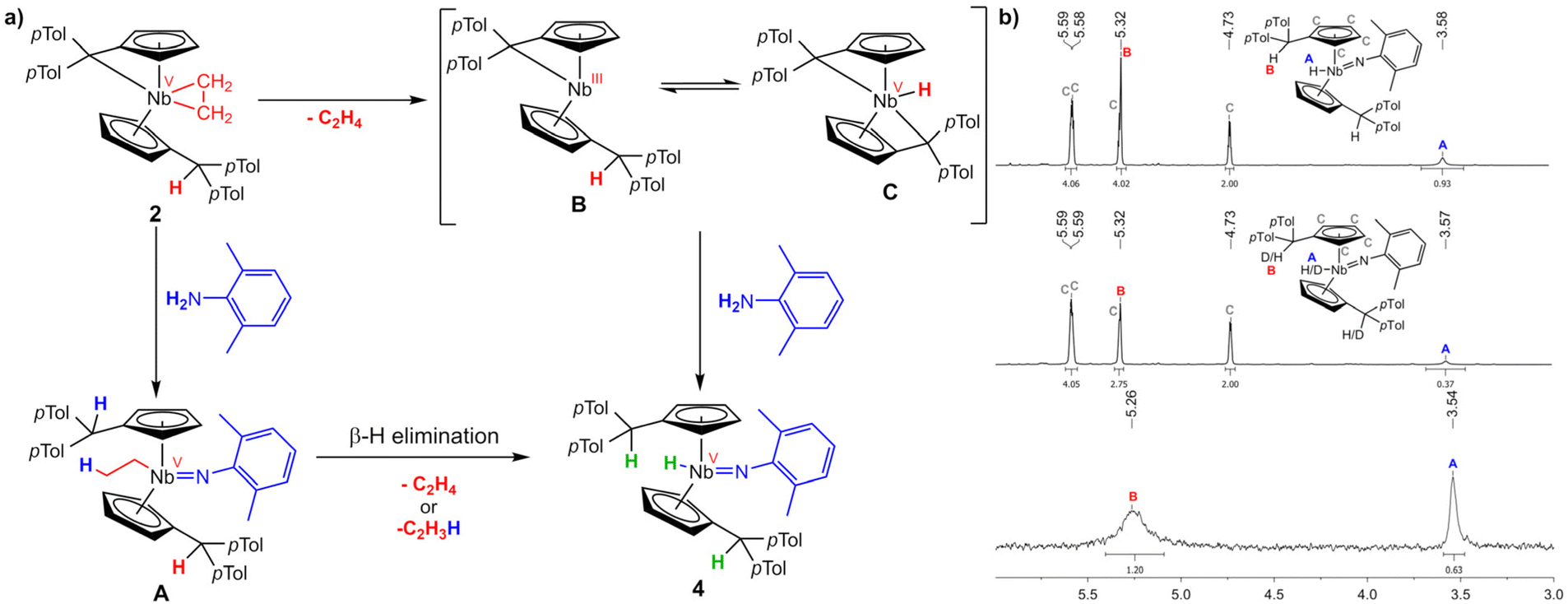
At least two principal reaction schemes for the formation of the niobocene imido hydride 4 and niobaaziridine 5 are possible, exemplary shown for 4 in Scheme 5a. A redox-neutral pathway where the pentafulvene and the ethylene moieties activate the E–H bonds, reforming the ethyl ligand (A), subsequently followed by β-hydride elimination, forming ethylene and the product. The second possibility is a redox route, with initial loss of the ethylene, forming an intermediate NbIII complex (B). This complex could be subject to an equilibrium with an bis(pentafulvene)niobium hydride C, formed by oxidative addition of one CexoH to the metal center. To distinguish whether the niobium hydride is formed by β-C–H activation or by oxidative addition, firstly, we used deuterium labeled 2,6-xylidene to monitor the deuterium incorporation into the product complex 4. Synthesizing complex 4-d2 by analogously reacting 2 with 2,6-xylidene-d2 and comparing the 1H NMR spectra (Scheme 5b), it becomes clear that the deuterium is fully incorporated into the target complex and not lost by β-hydride elimination of ethylene-d1. However, the deuteration of the Cexo position exceeds expectations for a mechanism viaB (integral lacks 1.25H vs. expected 1H) while the hydride deuteration is 0.63H to low. Hence, a mechanism via an equilibrium of B and C is plausible.
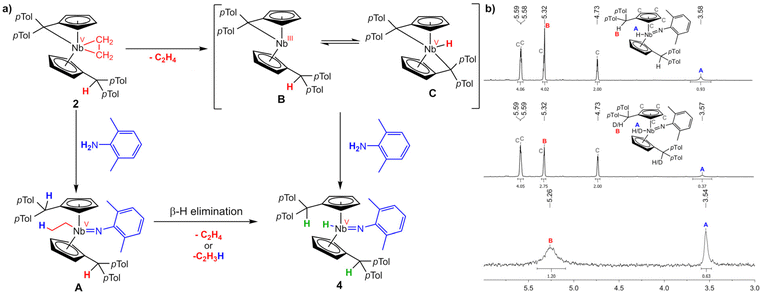 |
| | Scheme 5 (a) Supposed reaction pathways for the formation of 4. β-H elimination pathway viaA and redox route viaB and C. (b) top: excerpt of the 1H NMR spectrum of 4 (500 MHz, C6D6, 305 K); middle: excerpt of the 1H NMR spectrum of 4-d2 (500 MHz, C6D6, 305 K); bottom: excerpt of the 2H NMR spectrum of 4-d2 (77 MHz, C6H6, 305 K). | |
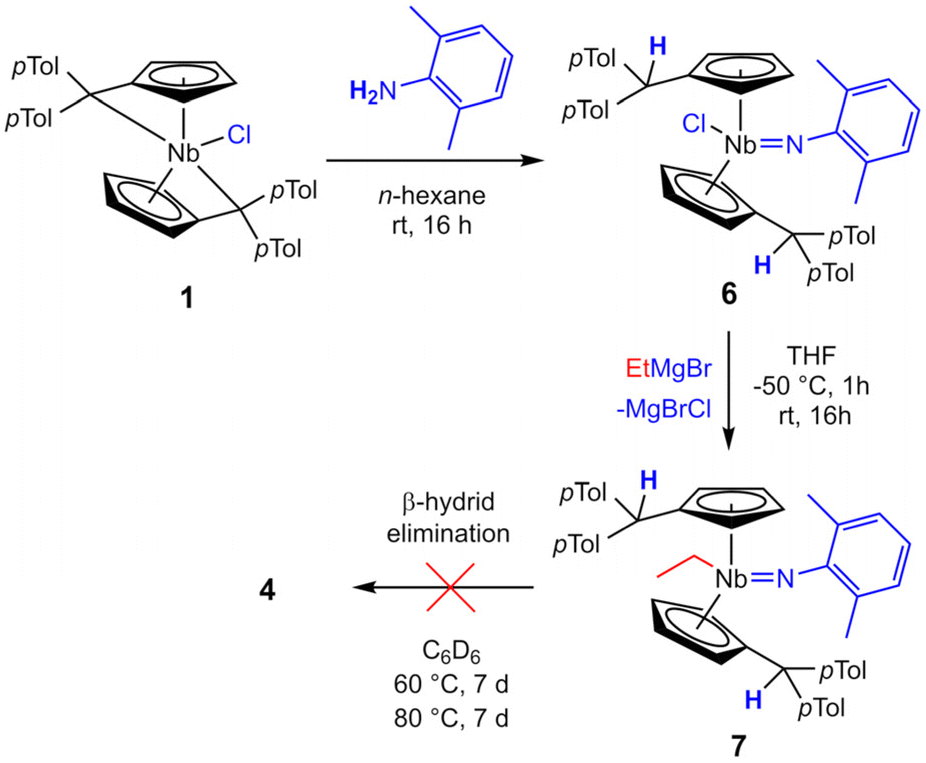
The β-hydride elimination pathway A can be further disproven. Reacting 1 with one equivalent of 2,6-dimethylaniline in n-hexane yields immediate precipitation of a yellow solid. Separation by filtration, washing with n-hexane and drying in high vacuum yields 6 as a yellow, air and moisture sensitive microcrystalline solid in good isolated yield (87%). Complex 6 is moderately soluble in n-hexane and well soluble in toluene and THF. The Lewis structure of 6 is identical to 4 apart from the hydride being formally exchanged for a chloride. Therefore, by salt metathesis, an ethyl ligand can easily be introduced (Scheme 6). Dropwise addition of one equivalent of ethyl magnesium bromide in diethyl ether to a solution of 6 in THF at −50 °C, followed by warming to room temperature and stirring for 16 h yields imido ethyl niobocene 7. Drying and subsequent extraction with n-hexane separates 7 from the formed salts. After removal of the solvent, complex 7 is obtained as an orange solid in good isolated yield (80%). Compound 7 is air and moisture sensitive, well soluble in aliphatic and aromatic solvents and melts at 68 °C. Niobium imido complexes 6 and 7 were fully characterized by NMR spectroscopy. They exhibit the same CS symmetry as compounds 3, 4, and 5. In the 1H NMR spectra one signal for each of the four Cp′ protons of the two chemically identical ligands are found (6: δ = 5.17–5.84 ppm; 7: δ = 4.98–5.85 ppm). The double N–H activation in the formation of 6 yields two identical protonated Cexo positions for each complex (6: δ = 5.87 ppm; 7: δ = 5.18 ppm). Plainly, the main difference in the 1H NMR spectra is the introduced ethyl ligand of 7 and its typical NMR signal pattern (δ = 1.40 (t), 1.93 (q) ppm), demonstrating the successful substitution of the chlorido ligand for the ethyl moiety. The molecular structures of compounds 6 and 7 were proven by single crystal X-ray diffraction (Fig. 5). The single crystals were obtained from saturated n-hexane solutions, for 6 at room temperature and for 7 at 4 °C. Complex 6 crystallizes in the orthorhombic space group Pbca. Niobocene imido chlorides have been characterized by single crystal X-ray diffraction previously. The observed triple bond character of the Nb–N bond (1.798(2) Å) and the consequently widened Nb–N–C angle (161.3(2)°), as well as the Nb–Cl bond length (2.4398(6) Å) agree well with those examples.44,51 Complex 7 crystallizes in the orthorhombic space group Cmc21. The molecular structure of 7 proves the NMR spectroscopy-assigned constitution despite crystallizing in a symmetrically disordered fashion. While the para-tolyl substituents share common positions, the Cp rings and the ethyl and imido ligands are disordered over two positions (Fig. S22†). To investigate if 7 is an intermediate in the formation of 4, complex 7 was thermally treated. No conversion of 4 or liberation of ethylene was observed by NMR spectroscopy upon heating a portion of 7 in C6D6 to 60 °C or subsequently 80 °C for one week each (Fig. S13†). Thus, niobocene imido hydride 4 is not obtained by β-H elimination, but by Rosenthal reagent like reductive ethylene liberation, forming an NbIII intermediate.
 |
| | Scheme 6 Synthesis of imido ethyl niobocene 7via imido niobocene chloride 6. | |
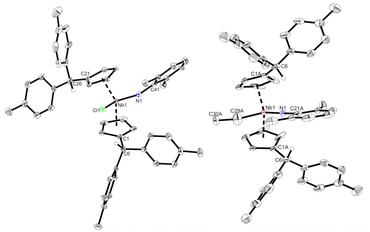 |
| | Fig. 5 Molecular structures of compounds 6 (left) and 7 (right). Thermal ellipsoids are drawn at the 50% probability level. Most hydrogen atoms and the distortion of 7 are omitted for clarity. | |
To investigate the formation of niobaaziridine 5, its synthesis was repeated using deuterium-marked amines (compare ESI†). Two conclusions can be drawn from this. Firstly, using N-(methyl-d3)-aniline selectively yields deuterium incorporation at the hydride position in the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, compared with the niobaaziridine methylene group. Hence, it is clear that the C–H bond is selectively activated by the oxidative addition to the niobium center. Secondly, using N-d-N-methylaniline yields selective deuterium incorporation at the Cexo, supporting the above assignment by showing that the N–H bond is split by the fulvene moiety. Moreover, the found H:D ratio at the Cexo position exceeds the expected 1:1 ratio, indicating an exchange of the hydrogen at the already protonated Cexo position with the amine deuterium. This could happen via an oxidative addition of the CexoH in the fashion of C and subsequent exchange or be due to the deprotonation of the amine underlying an equilibrium before the C–H bond is activated.
:2 ratio, compared with the niobaaziridine methylene group. Hence, it is clear that the C–H bond is selectively activated by the oxidative addition to the niobium center. Secondly, using N-d-N-methylaniline yields selective deuterium incorporation at the Cexo, supporting the above assignment by showing that the N–H bond is split by the fulvene moiety. Moreover, the found H:D ratio at the Cexo position exceeds the expected 1:1 ratio, indicating an exchange of the hydrogen at the already protonated Cexo position with the amine deuterium. This could happen via an oxidative addition of the CexoH in the fashion of C and subsequent exchange or be due to the deprotonation of the amine underlying an equilibrium before the C–H bond is activated.
Conclusions
In this study, the β-C–H activation at an ethyl substituent by a pentafulvene ligand was employed to form the niobium ethylene complex 2. NMR experiments, single crystal X-ray diffraction and quantum chemical calculations show strong back-donation from the metal center, yielding a niobacyclopropane structure. The intermediate bis(pentafulvene) complex, formed by salt metathesis, can be intercepted with water forming the terminal oxo ethyl complex 3, supporting a stepwise reaction. The remaining pentafulvene and the ethylene ligand in complex 2 show cooperative reaction patterns, splitting E–H bonds. While the pentafulvene Cexo is reliably protonated, the ethylene is eliminated to reveal a NbIII center, available for oxidative additions. By reaction with 2,6-xylidine or N-methylaniline, niobium imido hydride 4 and niobaaziridine hydride 5 are formed. Deuterium labeling of the used amines suggests a certain fluctuation of the deuterium incorporation, but dismissing its loss by an elimination route. The synthesis of niobocene imido ethyl complex 7 from the respected chloride 6 and subsequently showing its excellent stability excludes the possible β-hydride elimination pathway in the formation of 4. Thus, the initial loss of ethylene and subsequent oxidative addition of the respected E–H bond is favorable as a possible mechanism. Further, for the niobaaziridine formation, it was shown, that the pentafulvene activates the N–H bond, and the C–H bond is added oxidatively to the niobium center.
Experimental section
General considerations
All reactions were carried out under an inert atmosphere of argon or nitrogen with rigorous exclusion of oxygen and moisture using standard glovebox or Schlenk techniques. Solvents and liquid educts were dried according to standard procedures. The Solvents were distilled over Na/K alloy and benzophenone under a nitrogen atmosphere. Amines were distilled from CaH2 prior use. NMR spectra were recorded on a Bruker AVANCE III 500 spectrometer (1H 500 MHz; 13C 126 MHz; 15N 51 MHz) or Bruker Fourier 300 spectrometer (1H 300 MHz; 13C 75 MHz). The NMR chemical shifts were referenced to the signals of the residual protons of the solvent. Given chemical shifts of 15N result from15N,1H HMBC NMR experiments with nitromethane as external standard (δ = 378.9 vs. NH3). IR spectra were recorded on a Bruker Tensor 27 spectrometer using an attenuated total reflection (ATR) method. Elemental analyses were carried out on a Euro EA 3000 Elemental Analyzer. The carbon value in the elemental analyses is lowered by carbide formation. Melting points were determined using a “Mel-Temp” from Laboratory Devices, Cambridge, or a Mettler Toledo MP30. The bis(η5:η1-(di-p-tolyl)pentafulvene)niobium chloride (1)34 and the used deuterium labeled N-methylanilines52,53 were synthesized according to known procedures. Visualizations of quantum chemical calculation results were created with VESTA 354 and the contour plot was created with Multiwfn.55 Further exact details of supplementary experiments, crystallographic data, NMR spectra and details for the quantum chemical calculations are given in the ESI.†
Synthesized compounds
Niobium ethylene complex 2.
A 1.20 g (1.86 mmol) portion of 1 was dissolved in 20 mL of tetrahydrofuran and cooled to −70 °C. 0.62 mL of an ethyl magnesium bromide solution (3.0 M in diethyl ether, 1.86 mmol) were added dropwise. The reaction mixture was stirred at this temperature for 1 h, allowed to warm to room temperature and stirred for an additional 16 h. The solvents were removed under high vacuum. The residue was mixed with sand and continuously extracted with 40 mL of n-pentane (details in ESI†). The solvent was decanted and the residue dried in vacuum to receive 2 as a yellow powder. Recrystallization from n-hexane at 4 °C gave crystals suitable for XRD. Yield: 742 mg, 62%. 1H NMR (500 MHz, C6D6, 305 K) δ = −(0.28–0.18) (m, 1H, Nb(CH2)2), 0.92–1.00 (m, 1H, Nb(CH2)2), 1.14–1.32 (m, 1H, Nb(CH2)2)*, 1.33–1.40 (m, 1H, Nb(CH2)2), 2.11 (s, 3H, pTol-CH3), 2.14 (s, 3H, pTol-CH3), 2.15 (s, 3H, pTol-CH3), 2.18 (s, 3H, pTol-CH3), 2.92–2.96 (m, 1H, Fv-CH), 3.37–3.41 (m, 1H, Cp-CH), 3.72–3.76 (m, 1H, Fv-CH), 4.04–4.08 (m, 1H, Fv-CH), 4.08–4.12 (m, 1H, Cp-CH), 4.26–4.30 (m, 1H, Cp-CH), 5.15 (s, 1H, CexoH), 5.33–5.38 (m, 1H, Fv-CH), 5.46–5.50 (m, 1H, Cp-CH), 6.90–7.07 (m, 12H, pTol-CH), 7.45–7.52 (m, 4H, pTol-CH) ppm. 13C{1H} NMR (126 MHz, C6D6, 305 K) δ = 20.98 (pTol-CH3), 21.01 (pTol-CH3), 21.04 (pTol-CH3), 21.10 (pTol-CH3), 27.16 (Nb(CH2)2), 29.37 (Nb(CH2)2), 50.67 (Cp-CexoH), 80.92 (Fv-CH), 90.03 (Cp-CH), 95.33 (Cp-CH), 97.28 (Fv-CH), 98.16 (Cp-CH), 99.44 (Fv-Cexo4), 100.52 (Fv-CH), 105.97 (Fv-CH), 114.35 (Cp-CH), 116.51 (Fv-Cipso4), 118.09 (CpCipso4), 128.12 (pTol-CH), 128.30 (pTol-CH), 128.60 (pTol-CH), 128.83 (pTol-CH), 128.98 (pTol-CH), 129.21 (pTol-CH), 129.54 (pTol-CH), 130.85 (pTol-CH), 133.97 (pTol-C4), 134.14 (pTol-C4), 135.86 (pTol-C4), 136.02 (pTol-C4), 142.30 (pTol-C4), 142.80 (pTol-C4), 143.95 (pTol-C4), 144.94 (pTol-C4) ppm. IR (ATR): ṽ = 3020, 2963, 2919, 2859, 1607, 1579, 1508, 1449, 1410, 1261, 1108, 1036, 1021, 805, 762, 749, 728, 573 cm−1. Mp: 110 °C (dec.). EA: calcd for C42H41Nb: C 78.98, H 6.47. Found: C 78.14, H 6.83.
Niobocene oxo complex 3.
A 400 mg (0.62 mmol) portion of 1 was dissolved in 20 mL of THF and cooled to −70 °C. 0.21 mL of an ethyl magnesium bromide solution (3.0 M in diethyl ether, 1.86 mmol) were added dropwise. Within 30 min, the solution was warmed to −30 °C and 11 μL distilled and degassed water (11 mg, 0.61 mmol) were added. The solution was allowed to get to room temperature and stirred for 16 h. The solvent was removed in vacuum and the residue was extracted with 50 mL of n-hexane. The solvent was removed in vacuum and the product was recrystallized from n-hexane to give 3 as colorless crystals suitable for XRD. Yield: 20 mg, 5%. 1H NMR (500 MHz, C6D6, 305 K): δ = 1.56 (t, 3JHH = 7.5 Hz, 3H, Et-CH3), 1.85 (q, 3JHH = 7.5 Hz, 2H, Et-CH2), 2.10 (s, 6H, pTol-CH3), 2.12 (s, 6H, pTol-CH3), 4.56–4.62 (m, 2H, Cp-CH), 5.26–5.32 (m, 2H, Cp-CH), 5.36–5.40 (m, 2H, Cp-CH), 5.67–5.71 (m, 2H, Cp-CH), 5.79 (s, 2H, CexoH) 6.94–6.98 (m, 4H, pTol-CH), 6.98–7.02 (m, 4H, pTol-CH), 7.31–7.35 (m, 4H, pTol-CH), 7.37–7.42 (m, 4H, pTol-CH) ppm. 13C{1H} NMR (126 MHz, C6D6, 305 K) δ = 20.62 (Et-CH3), 20.99 (pTol-CH3), 21.02 (pTol-CH3), 29.90 (Et-CH2), 50.97 (CexoH), 100.85 (Cp-CH), 107.74 (Cp-CH), 109.39 (Cp-CH), 114.22 (Cp-CH), 129.33 (pTol-CH), 129.37 (pTol-CH), 129.71 (pTol-CH), 130.21 (pTol-CH), 134.57 (Cipso4), 135.82 (pTol-p-C4), 135.86 (pTol-p-C4), 142.12 (pTol-ipso-C4), 142.22 (pTol-ipso-C4) ppm. IR (ATR): ṽ = 2963, 2919, 2856, 1510, 1453, 1412, 1260, 1088, 1019, 859, 796, 762, 704, 575 cm−1. Mp: 83 °C (dec.). EA: calcd for C42H43NbO: C 76.62, H 6.43. Found: C 74.14, H 6.74.
Niobocene imido hydride 4.
200 mg (0.31 mmol) portion of 2 was dissolved in 4 mL of toluene and 34 μL (0.31 mmol) of 2,6-dimethylaniline were added. The solution was stirred at 60 °C for 2 d before the solvents were removed in vacuum. The residue was dissolved in 4 mL of n-hexane, stored at −18 °C over night, before decantation and drying of the yellow solid in vacuum. Recrystallization from n-hexane gave crystals suitable for XRD. Yield: 194 mg, 87%. 1H NMR (500 MHz, C6D6, 305 K): δ = 2.06 (s, 6H, pTol-CH3), 2.07 (s, 6H, pTol-CH3), 2.37 (s, 6H, Ph–CH3), 3.58 (s, 1H, NbH), 4.71–4.75 (m, 2H, Cp-CH), 5.29–5.34 (m, 4H, Cp-CH, CexoH), 5.56–5.62 (m, 4H, Cp-CH), 6.76 (t, 1H, 3JHH = 7.4 Hz, Ph-p-CH), 6.87–6.94 (m, 8H, pTol-CH), 7.01–7.05 (m, 4H, pTol-CH), 7.12–7.21 (m, 6H, pTol-CH, Ph-m-CH)* ppm. *signal overlaps with signal of solvent residue protons. 13C{1H} NMR (126 MHz, C6D6, 305 K) δ = 19.95 (Ph–CH3), 20.96 (pTol-CH3), 20.98 (pTol-CH3), 51.82 (CexoH), 99.83 (Cp-CH), 104.05 (Cp-CH), 105.72 (Cp-CH), 105.83 (Cp-CH), 119.29 (Ph-p-CH), 124.14 (Ph-o-C4), 127.94 (Ph-m-CH), 129.10 (pTol-CH), 129.22 (pTol-CH), 129.27 (pTol-CH), 129.46 (Cipso4), 129.48 (pTol-CH), 135.75 (pTol-p-C4), 135.86 (pTol-p-C4), 141.97 (pTol-ipso-C4), 142.76 (pTol-ipso-C4), 156.57 (Ph-ipso-C4) ppm. IR (ATR): ṽ = 2963, 2918, 1617, 1586, 1509, 1455, 1406, 1312, 1260, 1091, 1019, 954, 866, 797, d760, 733, 703, 575 cm−1. Mp: 126 °C (dec.). EA: calcd for C48H48NNb: C 78.78, H 6.61, N 1.91. Found: C 74.34, H 6.70, N 1.74.
Niobaaziridine hydride 5.
A 400 mg (0.63 mmol) portion of 2 was dissolved in 5 mL of toluene and 201 mg (3 equiv., 1.88 mmol) N-methylaniline were added. The solution was stirred at 60 °C for 4 d, replacing the atmosphere in the flask every day. The solvents were removed in vacuum, and 3 mL of n-hexane were repeatedly added and removed in vacuum to remove the excess amine as best as possible. Yield: compare NMR spectra. 1H NMR (500 MHz, C6D6, 305 K): δ = 1.22(s, 1H, NbH), 1.47 (s, 2H, NbCH2), 2.08 (s, 6H, pTol-CH3), 2.09 (s, 6H, pTol-CH3), 4.49–4.54 (m, 2H, Cp-CH), 4.54–4.59 (m, 2H, Cp-CH), 4.77–4.81 (m, 2H, Cp-CH), 4.81–4.86 (m, 2H, Cp-CH), 4.90 (s, 2H, CexoH), 6.87–6.95 (m, 10H, pTol-CH, Ph-CH), 7.06–7.09 (m, 4H, pTol-CH), 7.12–7.16 (m, 4H, pTol-CH)*, 7.45–7.50 (m, 2H, Ph-CH), 7.52–7.59 (m, 1H, Ph-CH) ppm. *overlapped by C6D5H signal. 13C{1H} NMR (126 MHz, C6D6, 305 K) δ = 17.47 (NbCH2), 20.98 (pTol-CH3), 20.99 (pTol-CH3), 50.82 (Cexo4), 92.07 (Cp-CH), 93.82 (Cp-CH), 99.65 (Cp-CH), 101.82 (Cp-CH), 117.28 (Ph-CH), 120.05 (Ph-CH) 123.39 (Cipso4), 129.15 (pTol-CH), 129.22 (pTol-CH), 129.42 (pTol-CH, Ph-CH), 129.49 (pTol-CH), 135.88 (pTol-p-C4), 135.91 (pTol-p-C4), 142.00 (pTol-ipso-C4), 142.52 (pTol-ipso-C4), 157.22 (Ph-ipso-C4) ppm. 15N-1H HMBC NMR (51 MHz, 500 MHz, C6D6, 305 K) δ = 119 ppm. IR (ATR): ṽ = 2963, 1587, 1509, 1487, 1343, 1260, 1089, 1017, 796, 762, 692, 574 cm−1. Mp: 115 °C (dec.). EA: calcd for C47H46NNb: C 78.65, H 6.64, N 1.95. Found: C 75.00, H 6.77, N 1.79.
Niobocene imido chloride 6.
An 800 mg (1.24 mmol) portion of 1 was dissolved in 15 mL of n-hexane and 150 mg (1.24 mmol) of 2,6-dimethylaniline were added. A yellow suspension formed immediately and was stirred for 16 h at room temperature to ensure full conversion. The solvent was separated by filtration, and the residue was washed with 20 mL of n-hexane and dried in vacuum. 6 was isolated as a yellow powder. Crystals suitable for XRD were obtained from a hot saturated n-hexane solution at room temperature. Yield: 824 mg, 87%. 1H NMR (500 MHz, C6D6, 305 K): δ = 2.04 (s, 6H, pTol-CH3), 2.05 (s, 6H, pTol-CH3), 2.27 (s, 6H, Ph–CH3), 5.17–5.21 (m, 2H, Cp-CH), 5.52–5.56 (m, 2H, Cp-CH), 5.61–5.66 (m, 2H, Cp-CH), 5.80–5.84 (m, 2H, Cp-CH), 5.87 (s, 2H, CexoH), 6.67 (t, 3JHH = 7.4 Hz, 1H, Ph-p-CH), 6.83–6.86 (m, 4H, pTol-CH), 6.87–6.93 (m, 6H, pTol-CH, Ph-m-CH), 7.06–7.11 (m, 4H, pTol-CH), 7.22–7.27 (m, 4H, pTol-CH) ppm. 13C{1H} NMR (126 MHz, C6D6, 305K) δ = 19.99 (Ph–CH3), 20.96 (pTol-CH3), 21.00 (pTol-CH3), 51.08 (Cexo4), 103.55 (Cp-CH), 111.27 (Cp-CH), 112.27 (Cp-CH), 119.07 (Cp-CH), 121.75 (Ph-CH), 127.92 (Ph-CH)*, 129.28 (pTol-CH), 129.36 (pTol-CH), 129.42 (pTol-CH), 129.60 (pTol-CH), 135.29 (Cipso4), 135.78 (pTol-p-C4), 136.08 (pTol-p-C4), 141.32 (pTol-ipso-C4), 141.63 (pTol-ipso-C4), 157.22 (Ph-ipso-C4) ppm. *Assigned by 13C{1H}-dept135; Ph-o-C4 could not be assigned. IR (ATR): ṽ = 3019, 2919, 2859, 2360, 1586, 1509, 1456, 1405, 1377, 1300, 1245, 1186, 1110, 1094, 1035, 1021, 982, 959, 847, 806, 760, 73, 645, 603, 575 cm−1. Mp: 146 °C (dec.). EA: calcd for C48H47ClNNb: C 75.24, H 6.18, N 1.83. Found: C 74.2, H 6.63, N 1.57.
Niobocene imido ethyl 7.
A 200 mg (0.26 mmol) portion of 6 was dissolved in 5 mL of THF and cooled to −50 °C. 0.09 mL (0.27 mmol) of ethyl magnesium bromide solution (3.0 M in diethyl ether) were added dropwise. The solution was allowed to heat to room temperature and stirred for 16 h. The solvents were evaporated, and the residue was extracted twice with 15 mL of n-hexane. The solvent was removed, and the yellow product was dried in vacuum. 7 was isolated as a yellow powder. Crystals suitable for XRD were obtained from an at room temperature saturated n-hexane solution at 4 °C. Yield: 164 mg, 80%. 1H NMR (500 MHz, C6D6, 305 K): δ = 1.40 (t, 3JHH = 7.4 Hz, 3H, CH2–CH3), 1.93 (q, 3JHH = 7.2 Hz, 3H, CH2–CH3), 2.04 (s, 12H, pTol-CH3), 2.32 (s, 6H, Ph–CH3), 4.98–5.05 (m, 2H, Cp-CH), 5.18 (s, 2H, CexoH), 5.33–5.39 (m, 2H, Cp-CH), 5.59–5.63 (m, 2H, Cp-CH), 5.79–5.85 (m, 2H, Cp-CH), 6.64 (t, 3JHH = 7.5 Hz, 1H, Ph-p-CH), 6.86–6.89 (m, 4H, pTol-CH), 6.91–6.94 (m, 4H, pTol-CH), 6.97–7.01 (m, 4H, pTol-CH), 7.14–7.19 (m, 6H, pTol-CH, Ph-m-CH)* ppm. *Overlapped by solvent residue proton signal. 13C{1H} NMR (126 MHz, C6D6, 305 K) δ = 20.29 (CH2–CH3), 20.61 (Ph–CH3), 20.95 (pTol-CH3), 20.97 (pTol-CH3), 23.62 (CH2–CH3), 51.61 (Cexo4), 102.81 (Cp-CH), 104.50 (Cp-CH), 109.13 (Cp-CH), 115.61 (Cp-CH), 119.87 (Ph-p-CH), 127.14 (Ph-o-C4), 128.42 (Ph-CH), 129.06 (pTol-CH), 129.21 (pTol-CH), 129.30 (pTol-CH), 129.47 (pTol-CH), 131.17 (Cipso4), 135.66 (pTol-p-C4), 135.91 (pTol-p-C4), 142.10 (pTol-ipso-C4), 143.10 (pTol-ipso-C4), 156.44 (Ph-C4)ppm. IR (ATR): ṽ = 2919, 2856, 1509, 1456, 1404, 1299, 1262, 1110, 1094, 1051, 1021, 953, 806, 761, 733, 645, 574 cm−1. Mp: 68 °C. EA: calcd for C50H52NNb: C 79.03, H 6.90, N 1.84. Found: C 77.23, H 7.09, N 1.58.
Author contributions
Conceptualization: S. d. G.; quantum chemical calculations: K. S.; XRD: M. S.; investigation: S. d. G., C. V. H., N. B.; supervision: R. B.; visualization: S. d. G.; writing – original draft: S. d. G.; writing – review & editing: S. d. G., R. B., M. S.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support by the DFG Research Training Group 2226 is kindly acknowledged.
References
- E. P. Beaumier, A. J. Pearce, X. Y. See and I. A. Tonks, Nat. Rev. Chem., 2019, 3, 15–34 CrossRef PubMed.
- A. Ohff, S. Pulst, C. Lefeber, N. Peulecke, P. Arndt, V. V. Burkalov and U. Rosenthal, Synlett, 1996, 111–118 CrossRef CAS.
- E.-i. Negishi, F. E. Cederbaum and T. Takahashi, Tetrahedron Lett., 1986, 27, 2829–2832 CrossRef CAS.
- T. L. Gianetti, R. G. Bergman and J. Arnold, Polyhedron, 2014, 84, 19–23 CrossRef CAS.
- F. A. Cotton and M. Shang, Inorg. Chem., 1990, 29, 508–514 CrossRef CAS.
- C. Felten, D. Rodewald, W. Priebsch, F. Olbrich and D. Rehder, J. Organomet. Chem., 1994, 480, 51–63 CrossRef CAS.
- J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2003, 125, 4020–4021 CrossRef CAS PubMed.
- J. S. Figueroa, N. A. Piro, D. J. Mindiola, M. G. Fickes and C. C. Cummins, Organometallics, 2010, 29, 5215–5229 CrossRef CAS.
- G. W. Parshall and F. N. Tebbe, J. Am. Chem. Soc., 1971, 93, 3793–3795 CrossRef CAS.
- N. M. Doherty and J. E. Bercaw, J. Am. Chem. Soc., 1985, 107, 2670–2682 CrossRef CAS.
- A. Antinolo, F. Carrillo, S. Garcia-Yuste and A. Otero, Organometallics, 1994, 13, 2761–2766 CrossRef CAS.
- A. Antiñolo, F. Carrillo-Hermosilla, I. del Hierro, A. Otero, M. Fajardo and Y. Mugnier, Organometallics, 1997, 16, 4161–4166 CrossRef.
- A. H. Klazinga and J. H. Teuben, J. Organomet. Chem., 1980, 194, 309–316 CrossRef CAS.
- P. J. Chirik, D. L. Zubris, L. J. Ackerman, L. M. Henling, M. W. Day and J. E. Bercaw, Organometallics, 2003, 22, 172–187 CrossRef CAS.
- P. Nicolás, P. Royo, M. V. Galakhov, O. Blacque, H. Jacobsen and H. Berke, Dalton Trans., 2004, 2943–2951, 10.1039/B406747A.
- A. S. Veige, T. S. Kleckley, R. M. Chamberlin, D. R. Neithamer, C. E. Lee, P. T. Wolczanski, E. B. Lobkovsky and W. V. Glassey, J. Organomet. Chem., 1999, 591, 194–203 CrossRef CAS.
- A. S. Veige, P. T. Wolczanski and E. B. Lobkovsky, Angew. Chem., Int. Ed., 2001, 40, 3629–3632 CrossRef CAS.
- K. F. Hirsekorn, A. S. Veige, M. P. Marshak, Y. Koldobskaya, P. T. Wolczanski, T. R. Cundari and E. B. Lobkovsky, J. Am. Chem. Soc., 2005, 127, 4809–4830 CrossRef CAS PubMed.
- M. J. Humphries, R. E. Douthwaite and M. L. H. Green, J. Chem. Soc., Dalton Trans., 2000, 2952–2959, 10.1039/B004062M.
- T. Kurogi and D. J. Mindiola, Organometallics, 2020, 39, 4474–4478 CrossRef CAS.
- J. S. Yu, L. Felter, M. C. Potyen, J. R. Clark, V. M. Visciglio, P. E. Fanwick and I. P. Rothwell, Organometallics, 1996, 15, 4443–4449 CrossRef CAS.
- P. Oulié, C. Dinoi, C. Li, A. Sournia-Saquet, K. Jacob, L. Vendier and M. Etienne, Organometallics, 2017, 36, 53–63 CrossRef.
- R. Beckhaus, Coord. Chem. Rev., 2018, 376, 467–477 CrossRef CAS.
- S. Fortier and A. Gomez-Torres, Chem. Commun., 2021, 57, 10292–10316 RSC.
- M. Manßen, S. de Graaff, M.-F. Meyer, M. Schmidtmann and R. Beckhaus, Organometallics, 2018, 37, 4506–4514 CrossRef.
- M. Manßen, N. Lauterbach, T. Woriescheck, M. Schmidtmann and R. Beckhaus, Organometallics, 2017, 36, 867–876 CrossRef.
- H. Ebert, V. Timmermann, T. Oswald, W. Saak, M. Schmidtmann, M. Friedemann, D. Haase and R. Beckhaus, Organometallics, 2014, 33, 1440–1452 CrossRef CAS.
- M. Fischer, K. Schwitalla, S. Baues, M. Schmidtmann and R. Beckhaus, Dalton Trans., 2019, 48, 1516–1523 RSC.
- M. Manßen, C. Adler and R. Beckhaus, Chem. – Eur. J., 2016, 22, 4405–4407 CrossRef PubMed.
- M. Manßen, I. Weimer, C. Adler, M. Fischer, M. Schmidtmann and R. Beckhaus, Eur. J. Inorg. Chem., 2018, 2018, 131–136 CrossRef.
- M. Fischer, L. Vincent-Heldt, M. Hillje, M. Schmidtmann and R. Beckhaus, Dalton Trans., 2020, 49, 2068–2072 RSC.
- S. de Graaff, A. Chandi, M. Schmidtmann and R. Beckhaus, Organometallics, 2021, 40, 3298–3305 CrossRef CAS.
-
S. Bienz, L. Bigler and T. Fox, Spektroskopische Methoden in der organischen Chemie, Georg Thieme Verlag, Stuttgart, 9th edn, 2016, pp. 172–176, DOI:10.1055/b-004-129729.
- M. Manßen, A. Dierks, S. de Graaff, M. Schmidtmann and R. Beckhaus, Angew. Chem., Int. Ed., 2018, 57, 12062–12066 CrossRef PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19, 10.1039/P298700000S1.
- N. C. Craig, P. Groner and D. C. McKean, J. Phys. Chem. A, 2006, 110, 7461–7469 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- P.-f. Fu, M. A. Khan and K. M. Nicholas, J. Organomet. Chem., 1996, 506, 49–59 CrossRef CAS.
- A. Antiñolo, J. M. de Ilarduya, A. Otero, P. Royo, A. M. M. Lanfredi and A. Tiripicchio, J. Chem. Soc., Dalton Trans., 1988, 2685–2693, 10.1039/DT9880002685.
- R. Mercier, J. Douglade, J. Amaudrut, J. Sala-Pala and J. E. Guerchais, J. Organomet. Chem., 1983, 244, 145–151 CrossRef CAS.
- A. N. Chernega, M. L. H. Green and A. G. Suárez, J. Chem. Soc., Dalton Trans., 1993, 3031–3034, 10.1039/DT9930003031.
- A. Antiñolo, M. Fajardo, C. López-Mardomingo, I. López-Solera, A. Otero, Y. Pérez and S. Prashar, Organometallics, 2001, 20, 3132–3138 CrossRef.
- W. A. Herrmann, W. Baratta and E. Herdtweck, J. Organomet. Chem., 1997, 541, 445–460 CrossRef CAS.
- G. I. Nikonov, L. G. Kuzmina and J. A. K. Howard, J. Chem. Soc., Dalton Trans., 2002, 3037–3046, 10.1039/B110998G.
- K. Y. Dorogov, A. V. Churakov, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, Eur. J. Inorg. Chem., 2004, 2004, 771–775 CrossRef.
- A. Neshat and J. A. R. Schmidt, Organometallics, 2010, 29, 6219–6229 CrossRef CAS.
- J. A. Labinger and K. S. Wong, J. Organomet. Chem., 1979, 170, 373–384 CrossRef CAS.
- A. Antiñolo, I. López-Solera, I. Orive, A. Otero, S. Prashar, A. M. Rodríguez and E. Villaseñor, Organometallics, 2001, 20, 71–78 CrossRef.
- J. W. Brandt, E. Chong and L. L. Schafer, ACS Catal., 2017, 7, 6323–6330 CrossRef CAS.
- R. Fusco and F. Sannicolo, J. Org. Chem., 1984, 49, 4374–4378 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Kevin
Schwitalla
,
Carolin V.
Haaker
,
Nina
Bengen
,
Marc
Schmidtmann
and
Rüdiger
Beckhaus
,
Kevin
Schwitalla
,
Carolin V.
Haaker
,
Nina
Bengen
,
Marc
Schmidtmann
and
Rüdiger
Beckhaus