
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal stability and structural studies on the mixtures of Mg(BH4)2 and glymes†
Noemi
Leick
 a,
Ba L.
Tran
*c,
Mark E.
Bowden
c,
Thomas
Gennett
ab and
Tom
Autrey
c
a,
Ba L.
Tran
*c,
Mark E.
Bowden
c,
Thomas
Gennett
ab and
Tom
Autrey
c
aNational Renewable Energy Laboratory, 15013 Denver W Pkwy, Golden, Colorado 80401, USA
bChemistry Department, Colorado School of Mines, 1012 14th Street, Golden, Colorado 80401, USA
cPacific Northwest National Laboratory, Richland, Washington 99354, USA. E-mail: ba.tran@pnnl.gov
First published on 20th April 2022
Abstract
Coordination complexes of Mg(BH4)2 are of interest for energy storage, ranging from hydrogen storage in BH4 to electrochemical storage in Mg based batteries. Understanding the stability of these complexes is crucial since storage materials are expected to undergo multiple charging and discharging cycles. To do so, we examined the thermal stabilities of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of Mg(BH4)2 with different glymes by DSC–TGA, TPD-MS and powder XRD analysis. Despite their structural similarities, these mixtures show diverse phase transitions, speciations and decomposition pathways as a function of linker length.
:1 mixtures of Mg(BH4)2 with different glymes by DSC–TGA, TPD-MS and powder XRD analysis. Despite their structural similarities, these mixtures show diverse phase transitions, speciations and decomposition pathways as a function of linker length.
Introduction
The generation of H2 from renewable energy provides a pathway toward a decarbonized, sustainable future.1 Overcoming the intermittency of renewable sources by safe, long-duration storage of H2 in a compact and efficient form compared to gaseous or cryogenic liquid H2 remains a major challenge.2 A reversible cycle between Mg(BH4)2 and MgB2 has attracted considerable attention due to a high gravimetric storage density (ca. 14.7 wt% H2) and an ideal thermodynamic range for H2 release and H2 uptake at moderate pressure and temperature, ΔG° ca. 5 kJ mol−1 H2.3–7 Notwithstanding these attributes, a major drawback is the slow rate of H2 release of neat, uncomplexed solid Mg(BH4)2 requiring temperatures >250 °C.8Introducing organic additives and transition metals have helped lower the temperature of dehydrogenation.9–12 We have shown that adding simple ethereal additives, such as THF and glymes to Mg(BH4)2 can promote dehydrogenation of Mg(BH4)2 at temperatures <200 °C with high selectivity of B10H102− over B12H122− and B3H8−.13,14 The product selectivity of B10H102− and B12H122− in the dehydrogenation of Mg(BH4)2 is highly dependent on the glyme-to-Mg(BH4)2 ratio and the identity of the glyme. In addition to H2 storage, various Mg(BH4)2·glyme mixtures have also garnered interest for Mg batteries because of the stability of BH4− anion under reductive conditions, higher natural abundance of Mg and B, increased safety by minimizing dendrite formation and higher energy density of Mg- compared to Li-batteries.15–18 Moreover, Mg complexes containing boron clusters19 or different anions stabilized by glymes20–22 have also been examined for battery electrolytes.
A molecular understanding of the interaction between glymes and Mg(BH4)2, and potential degradation pathways of these mixtures are experimentally lacking, yet crucial for materials development.23 In this report, we investigated the thermal stability and speciation of the mixtures of glymes and Mg(BH4)2 by examining the series of 1:1 Mg(BH4)2·glyme mixtures by differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and temperature programmed desorption coupled with quadrupole mass spectrometry (TPD-MS). The combination of these experimental approaches provides insight into both the physical and chemical changes as well as the volatile and non-volatile products formed.
Results and discussion
Phase transitions of the 1:1 mixtures of Mg(BH4)2·Gn (n = 1: monoglyme, 2: diglyme, 3: triglyme and 4: tetraglyme), determined by DSC and TGA, are presented in Fig. 1. The Mg(BH4)2·G1 data, collected in a closed system, shows an exotherm at ∼60 °C and intense endotherms at ∼100 °C and ∼110 °C, which most likely results from melting of the mixture. A similar melting process at 100 °C was observed for Mg(BH4)2·0.5THF (Fig. S1†). The collection of the DSC data of Mg(BH4)2·G1 in an open system shows similar exothermic and endothermic phase transitions that those of the closed system (Fig. S2†). We further compared the phase transitions of 1:1 mixture of Mg(BH4)2·G1 to those of pure Mg(BH4)212,24 (Fig. S3†) and Mg(BH4)2·1.5G1,25 which are the major species in the 1:1 mixture of Mg(BH4)2·G1 evidenced by powder XRD analysis (see below). DSC data of Mg(BH4)2 features intense endotherms above 160 °C and ∼195 °C while that of pure Mg(BH4)2·1.5G1 shows an endotherm at 135 °C, which has been attributed to partial loss of G1, and two subsequent exotherms at 250 °C and 500 °C. From these comparisons, the 1:1 Mg(BH4)2·G1 mixture exhibits drastically different phase transitions at much lower temperatures than that of pure Mg(BH4)2 and Mg(BH4)2·1.5G1.
 | ||
| Fig. 1 Mass normalized heat flow as a function of temperature obtained from the closed system of DSC (a) and TGA (b) data for 1:1 mixtures of glymes–Mg(BH4)2. G1 = monoglyme, G2 = diglyme, G3 = triglyme, G4 = tetraglyme. Collection parameters: 5 °C min−1 to 180 °C. | ||
In contrast to G1, the DSC data indicates that Mg(BH4)2·G2 is stable with no phase transitions. The DSC data for Mg(BH4)2·G3 and Mg(BH4)2·G4 are also drastically different. A large endotherm ascribed to melting was displayed by Mg(BH4)2·G3, whereas no phase transitions were observed for Mg(BH4)2·G4. This was attributed to Mg(BH4)2·G4 having already become molten before the DSC trace began, and has been confirmed by heating a preparative scale Mg(BH4)2·G4 to 40 °C.
TGA data shows that both Mg(BH4)2·G3 and Mg(BH4)2·G4 underwent more mass loss compared to that of Mg(BH4)2·G1 and Mg(BH4)2·G2 upon ramping to 180 °C. Mg(BH4)2·G1 and Mg(BH4)2·G2 showed negligible mass loss (<2%) compared to that of 9% and 19% for Mg(BH4)2·G3 and Mg(BH4)2·G4, respectively. Holding at 180 °C for 0.5 h after the ramp gave significant mass losses of 48% was observed for Mg(BH4)2·G4 and 23% for Mg(BH4)2·G3 (Fig. S4†). In contrast, Mg(BH4)2·G1 and Mg(BH4)2·G2 showed minor mass losses of <5% upon ramping up to 180 °C and holding for 0.5 h (Fig. S4†). The notable mass increase in the TGA data of Mg(BH4)2·G4 has been reproduced and likely results from the solubility of Mg(BH4)2 in G4 to give a viscous Mg(BH4)2·G4 that can rapidly interact with ambient species such O2 compared to the more crystalline samples of Mg(BH4)2–G1, Mg(BH4)2–G2 and Mg(BH4)2–G3.
We further examined the speciation of 1:1 Mg(BH4)2·G1 by powder XRD and TPD-MS because this mixture selectively produced B10H102− over B12H122−.26 Additionally, G1 is commonly employed to ligate Mg2+ ions in the development of battery materials.15,20,27–29 Powder XRD measurements of 1:1 Mg(BH4)2·G1 were collected at 25 °C, 60 °C, 70 °C (Fig. 2) and 120 °C. The preheated mixture of 25 °C shows the complex of [Mg(G1)3][Mg(BH4)4] (referred to as Mg(BH4)2·1.5G1 herein)25 as the major species with minor γ-Mg(BH4)2 starting material. After heating at 60 °C for 1 h, the XRD pattern contained Mg(BH4)2·1.5G1 with significant amounts of unidentified peaks primarily at 8.5 Å and 6.3 Å. We have also confirmed that the unidentified peaks are not associated with α-Mg(BH4)2 (Fig. S5†). The formation of crystalline products from Mg(BH4)2·1.5G1 is consistent with the exotherm at 60 °C in the DSC trace. Further heating at 60 °C for 6 days transformed the mixture predominantly to Mg(BH4)2·1.5G1 (Fig. 2A and Fig. S6†) rather than the new unidentified crystalline product observed at 60 °C at shorter heating time. The reappearance of Mg(BH4)2·1.5G1 at 60 °C (6 days) and 70 °C (16 h) was confirmed by a Rietveld fit of the pattern which showed excellent agreement with that calculated from the structure published by Grochala and co-workers (Fig. S7†).25
 | ||
| Fig. 2 (A) Summary of powder XRD measurements of the 1:1 Mg(BH4)2·G1 mixture at 25–70 °C. (B) Powder XRD data of 1:1 Mg(BH4)2·G1 mixture at 25–70 °C showing the formation of Mg(BH4)2·1.5G1 (#). | ||
To determine whether Mg(BH4)2·1.5G1 exists after the melt, at temperature higher than 100 °C as suggested by DSC data, we heated the 1:1 Mg(BH4)2·G1 mixture to 120 °C for 1 h leading to melting of the sample to produce a clear glass. Cooling to 25 °C under inert atmosphere gave a transparent product with no evidence of crystalline material. This melting event of 1:1 Mg(BH4)2·G1 is also corroborated by TPD-MS data (Fig. S8†), in which a major melting linked with the release of more G1 occurs at ∼100 °C. Additionally, the m/z = 2 signal (H2) increases but also reveals a noisy signal that usually indicates H2 transport through a viscous medium, not a solid. Taken together, powder XRD and TPD-MS data support the phase transitions observed in the DSC analysis of 1:1 Mg(BH4)2·G1.
In a separate study, XRD analysis of single crystals obtained from the reaction of Mg(BH4)2 and 21 equiv. of G1 at 25 °C also showed formation of Mg(BH4)2·1.5G1.25 The formation of Mg(BH4)2·1.5G1, as characterized by solid-state techniques, appears highly favourable for stoichiometric and excess G1 relative to Mg(BH4)2. It is unclear if Mg(BH4)2·1.5G1 is also the predominant species in a solution of Mg(BH4)2 and G1. The isolation and single crystal XRD characterization of Mg(BH4)2·1.5G1 from a solution of Mg(BH4)2 and G1 does not necessarily supplant the proposed solution structure of two neutral [(G1)Mg(BH4)2] units bridged by G1 in the development of Mg-battery electrolyte.27 These cationic–anionic species should be taken into consideration since similar species to that of Mg(BH4)2·1.5G1 have been structurally characterized from the mixtures of (hexafluoroisopropyloxy)borate Ca with G1 and perfluorinated pinacolatoborate Mg with G2 for the development of battery electrolytes.20,30 We further showed that a 0.5 M of Mg(BH4)2 in G1 is adequately soluble at ambient temperature as evidenced by observation of strong 11B resonance for BH4− against a PhBpin internal standard (Fig. S9†).
Generally, we have observed that additives which induce a melting event, such as THF or G1, are effective for both lowering the temperature and increasing the activity of dehydrogenation. G2 is an anomalously poor additive for the dehydrogenation of Mg(BH4)226 and the lack of a melting event may explains the sluggish dehydrogenation of Mg(BH4)2·G2 compared to that of Mg(BH4)2·G1 or Mg(BH4)2·0.5THF by DSC. Therefore, we analysed the preparative-scale thermolysis of 1:1 Mg(BH4)2·G2 by powder XRD at 100–170 °C (Scheme 1).
 | ||
| Scheme 1 Powder XRD studies indicate high thermal stability of [(G2)Mg(BH4)] to support the lack of phase transitions in the DSC data for 1:1 Mg(BH4)2·G2. | ||
The powder XRD data is presented in Fig. S10 of the ESI.† Indeed, heating the 1:1 Mg(BH4)2·G2 mixture at 100 °C and 170 °C for 1 h did not induce a melt. The crystalline solids of the predominant mononuclear [(G2)Mg(BH4)2] species, which has been structurally characterized, remained stable at 25 °C, 100 °C and 170 °C.31 Moreover, XRD studies clearly show that the complexation of Mg(BH4)2 with G1 or G2 produce fundamentally different species (Fig. 2A and Scheme 1). Additional 11B NMR analysis of the dehydrogenation of 1:1 Mg(BH4)2·G2 and 1:1 Mg(BH4)2·G1 at 180–200 °C for 8 h (Fig. S11†) further highlight the effect of G2 and G1 on speciation and thermal stability. The formation of Mg(B10H10) (30%) was observed for 1:1 Mg(BH4)2·G1 at 180 °C for 8 h whereas 1:1 Mg(BH4)2·G2 requires heating at 200 °C for 8 h to form trace amounts (<5%) of Mg(B10H10) (Fig. S11†).
The mass losses from Mg(BH4)2·G3 and Mg(BH4)2·G4 are not simply volatile G3 (MP = 216 °C) and G4 (MP = 275 °C) as evidenced by their different mass spectrometry (MS) fragmentation patterns compared to free G4 or G3 (Fig. 3 and Fig. S12†). This observation suggests that the complexation to the Mg2+ ion plausibly mediates the decomposition of G3 and G4. Therefore, to understand potential decomposition pathways of these mixtures, we analysed the volatiles at different temperatures by MS. A summary of the MS data of Mg(BH4)2·G1 and Mg(BH4)2·G4 is shown in Fig. 3 and those of Mg(BH4)2·G2 and Mg(BH4)2·G3 are in Fig. S12.† It appears that the volatile products may depend on the linker in the glyme.
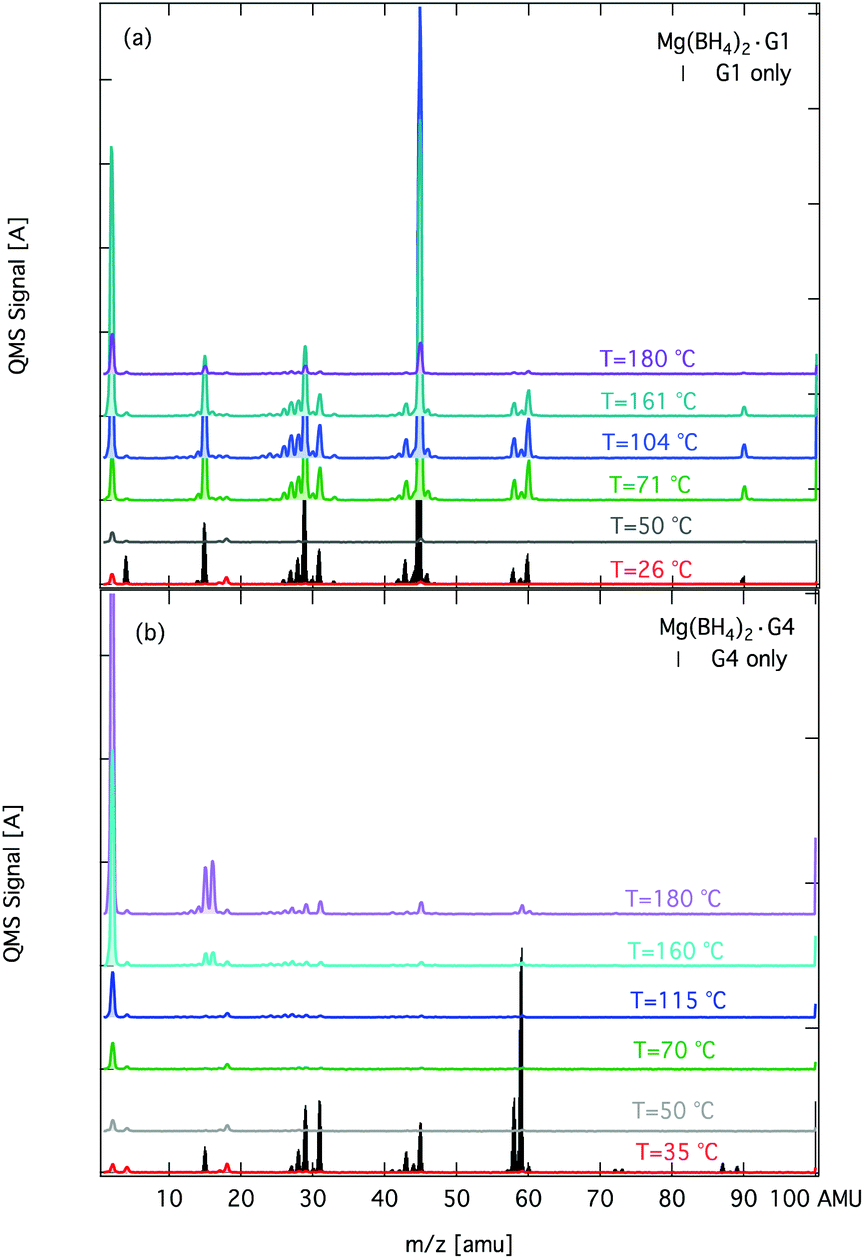 | ||
| Fig. 3 Signal from the QMS in the m/z range of 1–100 amu recorded during heating of (a) Mg(BH4)2·G1 and (b) Mg(BH4)2·G4. For comparison, the fragmentation pattern of G1 and G4 were recorded and are shown at 26 °C and 47 °C, respectively. The samples were heated at 5 °C min−1 to 180 °C. | ||
Specifically, the fragmentation pattern of the volatiles from the thermolysis of Mg(BH4)2·G1 resembles that of free G1 at 26 °C. In stark contrast, the fragmentation of volatiles of Mg(BH4)2·G4 is different from that of free G4 at 35 °C. For example, the fragmentation pattern of Mg(BH4)2·G4 at 50 °C is distinct from that of free G4 (Fig. 3). We observe the appearance of m/z = 15, 16 above 160 °C, which potentially corresponds to methane release from the demethylation of G4 in the Mg(BH4)2·G4 complex. The fragmentation pattern of Mg(BH4)2·G3 is a mix between that of Mg(BH4)2·G1 and Mg(BH4)2·G4 in that the volatiles consist of glyme units and methane at 183 °C (Fig. S12†). The results of MS analysis suggest that longer glymes, like G3 and G4, can undergo irreversible demethylation.
11B NMR analysis after the thermolysis of 1:1 Mg(BH4)2·G4 at 180 °C for 8 h produced Mg(B10H10), Mg(B12H12) and unreacted Mg(BH4)2 without evidence of B–O products (Fig. S13†). The lack of a B–O product from Mg(BH4)2 and a linear ether (i.e. G4) compared to that of B–O product formation in a cyclic ether (i.e. THF) by a plausible ring opening further highlights the different chemical reactivity of the different ethereal additives. This also suggests that Mg-alkoxide species may form instead of B–O products.32 Unfortunately, subsequent powder XRD analysis of the post-reaction mixture was uninformative because the mixture was amorphous (Fig. S14†). It is plausible that the dynamic flexibility of the longer glymes compared to that of shorter glymes can lead to different conformations, thus contributing to different degradation pathways such as demethylation. The demethylation of Mg(BH4)2·G3 and Mg(BH4)2·G4 at elevated temperatures may pose limitations on the use of these mixtures for multiple cycling processes required for an energy storage material.
Summary
By combining a suite of analytical techniques, we have shown that the 1:1 mixtures of Mg(BH4)2·Gn (n = 1–4) display notably different phase transitions and speciations, ranging from melts at low temperatures to compounds, such as Mg(BH4)2·G2, which are highly stable up to 180 °C. TPD-MS analysis provided evidence of H2 release, different fragmentation, and volatiles as a function of the glyme chain length. Observation of irreversible methane release from mixtures of Mg(BH4)2·G4 and Mg(BH4)2·G3 at operating conditions for Mg(BH4)2 dehydrogenation, which are absent for Mg(BH4)2·G1 and Mg(BH4)2·G2, offers insight into the chemical stabilities of employing Mg(BH4)2·glymes mixtures for the development of additives for energy storage materials.
Experimental section
General considerations
All materials were purchased, stored in a N2 glovebox and used as is unless otherwise noted. Mg(BH4)2 (1.0 g, 95%) was sourced from Sigma Aldrich. All glymes (G1, G2, G3, G4) were dried over CaH2 with vigorous stirring over 48 h, purified by distillation and stored over activated 4 Å molecular sieves in a nitrogen glove box. The heat flow measurements of the glymes only and the Mg(BH4)2–glyme mixtures were performed using differential scanning calorimetry (DSC) on a Q20 TA Instrument DSC. The samples were heated from 25 to 180 °C at a rate of 5 °C min−1 in a N2 atmosphere. The samples were enclosed in hermetically sealed Tzero aluminium pans prepared in a He glovebox, which necessitated the use of a reference pan prepared in a He atmosphere as well. For the collection of the open-system DSC–TGA data, the samples were prepared in an uncapped crucible inside an N2 glovebox, loaded in the instrument and flushed with N2. The data were obtained by heating the samples under N2 gas from 25 to 180 °C at a rate of 5 °C min−1. The samples studied for this work were analyzed on a calibrated, custom-built thermal programmed desorption (TPD) system equipped with a Stanford Research Systems RGA 100, with m/z = 1–100 amu, sampling rate of 4 seconds and 70 eV ionization energy. The samples were heated at a rate of 5 °C min−1 from 26 to 180 °C utilizing a Digi-Sense temperature controller. The quantity of material was adjusted to stay within the calibrated mass spectrometer's linear response region. In a typical analysis, 2–10 mg of sample was placed inside a quartz tube mounted to the TPD system. All experimental parameters were controlled via a LabView™ interface with the RGA, heating system, and pressure gauges. Typical initial pressures before heating the sample are at 10−8 Torr, with a flat baseline, i.e. no water, hydrogen or air signals above background. Further information on the system was previously published.33For powder XRD characterization, the solids were transferred to a glass capillary (500 μm diameter, 10 μm wall thickness, Charles Supper Co., MA) and sealed using wax and a wax pen inside an N2 glovebox. A Rigaku D/Max Rapid II micro diffraction system with a rotating Cr target (λ = 2.2910 Å) operated at 35 kV and 25 mA was used to collect the diffraction patterns. A parallel X-ray beam collimated to 300 μm diameter was directed onto the specimen and the diffracted intensities were recorded on a large 2D image plate over a 10 minute exposure. The 2D images were integrated between 10 and 150° 2θ to give standard powder traces. 11B NMR characterization of the boron products in the dehydrogenation reactions were recorded in 2:1 D2O:THF. All additional 11B NMR, DSC, TGA, TPD, MS and GC-MS data are provided in the ESI.†
General procedure for the preparation and thermolysis of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 Mg(BH4)2:glymes mixtures
:1 Mg(BH4)2:glymes mixtures
Inside a nitrogen-filled glovebox, to a 3 mL oven-dried scintillation vial was added solid Mg(BH4)2 (20.0 mg, 0.370 mmol) and glyme (0.370 mmol) by microsyringe. The resulting solid mixture was thoroughly mixed and repeatedly smeared against the wall using a spatula. The resulting mixture was transferred to a 25 mL Schlenk tube. The closed Schlenk tube was placed in a preheated aluminum block at the different temperatures and reaction time for the different experiments. The resulting mixture was returned to the glovebox and all manipulation and preparation of the sample for subsequent characterization were performed under inert atmosphere inside the glovebox.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the Hydrogen Materials-Advanced Research Consortium (HyMARC), established as part of the Energy Materials Network under the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Hydrogen and Fuel Cell Technologies Office (Contract No. DE-AC36-08GO28308), Environmental Molecular Sciences Laboratory (award 10.46936/cpcy.proj.2020.51656/60000242), a DOE Office of Science User Facility sponsored by the Biological and Environmental Research under Contract No. DE-AC05-76RL01830 for research support. The authors thank C. M. Jensen for insightful discussions. Pacific Northwest National Laboratory is operated by Battelle for the U.S. Department of Energy.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- B. Sakintuna, F. Lamaridarkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS.
- Y. Filinchuk, B. Richter, T. R. Jensen, V. Dmitriev, D. Chernyshov and H. Hagemann, Angew. Chem., Int. Ed., 2011, 50, 11162–11166 CrossRef CAS PubMed.
- O. Zavorotynska, A. El-Kharbachi, S. Deledda and B. C. Hauback, Int. J. Hydrogen Energy, 2016, 41, 14387–14403 CrossRef CAS.
- G. Soloveichik, Y. Gao, J. Rijssenbeek, M. Andrus, S. Kniajanski, R. Bowmanjr, S. Hwang and J. Zhao, Int. J. Hydrogen Energy, 2009, 34, 916–928 CrossRef CAS.
- Y. Zhang, E. Majzoub, V. Ozoliņš and C. Wolverton, J. Phys. Chem. C, 2012, 116, 10522–10528 CrossRef CAS.
- I. H. Nayyar, B. Ginovska, M. Bowden, G. Edvenson, B. Tran and T. Autrey, J. Phys. Chem. A, 2022, 126, 444–452 CrossRef CAS PubMed.
- N. P. Stadie, E. Callini, B. Richter, T. R. Jensen, A. Borgschulte and A. Züttel, J. Am. Chem. Soc., 2014, 136, 8181–8184 CrossRef CAS PubMed.
- J. Chen, Y. S. Chua, H. Wu, Z. Xiong, T. He, W. Zhou, X. Ju, M. Yang, G. Wu and P. Chen, Int. J. Hydrogen Energy, 2015, 40, 412–419 CrossRef CAS.
- S. Zhao, B. Xu, N. Sun, Z. Sun, Y. Zeng and L. Meng, Int. J. Hydrogen Energy, 2015, 40, 8721–9731 CrossRef CAS.
- E. G. Bardají, N. Hanada, O. Zabara and M. Frichtner, Int. J. Hydrogen Energy, 2011, 36, 12313–12318 CrossRef.
- N. Leick, N. A. Strange, A. Schneemann, V. Stavila, K. Gross, N. Washton, A. Settle, M. B. Martinez, T. Gennett and S. T. Christensen, ACS Appl. Energy Mater., 2021, 4, 1150–1162 CrossRef CAS.
- M. Chong, T. Autrey and C. Jensen, Inorganics, 2017, 5, 89 CrossRef.
- M. Chong, A. Karkamkar, T. Autrey, S. Orimo, S. Jalisatgi and C. M. Jensen, Chem. Commun., 2011, 47, 1330–1332 RSC.
- Y. Shao, N. N. Rajput, J. Hu, M. Hu, T. Liu, Z. Wei, M. Gu, X. Deng, S. Xu, K. S. Han, J. Wang, Z. Nie, G. Li, K. R. Zavadil, J. Xiao, C. Wang, W. A. Henderson, J.-G. Zhang, Y. Wang, K. T. Mueller, K. Persson and J. Liu, Nano Energy, 2015, 12, 750–759 CrossRef CAS.
- T. Burankova, E. Roedern, A. E. Maniadaki, H. Hagemann, D. Rentsch, Z. Lodziana, C. Battaglia, A. Remhof and J. P. Embs, J. Phys. Chem. Lett., 2018, 9, 6450–6455 CrossRef CAS PubMed.
- D. Samuel, C. Steinhauser, J. G. Smith, A. Kaufman, M. D. Radin, J. Naruse, H. Hiramatsu and D. J. Siegel, ACS Appl. Mater. Interfaces, 2017, 9, 43755–43766 CrossRef CAS PubMed.
- R. Mohtadi, M. Matsui, T. S. Arthur and S.-J. Hwang, Angew. Chem., Int. Ed., 2012, 51, 9780–9783 CrossRef CAS PubMed.
- R. Mohtadi, Molecules, 2020, 25, 1791–1816 CrossRef CAS PubMed.
- J. Luo, Y. Bi, L. Zhang, X. Zhang and T. L. Liu, Angew. Chem., Int. Ed., 2019, 58, 6967–6971 CrossRef CAS PubMed.
- S. Hebié, H. P. K. Ngo, J.-C. Leprêtre, C. Iojoiu, L. Cointeaux, R. Berthelot and F. Alloin, ACS Appl. Mater. Interfaces, 2017, 9, 28377–28385 CrossRef PubMed.
- N. Sa, N. N. Rajput, H. Wang, B. Key, M. Ferrandon, V. Srinivasan, K. A. Persson, A. K. Burrell and J. T. Vaughey, RSC Adv., 2016, 6, 113663–113670 RSC.
- N. N. Rajput, X. Qu, N. Sa, A. K. Burrell and K. A. Persson, J. Am. Chem. Soc., 2015, 137, 3411–3420 CrossRef CAS PubMed.
- I. Saldan, Int. J. Hydrogen Energy, 2016, 41, 11201–11224 CrossRef CAS.
- W. Wegner, T. Jaron, M. A. Dobrowolski, L. Dobrzycki, M. K. Cyranski and W. Grochala, Dalton Trans., 2016, 45, 14370–14377 RSC.
- B. L. Tran, T. N. Allen, M. E. Bowden, T. S. Autrey and C. M. Jensen, Inorganics, 2021, 9, 41 CrossRef CAS.
- Y. Shao, T. Liu, G. Li, M. Gu, Z. Nie, M. Engelhard, J. Xiao, D. Lv, C. Wang, J. G. Zhang and J. Liu, Sci. Rep., 2013, 3, 3130 CrossRef PubMed.
- S. Hou, X. Ji, K. Gaskell, P.-f. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- Y. Bi, S. He, C. Fan, J. Luo, B. Yuan and T. L. Liu, J. Mater. Chem. A, 2020, 8, 12301–12305 RSC.
- K. V. Nielson and T. L. Liu, Angew. Chem., Int. Ed., 2020, 59, 3368–3370 CrossRef CAS PubMed.
- M. V. Solovev, O. V. Chashchikhin, P. V. Dorovatovskii, V. N. Khrustalev, A. S. Zyubin, T. S. Zyubina, O. V. Kravchenko, A. A. Zaytsev and Y. A. Dobrovolsky, J. Power Sources, 2018, 377, 93–102 CrossRef CAS.
- See the ESI.† Acidification and GC-MS characterization of the post-thermolysis reaction of 1:1 Mg(BH4)2·G4 showed G4 and three new species (Table S1, pp. S12–S14†) containing similar fragmentation patterns.
- K. E. Hurst, M. J. Heben, J. L. Blackburn, T. Genett, A. C. Dillon and P. A. Parilla, Rev. Sci. Instrum., 2013, 84, 025103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of sample preparation, XRD, TGA, and TPD-MS data collection. See DOI: https://doi.org/10.1039/d2dt01106a |
| This journal is © The Royal Society of Chemistry 2022 |