
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of substituents in aryl groups on the structure, thermal transitions and electrorheological properties of zinc bis(diarylphosphate) hybrid polymers†
Maciej
Dębowski
 *,
Piotr A.
Guńka
*,
Konrad
Żurawski
,
Magdalena
Zybert
,
Beata
Modzelewska
,
Andrzej
Ostrowski
,
Janusz
Zachara
and
Zbigniew
Florjańczyk
*,
Piotr A.
Guńka
*,
Konrad
Żurawski
,
Magdalena
Zybert
,
Beata
Modzelewska
,
Andrzej
Ostrowski
,
Janusz
Zachara
and
Zbigniew
Florjańczyk
Faculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warszawa, Poland. E-mail: maciej.debowski@pw.edu.pl; piotr.gunka@pw.edu.pl
First published on 30th March 2022
Abstract
The structures and thermal properties of three new hybrid one-dimensional (1D) polymers based on zinc bis(diarylphosphate)s containing p-substituted phenyl rings are reported. The crystal structures of Zn[O2P(p-OC6H4NO2)2]2 (1), Zn[O2P(p-OC6H4OMe)2]2 (2) and Zn[O2P(p-OC6H4CO2Et)2]2 (3) differ from that of their unsubstituted analogue, Zn[O2P(OPh)2]2 (ZnDPhP). Compounds 1 and 3 consist of tetrahedrally coordinated zinc cations connected by double bridges of phosphate groups (2+2 bridging mode) and form polymeric chains that are packed in a distorted hexagonal lattice with six closest neighbours. In compound 2 zinc cations are linked by alternating single and triple phosphate bridges (3+1 bridging mode) and the resulting chains, having only four closest neighbors, are packed in a distorted tetragonal manner. DFT computations revealed that the 2+2 bridging mode, even at the highest energy conformation, is more stable than the 3+1 one. Simultaneous Thermal Analysis, Raman spectroscopy and powder XRD (PXRD) studies show that pyrolysis of the studied hybrid polymers begins above 260 °C, leading to a mixture of zinc condensed phosphates and carbonaceous deposits that may have electron-conducting properties. DSC and PXRD studies provide evidence that crystalline domains in 2 and 3 rearrange and/or disappear at a much lower temperature (ca. 150 °C) leading to an isotropic liquid (in the case of 3) or an amorphous solid material (in the case of 2). Electrorheological measurements indicate that 1–3 are polarized in an external electric field, and the type of electrorheological effect depends on the type of functional group attached to the phenyl ring; this feature can be utilized in designing new electrorheological devices.
Introduction
Hybrid inorganic–organic polymers based on metal phosphates and phosphinates have been studied for a long time due to their interesting structural features, unique properties and diverse applications.1–4 One group of such compounds encompasses zinc diorganophosphinates and zinc bis(diorganophosphate)s, Zn(O2PR2)2 and Zn[O2P(OR)2]2, respectively, which tend to crystallize in the form of one-dimensional (1D) rods or fibers consisting of infinite chains. They are built up from Zn2+ cations joined by bidentate bridging phosphoorganic ligands.5–14 A wide range of homopolymers comprising only one type of bridging unit, as well as copolymers containing two different phosphorus ligands have already been investigated, and some of their structural features together with literature references are listed in Table 1. Two modes of Zn2+ cation bridging have been observed: double PO2 bridges, or an arrangement of alternating triple and single PO2 bridges (see Chart 1). However, structural analysis has revealed no clear correlation between the type of organic substituent in the phosphate/phosphinate anion and the Zn2+ bridging mode adopted in the polymer. One can only assume that the presence of short alkyl substituents (Me, Et) or phenyl groups promotes the formation of 2+2 structures, while longer or bulky alkyl groups result in the 3+1 bridging mode. It is noteworthy that for some zinc phosphinates with alkyl substituents, only fiber diffraction patterns were obtained and the bridging mode between zinc cations has been inferred from the periodicity along the Zn(O2PR2)2 chain, i.e. the distance between symmetrically equivalent zinc cations.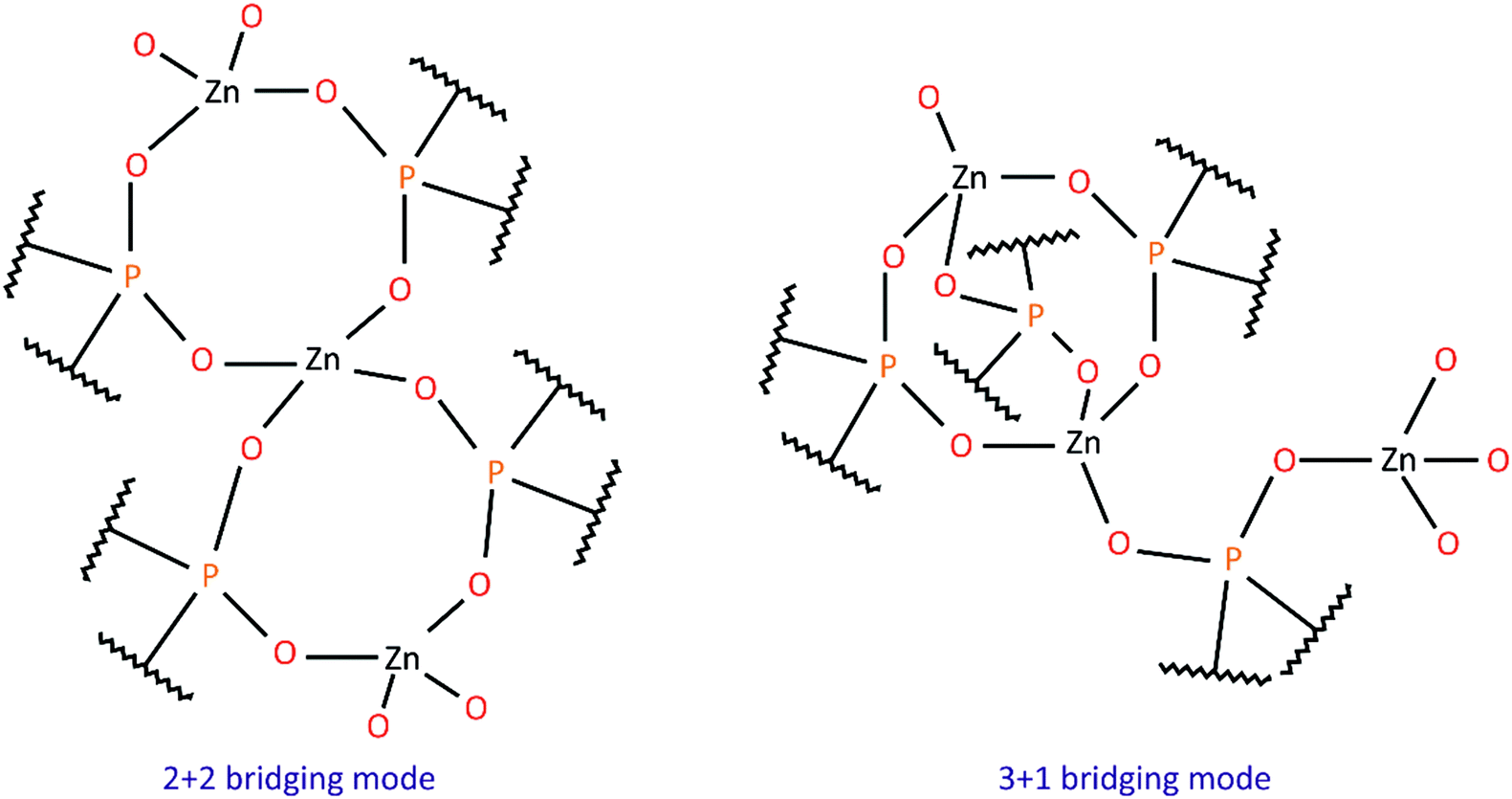 | ||
| Chart 1 Bridging modes between Zn2+ cations in zinc phosphates and phosphinates. | ||
| R (, R′) | Bridging mode | Zn⋯Zn distance (Å) | Ref. |
|---|---|---|---|
| a Copolymers Zn(R12PO2)2−2x(R22PO2)2x where R1 = nBu and R2 = n-C6H13 or n-C10H21. b Determined from fiber diffraction. | |||
| Zn(O 2 PR 2 ) 2 or Zn(O 2 PRR’) 2 | |||
| n Bu | 3+1 | 9.90 | 6 |
| n-C6H13 | 3+1 | 9.90b | 6 |
| n-C10H21 | 3+1 | 9.90b | 6 |
| Copolymersa | 3+1 | 9.87 | 10 |
| n Bu, Ph | 3+1 | 10.16 | 11 |
| Me, Ph | 2+2 | 9.229 | 12 |
| H, Ph | 2+2 | 8.272 | 13 |
| (CH3)(n-C3H7)CH | 2+2 | 9.16b | 14 |
Metal diarylphosphates exhibit greater structural stability, while derivatives bearing long alkyl groups are conformationally unstable even at low temperatures, making it impossible to reliably determine the positions of carbon atoms in their structure.15 The results of our recent studies have shown that metal diorganophosphates may undergo reversible structural transformations in the solid state at temperatures below 200 °C accompanied by changes in the conformation of their organic substituents.9,15
Recently, we have reported on the synthesis, structure and thermally-induced structural transformations occurring in zinc bis(diphenylphosphate) (ZnDPhP). Its crystals are composed of hexagonally packed, homochiral helices of ZnO4 tetrahedra connected by double O2P(OPh)2 bridges. On heating, the ZnDPhP structure undergoes thermal expansion in a direction perpendicular to its Z-axis, while at ca. 160 °C reversible translational movement of the chains along each other (parallel to the Z-axis) also occurs.9
The results of electrorheological measurements showed that ZnDPhP is also susceptible to a reversible polarization in an external electric field, which makes it a promising candidate for application in the field of so-called smart materials.9 This encouraged us to modify the structure of the aromatic group in zinc bis(diarylphosphate) by introducing either electron withdrawing or electron donating substituents in the para position with respect to the phosphoester linkage, and investigate the effect of these substituents on the structure of polymeric chains formed by zinc bis(diarylphosphate)s, the way they are organized into crystalline domains, as well as some physicochemical properties of the resulting materials. Herein, we report the results of structural and thermal studies, as well as DFT calculations, concerning 3 new zinc bis(diarylphosphate)s with p-OC6H4NO2 (1), p-OC6H4OCH3 (2) and p-OC6H4CO2Et (3) organic groups. Some preliminary findings on their electrorheological properties are also discussed.
Experimental
Materials
All chemicals, except for phosphoric acid esters, were purchased from commercial sources and used without further purification: zinc acetate dihydrate (99%, Merck KGaA), zinc oxide (≥99%, Merck KGaA), sodium hydroxide (pure, POCh Gliwice), phosphorus(V) oxychloride (99%, Sigma-Aldrich), benzyltriethylammonium chloride (99%, Sigma-Aldrich), 4-nitrophenol (≥99%, Merck Schuchardt OHG), 4-methoxyphenol (≥99%, Merck Schuchardt OHG), ethyl 4-hydroxybenzoate (99%, Sigma-Aldrich), and methyl-terminated polydimethylsiloxane (PDMS100; POLSIL®OM 100, viscosity 1 × 10−4 m2 s−1 at 25 °C, Chemical Plant “Silikony Polskie” Ltd). Before use, PDMS100 was vacuum-dried (1.33 × 10−6 mbar) at 120 °C for 10 h.Synthetic protocols
Phosphoric acid esters containing aryl substituents were synthesized from POCl3 and the respective phenols – a detailed description of their syntheses and basic spectral properties can be found in the ESI.†For the purpose of X-ray crystallography measurements, 1 was recrystallized from water.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) mixture. After stirring for 1 hour the dispersion was centrifuged and the separated clear liquid phase was concentrated on a rotary evaporator at 60 °C. The crude solid product was purified by washing with acetone and dried at 60 °C in a vacuum oven resulting in 2.00 g of compound 2 (reaction yield: 48%).
:4 v/v) mixture. After stirring for 1 hour the dispersion was centrifuged and the separated clear liquid phase was concentrated on a rotary evaporator at 60 °C. The crude solid product was purified by washing with acetone and dried at 60 °C in a vacuum oven resulting in 2.00 g of compound 2 (reaction yield: 48%).
For the purpose of X-ray crystallography measurements, 2 was recrystallized from a water/acetone (1:4 v/v) mixture.
Methods of characterization
In the case of triaryl phosphates, elemental analyses for C, H and N were performed using a Euro EA elemental analyser made by EuroVector Instruments & Software.
| Identification code | 1 | 2 | 3 |
|---|---|---|---|
| Empirical formula | C24H16N4O16P2Zn | C28H28O12P2Zn | C36H36O16P2Zn |
| Formula weight | 743.72 | 683.81 | 851.96 |
| T/K | 100.0(2) | 292(2) | 295(1) |
| Crystal system | Orthorhombic | Monoclinic | Triclinic |
| Space group | Pna21 | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 7.54290(10) | 48.8388(9) | 8.8426(2) |
| b/Å | 12.0207(2) | 9.85200(12) | 14.6002(3) |
| c/Å | 30.3806(5) | 25.1299(4) | 17.1931(3) |
| α/° | 90 | 90 | 112.6738(18) |
| β/° | 90 | 99.8669(16) | 95.5240(16) |
| γ/° | 90 | 90 | 102.3836(19) |
| V/Å3 | 2754.64(7) | 11912.7(3) |
1960.67(8) |
| Z | 4 | 16 | 2 |
| ρ calc/g cm−3 | 1.793 | 1.525 | 1.443 |
| μ/mm−1 | 1.098 | 0.995 | 0.779 |
| F(000) | 1504.0 | 5632.0 | 880.0 |
| Crystal size/mm3 | 0.99 × 0.12 × 0.11 | 0.31 × 0.12 × 0.094 | 0.98 × 0.20 × 0.12 |
| Radiation | Mo Kα (λ = 0.71073) | Mo Kα (λ = 0.71073) | Mo Kα (λ = 0.71073) |
| 2θ range for data collection/° | 6.38 to 52.04 | 6.51 to 60.00 | 6.65 to 60.00 |
| Index ranges | −9 ≤ h ≤ 9 | −68 ≤ h ≤ 68 | −12 ≤ h ≤ 12 |
| −14 ≤ k ≤ 14 | −13 ≤ k ≤ 13 | −20 ≤ k ≤ 20 | |
| −37 ≤ l ≤ 37 | −35 ≤ l ≤ 34 | −24 ≤ l ≤ 24 | |
| Reflections collected | 50852 |
76595 |
116037 |
| Independent reflections | 5436 [Rint = 0.1039, Rsigma = 0.0365] | 17317 [Rint = 0.0346, Rsigma = 0.0246] |
11434 [Rint = 0.0493, Rsigma = 0.0236] |
| Data/restraints/parameters | 5436/25/425 | 17317/0/857 |
11434/0/500 |
| Goodness-of-fit on F2 | 1.033 | 1.035 | 1.032 |
| Final R indices [I ≥ 2σ(I)] | R 1 = 0.0901 | R 1 = 0.0316 | R 1 = 0.0381 |
| wR2 = 0.2304 | wR2 = 0.0800 | wR2 = 0.0934 | |
| Final R indices [all data] | R 1 = 0.0910 | R 1 = 0.0376 | R 1 = 0.0563 |
| wR2 = 0.2321 | wR2 = 0.0838 | wR2 = 0.1048 | |
| Residual density: max/min/e Å−3 | +1.79/−1.34 | +0.49/−0.40 | +0.55/−0.34 |
| Flack parameter | 0.50(3) | — | — |
Results and discussion
Crystal structures
The results of X-ray crystallography show that compounds 1–3 are linear coordination polymers and crystallize in Pna21, C2/c and P space groups, respectively. Zn2+ cations are tetrahedrally coordinated by four diarylphosphate anions, each of which is shared by two Zn2+ cations (see Fig. 1 for asymmetric units). Polymeric chains are formed by connecting adjacent Zn2+ cations with double O–P–O bridges coming from different diarylphosphate anions in compounds 1 and 3. This mode of cation linking is analogous to that observed in Zn[O2P(OPh)2]29 and different from the one present in polymer 2 where alternating triple and single O–P–O bridges occur (see Fig. 2). Consequently, the linear chains in 2 exhibit the symmetry of the p1 rod group while the symmetry of those in 1 and 3 is higher and corresponds to the pc11 and p rod groups, respectively.
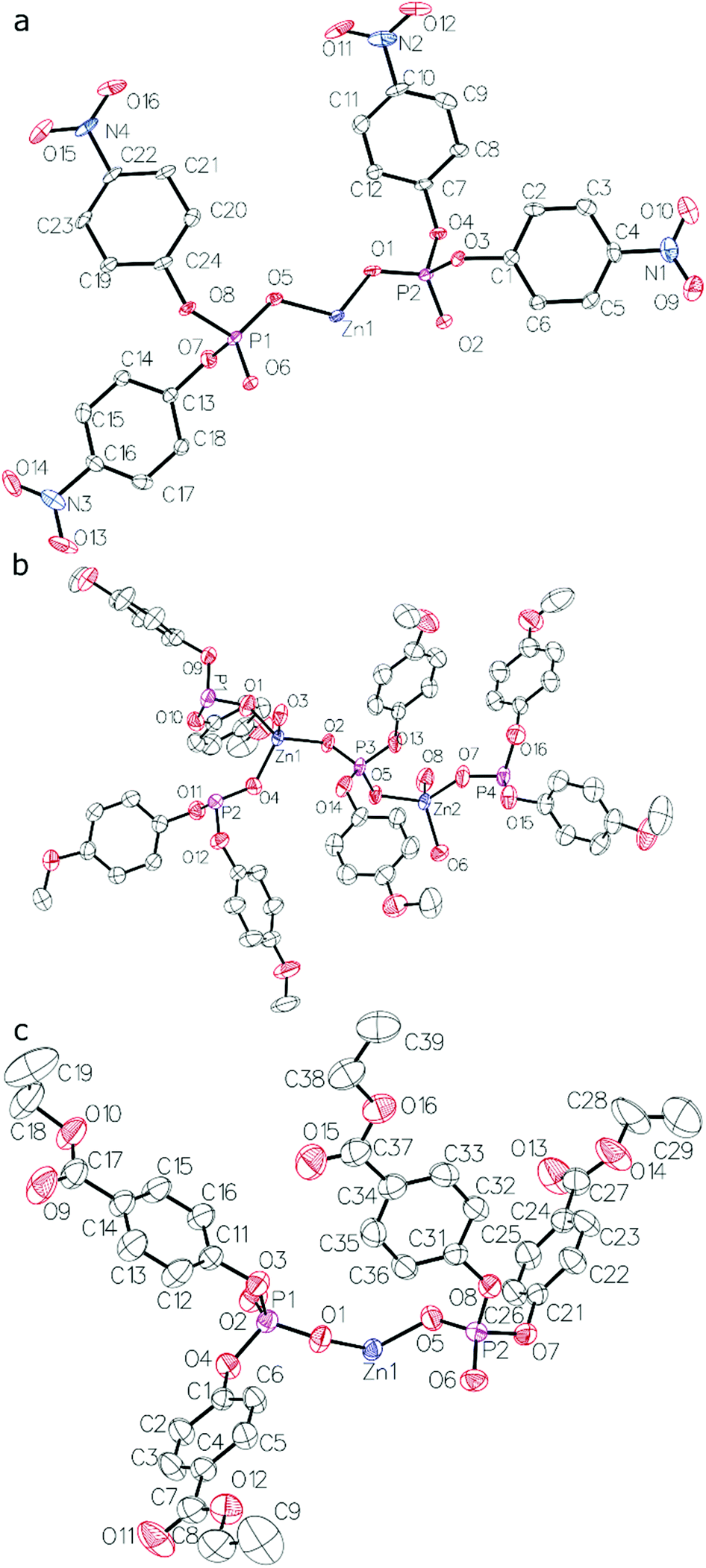 | ||
| Fig. 1 Asymmetric units of crystal structures of compounds: (a) 1, (b) 2 (only the major disorder component of one of the p-C6H4OMe groups is shown) and (c) 3. Thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
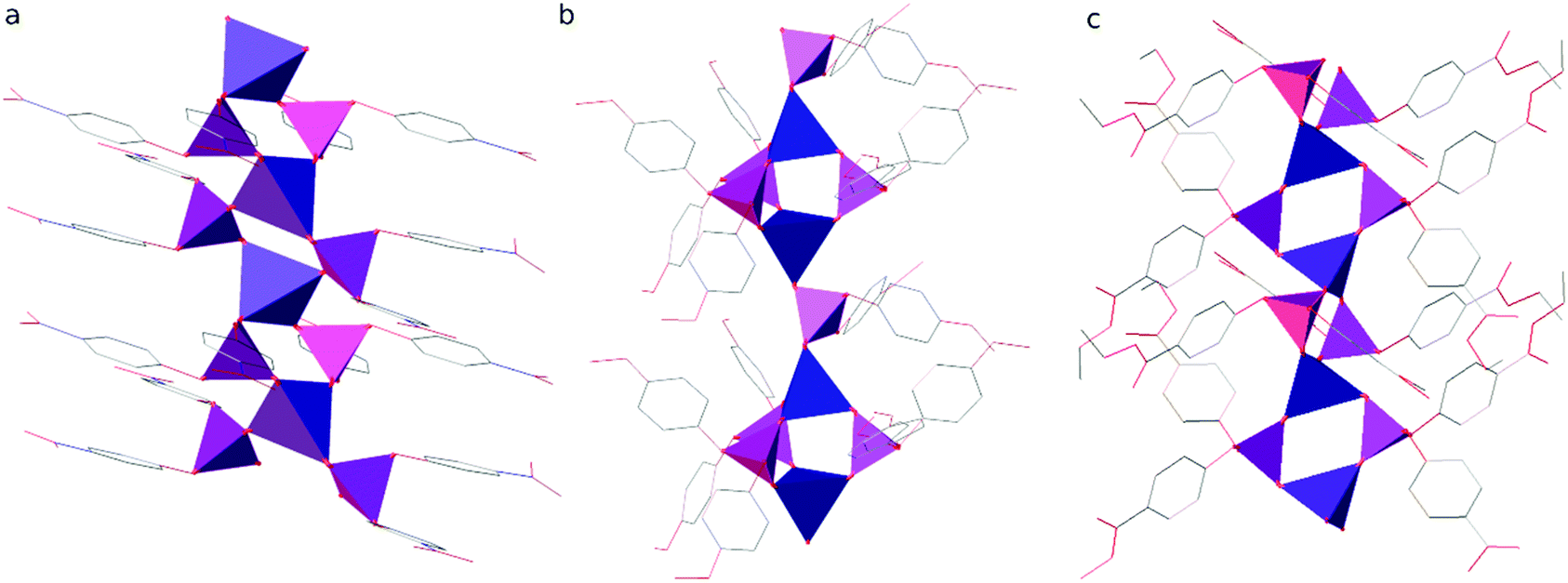 | ||
| Fig. 2 Polymeric rods present in the crystal structures of compounds (a) 1, (b) 2 and (c) 3. Zinc and phosphorus are depicted as blue and violet tetrahedra, respectively. Aryl substituents are represented in a wireframe model with carbon, nitrogen and oxygen coloured grey, blue and red, respectively. Hydrogen atoms are omitted for clarity. | ||
The distance between the adjacent Zn centers strictly depends on the number of diorganophosphate bridging units: in the case of the 2+2 bridging mode it is in the range of 4.329–4.425 Å, whereas for the alternating single and triple O2P(OR)2 bridges it becomes 6.041 and 3.913 Å, respectively. The coordination around phosphorus is distorted tetrahedral since the P–O bond lengths range from 1.474 to 1.502 Å for oxygen ligands bonded to zinc and are distinctly shorter than those involving oxygen atoms bonded to aryl groups (1.573–1.606 Å). The O–P–O bond angles in the bridges between Zn2+ cations are larger (117.4–120.3°) than other O–P–O angles (100.6–112.2°). ZnO4 tetrahedra are also distorted with the Zn–O bond lengths ranging from 1.904 to 1.960 Å.
The deviation in the geometry of the PO4 and ZnO4 tetrahedra from that of a regular tetrahedron, as well as the resulting structural stresses within 1–3 chains can be quantified by means of the bond-valence vector (BVV) model.33 In Table S5 (ESI†) the lengths of the resultant bond-valence vectors for each crystallographically independent phosphorus (|vP|) and zinc (|vZn|) center in compounds 1–3 are compared to an analogous set of data for ZnDPhP already published in ref. 9. The calculated values of |vP| and |vZn| are rather low and do not exceed 0.08 valence units. Taking that observation into account, as well as the estimated error of the BVV model one can assume that the type of organic substituent connected to the phenyl ring has a very limited impact on the strains generated within the PO4 and ZnO4 tetrahedra. The greatest deformations occur in the case of 1, however, they are on a comparable level with those observed in ZnDPhP, though the strains of ZnO4 tetrahedra in 1 are more pronounced.
Analysis of the C–O–P–O torsion angles indicates that the aromatic substituents in compounds 1–3 adopt different conformations than those present in ZnDPhP. In the latter, the phenyl groups connected to two crystallographically non-equivalent phosphorus centers are in a gauche–trans (G−T) conformation (based on the data deposited in the Cambridge Crystallographic Data Center with CCDC number 1472713 dihedral angles of either −71.7° and 152.4°, or −74.3° and −164.8°),34 according to the stereochemical convention recommended by IUPAC.35 Our studies on compounds 1–3 show that the presence of a substituent at the para position of the aromatic ring promotes the formation of gauche conformers (torsion angle moduli between 55° and 74°). As can be seen from the data presented in Table S4 (ESI†), in 1 and 3 the organic substituents are attached to two crystallographically non-equivalent phosphorus atoms in G+G+/G+G+ and G−G−/G−T conformations, respectively. In compound 2, three of four phosphorus centers have their organic substituents in a gauche–gauche conformation: G−G− in the case of phosphorus forming a single bridge, and G+G+ for the phosphorus ligands forming a triple bridge; the remaining one is characterized by a G−T mode. It is worth noting that the latter is also the most structurally strained, according to the BVV calculations. A very similar relationship between the deformation of the PO4 tetrahedra and conformational arrangement of the organic substituents has been previously observed for aluminum tris(diethylphosphate).36 However, at this stage of investigation it is too early to decide whether the conformations of organic groups in compounds 1–3 correspond solely to the lowest energy state of a single zinc bis(diarylphosphate) chain, or if they are forced by intermolecular interactions.
The polymeric chains in compound 1 are linked into layers parallel to the (001) lattice planes by interdigitated p-C6H4NO2 groups (see Fig. 3 and Table S1 in the ESI† for details) and C–H⋯O hydrogen bonds (C5–H5⋯O12i and C17–H17⋯O15i, where i = −½ + x, ½ − y, z; for details see Table S2 in the ESI†). It is noteworthy that those chains are stiffened by π-stacking between other p-C6H4NO2 groups (see Table S3 in the ESI† for details). The layers interact via C–H⋯O hydrogen bonds (C2–H2⋯O13iii, C9–H9⋯O14iv and C23–H23⋯O10v, where iii = ½ − x, −½ + y, ½ + z and iv = −x, 1 − y, ½ + z and v = ½ − x, −½ + y, −½ + z; see Table S2 in the ESI† for details) and dispersion interactions to form a 3D structure. Compound 1 crystallizes in the form of elongated columns with the largest dimension parallel to the crystallographic X axis and the other two dimensions very similar to each other, which indicates that the interactions within the layers and between them are very similar in terms of energy.
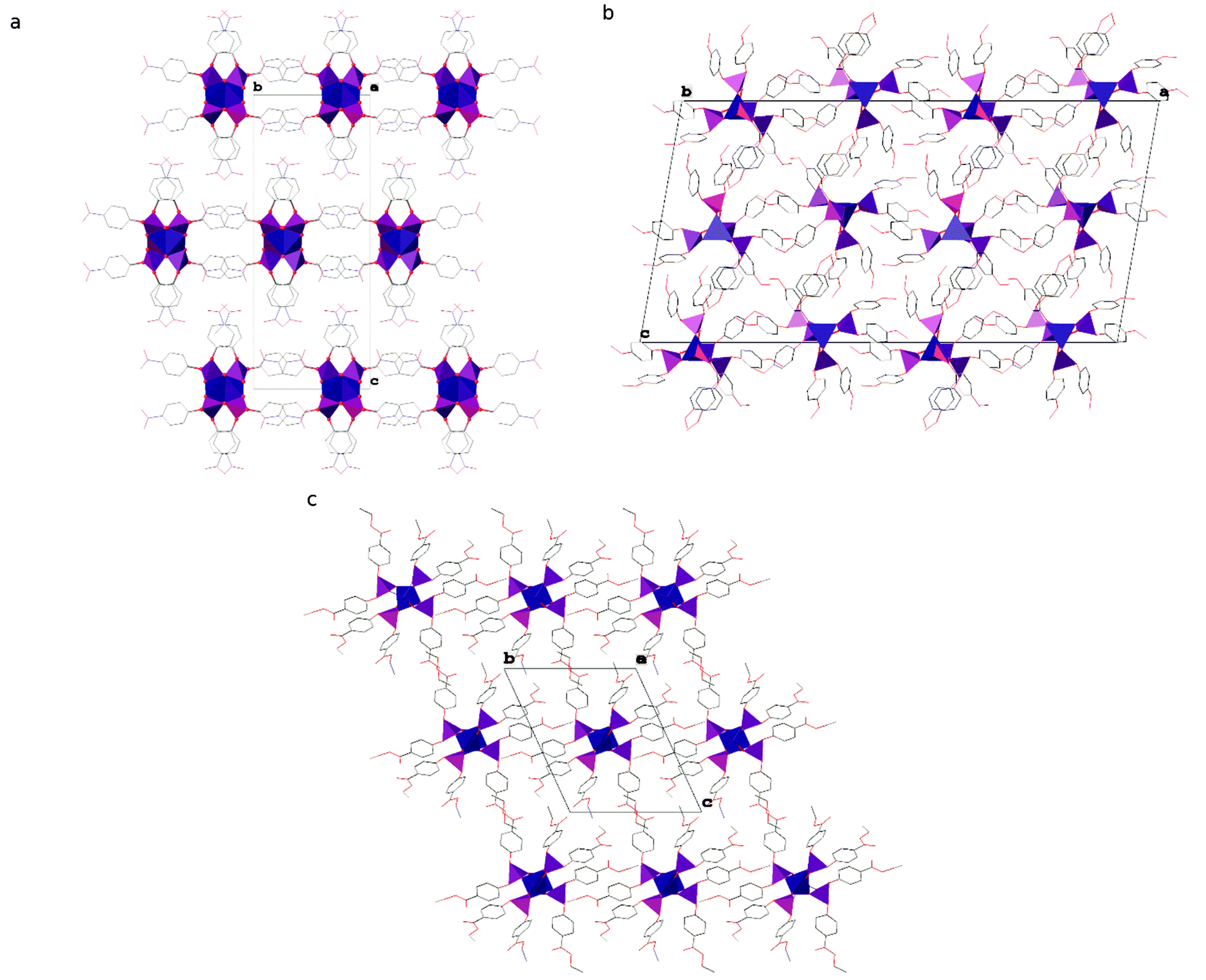 | ||
| Fig. 3 Packing diagrams of crystal structures of compounds (a) 1, (b) 2 and (c) 3. Zinc and phosphorus are depicted as blue and violet polyhedra, respectively. Aryl substituents are represented in a wireframe model with carbon, nitrogen and oxygen coloured grey, blue and red, respectively. Hydrogen atoms are omitted for clarity. | ||
The chains of Zn[O2P(OC6H4OMe)2]2 in the crystal structure of compound 2 interact via π-stacking between phenyl rings to form double chains running along the crystallographic [010] direction (ring of C43C44C45C46C47C48; Cg⋯Cgviii = 3.8559(13) Å, dihedral angle of 0.00(10)° and slippage of 1.881 Å, where viii = ½ − x, 2.5 − y, 1 − z and Cg stands for the centroid). The double chains interact via dispersion interactions assisted by weak directional C–H⋯O hydrogen bonds to form the crystal structure (see Fig. 3). Interestingly, 2 is the only one among the studied compounds, in which each polymeric chain is surrounded by only 4 closest chains. In other compounds distorted hexagonal packing is observed, with 6 closest chains neighbouring a central one.
A detailed analysis of the crystal structure of compound 3 revealed that there are no significant directional interactions between polymeric chains of Zn[O2P(OC6H4CO2Et)2]2 in addition to isotropic dispersion interactions and, consequently, every chain is surrounded by 6 closest neighbours (see Fig. 3).
DFT computations
Interestingly, zinc bis(diorganophosphate)s form either doubly bridged chains or chains with alternating single and triple bridges (see Table 1). In order to understand this, we have optimized and computed energies of two hypothetical chains of Zn[O2P(OMe)2]2, one with double and the other with alternating single and triple O2P(OMe)2 bridges. We have also optimized the structures of hypothetical Zn[O2P(OPh)2]2 chains whose structures have been derived from 1–3 and from the previously published ZnDPhP9 (see details in the “Methods of characterization” section).
The general observation from the DFT calculation results (Table 3) is that the 2+2 bridged chains exhibit lower energies than the 3+1 bridged ones. However, the 2+2 bridging mode leads to a wide variety of chain conformations, which can vary in energy by as much as 110 kJ mol−1 in the case of the hypothetical structure of zinc bis(diphenylphosphate). We suspect that the large differences in the individual chain energies must be compensated for in crystal structures by two factors. Firstly, it must be stressed that the models were obtained by removing functional groups from the phenyl rings. The interactions between these groups may stabilize such chain conformations and may actually even be the driving force for the adoption of the conformations unfavorable for unsubstituted phenyl rings. Secondly, intermolecular interactions between the chains in crystal structures may further stabilize the conformations. Our studies on the thermal properties of compounds 1–3 prove that such strong interactions, as well as modification of their strength, can affect other properties of zinc bis(diarylphosphate)s.
| Source model | Bridging mode | Relative energy (kJ mol−1) |
|---|---|---|
| (Zn[O 2 P(OMe) 2 ] 2 ) | ||
| YISDAY | 2+2 | 0 |
| NAKQAR | 3+1 | 67 |
| (Zn[O 2 P(OPh) 2 ] 2 ) | ||
| OMIGAV | 2+2 | 0 |
| 1 | 2+2 | 4.0 |
| 3 | 2+2 | 107 |
| 2 | 3+1 | 135 |
Thermal properties
The results of STA analysis (Fig. S16A and S17 and S18, Table S6 in the ESI†) indicate that thermal decomposition of the investigated zinc bis(diarylphosphate)s begins at 260–290 °C. They are more thermally labile than their analogue with unsubstituted phenyl rings (ZnDPhP),9 which is especially evident when such criteria for thermal stability of materials as T98%, T95% (temperatures corresponding to 2% and 5% weight loss, respectively) or Tonset (the extrapolated onset temperature of degradation) are considered. Generally, in the case of 1–3 these temperatures are ca. 24–100 °C lower than those characteristic for ZnDPhP. This difference becomes even smaller, reaching values of 0–23 °C, if a bend point on the respective TG curves is analyzed (Tb in Table S6, ESI†) indicating that on heating under the same process conditions zinc bis(diarylphosphate)s start losing their weight at almost the same temperature region independent of the functional groups attached to the phenyl ring. However, the overall shape of thermogravimetric curves, especially the number and width of peaks observed on the dTG curves, suggests that the thermal decomposition of 1–3 proceeds faster and in a less complicated way than that of ZnDPhP.9 From that point of view polymer 1 stands out among zinc bis(diarylphosphate)s investigated so far, since within the tested temperature range it undergoes pyrolysis in a single, quick step (the extrapolated onset and endset temperatures differing by less than 0.5 °C, see Table S6 in the ESI†). Moreover, the evolution of heat is observed during this process, as indicated by a sharp exothermic peak located at ca. 384 °C in the DTA curve of 1 (see Fig. S16A in the ESI†) – in the case of 2 and 3 only the endotherms appear (see Fig. S17 and S18 in the ESI,† respectively). Such behaviour of 1 is understandable given that the nitro group is one of the most common explosophores.Mass spectrometry of the evolved gases revealed the complete lack of any QMS peaks above the m/z value of 45 during the pyrolysis of compounds 1–3. A common feature of their QMS spectra (Fig. S16B and C, S19 and S20 in the ESI†) is the presence of signals characteristic of carbon dioxide (m/z = 44), carbon monoxide (m/z = 28, also detected in the mass spectrum of CO2), and water (m/z = 17 and 18),37 which could be identified as main gaseous products during thermal degradation of 1–3. It should be noted that some additional OMS peaks are also introduced into mass spectra due to the specific thermal transformations of functional groups attached to the aromatic ring. For example, in the case of pyrolysis of 1 one can observe the signal of the nitrosonium cation NO+ (m/z = 30) that can be ascribed to the fragmentation of either nitric or nitrous oxide,37 while the QMS spectrum of 2 contains an easily distinguishable peak of the methyl cation (m/z = 15). The absence of any QMS signals derived from volatile phenol or phosphorus-containing species (e.g., PO, PO2, HOPO, or HOPO2)38 and a substantially high carbon content in the solid residue of pyrolysis (22.00, 35.94 and 29.00 wt% for pyrolyzates of 1, 2 and 3, respectively), suggest that the C–O bond splitting is more favored than the P–O bond cleavage, while subsequent condensation into polyaromatic moieties proceeds more efficiently in comparison with ZnDPhP pyrolysis. Interestingly, these differences have a limited impact on the structure of the solid residues from the pyrolysis of 1–3, with their carbonaceous components being the least affected. Based on the similarities in the shape of Raman spectra (see Fig. S21 in the ESI†) one can assume that it consists of nanographene39 or graphene oxide in its pristine or reduced form,39,40 just like in the case of the ZnDPhP pyrolyzate.9 A complementary PXRD analysis (Fig. S22–S24 in the ESI†) revealed the presence of crystalline domains of zinc condensed phosphates, mainly a mixture of zinc metaphosphates and/or zinc pyrophosphates. It should be noted, however, that due to a significant amount of the amorphous phase, revealed by a characteristic broad halo occurring around 2θ = 24° on the PXRD baseline, as well as rather small dimensions of the crystallites forming the samples resulting in substantial widening of PXRD reflections, not all crystalline phases could be identified in the pyrolyzates of compounds 1–3.
A distinct differentiation in the behavior of compounds 1–3 also occurs below their thermal stability limit. In the respective DTA and DSC curves (Fig. S16–S18 and S25–S27 in the ESI,† respectively) single endothermic signals can be detected at ca. 245 °C (for 1) and ca. 150 °C (for 2 and 3). Optical observations and thermogravimetry indicate that they originate from processes occurring either in the solid state (for 1 and 2), or involving the solid → liquid phase transition (3). The origin of those peaks and their exact position on the temperature scale can be explained in terms of the energy of intermolecular interactions existing between neighboring Zn[O2P(OR)2]2 chains, just as in the case of previously reported ZnDPhP.9 Strong interchain interactions (e.g., π-stacking and hydrogen bonding) lead to a less labile and more compact crystal structure of compound 1, in which one has to overcome a larger energy barrier (apply higher temperature) to initiate any translational movement of the polymeric chains than in compounds 2 or 3. Since no visual change in the physical state of 1 was observed up to the decomposition temperature, one can assume that during the endotherm centered at 245 °C on the DSC curve (Fig. S25 in the ESI†) only a limited reorganization of the polymeric chains is possible – the interactions between organic groups are strong enough, even if weakened by temperature, to prevent full translation of the adjacent chains of 1. It should be noted that this endotherm is accompanied by an exothermic peak appearing at ca. 201 °C on the cooling curve, which is a strong indication of the reversibility of structural reorganization within 1. We were unable to precisely determine whether the original structure of 1 was restored, or some new crystal phase formed during cooling. However, the former explanation seems to be more likely because during a subsequent heating run, one can observe another endotherm which closely resembles (in terms of the enthalpy value and position) the one observed in the 1st heating DSC curve. The same behavior was previously reported in the case of ZnDPhP, for which a complete restoration of the original structure was proved by means of PXRD analysis.9
In contrast, much weaker interactions must exist between chains of compound 3 since they require less thermal energy to initiate translation – one can assume that the energy of these interactions must be on a comparable level with those in ZnDPhP since transformation occurs at almost the same temperature (ca. 150 °C in 3versus ca. 160 °C in ZnDPhP). However, a detailed analysis by means of DSC (Fig. S26 in the ESI†) and PXRD (Fig. S15 in the ESI†) suggests that the presence of the sterically bulky ethoxycarbonyl groups attached to the phenyl ring makes it more difficult for polymeric chains of 3 to rebuild their interactions, thus it becomes an isotropic liquid once the translation movement is possible. Such a hypothesis is also supported by the fact that the melted sample of 3 undergoes complete amorphization, even when cooled at a moderate rate (e.g., 10 °C min−1), rather than rearranging itself once again into a crystalline phase – in the DSC curve of the 2nd heating run one can only observe glass transition temperature (ca. 8 °C) and no other thermal effects. However, it should be noted that one cannot exclude the possibility of slow recrystallization of 3 from its glass state in a time span larger than that of a standard DSC measurement.
The energy of interchain interactions in 2 is on a similar level as in 3 or ZnDPhP, because, when heated, its crystalline sample gives rise to an endothermic signal positioned on the DSC curve (Fig. S27 in the ESI†) at almost the same temperature (ca. 150 °C) as in the latter two. However, the behavior of 2 after that transition distinguishes itself from 3 since the former remains solid (as indicated by visual observation), and on cooling undergoes only partial amorphization. Interestingly, a sample of compound 2 previously subjected to thermal treatment (e.g., heating to 200 °C and then cooling to room temperature), during the subsequent DSC heating run exhibits effects usually detected in any semicrystalline organic polymer, e.g. an inflection of the heating curve (at ca. 3 °C) ascribed to glass transition, an exotherm located at ca. 73 °C (the equivalent of cold crystallization), and a large endothermic peak with maximum few tens of degrees Celsius above the latter (the equivalent of melting, however, in the case of 2 no such transformation was visually observed). As can be seen in Fig. S27 (ESI†), both the position and thermal effect of this endothermic signal change after the first DSC heating run of 2: the maximum temperature and enthalpy decrease from 150.4 °C to 127.5 °C and 75.23 kJ mol−1 to 55.82 kJ mol−1, respectively. Moreover, this solid–solid high-temperature transformation is irreversible, since it is detected solely on the 1st heating curve, with no corresponding exotherm appearing during cooling. It is worth noting that the 2nd and 3rd heating runs do not alter the shape of the respective DSC curves significantly. A plausible explanation of the aforementioned phenomena might be the existence of two different modes of arrangement of polymeric chains of 2 in the solid state – a fact similar to the one previously reported for ZnDPhP.9 It seems that the conditions prevailing during the synthesis (e.g., the reaction carried out in a polar solvent and proceeding via the self-assembly of small building blocks: Zn2+ cations and diarylphosphate anions) favor the formation of a metastable structure (the one identified in the studied single crystal of 2), while the generation of the other crystal phase is hindered due to some kinetic factors. However, after crossing a certain energy barrier above which translational movement of the polymeric chains becomes possible, the pristine, metastable phase is destroyed (thus amorphization of the sample), while some part of it reorganizes into a more thermodynamically stable structure of 2. The formation of this second crystalline phase looks to be much more preferred if there are only polymeric chains of 2 irregularly dispersed in the system, as it is the sole product of the exothermic process occurring upon heating the previously amorphized sample. At this point it should be noted that the concept of a dual thermal transformation of the as-synthesized crystal phase of 2 could explain the observed discrepancies between the values of enthalpy of the endothermic and exothermic transitions proceeding on the DSC 2nd heating run: if the domains responsible for the 127.5 °C endotherm were formed solely during the preceding exothermic process both enthalpies should be equal, whereas in fact the one characterizing an endotherm is higher by ca. 18 kJ mol−1 – this suggests that a part of them had to be present within the sample before the exothermic process.
The results of the PXRD analysis of 2 before and after thermal treatment (Fig. S28 in ESI†) generally support the conclusions drawn from DSC tests. As can be seen, the diffraction signals ascribed to the crystalline domains of the pristine phase of 2 are becoming wider or disappear (a clear sign of amorphization) after conditioning the sample at 160 °C followed by its rapid cooling to room temperature. On the other hand, one can also observe the emergence of some new reflections suggesting new structural order within the crystalline fraction of the sample. It is worth noting that these new reflections are situated very close to the positions of those related to the pristine phase of 2 which suggests that there is a great similarity in the structure of both phases. Keeping such a sample for a long time (up to 4 days) at room temperature does not significantly alter its internal structure and the shape of PXRD patterns (see Fig. S29 in the ESI†), probably due to the proximity to the glass transition point and the resulting low mobility of polymeric chains. Only conditioning above the temperature of the DSC exotherm could increase the crystallinity of the sample (see Fig. S28 in the ESI†).
Electrorheological properties
The results of our previous studies showed that the presence of the electron-rich phenyl rings within the polymeric chains of Zn[O2P(OR)2]2 can endow their particles with interesting properties, e.g., the ability of spatial organization under an external electric field (EEF).9 We suspected that the attachment of substituents exhibiting different inductive and/or mesomeric effects should influence this behavior by changing electron cloud distribution within the phenyl group. This prompted us to carry out some preliminary studies on the ER properties of the 20 wt% dispersions of compounds 1–3 in silicone oil.As can be seen from data presented in Fig. 4 (flow and viscosity curves), particles of compounds 1–3 are susceptible to reorientation in EEF, however, they differ in terms of the direction in which the changes in viscosity or shear stress take place. In the case of 1, which contains strongly electron-withdrawing NO2 groups, the application of EEF gives rise to a positive ER effect, since the measured values of both shear stress and viscosity are increased: Table 4 shows that at shear rates above 100 s−1 the activation of EEF results in an averaged relative change in the viscosity of this dispersion (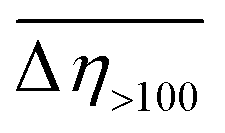 ) of ca. 69%. Such behavior clearly distinguishes compound 1 from ZnDPhP9 as well as the other two investigated compounds that contain OMe or CO2Et functionalities at the phenyl ring, all of which decrease their viscosity under the influence of EEF (a negative ER effect). Table 4 suggests that the dispersion of 2 is more susceptible to EEF than the one based on 3, since in their cases the calculated values of
) of ca. 69%. Such behavior clearly distinguishes compound 1 from ZnDPhP9 as well as the other two investigated compounds that contain OMe or CO2Et functionalities at the phenyl ring, all of which decrease their viscosity under the influence of EEF (a negative ER effect). Table 4 suggests that the dispersion of 2 is more susceptible to EEF than the one based on 3, since in their cases the calculated values of 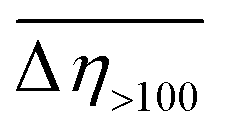 are equal to −23% and −8%, respectively.
are equal to −23% and −8%, respectively.
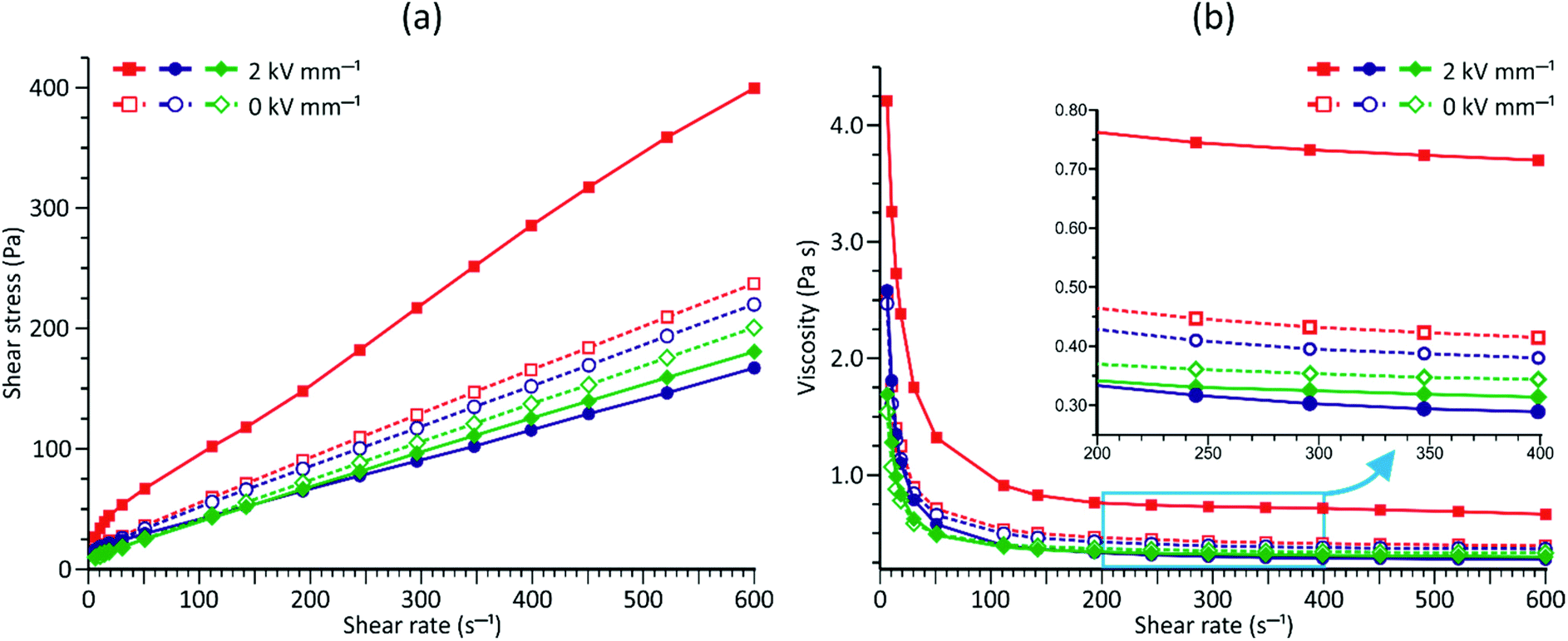 | ||
| Fig. 4 Electrorheology. ER flow (a) and viscosity (b) curves measured at E = 0 kV mm−1 (blank symbols) or E = 2 kV mm−1 (filled symbols) for PDMS100 dispersions containing 20 wt% of: 1 (red lines and squares), 2 (blue lines and circles), and 3 (green lines and diamond symbols). ER measurements conducted at 20 °C. | ||
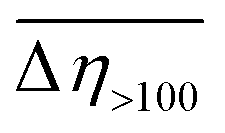 ) in the PDMS100-based dispersions containing 20 wt% of 1–3
) in the PDMS100-based dispersions containing 20 wt% of 1–3
| Dispersion of compound | |||
|---|---|---|---|
| 1 | 2 | 3 | |
a
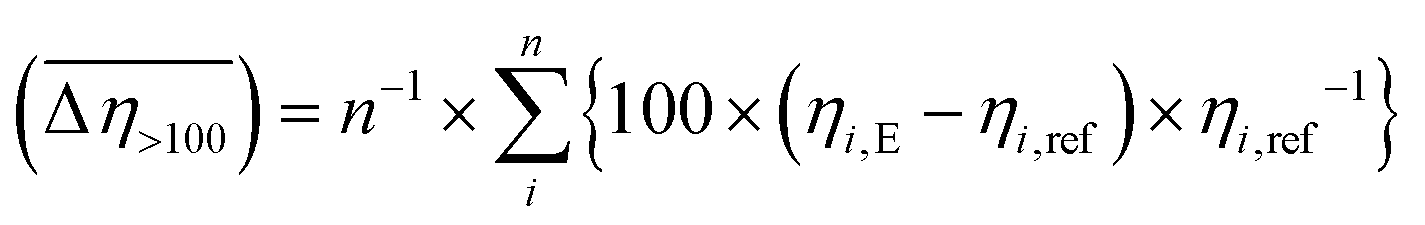 where: ηi,E is the EEF-induced viscosity measured at a given shear rate i (i > 100 s−1) and E = 2 kV mm−1; ηi,ref is the reference viscosity determined at the same shear rate i but without EEF (E = 0 kV mm−1); n is the total number of shear rate values tested at a given E. where: ηi,E is the EEF-induced viscosity measured at a given shear rate i (i > 100 s−1) and E = 2 kV mm−1; ηi,ref is the reference viscosity determined at the same shear rate i but without EEF (E = 0 kV mm−1); n is the total number of shear rate values tested at a given E.
|
|||
 (%)
(%) |
69(3) | −23(1) | −8(1) |
Conclusions
The crystal structures of three new zinc bis(diarylphosphate)s have been determined. They consist of polymeric chains of tetrahedrally coordinated zinc cations bridged by O2P(OC6H4R)2− anions. Zinc bis(diarylphosphate) containing a p-methoxyphenyl group stands out among the studied compounds as it exhibits the 3+1 bridging mode of phosphate anions and within its structure each polymeric chain is surrounded by 4 closest neighbours. Conversely, the nitro- and ethoxycarbonyl-substituted compounds exhibit the 2+2 bridging mode of anions and distorted hexagonal packing of 6 neighbouring chains. The presence of substituents in the para positions of the phenyl ring affects the spatial organization of organic groups around phosphorus centers in compounds 1–3, while it has only a limited effect on the geometry and structural strains within the PO4 and ZnO4 tetrahedra. DFT computations of the individual chains of compounds 1–3 revealed that the 2+2 bridged chains exhibit lower energies than the 3+1 bridged ones. Since the latter mode is adopted in the case of compound 2 having small alkoxy functionalities rather than the one with more bulky ester groups, it seems that steric crowding introduced by the organic groups attached to the phenyl ring has no effect on the way in which the ZnOPO polymer backbone in zinc bis(diarylphosphate)s is formed. This finding contrasts with the results of previous studies on disubstituted zinc phosphates and phosphinates (see Table 1); however, this difference may result simply from a larger distance between the center of steric hindrance and the Zn–O–P–O–Zn bridges. We think that the presence of the 3+1 system in compound 2 may be ascribed to the strong electron-donor effect of the OCH3 group, but this hypothesis needs further verification.Additional organic substituents attached to aromatic rings significantly affect the interactions between neighbouring chains and the way they self-organize into crystalline domains. Particularly strong interactions existing between the C6H4NO2 groups of 1 (e.g., π-stacking and hydrogen bonding) are responsible for the formation of a crystalline phase more resistant to thermal transformations (structural reorganisation on heating) than the other two investigated compounds, or ZnDPhP. Nevertheless, the results of some current work carried out in our research group indicate that those processes could be responsible for the catalytic activity of 1–3 towards epoxide polymerization. However, this subject will be discussed in a future paper.
Compounds 1–3 are thermally stable up to 260 °C and upon pyrolysis decompose into a mixture of zinc condensed phosphates and large amounts of carbonaceous deposits; our previous studies on the ZnDPhP pyrolyzates9 suggest that they could be utilized as antistatic additives in the formulation of electron-conductive polymer composites. It is worth noting that high thermal stability in combination with the presence of functional groups having affinity for strong interactions with polymer chains could also lead to the future application of zinc bis(diarylphosphate)s as nucleating agents in polymer composites; this hypothesis needs experimental verification since the extent of their influence will strongly depend on the type of polymer matrix.
The results of some preliminary electrorheological measurements indicate that, when dispersed in silicone oil, the obtained zinc bis(diarylphosphate)s undergo polarization in an external electric field and, depending on the type of additional functional group within the phenyl ring, show a positive or negative electrorheological effect; this phenomenon makes them promising candidates for application in electrorheological devices, e.g. dampers, valves, engine mounts, hydraulic actuators, squeeze mounts, or haptic medical devices.
Author contributions
Conceptualization, P. A. G., M. D., J. Z. and Z. F.; formal analysis, P. A. G. and M. D.; funding acquisition, Z. F., M. D. and P. A. G.; investigation, M. D., K. Ż., B. M., M. Z., A. O. and P. A. G.; methodology, P. A. G. and M. D.; project administration, M. D. and P. A. G.; resources, M. D. and P. A. G.; supervision, Z. F. and J. Z.; validation, M. D. and P. A. G.; visualization, M. D. and P. A. G.; writing – original draft, P. A. G., M. D. and Z. F.; writing – review & editing, P. A. G. and M. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Centre, Poland within the OPUS-11 research scheme, project number 2016/21/B/ST5/00126. DFT computations were carried out using resources provided by Wrocław Center for Networking and Supercomputing (https://www.wcss.pl), grant no. 260. The authors would like to thank Dr Eng. Paweł Falkowski, Faculty of Chemistry, Warsaw University of Technology, for conducting pyrolytic tests.Notes and references
- B. P. Block, H. D. Gillman and P. Nannelli, The Synthesis and Characterization of the Poly(Metal Phosphinates). Technical report for The Office of Naval Research, 1977. https://apps.dtic.mil/sti/pdfs/ADA041511.pdf, (accessed January 2022) Search PubMed.
- A. Vioux, J. Le Bideau, P. H. Mutin and D. Leclercq, Top. Curr. Chem., 2004, 232, 145–174 CrossRef CAS , and the references therein.
- M. Taddei and F. Costantino, Metal Phosphonates and Phosphinates, MDPI, Basel, 2020, and the references therein Search PubMed.
- R. Murugavel, A. Choudhury, M. G. Walawalkar, R. Pothiraja and C. N. R. Rao, Chem. Rev., 2008, 108, 3549–3655 CrossRef CAS PubMed , and the references therein.
- W. T. A. Harrison, T. M. Nenoff, T. E. Gier and G. D. Stucky, Inorg. Chem., 1992, 31, 5395–5399 CrossRef CAS.
- F. Giordano, L. Randaccio and A. Ripamonti, Chem. Commun., 1967, 19–20 RSC.
- C. G. Lugmair, T. D. Tilley and A. L. Rheingold, Chem. Mater., 1997, 9, 339–348 CrossRef CAS.
- W. T. A. Harrison, T. M. Nenoff, T. E. Gier and G. D. Stucky, J. Mater. Chem., 1994, 4, 1111–1115 RSC.
- M. Dębowski, Z. Florjańczyk, A. Ostrowski, P. A. Guńka, J. Zachara, A. Krztoń-Maziopa, J. Chazarkiewicz, A. Iuliano and A. Plichta, RSC Adv., 2021, 11, 7873–7885 RSC.
- V. Giancotti, F. Giordano, L. Randaccio and A. Ripamonti, J. Chem. Soc. A, 1968, 757–763 RSC.
- F. Giordano, L. Randaccio and A. Ripamonti, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1057–1065 CrossRef CAS.
- R. Cini, P. Orioli, M. Sabat and H. D. Gillman, Inorg. Chim. Acta, 1982, 59, 225–230 CrossRef CAS.
- M. Shieh, K. J. Martin, P. J. Squattrito and A. Clearfield, Inorg. Chem., 1990, 29, 958–963 CrossRef CAS.
- V. Giancotti, F. Giordano and A. Ripamonti, Makromol. Chem., 1972, 154, 271–277 CrossRef CAS.
- M. Dębowski, K. Łokaj, A. Ostrowski, J. Zachara, P. Wiecińska, P. Falkowski, A. Krztoń-Maziopa and Z. Florjańczyk, Dalton Trans., 2018, 47, 16480–16491 RSC.
- CrysAlisPro Software system ver. 171.40.71a, Rigaku OD, Oxford, UK, 2019 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, B. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco and M. Llunell, CRYSTAL09, University of Torino, Torino, 2009 Search PubMed.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571–573 CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. E. Jaffe and A. C. Hess, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 7903–7909 CrossRef CAS PubMed.
- C. M. Zicovich-Wilson, A. Bert, C. Roetti, R. Dovesi and V. R. Saunders, J. Chem. Phys., 2001, 116, 1120–1127 CrossRef.
- C. Gatti, V. R. Saunders and C. Roetti, J. Chem. Phys., 1994, 101, 10686–10696 CrossRef CAS.
- M. Catti, A. Pavese, R. Dovesi and V. R. Saunders, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 9189–9198 CrossRef CAS PubMed.
- C. G. Broyden, Math. Comput., 1965, 19, 577–593 CrossRef.
- D. D. Johnson, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 12807–12813 CrossRef PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- J. Zachara, Inorg. Chem., 2007, 46, 9760–9767 CrossRef CAS PubMed.
- The Cambridge Structural Database (CSD), Cambridge Crystallographic Data Center. Record no 1472713.
- IUPAC Compendium of Chemical Terminology (the “Gold Book”) https://goldbook.iupac.org/terms/view/T06406, (accessed January 2022).
- M. Dębowski, K. Łokaj, A. Wolak, K. Żurawski, A. Plichta, J. Zachara, A. Ostrowski and Z. Florjańczyk, Dalton Trans., 2016, 45, 8008–8020 RSC.
- NIST Mass Spectrometry Data Center and W. E. Wallace, director, Mass Spectra, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, USA, can be found under, https://webbook.nist.gov/chemistry/ (accessed, March 2022), NIST MS number: 7 (water), 19 (carbon monoxide), 31 (nitric oxide, NO), 69 (carbon dioxide), 70 (nitrous oxide, N2O) Search PubMed.
- O. P. Korobeinichev, S. B. Ilyin, V. M. Shvartsberg and A. A. Chernov, Combust. Flame, 1999, 118, 718–726 CrossRef CAS.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liu and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- H. J. Kim, S.-M. Lee, Y.-S. Oh, Y.-H. Yang, Y. S. Lim, D. H. Yoon, C. Lee, J.-Y. Kim and R. S. Ruoff, Sci. Rep., 2014, 4, 5176, DOI:10.1038/srep05176.
Footnote |
| † Electronic supplementary information (ESI) available: Details of triaryl phosphate synthesis combined with their FTIR, 1H and 31P NMR spectral analyses; details of the crystal structure analysis of compound 1; selected torsion angles and calculations of bond-valence vectors in compounds 1–3; tabulated and graphical TGA and DTA data obtained in argon for compounds 1–3; FTIR, 1H, 13C and 31P NMR spectra as well as PXRD patterns and DSC traces of compounds 1–3; Raman spectra and PXRD patterns of the pyrolyzates of compounds 1–3; FTIR spectra and PXRD patterns of compounds 2 and 3 after conditioning at 160 °C. CCDC 2068271–2068273. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00492e |
| This journal is © The Royal Society of Chemistry 2022 |