
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and computational tuning of metalla-N-heterocyclic carbenes at palladium(II) and platinum(II) centers†
Maria V.
Kashina
 a,
Konstantin V.
Luzyanin
*b,
Eugene A.
Katlenok
a,
Alexander S.
Novikov
a and
Mikhail A.
Kinzhalov
*a
a,
Konstantin V.
Luzyanin
*b,
Eugene A.
Katlenok
a,
Alexander S.
Novikov
a and
Mikhail A.
Kinzhalov
*a
aSt Petersburg University, 7/9 Universitetskaya Nab., Saint Petersburg 199034, Russian Federation. E-mail: m.kinzhalov@spbu.ru
bDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: konstantin.luzyanin@liverpool.ac.uk
First published on 14th March 2022
Abstract
Palladium(II) and platinum(II) complexes featuring metalla-N-heterocyclic carbenes (7–12) were synthesised via metal-mediated coupling between equimolar cis-[MCl2(CNR)2] (R = 2,6-Me2C6H3 (Xyl), 2,4,6-Me3C6H3 (Mes)) and 2-aminopyridine or 2-aminopyrazine. Thiocyanate complexes 13–18 with two thiocyanate ligands were obtained through the ligand exchange in the parent compounds 7–12 with NH4CNS in acetone/CH2Cl2. Complexes 7–18 were isolated and characterised by HRESI+-MS, IR, 1H and 13C{1H} NMR spectroscopy and single-crystal X-ray diffraction (in the case of 11, 16, and 18). The UV-vis properties of 7–18 and the electrochemical properties of 7–12 were also evaluated. To study the electronic structure and bonding nature in the new compounds, the quantum theory of atoms in molecules (QTAIM) and Mayer bond order analysis together with the extended transition state with the natural orbitals for chemical valence (ETS-NOCV) method, were used. X-ray diffraction studies and theoretical considerations indicate that the thiocyanate derivatives 16 and 18 form supramolecular dimers by two symmetrical pairs M1⋯C5 and S1⋯C2 with short intermolecular contacts between an electron-rich MII-center and thiocyanate ligand on the one side and the electron-poor π-system of an azaheterocyclic ring on the other side. Representative carbenes 8, 11 and 12 were evaluated as photocatalysts for the hydrosilylation of diphenylacetylene with triethylsilane giving 1,2-(diphenylvinyl)triethylsilane in 98% yield under visible light irradiation (blue light, 445 nm).
Introduction
Transition metal complexes with heteroatom-stabilised carbenes, i.e. N-heterocyclic carbenes (NHCs)1 and acyclic diaminocarbenes (ADCs),2 underpin a broad range of applications in the catalysis of organic processes;1a,2a,3 they are studied as emissive materials in optoelectronics4 and potential anticancer drugs.2a,5 The ligand properties of diaminocarbenes are manifested through the formation of the M–Ccarbene σ-bond by transferring a pair of nonbonding electrons from the nucleophilic σ-orbital of the carbene carbon atom to the metal atom M. At the same time, the formation of a π-bond is also possible as a result of the interaction of a vacant electron-deficient pπ-orbital of a carbene carbon atom with a metal, whose orbital symmetry is suitable. The diaminocarbene ligand is usually an effective σ-donor and a weak π-acceptor, which is reflected in the catalytic and photophysical properties of the respective metal complexes. Tuning of the electron-donor capacity of diaminocarbenes, which is crucial for the success of their target applications, has been mainly accomplished by varying the substituents on the nitrogen atoms or the modification of the heterocyclic skeleton (Fig. 1A). It was shown that NHC complexes containing a metal atom within the NHC skeleton, named metalla-N-heterocyclic carbenes (MNHCs),6 exhibit a stronger electron-donor character by comparison with classical imidazole-2-ylidenes.7 In pursuit of our ongoing project on the chemistry of diaminocarbenes,3d,e,8 we have reported that platinum(II)–MNHC species (Fig. 1B) work as self-photosensitising transition-metal photocatalysts for the hydrosilylation of alkynes under visible light.9 In this catalytic reaction the platinum(II)–MNHC catalyst has a dual function: it serves as a light-absorbing species and a transition metal catalyst enabling the catalytic process.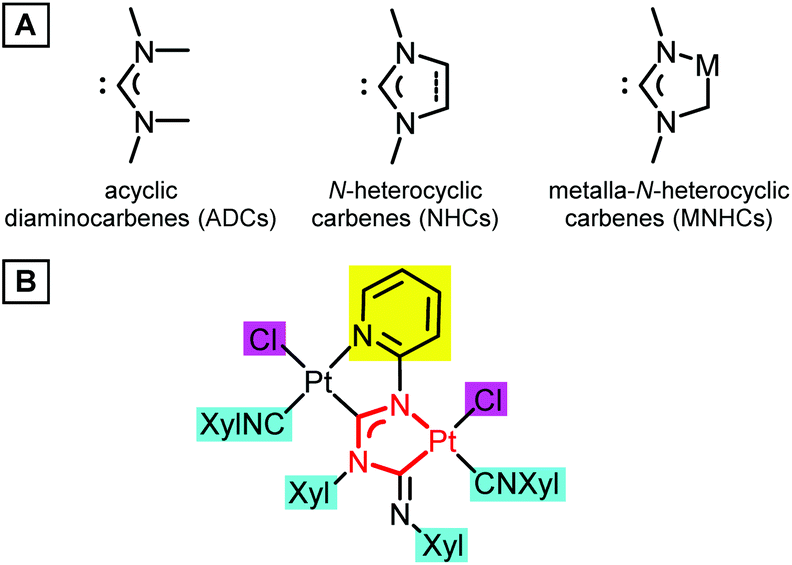 | ||
| Fig. 1 (A) Examples of cyclic, N-heterocyclic and metalla-N-heterocyclic diaminocarbenes. (B) The platinum(II)–MNHC catalyst exhibiting photocatalytic properties under visible light irradiation. | ||
Additional strategies for fine-tuning the properties of complexes of this type are linked to (i) the modification of the structure of the azaheterocyclic ring (highlighted with a yellow rectangle), (ii) substitution in the aryl rings (highlighted with a cyan rectangle), and (iii) the replacement of the anionic ligands at both metal centers (highlighted with a magenta rectangle). An interplay of these will define the application of the complex thus designed towards a specific goal.
The scenario of this work was as follows. We started by the preparation of a series of palladium(II)– and platinum(II)–MNHC complexes via the metal-mediated coupling of isocyanides with various 2-aminoazaheterocycles. The displacement of chlorides with thiocyanates led to the generation of previously unknown diaminocarbene-thiocyanate derivatives. The electronic structure and bonding nature in all new compounds were assessed by using the quantum theory of atoms in molecules (QTAIM), the natural bond orbital (NBO) analysis, and Mayer bond order analysis together with the extended transition state with the natural orbitals for chemical valence (ETS-NOCV) method. Analysis of the frontier molecular orbital character, energy gaps, and UV/vis spectra by density functional theory (DFT) and time-dependent DFT (TD-DFT) approaches provided insights into the excited states involved in the light absorption and electrochemical processes, in turn, related to the potential photocatalytic properties. Finally, we assessed the catalytic properties of the thus prepared carbene complexes in the photocatalytic hydrosilylation of alkynes under visible light irradiation.
Results and discussion
Preparation and characterisation of metalla-N-heterocyclic diaminocarbenes
For this study, a series of PdII and PtII MNHC species 7–12 were prepared via the nucleophilic addition of 2-aminoazaheterocycles 5 and 6 to metal-bound isocyanides in cis-[MCl2(CNR)2] (1–4, M = Pd, Pt). The reaction of 1–4 and azaheterocycles 5, 6 proceeds in CH2Cl2 at RT for ca. 1 d and the subsequent workup provided the binuclear diaminocarbene species 7–12 in good (84–95%) isolated yields (Scheme 1, Route A). This process occurs via the intermediate generation of the corresponding mononuclear M-ADC species and its nucleophilic addition to the unreacted isocyanide material facilitated by the deprotonation of the M-ADC intermediate by an excess of the aminoazaheterocycle used.9,10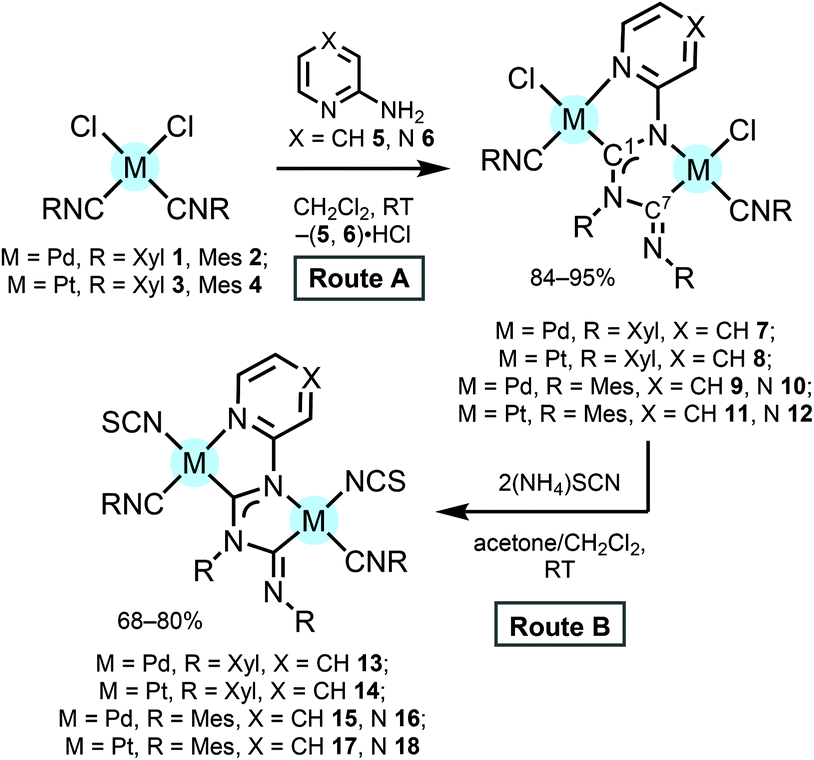 | ||
| Scheme 1 Preparation of 7–18. | ||
MNHC complexes 7–12 are yellow solids stable in the 20–80 °C temperature range. New complexes 9–12 were characterised by elemental analyses (C, H, and N), accurate-mass ESI+-MS, IR, 1D (1H, 13C{1H}, and 195Pt{1H}) NMR and 2D (1H,13C-HSQC and 1H,13C-HMBC) NMR spectroscopy (see the Experimental section for more details), while the authenticity of the known species 7 and 8 was confirmed by 1H NMR spectroscopy.9,10 The intense absorption bands at 1619–1667 cm−1 in the FTIR spectra of 7–12 were assigned to ν(Ccarbene–N) of the NCN moiety. For 7–12, the important 13C resonances of the C atoms from two carbene ligands are different and are detected at δC 168 and 182 ppm. This evidences that two NCN groups in 7–12 are substantially structurally different; one of them (having the downfield resonance at δC 168 ppm and assigned to C7 carbon) is of the amidine type, whereas another one (having the downfield resonance at δC 182 ppm and assigned to C1 carbon) is of the diaminocarbene type. These δC values for diaminocarbene carbon C1 are downfield relative to those (δC 157–159) reported for the series of palladium complexes featuring chelated NHCs, such as bis(imidazolylidene) palladium dichloride complexes,80 but are some upfield of the δC 197–201 range observed for the series of palladium complexes featuring chelating acyclic aminocarbene ligands, i.e. [PdCl{C(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(C6R2R3R4R5CON))N(H)R1}(CNR1)].11
C(C6R2R3R4R5CON))N(H)R1}(CNR1)].11
The thiocyanate anion NCS− enables several pathways to alter the properties of the metal centres due to its ambidentate nature, allowing the coordination via either S- or N-ends. To the best of our knowledge, only two examples of thiocyanate diaminocarbene complexes, viz. nickel(II)–NHC with N-coordinated thiocyanate12 and gold(I)–NHC with S-coordinated thiocyanate,13 have been reported. Thiocyanate species demonstrate better donor abilities and increased dipole moment of complex molecules relative to halide analogues which may lead to better catalytic activity of thiocyanate species, e.g. in the Kumada–Corriu coupling reaction.12 In addition, the better donor character of thiocyanate is related to the stabilisation of the HOMOs of the complexes and their red-shifted absorption in the UV-vis spectrum.14
The common method for the preparation of thiocyanate complexes from chloride substrates is a metathetic reaction with NaNCS or KSCN in an aqueous or alcohol solution, occasionally aided by AgNO3 to facilitate chloride subtraction.15 Metathetic reactions of 7–12 with a three-fold excess of NH4CNS in acetone/CH2Cl2 at RT for 24 h afforded yellow bis(N-coordinated thiocyanate) complexes 13–18 in good yields (Scheme 1, Route B). Owing to the poor solubility of 13–18 in the most common solvents, all 13–18 were characterized in solution by means of HR-MS (ESI+) and in the solid state by FTIR and solid-state 13C CP/MAS NMR. The structures of 16 and 18 were also elucidated by single-crystal XRD.
Complexes 13–18 gave satisfactory CHN data, which are consistent with the proposed formulae confirming the replacement of both chloride ligands with thiocyanate. The HRESI+-MS spectra of 13–18 exhibit sets of peaks with the characteristic isotopic distribution due to [M + H]+, [M + Na]+ and/or [M − NCS]+ ions. IR spectroscopy was used to identify the coordination modes of thiocyanate in metal thiocyanate complexes.16 As a general rule, the value of ν(CN) stretching for the thiocyanate of N-bonded complexes lies near 2050 cm−1 (strong band) and that of S-bonded complexes is close to 2100 cm−1 (weaker band).16c The IR spectra of 13–18 measured in KBr revealed two intense absorption bands in the range 2030–2090 cm−1 attributed to the ν(CN) stretching of the N-bonded thiocyanate. Additionally, the FTIR spectra of 13–18 displayed strong absorption overlapping bands at ca. 2200 cm−1 which are also present in the spectra of parent compounds 7–12 and are attributed to the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) of the isocyanide ligands.
N) of the isocyanide ligands.
UV-Vis absorption properties
All prepared complexes are yellow (10–18) or yellowish (7–9) solids, which makes them capable of absorbing light in the visible range. UV-vis absorption spectra of 7–18 were measured at ambient temperature in CH2Cl2 solution (Fig. 2) and in KBr pellets (Fig. S6 and S7 in the ESI;† UV-vis data for 7–18 are summarised in Table S4 (ESI)).†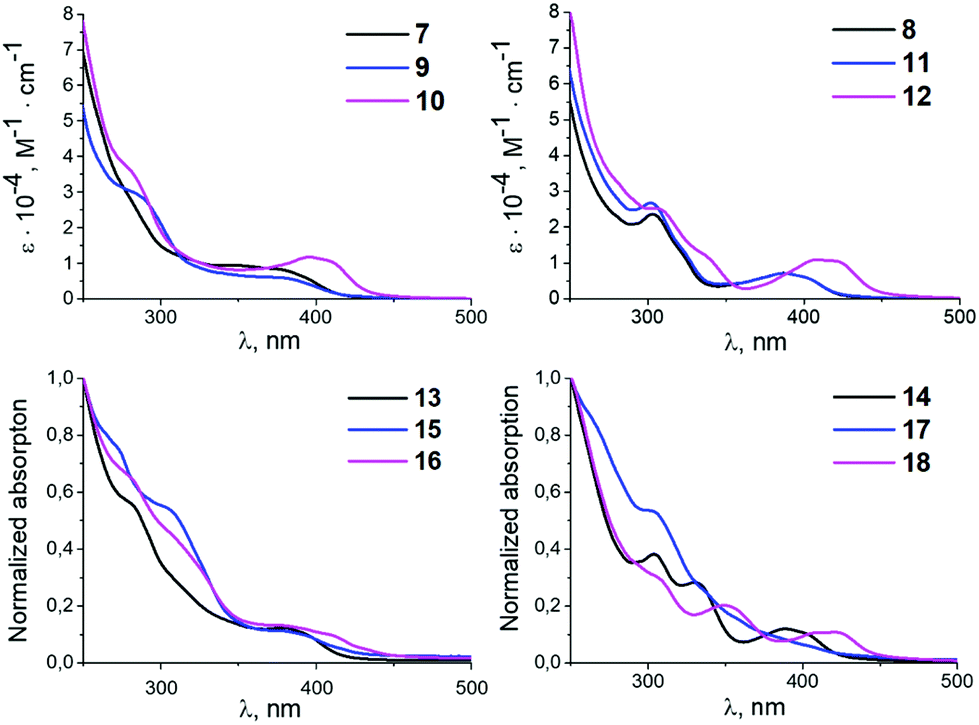 | ||
| Fig. 2 UV-vis absorption spectra for 7–18 in CH2Cl2 at RT (0.03 mM for 7–12 and saturated solutions for 13–18). | ||
Similar to other palladium17 and platinum17a,d,18 cyclometallated complexes, the UV-vis spectra of 7–12 contain strong absorption bands below 340 nm [ε = (1.15–3.59) × 104 M−1 cm−1] assigned to π–π* ligand centred transitions in the xylyl and mesityl moieties (Fig. 2).19 The wide intense absorption bands in the 340–450 nm range with characteristic molar extinction coefficients [ε = (0.60–1.13) × 104 M−1 cm−1] can be assigned to mixed 1L′LCT (ligand-to-ligand charge transfer) and 1ILCT (intraligand charge transfer) transitions based on TD-DFT calculations (see the section TD-DFT calculations). The energy of the 1L′LCT/1ILCT absorption band is affected by the change of the azaheterocyclic fragment.20 Complexes with pyridine rings 7–9 and 11 show blue-shifted 1L′LCT/1ILCT bands relative to their pyrazine counterparts 10 and 12 (e.g. 364–398 nm [ε = (0.60–0.83) × 103 M−1 cm−1] in 7–9 and 11vs. 402–414 nm [ε = (1.06–1.13) × 103 M−1 cm−1] in 10 and 12).
This could be explained by a decrease of the LUMO energy in the case of 10 and 12 with pyrazine species due to the additional intercalation electron negative nitrogen atom in the azaheterocyclic fragment. Variation of the aryl fragment of the CNR or the corresponding diaminocarbene ligands induces no significant changes in the positions of the 1L′LCT/1ILCT bands and only exerts minimal influence on the extinction coefficients (e.g. 364–393 nm [ε = (0.64–0.83) × 103 M−1 cm−1] in 7 and 8vs. 378–414 nm [ε = (0.60–1.13) × 103 M−1 cm−1] in 9–12).
The UV-vis spectra of 13–18 demonstrate similar absorption profiles to the spectra of the 7–12 (Fig. 2). Replacing both chloride ligands with thiocyanate ligands leads to minor changes in the absorption spectra. Complexes 7–18 are not fluorescent and/or phosphorescent at RT, and no emission bands were detected.
Frontier molecular orbital analysis
To gain insight into the electronic structures and optical properties of the obtained complexes time-dependent DFT calculations on 7, 8, 11, and 12 were undertaken. The theoretically predicted structures are in good agreement with experimental X-ray structures (Fig. S8 and Table S5 in the ESI†). In general, the computed bond lengths are close to the experimental ones indicating sufficient accuracy of the selected computational protocol.21 The main difference lies in slight elongation of coordination bonds (by 0.01–0.03 Å) and CN triple bonds (0.03–0.04 Å) in the optimized structures compared to experimental results, due to relaxation of the cyclic metal including systems.Fig. 3 shows the shape and energy of the frontier molecular orbitals for 7, 8, 11, and 12, while the FMO compositions are given in Table S8.† In all cases, the highest occupied molecular orbitals (HOMOs) are delocalized by the π molecular orbitals on the aryl ring connected with the exocyclic nitrogen atom of the diaminocarbene species (contribution of aryls >80%), while the contribution of the metal d orbitals is insignificant (<5%). The energies of the HOMOs of 7, 8, 11, and 12 are similar, while the lowest unoccupied molecular orbitals (LUMOs) are localised over the entire diaminocarbene (22–34%) and azaheterocyclic (38–54%) species. The large contribution (12–16%) from the metal d orbitals in the LUMO may be related to the absence of luminescence for 7–12 at RT due to the population of a metal-centred state.22
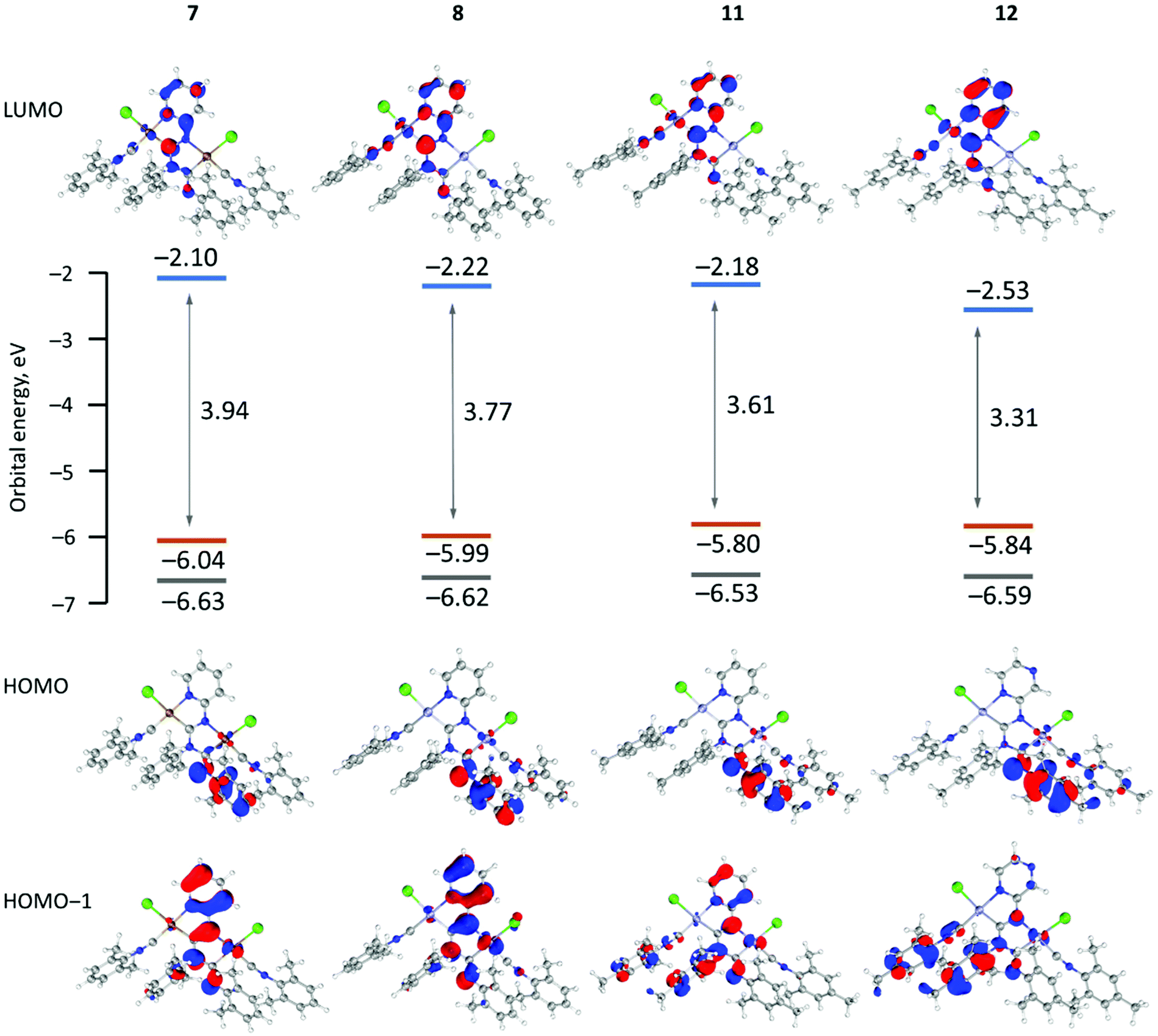 | ||
| Fig. 3 Calculated molecular orbital energy HOMO–LUMO gaps and surface plots of the HOMO−1, HOMO and LUMO for 7, 8, 11 and 12. | ||
The HOMO–LUMO energy difference varies considerably between 3.94 eV (7) and 3.31 eV (12). The small HOMO–LUMO gap of 12 with pyrazine species comes from the low-lying LUMO which is affected by the additional intercalation of the electron negative nitrogen atom in the azaheterocyclic fragment. There is a correlation between the HOMO–LUMO energy difference and the red-shifted absorption bands determined experimentally for 10 and 12 with pyrazine rings relative to their pyridine counterparts 7–9 and 11.
TD-DFT calculations
TD-DFT calculations were carried out to elucidate the nature of the transitions in the absorbance profile. The TD-DFT calculated UV-vis spectra of all complexes are in good agreement with the experimental ones. The calculated absorption spectra for 7, 8, 11, and 12 are reasonably consistent with the experimental spectral profiles (Fig. S9 and S11†). The absorption band in the 320–450 nm range results from the combination of the most intense transfers appearing at ca. 340–360 nm (f ca. 0.123–0.156) and ca. 393–410 nm (f ca. 0.031–0.038) associated correspondingly with the HOMO → LUMO (91–92%) and HOMO−1 → LUMO (91–94%) transitions (Tables S6 and S7 in the ESI†).Electrochemical studies
The electrochemical properties of 7–12 were examined using cyclic voltammetry (CV) in dichloromethane solution (Fig. 4). Palladium complexes 7, 9, and 10 exhibit two irreversible oxidation waves in the ranges of 0.48–0.58 and 0.83–0.85 V, which correspond to the multistep oxidation processes (subsequent oxidation processes are probably assigned to the next step of the oxidation of the mono-oxidized species23). Platinum complexes 8, 11, and 12 are oxidized at more positive potentials than the corresponding palladium counterparts and the first irreversible oxidation peak was observed in the range of 0.72–0.77 V.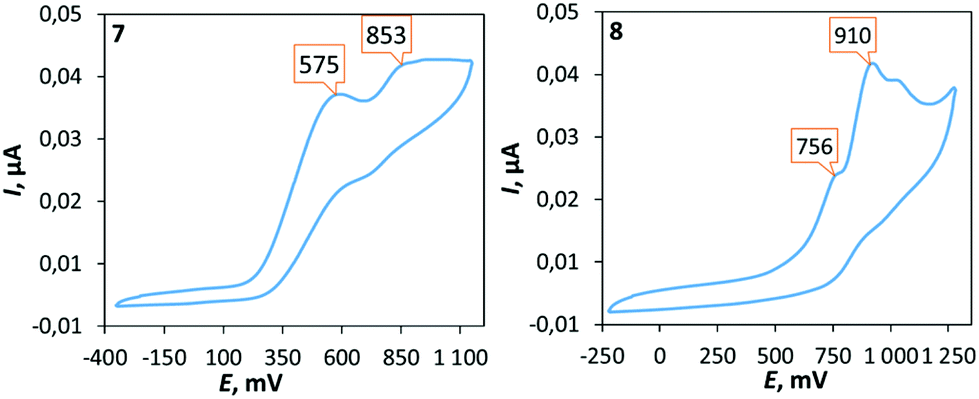 | ||
| Fig. 4 Cyclic voltammograms of 7 and 8 in CH2Cl2 at RT (potentials given in mV vs. Fc+/Fc). Oxidation potentials (mV) of 9: 483, 833; 10: 718, 968; 11: 566, 850; and 12: 740, 1028. | ||
It is worth noting that complexes with mesityl substituent 9–12 are oxidized easier than their corresponding analogues 7 and 8 with xylyl substituent. To interpret the electrochemical behaviour of the complexes, quantum-chemical calculations were performed for 7, 8, 11, and 12 and their mono-oxidized forms at the level of the DFT theory. The plot of the spin density calculated for the oxidized forms of 7, 8, 11, and 12 (Fig. 5 and S12†) demonstrates that the first oxidation process could be associated with the oxidation of the aryl fragment connecting to the exocyclic nitrogen atom.
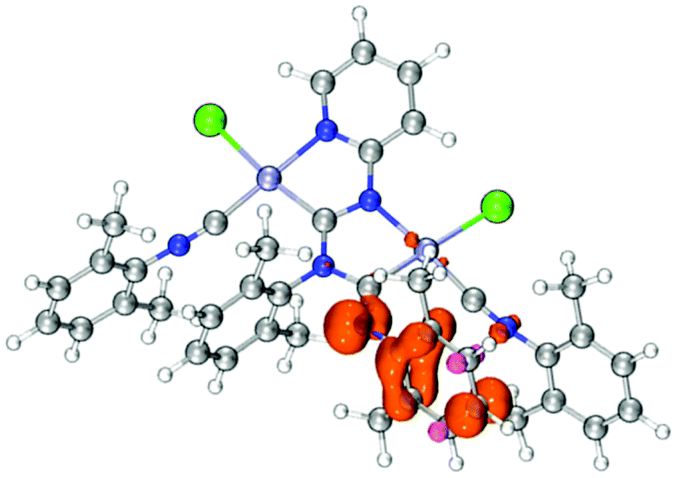 | ||
| Fig. 5 A plot of the spin density calculated for [8]+. | ||
X-Ray diffraction studies of chloride species
Single crystals of 11 were obtained by the slow evaporation of CH2Cl2 solution. Crystals of 11 are yellow and possess a low symmetry P1 space group (Fig. 6). In 11, both metal centres acquire the distorted square-planar coordination environment. The CNR ligands are at the cis-position to the C atoms of {![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (NR)N(R)
(NR)N(R)![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) (C4H3X)} (X = CH, N); the latter forms a dinuclear bicyclic framework.
(C4H3X)} (X = CH, N); the latter forms a dinuclear bicyclic framework.
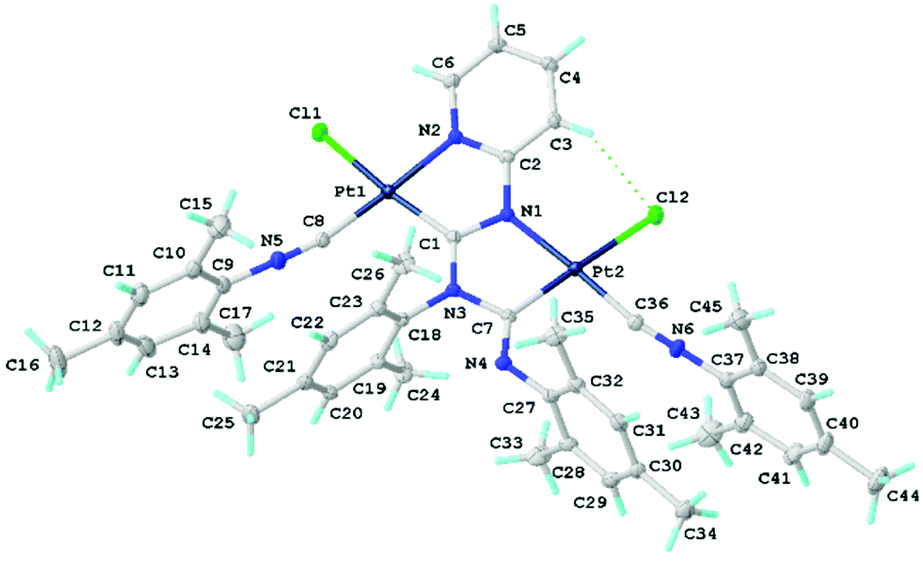 | ||
| Fig. 6 View of 11 with the atomic numbering schemes. Solvent molecule and hydrogen labels are omitted for simplicity. The crystal data, and selected bond lengths and angles are given in the ESI, section S1.† | ||
The bond lengths of the two coordinated CN groups are in the interval of 1.136(8)–1.145(8) Å, typical of the CN triple bonds in isocyanide PdII and PtII complexes.3e,24 The Pt–Ccarbene distances 1.983(6)–1.989(5) Å are slightly smaller than those of typical Pt–C bonds (1.995–2.023 Å)5d,25 reported for the relevant PtII–ADC complexes and are close to the Pt–C distances in Pt–NHC complexes (1.970–1.991 Å).26 On the other hand, the Pt–C bond lengths are sufficiently larger than those in Pt–NHCs, where the NHC is included in the extended rigid π-system (1.906–1.970 Å)27 and in Pt–CAAC complexes (1.942–1.967 Å).28 The bond lengths in the first carbene moiety C1–N1 (1.349(8) Å) and C1–N3 (1.341(7) Å) are similar and are between a single CN bond (e.g., 1.469(10) Å) in amines29 and a double CN bond (e.g., 1.279(8) Å) in imines,29 and the distances indicate a substantial delocalization of the electron density. In the second carbene, the endocyclic C7–N3 bond of the metallacycle is typically single (1.449(8) Å), while the exocyclic C7–N4 bond is typically double (1.264(8) Å) indicating a low degree of delocalisation in this NCN carbene fragment. The structure of this carbene moiety is, therefore, closer to the amino(imino)carbene or diaminocarbene-like structures reported previously.11,30 All other bond lengths in 11 are normal, and their values agree with those for the previously reported 7,10b89 and other related diaminocarbene and isocyanide species.8c,10a,31
DFT analysis of ligand properties in chloride species 7–12
To shed light on the nature of the bonding in these compounds, the quantum theory of atoms in molecules (QTAIM)32 and Mayer bond order analysis,33 as well as the extended transition state and the natural orbitals for chemical valence (ETS-NOCV) method,34 were used.Bader's theory of atoms in molecules provides a unique way to analyse the electron density, ρи.32 The most important property to assign a bond is the presence of a bond critical point (BCP) which is a point of electron and energy densities along the bond path. It is quite interesting to compare the M1–C1 bonds, which involve the metalla-N-heterocyclic carbene C1 atoms, with the almost equivalent M2–C7 bonds, which engage the only formal acyclic diaminocarbene C7 atom. The BCPs for both pairs of M–Ccarbene bonds show a moderate value of electron density (ρb 0.070–0.091e) and a positive value of the Laplacian (Table S9†). Since a covalent bond in transition metal complexes is characterized by a small value of ρb and small and positive values of ∇2ρ(r) due to the diffuse character of the electron distribution,35 the best descriptor to ascertain the type of interaction is the local energy electron density H(r).36 All of these M–Ccarbene have negative values of H(r) at the BCPs. The listed parameters indicate a significant degree of covalency. Another similar criterion was proposed by Espinosa37 on the basis of the ratio |V(r)|/G(r) for closed-shell interactions, |V(r)|/G(r) < 1 and for shared shell interactions, |V(r)|/G(r) > 2. The interaction is taken as an intermediate type if the ratio falls between these two limits.38 The BCPs of M–Ccarbene bonds have |V(r)|/G(r) values of 1.47–1.70 and it is taken as evidence for a partially covalent nature. At the same time, the values for the ρb bond and |V(r)|/G(r) for all M–Ccarbene bonds are higher than those for the M–Cl, M–CCNR and M–N bonds; all this indicates that M–Ccarbene is more covalent. The values for the local topological properties at the BCP of these two types of M–Ccarbene bonds are close enough (Table S9†), and are comparable to those obtained from the theoretical electronic energy density for the M–Ccarbene bonds of the other diaminocarbene complexes.36,39 The calculated Mayer bond orders (MBOs) of the M–Ccarbene bonds are similar for both M–Ccarbene bonds of each complex (0.5491 and 0.5512 for 7 and 0.9907 and 1.0678 for 8) and are larger than the MBOs of the M–CCNR bonds (0.2675 and 0.2753 for 7 and 0.9029 and 0.9058 for 8). The values of the topological parameters at BCPs of the Ccarbene–N bonds are all typical of covalent bonds between nonmetal atoms with some degree of delocalization.40 The MBOs for Ccarbene–N bonds in the N1–C1–N3 moiety are higher than 1 and very similar (1.2570–1.3101), which support the previous proposal that the unsaturation of the C atom of diaminocarbene is alleviated by π-donation of electron density from the filled pπ orbitals of the N atoms to the empty pπ orbital of the carbene carbon.41 The MBOs for Ccarbene–N bonds in the second N3–C7–N4 moiety are different (0.9715–0.9989 for C7–N3 and 1.6346–1.6752 for C7–N4) as evidenced by XRD data.
The coordination bonding in binuclear diaminocarbene complexes was rationalised by the extended transition state (ETS)42 together with the natural orbitals for chemical valence (NOCV)34,43 method. The ETS-NOCV scheme in this manner gives quantitative (ΔEorb) data on the strength of orbital interactions in chemical bonds.44 The ETS-NOCV calculations for 7 and 8 were carried out using PBE0/ZORA-def2-TZVP geometries with the ADF program package.45 The fragmentation pattern is shown in Fig. S13,† where the C1–N1, C7–N3 and M–L bonds were fragmented homolytically in the frozen geometry of the complex molecule for this purpose. The major contribution to the total orbital interaction comes from the three orbital terms ΔEorb(1) − ΔEorb(3) (90%), indicating a strong covalent bonding (Fig. 7). The strongest pairwise orbital interaction (ΔEorb(1), −470 kcal mol−1) comes from the ligand to metal σ-dative interactions, which is provided by 41% of total ΔEOrb. Then, the second term (ΔEorb(2), −362 kcal mol−1) is associated with the coupling of p orbitals in diaminocarbene moieties. The last ΔEorb(3) (−225 kcal mol−1) is due to the π-back-donation from metal centers to carbene ligands and comprises only 20% of the total ΔEOrb energy.
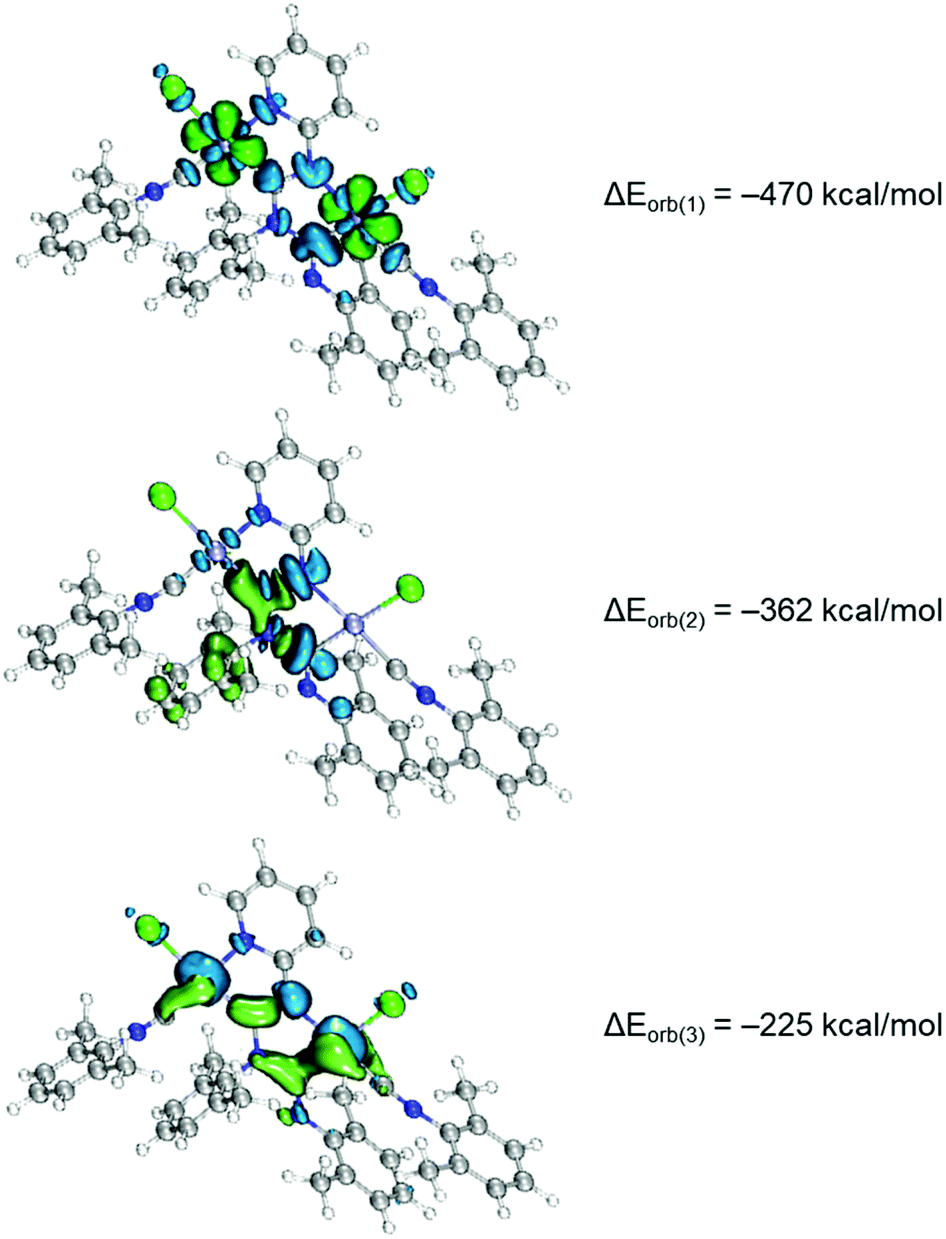 | ||
| Fig. 7 Shape of the deformation densities Δρ(1)–(3) that correspond to ΔEorb(1)–(3). The direction of the charge flow of the deformation densities is blue (loss of electron density) → green (gain of electron density). | ||
X-Ray diffraction studies of thiocyanate derivatives
Single crystals of 16 and 18 were obtained directly from the reaction mixture upon its slow evaporation (Fig. 8, 9, and S1†).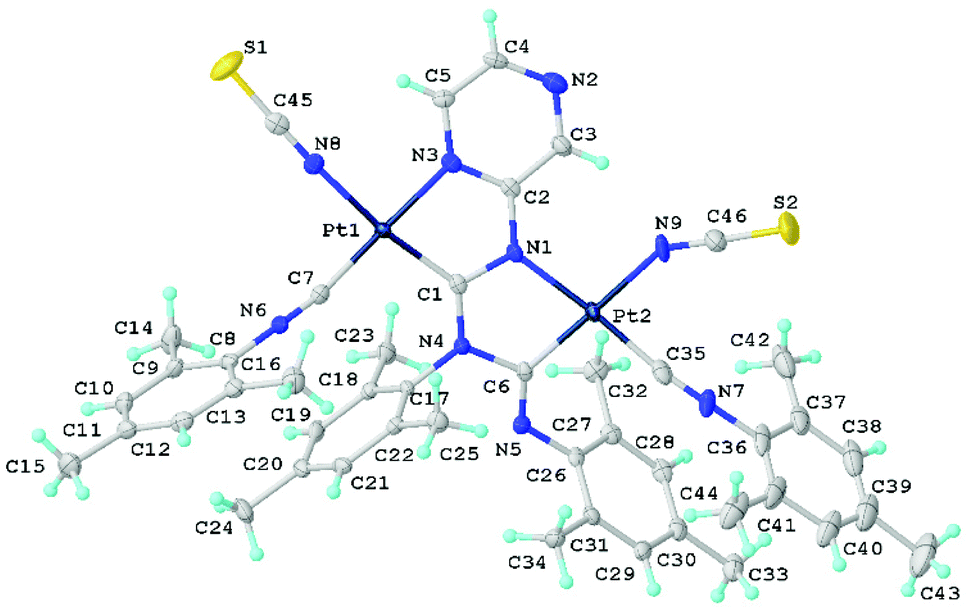 | ||
| Fig. 8 View of 18 with the atomic numbering schemes. Solvent molecule and hydrogen labels are omitted for simplicity. Crystal structure of 16 is given in section S1 in the ESI† alongside selected bond lengths and angles. | ||
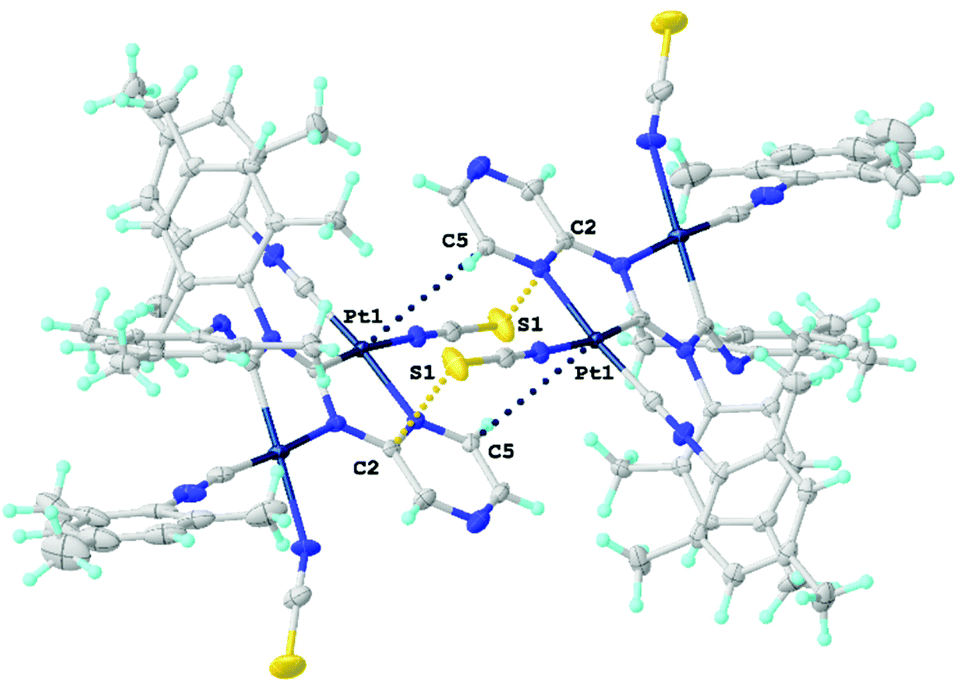 | ||
| Fig. 9 Views of dimers of 18. Dotted lines indicate the Pt1⋯C5 and S1⋯C2 contacts. Thermal ellipsoids are shown with 50% probability. | ||
Compounds 16 and 18 form isomorphs via the Pd/Pt atom exchange in the molecular crystals.5d,24d,e Both thiocyanates in 16 and 18 are bound to the metals by the N atom in agreement with the IR spectroscopy results. The M1–N8 and M2–N9 distances were within the range of typical values for Pd–N16b,46 and Pt–N16a,46 coordination bonds reported for relevant N-ligated thiocyanates. The M1–N8–C45 and M2–N9–C46 angles due to the thiocyanate binding are considerably different: one thiocyanate in 16 and 18 is coordinated to the metal in a somewhat linear fashion (M1–N8–C45 175.7(3)–178.5(3)°), while the other adopts a bent configuration (M2–N9–C46 angle in the range of 147.1(3)–159.1(4)°). Subsequent theoretical studies (section S4.1†) and analysis of the CCDC (section S5†) showed that the differences are associated with the effect of crystal packing.
The polycyclic organometallic skeleton of thiocyanate complexes 16 and 18 is almost identical to that of chloride species (Fig. S2 and S3†). Thiocyanate ligands are explored as potential crystal-driving supramolecular generators in biochemistry47 and materials48 studies. The versatile nature of the interacting centers in NCS is mainly associated with sulfur and the nitrogen atoms allow it to be involved in hydrogen bonding,49 halogen bonding,50 and π-interactions.51 The Hirshfeld surface analysis of the XRD structures of 16 and 18 indicates the domination of the contacts that involve hydrogen atoms, specifically, H–H, C–H, S–H, and N–H (section S6 in the ESI†). The H–H contacts provide the largest contributions to the molecular Hirshfeld surfaces, because the fraction of these atoms is maximal and the Hirshfeld surface analysis does not disclose the attractive or repulsive nature of these contacts. Apart from the contacts involving H atoms, 16 and 18 exhibit two pairs M1⋯C5 and S1⋯C2 short intermolecular contacts forming supramolecular dimers (Fig. 9). The distances between the M1⋯C5 and S1⋯C2 interacting atoms are slightly larger than the sum of Bondi's vdW radii (102–109%),52 but are comparable with the sum of Alvarez's (86–89% for M1⋯C5 and 98–99% for S1⋯C2)53 vdW radii, confirming the presence of noncovalent interactions. To further confirm the existence of noncovalent interactions between the MII-center and C5 atom and between the S-center of thiocyanato and C2 atom, a further computational study was carried out.
QTAIM analysis32 of model supramolecular associates (16)2 and (18)2 shows appropriate bond critical points (3, −1) (BCPs) for the discussed contacts (Fig. 10, 11, S15, S16 and Table S13†). In the case of (18)2, the formally bifurcated μ2-S⋯[PtII,C] contact (Fig. 11) and in the case of (16)2 a S1⋯Pd2 contact (Fig. S16†) are formed. The low magnitudes of electron density (0.005–0.008 a.u.), the positive values of the Laplacian of electron density (0.015–0.021 a.u.), and the positive energy density close to zero (0.001 a.u.) in the BCPs, as well as the balance between the Lagrangian kinetic energy G(r) and potential energy density V(r) at the BCPs (ratio |G(r)|/V(r) > 1), reveal that these contacts are purely noncovalent.37
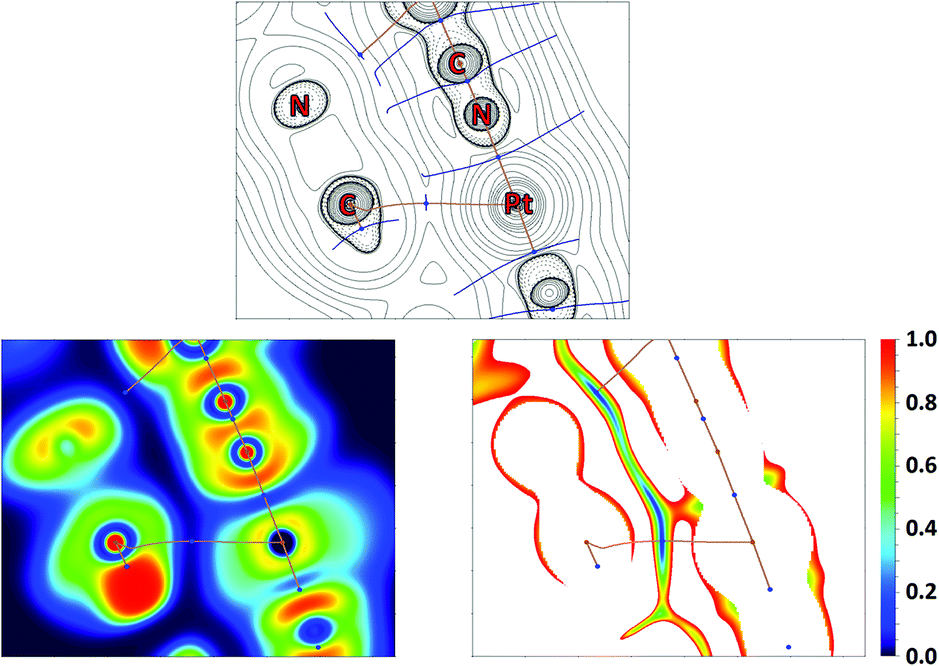 | ||
| Fig. 10 Contour line diagram of the Laplacian of electron density distribution ∇2ρ(r), bond paths, and selected zero-flux surfaces (left panel), visualization of the electron localization function (ELF, center panel) and reduced density gradient (RDG, right panel) analyses of Pt1⋯C5 intermolecular contacts in (18)2. Bond critical points (3, −1) are shown in blue, nuclear critical points (3, −3) in pale brown, and bond paths as pale brown lines, length units are Å, and the color scale for the ELF and RDG maps is presented in a.u. | ||
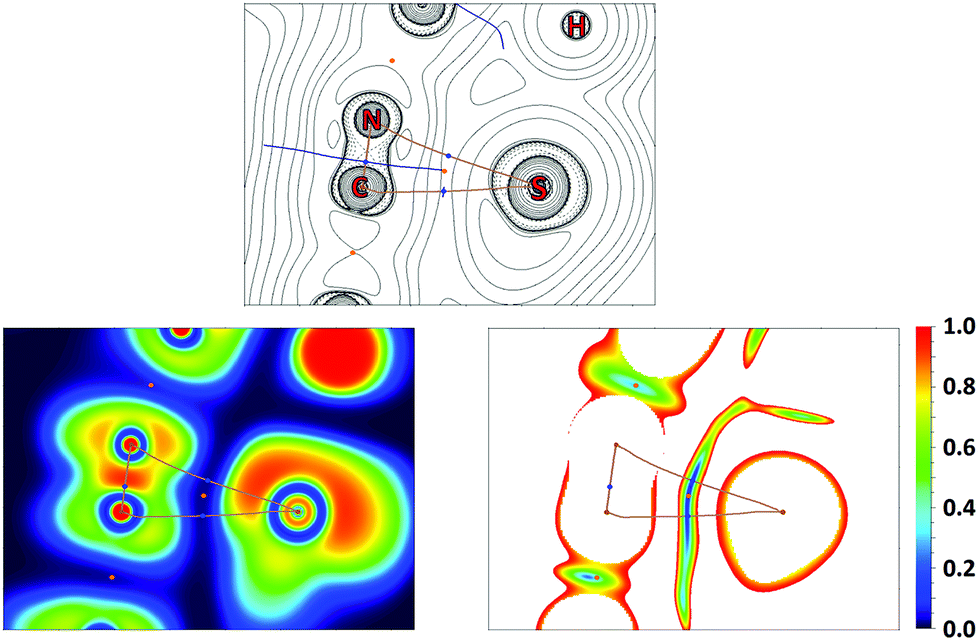 | ||
| Fig. 11 Contour line diagram of the Laplacian of electron density distribution ∇2ρ(r), bond paths, and selected zero-flux surfaces (left panel), visualization of the electron localization function (ELF, center panel) and reduced density gradient (RDG, right panel) analyses of μ2-S⋯[PtII,C] intermolecular contacts in (18)2. Bond critical points (3, −1) are shown in blue, nuclear critical points (3, −3) in pale brown, ring critical points (3, +1) in orange, and bond paths as pale brown lines, length units are Å, and the color scale for the ELF and RDG maps is presented in a.u. | ||
The Electron Localization Function54 (ELF) projections for the M1⋯C5 contacts were plotted along with CPs and bond paths (Fig. 10, 11 and S15, S16†). ELF projections show increased ELF areas around PdII and PtII atoms near the bond paths connecting the metal centers and azaheterocyclic species that can be interpreted as filled dz2 orbitals. The M⋯C bond paths that connect the metal centers and C5 atom from the azaheterocyclic species go through these dz2 and π*-orbitals. In the ELF projections plotted for the S1⋯C2 contacts, a sulfur lone pair is visible near the bond paths; also, the electron density depletion regions associated with the π-holes are easily identified in the ELF basins. Reduced density gradient (RDG) analyses confirm the fact that all discussed contacts are attractive.
The comparison of the crystal structures of the thiocyanate derivatives with those of the chloride species (Fig. S4 and S5†) revealed significant changes that occurred on anion exchange. While the crystal structure of the chloride species is predominantly held by C–H⋯X (X = Cl, N) hydrogen bonds with no involvement of the MII-center, the change from Cl to NCS favours the formation of M⋯C interactions.
Application of PtII–MNHC complexes as photocatalysts for hydrosilylation of alkynes
Representative platinum(II) diaminocarbene derivatives 8, 11 and 12 were evaluated as potential photocatalysts in the hydrosilylation of alkynes. As a model system, we have chosen the reaction of 1,2-diphenylacetylene with triethylsilane giving 1,2-(diphenylvinyl)triethylsilane under the photocatalytic conditions similar to those reported previously.9Without the addition of a catalyst, no reaction was observed in the 20–60 °C range with or without visible light irradiation (Table 1, entries 1–3) indicating that irradiation itself is not sufficient to initiate this process. Furthermore, at 40–60 °C only an insignificant catalytic effect of any studied complexes was observed after 6 h without light (catalyst loading: 0.1 mol%, entries 4–5, 7–8 and 9–12). Under blue LED irradiation all catalysts 8, 11, and 12 showed moderate to good activity (62–89% product yields after 6 h, entries 6, 9 and 13); complex 11 was the most active (entry 9; 89% product yield). Increasing the reaction time to 12 h led to nearly quantitative conversion of the starting 1,2-diphenylacetylene to the respective silylated product (entry 14). Application of 0.5 mol% of catalyst shortens the reaction time to 4 h (entry 15), suggesting that the system with higher catalyst loading can be used if shorter reaction time is preferred over a larger TON. Although the exact reasons for the increased catalytic activity of mesityl carbene species require further studies, some first attempts suggest the optimum combination of donor and steric properties of these derivatives.55
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Product yieldb | ||
| At 40 °C, no LED | At 60 °C, no LED | At 40 °C, with LED light | ||
| a Reaction conditions: 1,2-diphenylacetylene (5.0 × 10−4 mol), Et3SiH (7.5 × 10−4 mol), selected catalyst (5.0 × 10−7 mol, 0.1 mol%), toluene (2 mL), and time: 6 h; for entries with light irradiation a 3 × 2 W LED source with a 445 nm maximum was used. b Yield of 1,2-(diphenylvinyl)triethylsilane was determined by 1H NMR spectroscopy using 1,2-dimethoxyethane as the internal standard; the product was obtained as a mixture of E/Z isomers (E 55–75%). c Time: 12 h. d Catalyst loading: 0.5 mol%, 4 h. | ||||
| 1–3 | — | <5 | <5 | <5 |
| 4–6 | 8 | <5 | 5 | 62 |
| 7–9 | 11 | <5 | 6 | 89 |
| 10–13 | 12 | <5 | <5 | 81 |
| 14 | 11 | 98c | ||
| 15 | 11 | 98d | ||
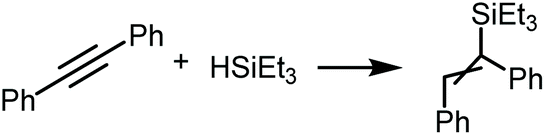
Conclusions
In the course of this study, a series of new binuclear palladium(II) and platinum(II) metalla-N-heterocyclic carbene (MNHC) type complexes were prepared via metal-mediated coupling between cis-[MCl2(CNR)2] (R = Xyl, Mes) and 2-aminopyridine or 2-aminopyrazine nucleophiles. Structural characterization of 7–12 thus achieved by HRESI+-MS, IR, and 1D (1H, 13C{1H}, 195Pt{1H}) and 2D (1H,13C-HSQC, 1H,13C-HMBC) NMR spectroscopy as well as by single-crystal XRD revealed the difference in diaminocarbene ligands where both Ccarbene–N bonds in the MNHC moiety are in agreement with the data for classical diaminocarbenes, while the second NCN fragment is an ADC-like carbene due to the rather double and single C–N bond character. QTAIM reveals that the M–Ccarbene bonds are characterized by low to moderate values of electron density ρb, positive values of the Laplacian ∇2ρ, and negative values of local electron energy densities, H(r), signifying that these M–Ccarbene bonds have a significant degree of covalency. In the UV-vis absorption spectrum of the MNHC species, the wide intense absorption in the 320–414 nm range is assigned to 1L′LCT/1ILCT transitions which are important for visible-light photocatalysis. The absorption characteristics are governed by the nature of the azaheterocyclic fragment and complexes with pyrazine and pyridine rings (10 and 12) show 20–40 nm red-shifted bands relative to their pyrazine counterparts (7–9 and 11). In contrast, the parent CNR fragments have more effect on the electrochemical properties of the MNHC compounds, and species with mesityl substituents (9–12) are oxidized at a lower potential than their corresponding xylyl analogues (7 and 8).Moreover, the first palladium(II) and platinum(II) diaminocarbene thiocyanate derivatives 13–18 were synthesized from the corresponding starting chlorides 7–12 by metathetic reaction with NH4NCS. In these complexes, both thiocyanate ligands are N-bound as confirmed by IR spectroscopy and single-crystal XRD. Replacing both chloride ligands with thiocyanate ligands leads to minor changes in absorption spectra, however, significantly reducing the solubility of compounds in organic solvents. Isomorphous crystal structures of the thiocyanate derivatives 16 and 18 exhibit two symmetrical pairs M1⋯C5 and S1⋯C2 short intermolecular contacts between the electron-rich MII-center and thiocyanate ligand on the one side, and the electron-poor π system of the azaheterocyclic ring on the other side; the observed noncovalent interactions provide supramolecular dimers.
The photocatalytic properties of the prepared platinum–diaminocarbenes 8, 11 and 12 as catalysts for hydrosilylation of alkynes with hydrosilanes were evaluated. The highest catalytic efficiency was achieved with complex 11 that is derived from the platinum-mediated coupling of mesityl isocyanide with 2-aminopyridine. The catalytic system uses blue LED irradiation for 6–12 h in toluene with a typical catalyst loading of 0.1 mol% and allows the transformation of 1,2-diphenylacetylene and triethylsilane into the respective 1,2-(diphenylvinyl)triethylsilane with up to 98% yields.
Together, the experimental and computational data provide an insight into the flexible character of the MNHC species. We explored the photocatalytic properties of the [M]–MNHCs derived from reactions of metal-activated RNCs and various 2-aminoazaheterocycles in order to develop useful catalytic systems and to establish catalyst structure–activity relationships for other visible light activated systems,56 including Pd-catalyzed cross-coupling reactions and Pt-catalyzed hydroarylation reactions.
Experimental
Materials and instrumentation
Solvents, organic and inorganic reagents were obtained from commercial sources and used as received. Isocyanide complexes cis-[MCl2(CNR)2] (M = Pd, R = Xyl 1,11 Mes 2;81 M = Pt, R = Xyl 3,57 Mes 458) were prepared as reported earlier. C, H, and N elemental analyses were carried out on a Euro EA 3028 HT CHNSO analyzer. Mass-spectra were acquired on a Bruker micrOTOF spectrometer equipped with an ESI source; a CH2Cl2/MeOH mixture was used as the solvent. The instrument was operated in positive ion mode using an m/z range of 50–3000. The capillary voltage of the ion source was set at −4500 V (ESI+) and the capillary exit at +(70–150) V. The nebulizer gas pressure was 0.4 bar and drying gas flow was 4.0 L min−1. The most intense peak in the isotopic pattern is reported. Infrared spectra were recorded on a Bruker Tensor 27 FTIR instrument (4000–2400 cm−1, resolution 2 cm−1) using KBr pellets. The UV/vis absorption spectra in CH2Cl2 solution were recorded on a Shimadzu UV-2500 spectrophotometer in a quartz cuvette with l = 1.0 mm and the complex's concentration was 0.03 mM. A Shimadzu UV-1800 spectrophotometer was used to measure the UV/vis absorbance of solid-state complexes. The 13C CP/MAS NMR spectra were acquired using a double-resonance 4 mm MAS Bruker probe at a resonance frequency of 101 MHz under 14 kHz MAS. The CP contact time in all experiments was 3.5 μs with a delay between acquisitions of 1 s and the number of scans collected was 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Suitable single crystals were selected and mounted on a MiTeGen tip via a crystallographic oil. Data were collected using a Rigaku XtaLAB Synergy, single source at home/near, HyPix diffractometer (monochromated CuKα radiation, λ = 1.54184 Å) at 100(0) K. In each case, the structure was solved with a ShelXT59 structure solution program using Intrinsic Phasing and refined with a ShelXL59 refinement package incorporated in the OLEX2 program package60 using Least Squares minimization. Empirical absorption correction was applied in the CrysAlisPro61 program complex using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. The CVs were measured using an Autolab PGSTAT302N voltammetric analyzer at a scan rate of 100 mV s−1. Glassy carbon and a platinum wire were used as the working and counter electrodes, respectively. The sample solutions (ca. 5 mM) were in 0.1 M [nBu4N](BF4)–CH2Cl2. Ag/AgNO3 (0.01 M AgNO3 in MeCN) was used as the reference electrode and the potential values were corrected against an Fc+/Fc couple (E1/2 = 0.52 V vs. Ag/AgNO3 in CH2Cl2).
000. Suitable single crystals were selected and mounted on a MiTeGen tip via a crystallographic oil. Data were collected using a Rigaku XtaLAB Synergy, single source at home/near, HyPix diffractometer (monochromated CuKα radiation, λ = 1.54184 Å) at 100(0) K. In each case, the structure was solved with a ShelXT59 structure solution program using Intrinsic Phasing and refined with a ShelXL59 refinement package incorporated in the OLEX2 program package60 using Least Squares minimization. Empirical absorption correction was applied in the CrysAlisPro61 program complex using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. The CVs were measured using an Autolab PGSTAT302N voltammetric analyzer at a scan rate of 100 mV s−1. Glassy carbon and a platinum wire were used as the working and counter electrodes, respectively. The sample solutions (ca. 5 mM) were in 0.1 M [nBu4N](BF4)–CH2Cl2. Ag/AgNO3 (0.01 M AgNO3 in MeCN) was used as the reference electrode and the potential values were corrected against an Fc+/Fc couple (E1/2 = 0.52 V vs. Ag/AgNO3 in CH2Cl2).
Synthetic work
9. Yellowish solid (37 mg, yield 78%). Anal. Calcd for C45H48N6Cl2Pd2: C, 56.5; H, 5.1; N, 8.8; found: C, 56.7; H, 5.4; N, 9.0. [M − Cl]+ calcd for C45H48N6ClPd2+ 921.1703, found 921.1706. IR (KBr, selected bands, cm−1): ν(C–H) 2920 (s), 2854 (s); ν(CN) 2189 (s); ν(Ccarbene–N) 1629 (s); δ(C–H from Mes) 855 (s). 1H NMR (CDCl3, 400.13 MHz, δ): 9.10 (d, 3JH,H = 8.5 Hz, 1H, H3), 9.06–8.99 (m, 1H, H5), 7.84 (ddd, 3JH,H = 8.8 Hz, 3JH,H = 7.4, 4JH,H = 1.7 Hz, 1H, H4), 7.03–6.94 (m, 1H, H6), 6.91 (s, 2H, m-H from Mes), 6.82 (s, 2H, m-H from Mes), 6.68 (s, 2H, m-H from Mes), 6.46 (s, 2H, m-H from Mes), 2.41 (s, 6H, 2Me), 2.31 (s, 3H, Me), 2.26 (s, 15H, 3Me), 2.23 (s, 6H, 2Me), 2.02 (s, 6H, 2Me), 1.47 (s, 3H, Me), 1.46 (s, 3H, Me). 13C{1H} NMR (101 MHz, CDCl3, δ): 195.0 (Ccarbene–N), 167.0 (Ccarbene–N), 162.8 (CN), 147.7 (p-C from Mes), 146.7, 141.5 (C3, C4), 140.7 (ipso-C from Mes), 140.0, 139.8, 138.9 (p-C from Mes), 136.2, 133.9, 133.9 (o-C from Mes), 133.7 (p-C from Mes), 128.5, 128.3, 128.1, 127.2 (m-C from Mes), 124.9, 124.2 (ipso-C from Mes), 118.3, 117.2 (C5, C6), 21.2, 21.1, 20.1, 20.0 (Me's), 19.5, 19.3 (2Me's), 18.4 (4Me).
10. Yellow solid (42 mg, yield 88%). Anal. Calcd for C44H47N7Cl2Pd2: C, 55.2; H, 5.0; N, 10.2; found: C, 54.8; H, 5.0; N, 10.6. [M + H]+ calcd for C44H48N7Cl2Pd2+ 958.1417, found 958.1412. IR (KBr, selected bands, cm−1): ν(C–H) 2916 (s), 2853 (s); ν(CN) 2199 (s), 2189 (s); ν(Ccarbene–N) 1632 (s); δ(C–H from Mes) 854 (s). 1H NMR (CDCl3, 400.13 MHz, δ): 10.37 (d, 4JH,H = 0.7 Hz, 1H, H3), 8.89 (dd, 3JH,H = 3.1 Hz, 4JH,H = 1.0 Hz, 1H, H5), 8.32 (d, 3JH,H = 3.3 Hz, 1H, H6), 6.93 (s, 2H, m-H from Mes), 6.83 (s, 2H, m-H from Mes), 6.69 (s, 2H, m-H from Mes), 6.48 (s, 2H, m-H from Mes), 2.40 (s, 6H, 2Me), 2.32 (s, 3H, Me), 2.26 (s, 3H, Me), 2.02 (c, 6H, 2Me), 2.26 (c, 3H, Me), 2.02 (s, 6H, 2Me), 1.47 (s, 6H, 2Me). 13C{1H} NMR (101 MHz, CDCl3, δ): 196.8 (Ccarbene–N), 163.2 (ipso-C from Mes), 161.8 (Ccarbene–N), 147.6 (p-C from Mes), 142.2 (C3), 140.6 (p-C from Mes), 140.2 (p-C from Mes), 139.1 (ipso-C from Mes), 137.9 (C5), 137.6 (C6), 135.9 (o-C from Mes), 134.0 (o-C from Mes), 133.9 (ipso-C from Mes), 133.9 (o-C from Mes), 128.6, 128.3, 128.2 (m-C from Mes), 127.0 (o-C from Mes), 124.7 and 123.9 (CN), 21.2, 21.1, 20.1, 20.0, 19.5 (Me's), 19.2 (2Me's), 18.4 (4Me's).
11. Yellow solid (47 mg, yield 83%). Anal. Calcd for C45H48N6Cl2Pt2: C, 47.7; H, 4.3; N, 7.4; found: C, 47.2; H, 4.3; N, 7.6. [M + H]+ calcd for C45H49Cl2N6Pt2+ 1134.2688, found 1134.2678. IR (KBr, selected bands, cm−1): ν(C–H) 2915 (s); ν(CN) 2194 (s); ν(Ccarbene–N) 1667 (s), 1619 (s); δ(C–H from Mes) 771 (s). 1H NMR (CDCl3, 400.13 MHz, δ): 9.36–9.07 (m, 2H, H5 + H3), 8.02–7.73 (m, 1H, H4), 7.05 (t, 3JH,H = 6.6 Hz, 1H, H6), 6.92 (s, 2H, m-H from Mes), 6.82 (s, 2H, m-H from Mes), 6.68 (s, 2H, m-H from Mes), 6.42 (s, 2H, m-H from Mes), 2.40 (s, 6H, 2Me), 2.32 (s, 3H, Me), 2.27 (s, 15H, 3Me), 2.25 (s, 6H, 2Me), 2.04 (s, 6H, 2Me), 1.45 (s, 3H, Me), 1.40 (s, 3H, Me). 13C{1H} NMR (101 MHz, CDCl3, δ): 183.6 (Ccarbene–N), 168.3 (Ccarbene–N), 153.0 (CN), 148.3 (p-C from Mes), 145.1, 141.7 (C3, C4), 140.1, (ipso-C from Mes), 139.9, 139.6, 138.6 (p-C from Mes), 136.2, 134.0, 133.9 (o-C from Mes), 133.1 (p-C from Mes), 128.5, 128.2, 128.1, 126.7 (m-C from Mes), 125.2, 124.6 (ipso-C from Mes), 117.7, 116.9 (C5, C6), 21.2, 21.1, 20.1, 19.9 (Me's), 19.5, 19.0 (2Me's), 18.4 (4Me's). 195Pt{1H} NMR (CDCl3, 86.02 MHz, δ): −3756, −3796.
12. Yellow solid (53 mg, yield 94%). Anal. Calcd for C44H47N7Cl2Pt2: C, 46.6; H, 4.2; N, 8.6; found: C, 45.8; H, 4.4; N, 8.4. [M + H]+ calcd for C44H48N7Cl2Pt2+ 1135.2640, found 1135.2644. IR (KBr, selected bands, cm−1): ν(C–H) 2915 (s); ν(CN) 2208 (s), 2190 (s); ν(Ccarbene–N) 1634 (s); δ(C–H from aryls) 835 (s). 1H NMR (CDCl3, 400.13 MHz, δ): 10.50 (s, 1H, H3), 9.10 (d, 3JH,H = 3.0 Hz, 1H, H6), 8.38 (d, 3JH,H = 3.4 Hz, 1H, H5), 6.94 (s, 2H, m-H from Mes), 6.83 (s, 2H, m-H from Mes), 6.70 (s, 2H, m-H from Mes), 6.44 (s, 2H, m-H from Mes), 2.39 (s, 6H, 2Me), 2.33 (s, 3H, Me), 2.28 (s, 3H, Me), 2.27 (s, 6H, 2Me), 2.25 (s, 6H, 2Me), 2.04 (s, 6H, 2Me), 1.47 (s, 3H, Me), 1.41 (s, 3H, Me). 13C{1H} NMR (101 MHz, CDCl3, δ): 185.05 (Ccarbene–N), 162.89 (ipso-C from Mes), 153.62 (C2), 148.11 (p-C from Mes), 141.98 (C3), 141.80 (p-C from Mes), 140.20 (p-C from Mes), 139.93 (ipso-C from Mes), 139.88 (C3), 138.73 (p-C from Mes), 137.06 (C5), 135.97 (o-C from Mes), 135.84 (o-C from Mes), 134.34 (C6), 134.00 (o-C from Mes), 133.30 (ipso-C from Mes), 132.64 (ipso-C from Mes), 129.08 (o-C from Mes), 128.58, 128.57, 128.26, 128.09 (m-C from Mes), 126.55 (o-C from Mes), 125.05 and 124.30 (CN), 21.19 (Me's), 21.07 (Me's), 20.06 (Me's), 19.90 (Me's), 19.42 (2Me's), 18.97 (2Me's), 18.32 (4Me's). 195Pt{1H} NMR (CDCl3, 86.02 MHz, δ): −3718, −3803.
:1, 2 mL) at RT. The reaction mixture was stirred overnight to give a yellow solution over the yellow or orange precipitate. These precipitates of 13–18 were separated by centrifugation, washed with two 2 mL portions of H2O and dried in air at RT.
13. Yellow solid (35 mg, yield 74%). Anal. Calcd for C43H40N8S2Pd2: C, 54.6; H, 4.3; N, 11.9; S, 6.8, found C, 54.3; H, 4.8; N, 11.5; S, 6.6. [M + Na]+ calcd for C43H40N8S2Pd2Na+ 969.0784, found 969.0792. IR (KBr, selected bands, cm−1): ν(CN) 2204 (s), ν(NCthiocyanate) 2064, 2087 (s); ν(Ccarbene–N) 1605, 1634 (s); δ(C–H from aryls) 778 (s). 13C CP MAS NMR: 136.75, 136.42, 134.39, 133.46, 132.51, 131.36, 130.76, 129.55, 128.80, 127.96, 127.16, 125.82 (C and CH from aryls), 118.49 and 117.77 (CN), 21.42, 20.78, 19.03, 18.30 (Me's).
14. Yellow solid (42 mg, yield 75%). Anal. Calcd for C43H40N8S2Pt2: C, 46.0; H, 3.6; N, 10.0; S, 5.7, found C, 45.7; H, 3.3; N, 9.8; S, 5.6. [M − NCS]+ calcd for C42H40N7SPt2+ 1063.2363, found 1064.2355. IR (KBr, selected bands, cm−1): ν(CN) 2202 (s), ν(NCthiocyanate) 2072, 2094 (s); ν(Ccarbene–N) 1611, 1627 (s); δ(C–H from aryls) 777 (s). 13C CP MAS NMR: 166.7, 151.0, 145.1, 144.5, 136.7, 133.7, 130.7, 128.7, 127.9, 126.8, 126.2 (C and CH from aryls), 118.4 and 117.0 (CN), 21.5, 20.9, 20.5, 19.0, 18.6, 18.4, 17.8 (Me's).
15. Yellow solid (34 mg, yield 68%). Anal. Calcd for C47H48N8S2Pd2: C, 56.3; H, 4.8; N, 11.2; S, 6.4, found C, 55.8; H, 4.3; N, 10.8; S, 6.6. [M − NCS]+ calcd for C46H48N7SPd2+ 944.1766, found 944.1754. IR (KBr, selected bands, cm−1): ν(CN) 2187 (s), ν(NCthiocyanate) 2071, 2100 (s); ν(Ccarbene–N) 1619 (s); δ(C–H from aryls) 763 (s). 13C CP MAS NMR: 166.2, 140.3, 136.1, 134.8, 129.3, 127.0 (C and CH from aryls), 118.5 and 115.6 (CN), 20.0, 19.0 (Me's).
16. Yellow solid (36 mg, yield 71%). Anal. Calcd for C46H47N9S2Pd2: C, 55.1; H, 4.7; N, 12.6; S, 6.4, found C, 54.8; H, 4.6; N, 12.8; S, 6.4. [M – NCS]+ calcd for C46H48N7SPd2+ 945.1718, found 945.1716. IR (KBr, selected bands, cm−1): ν(CN) 2193 (s); ν(NCthiocyanate) 2057; ν(Ccarbene–N) 1625 (s); δ(C–H from aryls) 855 (s). 13C CP MAS NMR: 140.1, 137.9, 135.5, 135.1, 134.2, 133.7, 132.7, 131.5, 128.8 (C and CH from aryls), 19.8, 19.3, 19.1 (Me's).
17. Yellow solid (54 mg, yield 77%). Anal. Calcd for C47H48N8S2Pt2: C, 48.0; H, 4.0; N, 9.5; S, 5.4, found C, 47.6; H, 3.7; N, 9.5; S, 5.6. [M + H]+ calcd for C47H49N8S2Pt2+ 1180.2814, found 1180.2820. IR (KBr, selected bands, cm−1): ν(CN) 2205 (s), ν(NCthiocyanate) 2030, 2062 (s); ν(Ccarbene–N) 1608 (s); δ(C–H from aryls) 854 (s). 13C CP MAS NMR: 196.81 and 190.60 (Ccarbene–N), 160.66, 154.03, 150.48, 141.46, 138.79, 135.41, 133.29, 130.58, 129.43, 127.34, 126.07, 124.60 (C and CH from aryls), 116.12 and 114.59 (CN), 20.81, 20.04, 18.11, 16.06 (Me's).
18. Yellow solid (47 mg, yield 80%). Anal. Calcd for C46H48N9S2Pt2: C, 46.8; H, 4.0; N, 10.7; S, 5.4, found C, 46.3; H, 3.6; N, 10.6; S, 5.5. [M + H]+ calcd for C46H48N9S2Pt2+ 1180.2750, found 1180.2754. IR (KBr, selected bands, cm−1): ν(CN) 2202 (s), ν(NCthiocyanate) 2055, 2097 (s); ν(Ccarbene–N) 1604, 1629 (s); δ(C–H from aryls) 845, 860 (s). 13C CP MAS NMR: 185.6 (Ccarbene–N), 161.9, 153.5, 147.5, 145.3, 142.2, 140.8, 140.2, 139.7, 138.0, 136.2, 135.7, 135.1, 134.9, 133.8, 131.2, 130.1, 128.9, 128.4, 128.0, 125.5, 122.7 (C and CH from aryls), 119.0 and 118.4 (CN), 24.1, 21.9, 21.4, 19.7, 19.1, 18.4, 17.1 (Me's).
Computational details
Author contributions
M.V.K. – conceptualisation, investigation (synthesis, physicochemical study and catalysis), and writing; K.V.L. – conceptualisation and writing; E.A.K. – investigation (electrochemical study and computational study of 7, 8, 11 and 12) and writing; A.S.N. – investigation (computational study of 16 and 18) and writing; M.A.K.– conceptualisation, investigation (catalysis), writing, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (project 21-73-10083). The DFT/TD-DFT calculations for studies on 7, 8, 11 and 12 were performed by E.A.K. in the framework of the Russian Science Foundation project 21-73-00056, while the electrochemical studies were performed in the framework of the Russian Foundation for Basic Research project 19-29-08026. Measurements were performed at the Center for Magnetic Resonance, Center for X-ray Diffraction Studies, Center for Chemical Analysis, Thermogravimetric and Calorimetric Research Centre, Centre for Optical and Laser Materials Research, Department of Cryogenic Engineering and Materials Research, and Computing Centre (all belong to St Petersburg University).References
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) H. V. Huynh, Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination, Chem. Rev., 2018, 118, 9457 CrossRef CAS PubMed.
- (a) M. A. Kinzhalov and K. V. Luzyanin, Synthesis and Contemporary Applications of Platinum Group Metals Complexes with Acyclic Diaminocarbene Ligands, Russ. J. Inorg. Chem., 2022, 67, 48 CrossRef CAS; (b) M. A. Kinzhalov and K. V. Luzyanin, Reactivity of acyclic diaminocarbene ligands, Coord. Chem. Rev., 2019, 399, 213014 CrossRef CAS.
- (a) V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Acyclic diaminocarbenes (ADCs) as a promising alternative to N-heterocyclic carbenes (NHCs) in transition metal catalyzed organic transformations, Coord. Chem. Rev., 2012, 256, 2029 CrossRef CAS; (b) L. M. Slaughter, Acyclic Aminocarbenes in Catalysis, ACS Catal., 2012, 2, 1802 CrossRef CAS; (c) E. Peris, Smart N-Heterocyclic Carbene Ligands in Catalysis, Chem. Rev., 2018, 118, 9988 CrossRef CAS PubMed; (d) S. A. Timofeeva, M. A. Kinzhalov, E. A. Valishina, K. V. Luzyanin, V. P. Boyarskiy, T. M. Buslaeva, M. Haukka and V. Y. Kukushkin, Application of palladium complexes bearing acyclic amino(hydrazido)carbene ligands as catalysts for copper-free Sonogashira cross-coupling, J. Catal., 2015, 329, 449–456 CrossRef CAS; (e) M. A. Kinzhalov, K. V. Luzyanin, V. P. Boyarskiy, M. Haukka and V. Y. Kukushkin, ADC-Based Palladium Catalysts for Aqueous Suzuki–Miyaura Cross-Coupling Exhibit Greater Activity than the Most Advantageous Catalytic Systems, Organometallics, 2013, 32, 5212 CrossRef CAS.
- (a) M. A. Kinzhalov, E. V. Grachova and K. V. Luzyanin, Tuning the luminescence of transition metal complexes with acyclic diaminocarbene ligands, Inorg. Chem. Front., 2022, 9, 417–439 RSC; (b) M. Elie, J. L. Renaud and S. Gaillard, N-Heterocyclic carbene transition metal complexes in light emitting devices, Polyhedron, 2018, 140, 158 CrossRef CAS; (c) R. Visbal and M. C. Gimeno, N-heterocyclic carbene metal complexes: photoluminescence and applications, Chem. Soc. Rev., 2014, 43, 3551 RSC.
- (a) I. Ott, in Advances in Inorganic Chemistry, ed. P. J. Sadler and R. van Eldik, Academic Press, 2020, vol. 75, p. 121 Search PubMed; (b) W. Liu and R. Gust, Update on metal N-heterocyclic carbene complexes as potential anti-tumor metallodrugs, Coord. Chem. Rev., 2016, 329, 191 CrossRef CAS; (c) S. A. Patil, A. P. Hoagland, S. A. Patil and A. Bugarin, N-heterocyclic carbene-metal complexes as bio-organometallic antimicrobial and anticancer drugs, an update (2015–2020), Future Med. Chem., 2020, 12, 2239 CrossRef CAS PubMed; (d) T. V. Serebryanskaya, M. A. Kinzhalov, V. Bakulev, G. Alekseev, A. Andreeva, P. V. Gushchin, A. Protas, A. Smirnov, T. L. Panikorovskii, P. Lippmann, I. Ott, C. M. Verbilo, A. Zuraev, A. S. Bunev, V. Boyarskiy and N. A. Kasyanenko, Water Soluble Palladium(II) and Platinum(II) Acyclic Diaminocarbene Complexes: Solution Behavior, DNA Binding, and Antiproliferative Activity, New J. Chem., 2020, 44, 5762 RSC.
- J. Ruiz, L. García, B. F. Perandones and M. Vivanco, A Fischer Carbene within an Arduengo Carbene, Angew. Chem., Int. Ed., 2011, 50, 3010 CrossRef CAS PubMed.
- J. Ruiz, L. Garcia, C. Mejuto, M. Vivanco, M. R. Diaz and S. Garcia-Granda, Strong electron-donating metalla-N-heterocyclic carbenes, Chem. Commun., 2014, 50, 2129 RSC.
- (a) A. A. Eremina, M. A. Kinzhalov, E. A. Katlenok, A. S. Smirnov, E. V. Andrusenko, E. A. Pidko, V. V. Suslonov and K. V. Luzyanin, Phosphorescent Iridium(III) Complexes with Acyclic Diaminocarbene Ligands as Chemosensors for Mercury, Inorg. Chem., 2020, 59, 2209 CrossRef CAS PubMed; (b) A. S. Mikherdov, A. S. Novikov, M. A. Kinzhalov, V. P. Boyarskiy, G. L. Starova, A. Y. Ivanov and V. Y. Kukushkin, Halides Held by Bifurcated Chalcogen–Hydrogen Bonds. Effect of μ(S,N–H)Cl Contacts on Dimerization of Cl(carbene)PdII Species, Inorg. Chem., 2018, 57, 3420 CrossRef CAS PubMed; (c) A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, M. S. Avdontceva and V. Y. Kukushkin, Ligation-Enhanced π-Hole⋯π Interactions Involving Isocyanides: Effect of π-Hole⋯π Noncovalent Bonding on Conformational Stabilization of Acyclic Diaminocarbene Ligands, Inorg. Chem., 2018, 57, 6722 CrossRef CAS PubMed; (d) A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, D. V. Dar'in, G. L. Starova and V. Y. Kukushkin, Difference in Energy between Two Distinct Types of Chalcogen Bonds Drives Regioisomerization of Binuclear (Diaminocarbene)PdII Complexes, J. Am. Chem. Soc., 2016, 138, 14129 CrossRef CAS PubMed; (e) M. A. Kinzhalov, A. S. Legkodukh, T. B. Anisimova, A. S. Novikov, V. V. Suslonov, K. V. Luzyanin and V. Y. Kukushkin, Tetrazol-5-ylidene Gold(III) Complexes from Sequential [2 + 3] Cycloaddition of Azide to Metal-Bound Isocyanides and N4 Alkylation, Organometallics, 2017, 36, 3974 CrossRef CAS; (f) B. G. M. Rocha, E. A. Valishina, R. S. Chay, M. F. C. Guedes da Silva, T. M. Buslaeva, A. J. L. Pombeiro, V. Y. Kukushkin and K. V. Luzyanin, ADC-metal complexes as effective catalysts for hydrosilylation of alkynes, J. Catal., 2014, 309, 79 CrossRef CAS.
- J. C. Gee, B. A. Fuller, H.-M. Lockett, G. Sedghi, C. M. Robertson and K. V. Luzyanin, Visible light accelerated hydrosilylation of alkynes using platinum–[acyclic diaminocarbene] photocatalysts, Chem. Commun., 2018, 54, 9450 RSC.
- (a) M. A. Kinzhalov, K. V. Luzyanin, V. P. Boyarskiy, M. Haukka and V. Y. Kukushkin, Coupling of C-amino aza-substituted heterocycles with an isocyanide ligand in palladium(II) complex, Russ. Chem. Bull., 2013, 62, 758 CrossRef CAS; (b) A. G. Tskhovrebov, K. V. Luzyanin, F. M. Dolgushin, M. F. C. Guedes da Silva, A. J. L. Pombeiro and V. Y. Kukushkin, Novel Reactivity Mode of Metal Diaminocarbenes: Palladium(II)-Mediated Coupling between Acyclic Diaminocarbenes and Isonitriles Leading to Dinuclear Species, Organometallics, 2011, 30, 3362 CrossRef CAS.
- K. V. Luzyanin, A. J. L. Pombeiro, M. Haukka and V. Y. Kukushkin, Coupling between 3-Iminoisoindolin-1-ones and Complexed Isonitriles as a Metal-mediated Route to a Novel Type of Palladium and Platinum Iminocarbene Species, Organometallics, 2008, 27, 5379 CrossRef CAS.
- R. Jothibasu, K.-W. Huang and H. V. Huynh, Synthesis of cis- and trans-Diisothiocyanato−Bis(NHC) Complexes of Nickel(II) and Applications in the Kumada−Corriu Reaction, Organometallics, 2010, 29, 3746 CrossRef CAS.
- M. V. Baker, P. J. Barnard, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Synthetic, structural and spectroscopic studies of (pseudo)halo(1,3-di-tert-butylimidazol-2-ylidine)gold complexes, Dalton Trans., 2005, 37–43 RSC.
- (a) F. De Angelis, S. Fantacci, A. Selloni and M. K. Nazeeruddin, Time dependent density functional theory study of the absorption spectrum of the [Ru(4,4′-COO−-2,2′-bpy)2(X)2]4− (X=NCS, Cl) dyes in water solution, Chem. Phys. Lett., 2005, 415, 115 CrossRef CAS; (b) M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, S. I. Gorelsky, A. B. P. Lever and M. Grätzel, Synthesis, spectroscopic and a ZINDO study of cis- and trans-(X2)bis(4,4′-dicarboxylic acid-2,2′-bipyridine)ruthenium(II) complexes (X=Cl−, H2O, NCS−), Coord. Chem. Rev., 2000, 208, 213 CrossRef CAS.
- (a) K.-H. Chen, T.-H. Lin, T.-E. Hsu, Y.-J. Li, G.-H. Chen, W.-J. Leu, J.-H. Guh, C.-H. Lin and J.-H. Huang, Ruthenium(II) complexes containing dehydroacetic acid and its imine derivative ligands. Synthesis, characterization and cancer cell growth anti-proliferation activity (GI50) study, J. Org. Chem., 2018, 871, 150 CrossRef CAS; (b) F. Nasouti and A. Eslami, Thermoanalytical Study of Linkage Isomerism in Coordination Compounds. Part 7. A DSC and DFT Investigation of Solid-state Linkage Isomerization in Bis(thiocyanato)-bipyridineplatinum(II) Complex, Z. Anorg. Allg. Chem., 2017, 643, 1131 CrossRef CAS; (c) M. A. Fernandes, G. Z. Mashabane, R. Weber and L. Carlton, Structural study of analogues of Wilkinson's compound [Rh(X)(PPh3)3] (X = NCO, NCS, N3, N(CN)2) and derivatives [Rh(NCO)(O2)(PPh3)3] and [Rh(η6-C6H5B(NCO)Ph2)(PPh3)2], Polyhedron, 2020, 181, 114468 CrossRef CAS.
- (a) S. Kishi and M. Kato, Thermal and Photo Control of the Linkage Isomerism of Bis(thiocyanato)(2,2′-bipyridine)platinum(II), Inorg. Chem., 2003, 42, 8728 CrossRef CAS PubMed; (b) A. J. Paviglianiti, D. J. Minn, W. C. Fultz and J. L. Burmeister, The conjunctive response to steric hindrance in dithiocyanato[bis(diphenylphosphino)alkyl or aryl]palladium(II) complexes: a new look at a classic series, Inorg. Chim. Acta, 1989, 159, 65 CrossRef CAS; (c) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part A: Theory and Applications in Inorganic Chemistry, John Wiley & Sons, Inc., 6th edn, 2008 Search PubMed.
- (a) J. Yasuda, K. Inoue, K. Mizuno, S. Arai, K. Uehara, A. Kikuchi, Y.-N. Yan, K. Yamanishi, Y. Kataoka, M. Kato, A. Kawai and T. Kawamoto, Photooxidation Reactions of Cyclometalated Palladium(II) and Platinum(II) Complexes, Inorg. Chem., 2019, 58, 15720 CrossRef CAS PubMed; (b) K. Karami, A. Ramezanpour, M. Zakariazadeh and C. Silvestru, Catalytic activity and facile recovery of a cyclometalated N-heterocyclic carbene palladium(II) complex immobilized by non-covalent interactions on reduced graphene oxide, Appl. Organomet. Chem., 2019, 33, e4907 CrossRef; (c) M. Iliş, M. Micutz and V. Cîrcu, Luminescent palladium(II) metallomesogens based on cyclometalated Schiff bases and N-benzoyl thiourea derivatives as co-ligands, J. Org. Chem., 2017, 836–837, 81 CrossRef; (d) K. Peng, D. Moreth and U. Schatzschneider, C^N^N Coordination Accelerates the iClick Reaction of Square-Planar Palladium(II) and Platinum(II) Azido Complexes with Electron-Poor Alkynes and Enables Cycloaddition with Terminal Alkynes, Organometallics, 2021, 40, 2584 CrossRef CAS.
- (a) V. Sivchik, R. K. Sarker, Z.-Y. Liu, K.-Y. Chung, E. V. Grachova, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, Improvement of the Photophysical Performance of Platinum-Cyclometalated Complexes in Halogen-Bonded Adducts, Chem. – Eur. J., 2018, 24, 11475 CrossRef CAS PubMed; (b) A. K.-W. Chan, M. Ng, Y.-C. Wong, M.-Y. Chan, W.-T. Wong and V. W.-W. Yam, Synthesis and Characterization of Luminescent Cyclometalated Platinum(II) Complexes with Tunable Emissive Colors and Studies of Their Application in Organic Memories and Organic Light-Emitting Devices, J. Am. Chem. Soc., 2017, 139, 10750 CrossRef CAS PubMed.
- D. A. Skoog, S. R. Crouch and F. J. Holler, Principles of instrumental analysis, Thomson Brooks/Cole, Belmont, CA, 6th edn., 2007 Search PubMed.
- N. Deibel, S. Hohloch, M. G. Sommer, D. Schweinfurth, F. Ehret, P. Braunstein and B. Sarkar, Electrochromic Platinum(II) Complexes Derived from Azobenzene and Zwitterionic Quinonoid Ligands: Electronic and Geometric Structures, Organometallics, 2013, 32, 7366 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, Benchmark Energetic Data in a Model System for Grubbs II Metathesis Catalysis and Their Use for the Development, Assessment, and Validation of Electronic Structure Methods, J. Chem. Theory Comp., 2009, 5, 324 CrossRef CAS PubMed; (b) R. Valero, R. Costa, I. d. P. R. Moreira, D. G. Truhlar and F. Illas, Performance of the M06 family of exchange-correlation functionals for predicting magnetic coupling in organic and inorganic molecules, J. Chem. Phys., 2008, 128, 114103 CrossRef PubMed; (c) R. Srivastava and L. R. Joshi, The effect of substituted 1,2,4-triazole moiety on the emission, phosphorescent properties of the blue emitting heteroleptic iridium(III) complexes and the OLED performance: a theoretical study, Phys. Chem. Chem. Phys., 2014, 16, 17284 RSC; (d) C. Adamo and D. Jacquemin, The calculations of excited-state properties with Time-Dependent Density Functional Theory, Chem. Soc. Rev., 2013, 42, 845 RSC.
- P. S. Wagenknecht and P. C. Ford, Metal centered ligand field excited states: Their roles in the design and performance of transition metal based photochemical molecular devices, Coord. Chem. Rev., 2011, 255, 591 CrossRef CAS.
- Y. B. Dudkina, D. Y. Mikhaylov, T. V. Gryaznova, A. I. Tufatullin, O. N. Kataeva, D. A. Vicic and Y. H. Budnikova, Electrochemical Ortho Functionalization of 2-Phenylpyridine with Perfluorocarboxylic Acids Catalyzed by Palladium in Higher Oxidation States, Organometallics, 2013, 32, 4785 CrossRef CAS.
- (a) M. V. Kashina, D. M. Ivanov and M. A. Kinzhalov, The Isocyanide Complexes cis-[MCl2(CNC6H4–4-X)2] (M = Pd, Pt; X = Cl, Br) as Tectons in Crystal Engineering Involving Halogen Bonds, Crystals, 2021, 11, 799 CrossRef CAS; (b) M. Bulatova, D. M. Ivanov, J. M. Rautiainen, M. A. Kinzhalov, K.-N. Truong, M. Lahtinen and M. Haukka, Studies of Nature of Uncommon Bifurcated I–I⋯(I–M) Metal-Involving Noncovalent Interaction in Palladium(II) and Platinum(II) Isocyanide Cocrystals, Inorg. Chem., 2021, 60, 13200 CrossRef CAS PubMed; (c) A. V. Buldakov, M. A. Kinzhalov, M. A. Kryukova, D. M. Ivanov, A. S. Novikov, A. S. Smirnov, G. L. Starova, N. A. Bokach and V. Y. Kukushkin, Isomorphous Series of PdII-Containing Halogen Bond Donors Exhibiting Cl/Br/I Triple Halogen Isostructural Exchange, Cryst. Growth Des., 2020, 3, 1975 CrossRef; (d) M. A. Kryukova, D. M. Ivanov, M. A. Kinzhalov, A. S. Novikov, A. S. Smirnov, N. A. Bokach and V. Y. Kukushkin, Four-Center Nodes: Supramolecular Synthons Based on Cyclic Halogen Bonding, Chem. – Eur. J., 2019, 25, 13671 CrossRef CAS PubMed; (e) M. V. Kashina, M. A. Kinzhalov, A. S. Smirnov, D. M. Ivanov, A. S. Novikov and V. Y. Kukushkin, Dihalomethanes as Bent Bifunctional XB/XB-Donating Building Blocks for Construction of Metal-involving Halogen Bonded Hexagons, Chem. - Asian J., 2019, 14, 3915 CrossRef CAS PubMed; (f) M. A. Kinzhalov, M. V. Kashina, A. S. Mikherdov, E. A. Mozheeva, A. S. Novikov, A. S. Smirnov, D. M. Ivanov, M. A. Kryukova, A. Y. Ivanov, S. N. Smirnov, V. Y. Kukushkin and K. V. Luzyanin, Dramatically Enhanced Solubility of Halide-Containing Organometallic Species in Diiodomethane: The Role of Solvent⋯Complex Halogen Bonding, Angew. Chem., Int. Ed., 2018, 57, 12785 CrossRef CAS PubMed.
- (a) B. G. M. Rocha, E. A. Valishina, R. S. Chay, M. F. C. Guedes da Silva, T. M. Buslaeva, A. J. L. Pombeiro, V. Y. Kukushkin and K. V. Luzyanin, ADC-metal complexes as effective catalysts for hydrosilylation of alkynes, J. Catal., 2014, 309, 79 CrossRef CAS; (b) R. S. Chay, B. G. M. Rocha, A. J. L. Pombeiro, V. Y. Kukushkin and K. V. Luzyanin, Platinum Complexes with Chelating Acyclic Aminocarbene Ligands Work as Catalysts for Hydrosilylation of Alkynes, ACS Omega, 2018, 3, 863 CrossRef CAS PubMed.
- (a) S. Diez-Gonzalez, N. Marion and S. P. Nolan, N-Heterocyclic Carbenes in Late Transition Metal Catalysis, Chem. Rev., 2009, 109, 3612 CrossRef CAS PubMed; (b) C. P. Newman, R. J. Deeth, G. J. Clarkson and J. P. Rourke, Synthesis of Mixed NHC/L Platinum(II) Complexes: Restricted Rotation of the NHC Group, Organometallics, 2007, 26, 6225 CrossRef CAS; (c) J. W. K. Seah, J. X. T. Lee, Y. Li, S. A. Pullarkat, N. S. Tan and P.-H. Leung, Chelating Phosphine–N-Heterocyclic Carbene Platinum Complexes via Catalytic Asymmetric Hydrophosphination and Their Cytotoxicity Toward MKN74 and MCF7 Cancer Cell Lines, Inorg. Chem., 2021, 60, 17276 CrossRef CAS PubMed.
- A. Tronnier, U. Heinemeyer, S. Metz, G. Wagenblast, I. Muenster and T. Strassner, Heteroleptic platinum(II) NHC complexes with a C^C* cyclometalated ligand – synthesis, structure and photophysics, J. Mater. Chem. C, 2015, 3, 1680 RSC.
- (a) S. Roy, K. C. Mondal, J. Meyer, B. Niepötter, C. Köhler, R. Herbst-Irmer, D. Stalke, B. Dittrich, D. M. Andrada, G. Frenking and H. W. Roesky, Synthesis, Characterization, and Theoretical Investigation of Two-Coordinate Palladium(0) and Platinum(0) Complexes Utilizing π-Accepting Carbenes, Chem. – Eur. J., 2015, 21, 9312 CrossRef CAS PubMed; (b) S. Roy, K. C. Mondal and H. W. Roesky, Cyclic Alkyl(amino) Carbene Stabilized Complexes with Low Coordinate Metals of Enduring Nature, Acc. Chem. Res., 2016, 49, 357 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- R. S. Chay, K. V. Luzyanin, V. Y. Kukushkin, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Novel Palladium–Aminocarbene Species Derived from Metal-Mediated Coupling of Isonitriles and 1,3-Diiminoisoindoline: Synthesis and Catalytic Application in Suzuki–Miyaura Cross-Coupling, Organometallics, 2012, 31, 2379 CrossRef CAS.
- (a) S. A. Katkova, M. A. Kinzhalov, P. M. Tolstoy, A. S. Novikov, V. P. Boyarskiy, A. Y. Ananyan, P. V. Gushchin, M. Haukka, A. A. Zolotarev, A. Y. Ivanov, S. S. Zlotsky and V. Y. Kukushkin, Diversity of Isomerization Patterns and Protolytic Forms in Aminocarbene PdII and PtII Complexes Formed upon Addition of N,N′-Diphenylguanidine to Metal-Activated Isocyanides, Organometallics, 2017, 36, 4145 CrossRef CAS; (b) M. A. Kinzhalov, S. A. Timofeeva, K. V. Luzyanin, V. P. Boyarskiy, A. A. Yakimanskiy, M. Haukka and V. Y. Kukushkin, Palladium(II)-Mediated Addition of Benzenediamines to Isocyanides: Generation of Three Types of Diaminocarbene Ligands Depending on the Isomeric Structure of the Nucleophile, Organometallics, 2016, 35, 218 CrossRef CAS; (c) E. A. Valishina, M. F. C. Guedes da Silva, M. A. Kinzhalov, S. A. Timofeeva, T. M. Buslaeva, M. Haukka, A. J. L. Pombeiro, V. P. Boyarskiy, V. Y. Kukushkin and K. V. Luzyanin, Palladium-ADC complexes as efficient catalysts in copper-free and room temperature Sonogashira coupling, J. Mol. Catal. A: Chem., 2014, 395, 162–171 CrossRef CAS; (d) M. A. Kinzhalov, V. P. Boyarskiy, K. V. Luzyanin, F. M. Dolgushin and V. Y. Kukushkin, Metal-mediated coupling of a coordinated isocyanide and indazoles, Dalton Trans., 2013, 42, 10394 RSC.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893 CrossRef CAS.
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, The Mayer bond order as a tool in inorganic chemistry, Dalton Trans., 2001, 2095 RSC.
- M. P. Mitoraj, A. Michalak and T. Ziegler, A Combined Charge and Energy Decomposition Scheme for Bond Analysis, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS PubMed.
- L. J. Farrugia, C. Evans and M. Tegel, Chemical Bonds without “Chemical Bonding”? A Combined Experimental and Theoretical Charge Density Study on an Iron Trimethylenemethane Complex, J. Phys. Chem. A, 2006, 110, 7952 CrossRef CAS PubMed.
- A. K. Guha, C. Das and A. K. Phukan, Heterocyclic carbenes of diverse flexibility: A theoretical insight, J. Org. Chem., 2011, 696, 586 CrossRef.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- A. Shahi and E. Arunan, Hydrogen bonding, halogen bonding and lithium bonding: an atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding, Phys. Chem. Chem. Phys., 2014, 16, 22935 RSC.
- (a) K. R. Geethalakshmi, X. Yang, Q. Sun, T. Y. Ng and D. Wang, The nature of interfacial binding of imidazole and carbene ligands with M20 nanoclusters (M = Au, Ag and Cu) – a theoretical study, RSC Adv., 2015, 5, 88625 RSC; (b) G. F. Caramori, L. C. Garcia, D. M. Andrada and G. Frenking, Ruthenium(II) complexes of N-heterocyclic carbenes derived from imidazolium-linked cyclophanes, Dalton Trans., 2014, 43, 14710 RSC.
- R. F. W. Bader, A Bond Path: A Universal Indicator of Bonded Interactions, J. Phys. Chem. A, 1998, 102, 7314 CrossRef CAS.
- L. E. Johnson and D. B. DuPré, QTAIM Analysis of Ligand Properties and Mechanisms of Tuning of 6-Membered Ring N-Heterocyclic Carbenes in Transition Metal Complexes through Ring-Substituent Variation, J. Phys. Chem. A, 2009, 113, 8647 CrossRef CAS PubMed.
- T. Ziegler and A. Rauk, On the calculation of bonding energies by the Hartree Fock Slater method, Theor. Chim. Acta, 1977, 46, 1 CrossRef CAS.
- M. Mitoraj and A. Michalak, Donor–Acceptor Properties of Ligands from the Natural Orbitals for Chemical Valence, Organometallics, 2007, 26, 6576 CrossRef CAS.
- (a) G. Frenking and S. Shaik, The Chemical Bond: Fundamental Aspects of Chemical Bonding, Wiley, 2014 CrossRef; (b) L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Energy decomposition analysis, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed; (c) L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chemical Bonding and Bonding Models of Main-Group Compounds, Chem. Rev., 2019, 119, 8781 CrossRef CAS PubMed.
- A. Bérces, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, L. Fan, T. H. Fischer, C. Fonseca Guerra, S. J. A. van Gisbergen, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, F. E. Harris, P. van den Hoek, H. Jacobsen, G. van Kessel, F. Kootstra, E. van Lenthe, D. A. McCormack, V. P. Osinga, S. Patchkovskii, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, P. Ros, P. R. T. Schipper, G. Schreckenbach, J. G. Snijders, M. Sola, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, O. Visser, E. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo, E. J. Baerends, J. Autschbach and T. Ziegler, TADF2017; SCM, Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands, 2017.
- X. Chang, K.-E. Lee, S. Il Jeon, Y.-J. Kim, H.-K. Lee and S. W. Lee, Bis(isothiocyanato)bis(phosphine) complexes of group 10 metals: reactivity toward organic isocyanides, Dalton Trans., 2005, 3722 RSC.
- L. Tchertanov, Understanding the Peculiarities of Azide and Thiocyanate Binding in Proteins: Use of the Small Molecule Structural Data, Supramol. Chem., 2000, 12, 67 CrossRef CAS.
- (a) C. Wechwithayakhlung, D. M. Packwood, J. Chaopaknam, P. Worakajit, S. Ittisanronnachai, N. Chanlek, V. Promarak, K. Kongpatpanich, D. J. Harding and P. Pattanasattayavong, Tin(II) thiocyanate Sn(NCS)2 – a wide band gap coordination polymer semiconductor with a 2D structure, J. Mater. Chem. C, 2019, 7, 3452 RSC; (b) P. Ghorai, P. Brandão, A. Bauzá, A. Frontera and A. Saha, Anion-reliant structural versatility of novel cadmium(II) complexes: Synthesis, crystal structures, photoluminescence properties and exploration of unusual O⋯S chalcogen bonding involving thiocyanate coligand, Inorg. Chim. Acta, 2018, 469, 189 CrossRef CAS.
- L. Tchertanov and C. Pascard, Statistical Analysis of Noncovalent Interactions of Anion Groups in Crystal Structures. III. Metal Complexes of Thiocyanate and their Hydrogen-Donor Accepting Function, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 904 CrossRef.
- P. Cauliez, V. Polo, T. Roisnel, R. Llusar and M. Fourmigué, The thiocyanate anion as a polydentate halogen bond acceptor, CrystEngComm, 2010, 12, 558 RSC.
- (a) J. Wilson, T. Maxson, I. Wright, M. Zeller and S. V. Rosokha, Diversity and uniformity in anion–π complexes of thiocyanate with aromatic, olefinic and quinoidal π-acceptors, Dalton Trans., 2020, 49, 8734 RSC; (b) J. Echeverría, Intermolecular Interactions between Thiocyanato Ligands in Metal Complexes, Cryst. Growth Des., 2021, 21, 1636 CrossRef.
- A. Bondi, van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- S. Alvarez, A cartography of the van der Waals territories, Dalton Trans., 2013, 42, 8617 RSC.
- (a) A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Phys. Chem., 1990, 92, 5397 CrossRef CAS; (b) B. Silvi and A. Savin, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371, 683 CrossRef CAS; (c) A. Savin, R. Nesper, S. Wengert and T. F. Fässler, ELF: The Electron Localization Function, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808 CrossRef CAS.
- T. K. Meister, J. W. Kück, K. Riener, A. Pöthig, W. A. Herrmann and F. E. Kühn, Decoding catalytic activity of platinum carbene hydrosilylation catalysts, J. Catal., 2016, 337, 157 CrossRef CAS.
- (a) M. Parasram and V. Gevorgyan, Visible light-induced transition metal-catalyzed transformations: beyond conventional photosensitizers, Chem. Soc. Rev., 2017, 46, 6227 RSC; (b) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Visible Light-Induced Transition Metal Catalysis, Chem. Rev., 2022, 122, 1543 CrossRef PubMed.
- F. Bonati and G. Minghetti, New isocyanide complexes of platinum(II), J. Org. Chem., 1970, 24, 251 CrossRef CAS.
- M. A. Kinzhalov, M. V. Kashina, A. S. Mikherdov, S. A. Katkova and V. V. Suslonov, Synthesis of Platinum(II) Phoshine Isocyanide Complexes and Study of Their Stability in Isomerization and Ligand Disproportionation Reactions, Russ. J. Gen. Chem., 2018, 88, 1180 CrossRef CAS.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2008, 42, 339 CrossRef.
- Agilent, C., CrysAlis PRO, Agilent Technologies Ltd, Yarnton, UK, 2014 Search PubMed.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (b) C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- (a) S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (b) S. Grimme, A. Jens, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS; (b) F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- J. D. Rolfes, F. Neese and D. A. Pantazis, All-electron scalar relativistic basis sets for the elements Rb–Xe, J. Comput. Chem., 2020, 41, 1842 CrossRef CAS PubMed.
- F. Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comput. Chem., 2003, 24, 1740 CrossRef CAS PubMed.
- D. A. Pantazis and F. Neese, All-Electron Scalar Relativistic Basis Sets for the Actinides, J. Chem. Theory Comput., 2011, 7, 677 CrossRef CAS.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- (a) T. A. Keith, AIMAll (Version 19.02.13), TK Gristmill Software, Overland Park KS, 2019 Search PubMed; (b) T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed; (c) T. Lu and F. Chen, Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm, J. Mol. Graphics Modell., 2012, 38, 314 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics Modell., 1996, 14, 33 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai and T. Vreven, Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Energy-adjustedab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- (a) M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, Perth, Australia, 2017 Search PubMed; (b) P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef CAS PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces, Chem. Commun., 2007, 3814 RSC.
- S. Sueki and Y. Kuninobu, Rhodium-catalysed synthesis of multi-substituted silylindenes from aryl alkynes and hydrosilanes via C–H bond activation, Chem. Commun., 2015, 51, 7685 RSC.
- M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, Olefin Isomerization and Hydrosilylation Catalysis by Lewis Acidic Organofluorophosphonium Salts, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed.
- F. E. Hahn and M. C. Jahnke, Heterocyclic Carbenes: Synthesis and Coordination Chemistry, Angew. Chem. Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- M. A. Kinzhalov, A. A. Zolotarev and V. P. Boyarskiy, Crystal structure of cis-[PdCl2(CNMes)2], J. Struct. Chem., 2016, 57, 822–825 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2133432, 2133433 and 2133435. For ESI and crystallographic data in CIF or other electronic format see DOI: http://doi.org/10.1039/d2dt00252c |
| This journal is © The Royal Society of Chemistry 2022 |