
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand isomerism fine-tunes structure and stability in zinc complexes of fused pyrazolopyridines†
Amelia M.
Swarbrook
,
Rohan J.
Weekes
 ,
Jack W.
Goodwin
and
Chris S.
Hawes
*
,
Jack W.
Goodwin
and
Chris S.
Hawes
*
School of Chemical and Physical Sciences, Keele University, Keele ST5 5BG, UK. E-mail: c.s.hawes@keele.ac.uk
First published on 15th December 2021
Abstract
Fused-ring pyrazoles offer a versatile platform for derivitization to give finely tuned and functional ligands in coordination assemblies. Here, we explore the pyrazolo[4,3-b]pyridine (HL1) and pyrazolo[3,4-c]pyridine (HL2) backbones and their N-substituted derivatives, using their coordination chemistry with zinc(II) in the solid state and in solution to examine the steric and electronic effects of varying their substitution pattern. The parent heterocycles HL1 and HL2 both generate robust and permanently porous isomeric MOFs on reaction with zinc and a dicarboxylate co-ligand. The subtle geometric change offered by the position of the backbone pyridyl nitrogen atom leads to substantial changes in the pore size and total pore volume, which is reflected in both their surface areas and CO2 uptake performance. Both materials are also unusually resilient to atmospheric water vapour by virtue of the strong metal–azolate bonding. The isomeric chelating ligands L3–L6, generated by N-arylation of the parent heterocycles with a 2-pyridyl group, each coordinate to zinc to give either mononuclear or polymeric coordination compounds depending on the involvement of the backbone pyridine nitrogen atom. While crystal packing influences based on the steric preferences of the ligands are dominant in the crystalline phase, fluorescence spectroscopy is used to show that the 2H isomers L4 and L6 show distinct coordination behaviour to the 1H isomers L3 and L5, forming competing [ML] and [ML2] species in soution. The first stability constant for L6 with zinc(II) is an order of magnitude larger than for the other three ligands, suggesting an improved binding strength based on the electron configuration in this isomer. These results show that careful control of remote substitution on fused pyrazole ligands can lead to substantial improvements in the stability of the resulting complexes, with consequences for the design of stable coordination assemblies containining labile metal ions.
Introduction
Ligand design is of paramount importance in the development of new metal–organic frameworks and discrete metallosupramolecular systems. Ligand-centred functionality is key to the operation of several important classes of chemical sensors,1 adsorbents,2 catalysts3 and electronic materials4 based on MOFs and related materials.5 On a more fundamental level however, the basic stability and operation of any coordination assembly is predicated on the nature of the metal–ligand interactions, as typically both the most labile and geometrically variable part of these assemblies.6 With the need to focus attention on cheap and abundant metal ions for constructing coordination assemblies with large scale industrial applications,7 the onus falls onto the ligand choice to maximise stability and coordination predictability, especially with labile metal nodes involving zinc(II).8Azole and azolate-based ligands have long been staples of coordination chemistry,9 with pyrazoles and pyrazolylborates being widely featured in transition metal coordination compounds from the mid-20th century and continuing their popularity into MOF chemistry from the early 2000s onwards.10 One major appeal of azoles as ligands, especially imidazole and pyrazole, is associated with the acidic N–H group within the ring which when deprotonated tends to give very strong coordination bonds from the corresponding azolate anion.11 This has been used to great effect in the well-known ZIF series of materials,12 but also in a variety of pyrazolate MOFs reported by Colombo and others which have shown exceptional stability to water.13 Far less widely explored are the fused pyrazoles. Some studies have reported coordination chemistry and MOF formation with indazoles,14 but pyrazoles fused with other carbocyclic or heterocyclic groups are very rare in coordination compounds.15 Ring fusion in these systems may not only provide a wealth of additional backbone functionalities and new bridging geometries, but ring fusion at the C3–C4 bond of the pyrazole ring also breaks the symmetry of the 1H and 2H tautomers.16 While selective N-substitution in these cases becomes challenging,17 the isomeric products of N-substitution exhibit both steric differences and changes to the backbone electronic configurations.18 As such, fused pyrazoles offer rich opportunities as ligand targets for optimising the properties of functional coordination assemblies. In this study, we selected the isomeric fused heterocycles pyrazolo[4,3-b]pyridine (HL1) and pyrazolo[3,4-c]pyridine (HL2), as shown in Fig. 1, as starting points for developing fused pyrazole-containing coordination assemblies. We aimed to establish the optimal substitution pattern for these systems, combining variations in the ring junction position with non-selective N-arylation to give four isomeric chelating ligands L3–L6, and set out to probe the balance of steric and electronic effects that dictate their coordination preferences.
 | ||
| Fig. 1 Structures of parent heterocycles HL1 and HL2 and isomeric chelating ligands L3–L6. | ||
Results and discussion
Synthesis and structures of complexes 1 and 2
The parent heterocycles HL1 and HL2 were first reacted with zinc(II) salts under a range of conditions in an attempt to generate homoleptic coordination polymers containing these species, but no crystalline material could be generated. Instead, we turned our attention to the well-known 4,4′-biphenyldicarboxylic acid (H2bpdc) as a co-ligand, in an attempt to generate mixed-ligand coordination polymers. The mixed-ligand strategy has been employed successfully in a range of azole and azolate-containing MOFs,19 and can offer improved stability in the resulting frameworks by disfavouring hydrolytically unstable carboxylate bridging modes.20 The reaction of either HL1 or HL2 with H2bpdc and zinc nitrate in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF:H2O gave crystals of the coordination polymers poly-[Zn2(L1)2(bpdc)] 1 or poly-[Zn2(L2)2(bpdc)] 2, respectively. Both species share very similar local structures, with the asymmetric unit of each containing one molecule of the deprotonated azolate ligand, one tetrahedral zinc(II) ion and half of a bpdc dianion, as shown in Fig. 2 and 3. One DMF molecule could be resolved within the structure of 1, while the solvation in 2 was fully disordered and the scattering contribution of the solvated regions to the diffraction data was accounted for with a solvent mask (ESI†).
:1 DMF:H2O gave crystals of the coordination polymers poly-[Zn2(L1)2(bpdc)] 1 or poly-[Zn2(L2)2(bpdc)] 2, respectively. Both species share very similar local structures, with the asymmetric unit of each containing one molecule of the deprotonated azolate ligand, one tetrahedral zinc(II) ion and half of a bpdc dianion, as shown in Fig. 2 and 3. One DMF molecule could be resolved within the structure of 1, while the solvation in 2 was fully disordered and the scattering contribution of the solvated regions to the diffraction data was accounted for with a solvent mask (ESI†).
 | ||
| Fig. 2 (A) Metal and ligand environment in the structure of 1. ADPs are rendered at the 50% probability level and hydrogen atoms are omitted for clarity. (B) The metal–azolate layer in the structure of 1, carboxylate groups not shown; (C) the bpdc linkages between metal–azolate layers in the structure of 1. Symmetry codes used to generate equivalent atoms: (i) +x, 1 − y, z − 1/2; (ii) +x, 1 − y, z + 1/2; (iii) 3/2 − x, 1/2 − y, 1 − z. | ||
 | ||
| Fig. 3 (A) Metal and ligand environment in the structure of 2. ADPs are rendered at the 50% probability level and hydrogen atoms and lattice DMF molecules are omitted for clarity. (B) The metal–azolate layer in the structure of 2, carboxylate groups not shown; (C) the bpdc linkages between metal–azolate layers in the structure of 2. Symmetry codes used to generate equivalent atoms: (i) +x, 3/2 − y, z − 1/2; (ii) +x, 3/2 − y, 1/2 + z; (iii) 1 − x, 1 − y, 2 − z. | ||
The extended structures of both complexes are best visualised by considering the contributions of the heterocycle and carboxylate ligands separately. The coordination of the azolates is equivalent between the two complexes, where both ligands bridge three zinc ions. The two Zn–N bonds for the azolate moiety are marginally but reliably shorter than those involving the pyridine donor (2.007(3) and 1.990(3) vs. 2.069(3) Å for 1, 2.012(2) and 2.027(2) vs. 2.053(2) Å for 2), consistent with the expected increase in binding strength upon deprotonation of the pyrazole group. The coordination through the azolate fragment gives the six-membered [Zn2pz2] ring with a slight tendency towards a chair conformation by distortion of the zinc ions out of the plane of the heterocycles. Bridging of these units through the pyridine nitrogen atoms gives densely packed corrugated two-dimensional sheets. Despite the isomerism of the two heterocycles, both sheets share very similar structures, and the [Zn2pz2] nodes share nearly identical centroid–centroid distances in the two compounds (7.40 vs. 7.36 Å for 1 and 2, respectively).
The remaining coordination site of the zinc ions is occupied by a monodentate carboxylate oxygen atom, and in both cases the bpdc linker acts to bridge the 2-dimensional layers into 3-dimensional networks. A clear variation in structure is observed between the two complexes when considering the carboxylate bridging. In both cases, the carboxylate groups occupy the pseudo-equatorial 1,4-positions of the chair-like [Zn2pz2] nodes. However, alternating nodes within each layer are related by a slightly larger interplanar angle in 1 compared to 2 (69.2(3) vs. 59.1(3)°), with the result that the carboxylate groups extend at a different angle from the corrugated surface of the 2-D layers in the two structures. In 1, the bpdc ligands are near-parallel to the normal vector of the metal–azolate layer (14.1°), while in 2 this angle is much larger, at 41.0°. The result is a considerable difference in both pore shape and interlayer distance in the two structures, as shown in Fig. 4; 1 is defined by linear slit-shaped channels of approximate inter-atomic dimensions 15 × 8 Å and a 17.66 Å interlayer distance. This is compared with 2 which contains narrower (ca. 9 × 9 Å) triangular channels criss-crossed by bpdc molecules, and a shorter inter-layer distance of 15.95 Å. The difference in co-ligand geometry also manifests as a variation in solvent-accessible volume, with 1 showing larger free volume than 2 when disregarding lattice solvent molecules (45% vs. 38%, probe radius 1.2 Å).
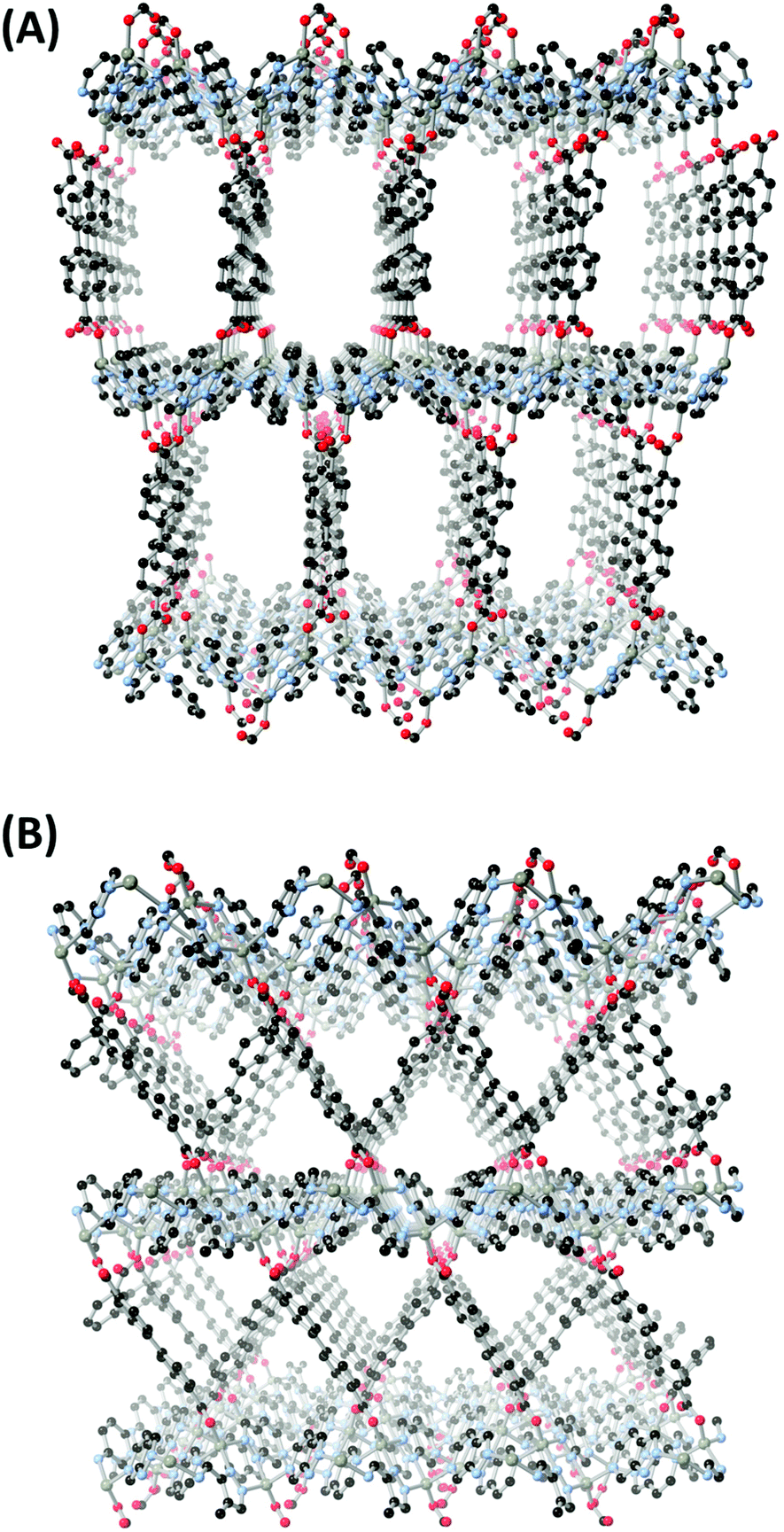 | ||
| Fig. 4 Comparison of the pore structures in MOFs 1 (A) and 2 (B). | ||
Taking the dinuclear [Zn2pz2] groups as six-connected nodes (linked to four others through L1/L2 ligands and two others through carboxylates), complex 1 adopts the rob topology, while complex 2 is described by the mab net (ESI, Fig. S1†). Both nets are formed by connecting (4,4) sql-type layers in the perpendicular direction with offset axial links, however while the layers are fully eclipsed in the highest symmetry form of rob, the mab net includes an additional half-cell offset of adjacent layers, leading to more oblique linkages between each plane. It is notable that no interpenetration is observed in these structures even despite the relatively large pore volumes and (in the case of 2) adopting a self-dual topology. This relates to the nature of the metal–azolate layers, which are close-packed and prohibit the threading of the bpdc groups within the loops comprised of four [Zn2pz2] nodes. As a polymeric secondary building unit, these azolate layers may prove useful in preventing interpenetration in other porous frameworks.
Gas adsorption in complexes 1 and 2
The considerable solvent-accessible volumes evident within the linear channels in complexes 1 and 2 prompted a study into the solvent exchange and gas uptake capabilities of these materials. The freshly isolated complex 1 exhibits a continuous multi-step mass loss immediately from room temperature to 220 °C of 25 wt% (calc. 22%) consistent with the lattice water and DMF molecules. The slight excess is due to surface solvent, the drying of which merges into the first onset of lattice desolvation. Mass loss in complex 2 initiates above 100 °C but similarly plateaus at approximately 220 °C with a total mass loss of 18 wt% (calc. 21%). Both materials undergo thermal decomposition with onset temperatures of 400–410 °C (ESI, Fig. S15 and S16†). The high desolvation temperature necessitated solvent exchange for both compounds before gas adsorption measurements. Immersion of the solids in methanol for 3 days, refreshing the solvent every 12 hours, effected complete exchange of the DMF guests within the pores, and post-exchange both materials reached complete desolvation below 100 °C by TGA. X-ray powder diffraction of the methanol-exchanged material revealed that crystallinity was retained in both cases. While the exchanged 2 gave a pattern near-identical to the fresh phase, the emergence of an additional reflection at 2θ = 6.0° in 1 with retention of all other major reflections may indicate a slight rearrangement accompanies solvent exchange in this material. Unfortunately, cracking in the individual crystallites precluded direct examination of the exchanged material with single crystal X-ray diffraction.The exchanged materials were activated at 100 °C under dynamic vacuum overnight prior to nitrogen and carbon dioxide adsorption measurements. Nitrogen adsorption (77 K) on both materials gave the expected type-I isotherms characteristic of microporous MOFs and no significant mesoporous features, as shown in Fig. 5. Complex 1 showed higher loading and a larger corresponding BET surface area of 860 m2 g−1, compared with 780 m2 g−1 for 2. Consistent with the larger and more accessible pores, 1 also exhibited a monodisperse pore size distribution centred at 8.5 Å diameter, while the median pore width in 2 fell below the measurement range at <5.5 Å diameter. Carbon dioxide adsorption isotherms were also carried out at 283, 293 and 303 K, and showed uniformly higher loading in 2 across all temperature ranges (Fig. 5). This finding is consistent with typical comparisons of narrow-pore versus large-pore materials,21 where narrower micropores tend to saturate at much lower partial pressures. While complex 1 reached a maximum loading of 40 cc(STP) g−1 at 1 atm without approaching saturation, complex 2 reached a loading of 80 cc(STP) g−1 (14 wt%) at 1 atm with a clear inflection in the adsorption branch indicating saturation of the micropores. The calculated enthalpy of adsorption for CO2 was essentially equivalent in both materials, at −32 kJ mol−1 for 1 and −31.5 kJ mol−1 for 2 at zero loading, with both trending monotonically downwards at higher loadings (ESI, Fig. S19†). The similarity in adsorption enthalpy is consistent with the near-identical pore surface chemistry in both materials.
 | ||
| Fig. 5 Comparison of gas adsorption isotherms between complexes 1 and 2, showing N2 isotherms at 77 K (top) and CO2 adsorption at 283 K (bottom). | ||
Interestingly, while zinc carboxylate MOFs are well known for poor stability to air or humidity exposure,22 especially after activation, X-ray powder diffraction of both 1 and 2 showed crystallinity was retained following gas adsorption and exposure to ambient air. After one week of air exposure, the dried samples were re-immersed in methanol for 24 hours and activated again under the same conditions (100 °C under dynamic vacuum overnight), and the nitrogen adsorption isotherms were re-measured. While both compounds showed a slight decline in uptake, the surface areas for the aged samples (690 and 680 m2 g−1 for 1 and 2, respectively) were still within 20% of their original values. This indicates that despite the carboxylate bridges, both materials show a surprisingly high stability to humidity, resisting the immediate degradation which is otherwise common in zinc carboxylate MOFs.
Synthesis and structures of L3–L6
Following the preparation of complexes containing the unsubstituted heterocycles L1 and L2, we turned our attention to their N-substituted chelating derivatives. Our intention was to expand upon the geometric differences shown between the two ligands in complexes 1 and 2 with studies of the combined steric and electronic influences in their covalently trapped 1H and 2H electronic configurations. Reacting the sodium salts of either HL1 or HL2 with 2-bromopyridine gave mixtures of L3/L4 or L5/L6, respectively. The pairs of isomers were separated chromatographically, and although the isolated yields of each were modest (7–16%), the total product recovery from each isomer of ca. 25% after chromatography is reasonable compared with that seen in other uncatalysed SNAr N-arylations of electron-deficient pyrazoles.23 The NMR spectra of the four isomers show clean separation of each compound and substantial variation in chemical shifts, indicating different degrees of bond localisation (ESI, Fig. S35†). This is exemplified in the pyrazole C–H resonance which tends to fall up to 1 ppm further downfield in the 2H isomers compared to the 1H, due to the predominance of the enamine-like resonance form. The substitution pattern also has a profound influence on the electronics of the fused pyridine ring, where the pyridyl C–H group nearest to the unsubstituted pyrazole nitrogen atom (H3 for L3/L4, H5 for L5/L6) exhibits a ca. 1 ppm downfield shift for the 1H isomer compared to the 2H.The structures of all four ligands were confirmed with single crystal X-ray diffraction, using crystals grown by slow evaporation from acetonitrile. All four isomers crystallised with a single molecule within the asymmetric unit and in the absence of any lattice solvent or guest molecules, as shown in Fig. 6. Each molecule adopts the expected trans-coplanar orientation of the two aromatic segments,24 with mean interplanar angles in the range 3.3–8.7°. The trends of partial bond localisation in the pyrazole ring and close to the ring junction suggested by the NMR are also evident in the crystallographic bond lengths. The 2H isomers L4 and L6 exhibit consistently longer ring junction C–C bond lengths compared to L3 and L5 (1.421(2) and 1.418(3) Å, vs. 1.404(2) and 1.399(2) Å respectively), and the pyrazole CH to ring junction C–C bond is likewise shorter in the 2H isomers compared to the 1H (1.389(2) and 1.385(3) Å vs. 1.418(3) and 1.423(3) Å for L4, L6, L3 and L5, respectively).
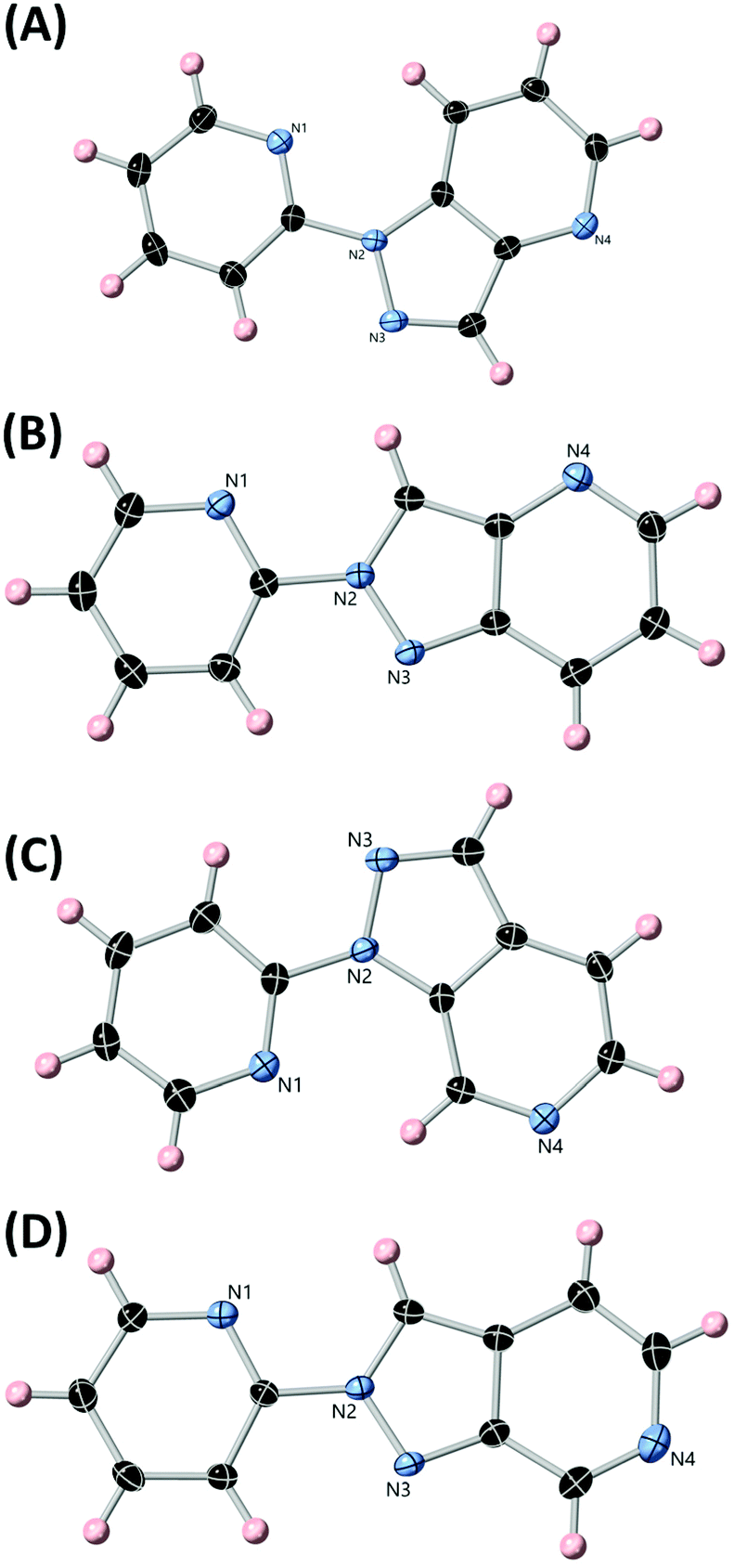 | ||
| Fig. 6 The structures of ligands L3, L4, L5 and L6 (A, B, C, D, respectively) with heteroatom labelling scheme. ADPs are rendered at the 50% probability level. | ||
The intermolecular interactions in all four of the ligand structures are, as expected, mainly characterised by face-to-face π⋯π interactions. The two 1H isomers both stack in linear head-to-tail columnar arrangements with efficient overlap between the pyridyl groups and the centre of the fused rings between adjacent layers. The mean interplanar distances of these interactions are 3.34 Å for L3 and 3.47 Å for L5. The 2H isomers tend to adopt slip-stacked columns with slightly smaller degrees of overlap between adjacent π systems, although the mean interplanar distances are similar (3.39 and 3.37 Å for L4 and L6, respectively).
Synthesis and structures of complexes 3–6
With the chelating ligands L3–L6 in hand and following crystallographic confirmation of the identity of each isomer, we turned our attention to generating zinc(II) complexes of each to establish the differences in coordination preference between the four isomers. Zinc was chosen as the ideal metal ion for these studies, as the d10 electron configuration of the dication not only tends towards more flexible ligand-directed coordination geometries, but can also retain or enhance the intrinsic photoluminescence of aromatic ligands for monitoring in solution by fluorescence spectroscopy.25 Single crystals of complexes 3–6 were prepared by the reaction of the corresponding ligands with zinc nitrate in acetonitrile, with single crystals either depositing from solution or following slow evaporation or the action of diethyl ether as an antisolvent.The diffraction data for poly-[Zn(L3)(NO3)2] 3 were solved and refined in the monoclinic space group P21/n, where the asymmetric unit contains a single zinc(II) ion coordinated by a molecule of L3 and two nitrato ligands, shown in Fig. 7. Both nitrato ligands coordinate in a weakly chelating mode, and while the Zn–O distances for O4 and O5 are similar (2.2755(15) and 2.2998(18) Å respectively), O2 is more tightly bound than O1 (2.1086(13) vs. 2.3262(12) Å respectively). Coordination of two symmetry-related molecules of L3 through all three nitrogen atoms gives a seven-coordinate distorted pentagonal bipyramidal coordination geometry, with the axial positions occupied by the pyridine nitrogen atoms N1 and N4. Both pyridine nitrogen atoms exhibit shorter Zn–N bonds than the pyrazole nitrogen atom N3 (2.1077(12), 2.1415(11) and 2.1888(11) Å, for N1, N4 and N3 respectively).
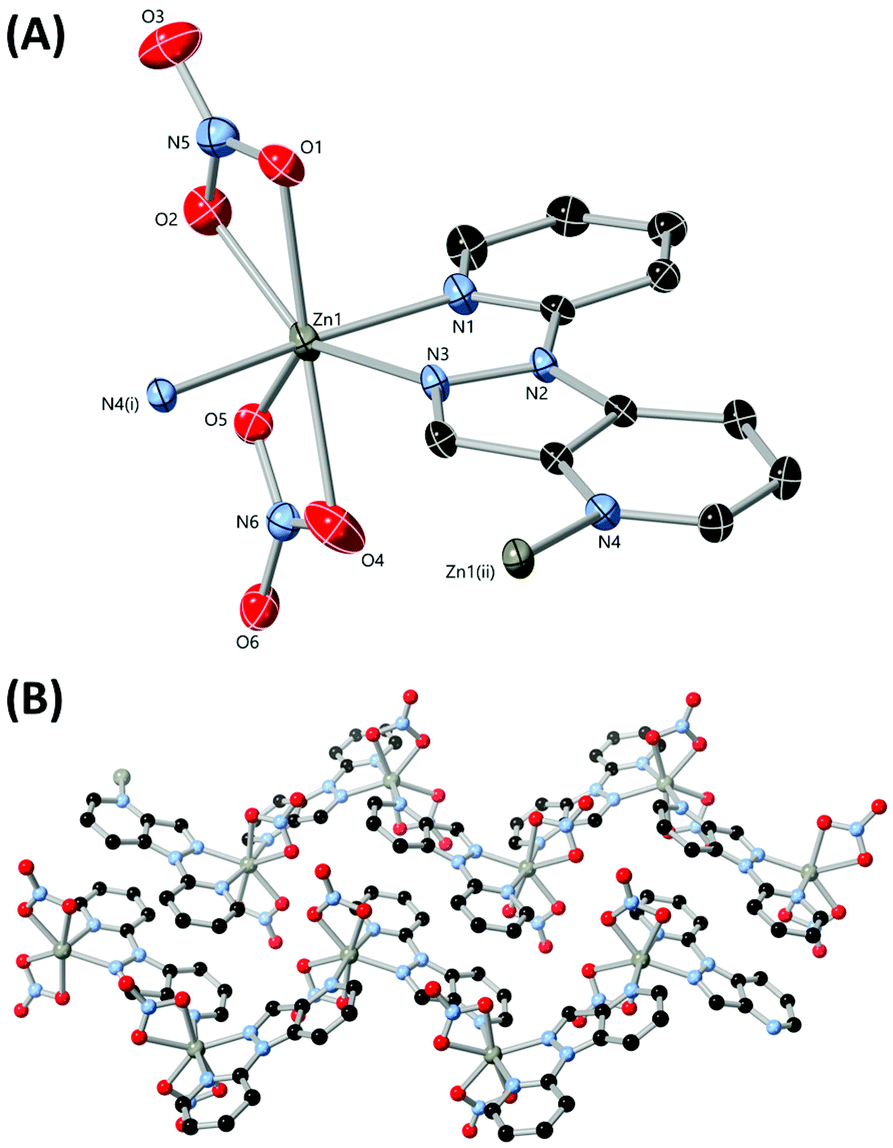 | ||
| Fig. 7 (A) The metal and ligand environment in the structure of complex 3 with heteroatom labelling scheme. Hydrogen atoms are omitted for clarity and ADPs are rendered at the 50% probability level. (B) Extended structure of 3 showing propagation of two adjacent chains and π⋯π interactions between ligand molecules. Symmetry codes used to generate equivalent atoms: (i) 1/2 − x, y − 1/2, 3/2 − z; (ii) 1/2 − x, 1/2 + y, 3/2 − z. | ||
Linkage of adjacent zinc ions through the bridging L3 gives a one-dimensional polymeric structure oriented parallel to the b axis. The Zn–Zn distance afforded by this bridging mode is 6.8589(2) Å, ca. 10% larger than the equivalent distance in complex 1 (6.1490(7) Å). Adjacent chains associate through a dimeric π⋯π stacking interaction between parallel L3 groups at a mean interplanar distance of 3.40 Å. Similar to other systems containing angular chelating heterocycles where nitrate anions are present26 this interaction is supported by a reciprocated pair of chelating C–H⋯O contacts, between the nitrato oxygen atom O1 and the two inward-facing pyridyl C–H groups C3 and C8. The C⋯O distances and C–H⋯O angles (3.645(2) and 3.239(2) Å, 161.6 and 138.4° for C3 and C8 respectively) are consistent with the expected values for typical C–H⋯O contacts.27
The diffraction data for [Zn(L4)(NO3)2(OH2)] 4 were solved and refined in the orthorhombic space group Pbcn, and the asymmetric unit contains a five-coordinate zinc(II) ion bound by one chelating L4 molecule, two monodentate nitrato ligands and an aqua ligand as shown in Fig. 8. As expected for the lower coordination number all metal–ligand bonds are shorter compared to those in complex 3, but notably the pyrazole and pyridine coordination bonds are statistically equivalent in length (2.102(3) and 2.103(3) Å for N3 and N1, respectively). The coordination geometry of the zinc ion is best described as distorted square planar (τ5 = 0.24)28 with the nitrato oxygen atom O1 occupying the axial position.
 | ||
| Fig. 8 (A) The metal and ligand environment in the structure of complex 4 with heteroatom labelling scheme. Selected hydrogen atoms are omitted for clarity and ADPs are rendered at the 50% probability level. (B) The hydrogen bonding modes present in the structure of 4, linking adjacent molecules via the aqua ligands. | ||
Adjacent complexes are linked through hydrogen bonding originating from the aqua ligand, involving the non-coordinating pyridine nitrogen atom N4 and a bifurcated interaction with the non-coordinating nitrato oxygen atoms O5 and O6. With each complex donating and accepting two hydrogen bonds each, the complex acts as a four-connected node in a 2-dimensional (4,4) sheet in the bc plane. By comparison, π⋯π interactions play a relatively minor role in the extended structure of 4, with only a partial overlap between the two pyridine rings evident (interplanar distance 3.34 Å) and the rest of the aromatic surface only engaging in more diffuse intermolecular contacts.
The reaction of L5 with zinc nitrate unavoidably gave two crystalline phases. The predominant phase [Zn(L5)(H2O)4](NO3)2·H2O 5α (∼94 wt% by elemental analysis) formed as colourless needles when the reaction solution was concentrated near dryness. However, under all reaction conditions a second phase [Zn2(L5)5(OH2)2]·2MeCN 5β was observed continuously depositing poorly crystalline plates in the reaction mixture. Modifying the reaction stoichiometry, water content or crystallisation method failed to produce either phase in an analytically pure form, most likely due to rapid equilibration of these species in solution, and so individual crystals of each phase were isolated from the reaction solution for structure determination.
The diffraction data for 5α were solved and refined in the monoclinic space group P21/c, and the asymmetric unit contains one six-coordinate zinc(II) ion coordinated by a chelating L5 molecule and four aqua ligands, with two non-coordinating nitrate anions and one water molecule also present within the asymmetric unit. The structure of 5α is shown in Fig. 9. As with 4, the Zn–N distances for the pyridine and pyrazole group are essentially equivalent (2.135(4) and 2.144(4) Å for N1 and N3, respectively). A minor torsion is also evident between the two coordinating heterocycles (14.9° interplanar angle).
 | ||
| Fig. 9 (A) The metal and ligand environment in the structure of 5α with heteroatom labelling scheme. Hydrogen atoms and the lattice water molecule are omitted for clarity and ADPs are rendered at the 50% probability level. (B) The hydrogen bonding environment in the structure of 5α showing the aqua ligands and lattice water molecule donating hydrogen bonds to the nitrate anions and non-coordinating pyridine nitrogen atom. | ||
The [Zn(L5)(H2O)4] dication contains eight hydrogen bond donors, and so unsurprisingly the extended structure of 5α is dictated by the extensive 3-dimensional hydrogen bonding network. The two nitrate anions each accept either three or four hydrogen bonds, while the lattice water molecule accepts two and donates two. Notably, the non-coordinating pyridine nitrogen atom accepts a hydrogen bond from the lattice water molecule (d(O⋯N) = 2.819(5) Å) rather than the more acidic aqua ligands. The π⋯π interactions in 5α are largely confined to a dimeric association of two L5 molecules involving only the fused pyrazolopyridine rings, with an interplanar distance of 3.30 Å.
The crystals of the minor phase 5β were generally poor quality and suffered from both weak scattering and heavy disorder; nonetheless, an indicative connectivity model could be obtained. The diffraction data were solved in the monoclinic space group P21/c. As shown in Fig. 10, the structure is a dinuclear complex of the formulation [μ2-L5{Zn(L5)2(NO3)(OH2)}2]2+, where one L5 species bridges two zinc ions, whose distorted octahedral coordination spheres are completed by two further monodentate L5 ligands alongside nitrato and aqua ligands. The presence of monodentate L5 ligands is unexpected given the expected thermodynamic advantage of chelating from these species, and suggests that the low solubility of this complex may play a more important role in its crystallisation than any particular prevalence in solution (as supported by solution studies, vide infra).
 | ||
| Fig. 10 The overall structure of complex 5β with labelling scheme for unique heteroatoms; the A suffixes represent the second disordered component for the nitrato ligands. Hydrogen atoms, lattice nitrate anions and the disorder in the central bridging L5 molecule are omitted for clarity (ESI, Fig. S2†). ADPs are rendered at the 50% probability level. | ||
The bridging L5 molecule and both zinc ions are subject to significant crystallographic disorder relating to overlap of the inversion centre with the bridging ligand (ESI†). Equivalent disorder was evident in lower symmetry solutions, and no obvious larger cells could be indexed, suggesting genuine distributed disorder of this group. Similar effects have been observed in other chelating pyrazolylpyridines,29 especially in cases where the crystal packing does not differentiate between either possible orientation of the ligand. The predominant interaction between adjacent complexes in the structure of 5β is a tetrameric face-to-face stack formed by the terminal monodentate ligands with association of two complexes. This interdigitation links the complexes into a 1-dimensional chain along c, with further interactions involving hydrogen bonding with the aqua ligand or more diffuse interactions with the external complex surfaces.
The diffraction data for poly-[Zn(L6)(NO3)2]·2MeCN 6 were solved and refined in the monoclinic space group C2/c. As shown in Fig. 11, the asymmetric unit contains one zinc(II) ion bound by an L6 molecule and two nitrato ligands, with additional coordination through a symmetry equivalent L6 molecule to generate a polymeric species. Two acetonitrile molecules are also present within the asymmetric unit. The coordination geometry of the zinc ion is distorted square pyramidal (τ5 = 0.22), with the vacant face weakly capped by an additional nitrato oxygen atom O1 at a long contact distance of 2.420(3) Å. The chelation through L6 shows similar bond lengths for the pyrazole and pyridine nitrogen atoms, slightly favouring the pyrazole (2.127(3) and 2.199(3) Å for N3 and N1 respectively), although a shorter bond of 2.074(3) Å is observed for the terminal coordination through N4. The bridging distance provided by L6 is 6.5203(8) Å, ca. 0.3 Å shorter than that observed in 3.
 | ||
| Fig. 11 (A) The metal and ligand environment in the structure of complex 6 with heteroatom labelling scheme. Hydrogen atoms and lattice acetonitrile molecules are omitted for clarity and ADPs are rendered at the 50% probability level. (B) The extended structure of complex 6 showing the packing of two adjacent chains. Symmetry codes used to generate equivalent atoms: (i) 3/2 − x, y − 1/2, 3/2 − z; (ii) 3/2 − x, 1/2 + y, 3/2 − z. | ||
The extended structure of 6 is a one-dimensional coordination polymer oriented parallel to the b axis. Adjacent chains are further linked along c through π⋯π interactions which overlap across the entire face of the ligand at an interplanar distance of 3.39 Å. The resulting sheets pack with alternating solvent channels containing the lattice acetonitrile molecules. The crystals of 6 were extremely sensitive to drying in ambient air and lost crystallinity almost immediately on removal from solvent, though remained stable when stored under acetonitrile in a capillary for X-ray powder diffraction. Elemental analysis of the dried material suggests that atmospheric water is taken up by the material following the loss of lattice solvent on drying.
Photophysics and solution-state coordination of L3–L6
The structural investigations of complexes 3–6 above reveal broadly similar coordination behaviour for all 4 ligands, including the predominance of a 1:1 M:L stoichiometry in all the major phases crystallised. However, the minor variations in coordination number and geometry relating to crystal packing and solubility effects makes the relative binding strength of each ligand difficult to discern from these data. As such, we turned our attention to studies of the coordination chemistry of these ligands with zinc(II) in solution, with the expectation that at micromolar concentrations the monomeric [ML] complexes would be prevalent and allow determination of association constants. Evaluation of the free ligands by UV-Visible absorption and fluorescence spectroscopy showed that all four ligands possess suitable absorbances near 300 nm, as shown in Fig. 12, most likely corresponding to n → π* transitions. Excitation into these bands gave fluorescence emission with maxima in the range 360–400 nm. The 2H isomers L4 and L6 were both red-shifted compared to their 1H equivalents, and showed considerably lower emission intensities than L3 and L5, while L6 was the only compound to show partially resolved vibronic structure in its emission band in acetonitrile. Excitation spectra were used to confirm the optical purity of all ligands and showed good agreement with the absorption spectra (ESI, Fig. S3–S6†).
 | ||
| Fig. 12 Combined absorption (A) and normalized emission (B) spectra for ligands L3–L6 in acetonitrile. The narrow band marked with * in the emission spectrum of L4 corresponds to a Raman scattering band from the solvent, visible for this compound due to its very low emission intensity. L3 10.7 μM, λex = 310 nm; L4 10.8 μM, λex = 326 nm; L5 10.2 μM, λex = 328 nm; L6 10.5 μM, λex = 287 nm. | ||
In all four cases, the addition of zinc nitrate aliquots in acetonitrile to the ligand solutions at 10–30 μM concentrations caused relatively minor changes to the absorption spectra, and required large excesses (20–30 eq.) of metal before the isotherms reached saturation (ESI, Fig. S7–S10†). As a result, the absorbances due to free zinc nitrate in solution overwhelmed the higher energy ligand absorbances. As such we focused on the fluorescence spectra, where the metal binding events were much clearer. The resulting titration profiles are shown in Fig. 13. For each ligand, the addition of zinc led to the growth of a new emission band at lower energy. For the 1H isomers L3 and L5, the new bands exhibited similar intensity to the free ligand once saturated, and growth of the bound species was accompanied by diminishing of the free ligand emission. Conversely, the emission intensity was substantially enhanced for L4 and L6 (by factors of 40 and 5, respectively) following binding.
 | ||
| Fig. 13 Emission profiles for ligands L3–L6 upon addition of zinc nitrate solution in acetonitrile, with representative binding isotherms (blue) and fits (green) inset. Arrows show the evolution of the emission profiles after the successive addition of zinc aliquots. (A) L3, initial concentration = 27 μM, λex = 310 nm; (B) L4, initial concentration = 14 μM, λex = 326 nm; (C) L5, initial concentration = 13 μM, λex = 328 nm; (D) L6, initial concentration = 13 μM, λex = 287 nm. | ||
The binding isotherms were subjected to global non-linear least squares fitting routines using ReactLab Equilibria,30 initially using a simple 1M:1L binding model (Table 1). The emission spectra calculated for each species using these global fits are shown in Fig. S11–S14, ESI.† This model showed good agreement with the data for L3 and L5, providing association constants logK1:1 of 3.46(9) and 3.61(11) respectively. No improvement in the fits were obtained by adding any additional species to either model, including the minor M2L5 species observed crystallographically for L5. The isotherms for the 2H isomers L4 and L6, however, yielded poor fits with large residuals when using only the 1:1 binding model. Examining the data it is clear that the emission spectra undergo two separate changes throughout the titration for these species, which are more evident in the simulated spectra for each species in solution obtained from the subsequent fits (ESI, Fig. S12 and S14†). For L4, the emission maximum of the new band is unaffected by the consumption of ligand due to its very low intensity, and gradually shifts from 404 nm to 393 nm as excess zinc(II) is added. The structured emission spectra of L6, however, made the variations throughout the titration more obvious. In the early part of the titration a single emission band with λmax = 400 nm emerged which, in the presence of larger excesses of zinc, was replaced with another structured emission band with λmax = 398 nm but with visible shoulders at 385 and 367 nm. The best global fits to these data were obtained with a simultaneous 1M:1L and 1M:2L binding model, giving logK1:1 values of 3.58(12) and 4.37(19), and logβ2:1 values of 8.70(8) and 9.64(4) for L4 and L6, respectively.
The spectroscopic data and model fits provide interesting insights into the coordination chemistry of these species, especially in the context of the structural study. Firstly, in the absence of crystal packing effects there is no discernible difference in solution binding affinity between L3 and L5. This suggests that variation in the fused ring substitution pattern is not significant for the 1H isomers in the absence of crystal packing effects and when coordination from the backbone pyridine nitrogen atom is disfavoured due to concentration. However, the switch from the 1H to the 2H substitution pattern causes the emergence of an ML2 species in addition to the expected ML complex. Given the first association constant for L4 is also equivalent to that for L3, this difference is likely steric rather than indicating any particular difference in binding strength between these two isomers. In the case of L6, however, the first binding event is an order of magnitude more favourable than any of the other three isomers. This indicates stronger individual binding from this species by virtue of both the substitution pattern and the resulting electronic configuration, which acts in addition to the steric influence that favours a second coordination event (as an isostere of L4).
Experimental
Full details of instrumentation and data collection methods are provided as ESI.†Synthesis of L3/L4
HL1 (1.67 g, 14.0 mmol) was dissolved in anhydrous DMF (28 mL) under a nitrogen atmosphere at 0 °C, and sodium hydride (60% dispersion in mineral oil, 690 mg, 17.3 mmol) was added gradually over 30 minutes. After gas evolution ceased 2-bromopyridine (1340 μL, 14.0 mmol) was added in one portion, and the suspension heated to 120 °C. After 72 hours, the reaction was cooled to room temperature before being added to water (100 mL). The product was extracted into ethyl acetate and the combined organic layers washed with water, dried with anhydrous MgSO4 and evaporated to dryness. The orange oil was purified by flash chromatography (silica gel, gradient 50:50 to 100:0 ethyl acetate:hexane) to yield L3 and L4 as off-white and pale-yellow crystalline solids respectively. Single crystals of both L3 and L4 for X-ray diffraction were obtained by slow evaporation of acetonitrile solution.
Synthesis of L5/L6
HL2 (1.07 g, 8.98 mmol) was dissolved in anhydrous DMF (20 mL) under a nitrogen atmosphere at 0 °C. Sodium hydride (60% dispersion in mineral oil, 443 mg, 11.1 mmol) was added gradually over 30 minutes, followed by 2-bromopyridine (800 μL, 8.98 mmol). The resulting suspension was heated at 120 °C for 72 hours. The reaction mixture was cooled to room temperature before being added to water (100 mL). The product was extracted into ethyl acetate, and the combined organic layers washed with water, dried with anhydrous MgSO4 and evaporated to dryness. The orange oil was purified by flash chromatography (silica gel, gradient 50:50 to 100:0 ethyl acetate:hexane) to yield L5 and L6, as pale-yellow crystalline solids. Single crystals of both L5 and L6 for X-ray diffraction were obtained by slow evaporation of acetonitrile solution.
Synthesis of poly-[Zn2(L1)2(bpdc)]·1.5DMF·3H2O 1
To an 8 mL scintillation vial with a PTFE-lined cap was added HL1 (10 mg, 84 μmol), zinc nitrate hexahydrate (25 mg, 84 μmol) and 4,4′-biphenyldicarboxylic acid (6 mg, 25 μmol), along with 3 mL of a 2:1 DMF/H2O mixture. The resulting suspension was dispersed by sonication and heated at 100 °C for 18 hours, giving the product as a white crystalline solid. Yield 8.6 mg (44%). MP > 300 °C; found C, 47.13; H, 3.80; N, 13.99; calc. for C26H16N6O4Zn2·1.5DMF·3H2O C, 47.52; H, 4.25; N, 13.63. vmax/cm−1 3151w, 3091w, 2922w, 2852w, 1663s, 1608s, 1566w, 1550w, 1357s, 1240m, 1197w, 1174w, 1133w, 1080s, 996m, 926m, 846m, 803m, 766s, 686m, 660m.
Synthesis of poly-[Zn2(L2)2(bpdc)]·2DMF·H2O 2
To an 8 mL scintillation vial with a PTFE-lined cap was added HL2 (10 mg, 84 μmol), zinc nitrate hexahydrate (25 mg, 84 μmol) and 4,4′-biphenyldicarboxylic acid (6 mg, 25 μmol), along with 3 mL of a 2:1 DMF/H2O mixture. The resulting suspension was dispersed by sonication and heated at 100 °C for 18 hours, giving the product as a white crystalline solid. Yield 8.3 mg (43%). MP > 300 °C; found C, 49.88; H, 3.90; N, 14.68; calc. for C26H16N6O4Zn2·2DMF·H2O C, 49.82; H, 4.18; N, 14.53%; vmax/cm−1 3131w, 3107w, 3072w, 3046w, 2919m, 2852w, 1671s, 1597s, 1547m, 1470w, 1365s sh, 1256m, 1208w, 1139w, 1185s, 1134s, 999m, 934w, 853s, 771s, 689m, 653m, 603s.
Synthesis of poly-[Zn(L3)(NO3)2] 3
A solution of L3 (11 mg, 56 μmol) dissolved in acetonitrile (3 mL) was added to a solution of zinc nitrate hexahydrate (13 mg, 44 μmol) dissolved in acetonitrile (3 mL). To this solution was added diethyl ether (40 mL) and sealed for 5 days, yielding colourless crystals, which were filtered and washed with acetonitrile. We found the crystals of 3 to be relatively hygroscopic; air-dried samples retained significant amounts of moisture, and samples dried in vacuo tended to regain atmospheric water on their surfaces. 3 (4.9 mg, 29%) MP. 248–252 °C (decomp). Found C, 33.95; H, 2.98; N, 21.30; calc. for C11H8N6O6Zn C, 34.26; H, 2.09; N, 21.79; vmax/cm−1 3243br, 3135w, 1673w, 1612m, 1580m, 1470m, 1416s, 1316vs, 1196m, 1150w, 1096w, 1022m, 999s, 919s, 860m, 820m, 770w, 736m, 695w, 635m.Synthesis of [Zn(L4)(NO3)2(OH2)] 4
A solution of L2 (11 mg, 56 μmol) dissolved in acetonitrile (3 mL) was added to a solution of zinc nitrate hexahydrate (13 mg, 44 μmol) dissolved in acetonitrile (3 mL). The solution was sealed, and left to stand for 2 days yielding colourless crystals, which were filtered, washed with acetonitrile, and dried in air. 4 (11 mg, 61%). MP. 242–246 °C (decomp); found C, 32.91; H, 2.67; N, 21.16; calc. for C11H10N6O7Zn C, 32.73; H, 2.50; N, 20.82%; vmax/cm−1 3417br, 3129w, 3107w, 1667w, 1618m, 1575w, 1474m, 1449m, 1398m, 1297vs, 1218m, 1162w, 1131w, 1075w, 1021m, 988w, 933w, 808s, 784s, 708w.Synthesis of [Zn(L5)(H2O)4](NO3)2·H2O 5α and [Zn2(L5)5(OH2)2]·2MeCN 5β
A solution of L5 (10 mg, 51 μmol) was dissolved in acetonitrile (1.5 mL), and a solution of zinc nitrate hexahydrate (14 mg, 47 μmol) in acetonitrile (500 μL) was added. The resulting mixture was mixed until fully dissolved and allowed to concentrate by slow evaporation to near-dryness, at which point the crystalline material (comprising predominantly 5α and a small but unavoidable quantity of 5β) were isolated by filtration. The presence of both phases was confirmed by X-ray powder diffraction (ESI†), and elemental analysis suggested a molar ratio of ca. 98:2 5α:5β corresponding to a ratio by mass of 94:6 5α:5β. Yield 2.5 mg (11%). MP 251–254 °C; found C, 28.68; H, 3.48; N, 17.73; calculated for {0.98(C11H18N6O11Zn)·0.02(C59H50N26O14Zn2)} C, 28.97; H, 3.79; N, 18.08%; vmax/cm−1 3208m br, 3110m, 1662w, 1614m, 1577w, 1563w, 1482m, 1465m, 1439m, 1437m, 1333s, 1207s, 1154m, 1121m, 1100m, 1036s, 997s, 891m, 824s, 770s, 734s, 624m.
Synthesis of poly-[Zn(L6)(NO3)2]·2MeCN 6
A solution of L6 (10 mg, 51 μmol) dissolved in acetonitrile (6 mL) was added to a solution of zinc nitrate hexahydrate (12 mg, 40 μmol) dissolved in acetonitrile (6 mL). The solution was sealed, and left for 10 days yielding colourless crystals, which were filtered, washed with acetonitrile, and dried in air. The crystals rapidly lose solvent on removal from the mother liquor, and elemental analysis suggests this process is accompanied by minor uptake of atmospheric water. 6 (6.1 mg, 39% dry basis). MP 262–266 °C (decomp). Found C, 33.87; H, 2.23; N, 21.75; calculated for C11H8N6O6Zn·1/3H2O C, 33.74; H, 2.23; N, 21.46% vmax/cm−1 3144w, 1608w, 1502w, 1470m, 1442m, 1305s, 1283s, 1224m, 1159w, 1114w, 1078w, 1023m, 989m, 842w, 782s, 716m, 654m, 633w.Conclusions
The pyrazolo[4,3-b]pyridine (HL1) and pyrazolo[3,4-c]pyridine (HL2) platforms make versatile starting points for constructing multifunctional heterocyclic ligands for coordination assemblies. Using the parent heterocycles two new permanently porous zinc(II) MOFs 1 and 2 were prepared using a mixed-ligand strategy. The minor geometric changes in the heterocycle backbones led to profound changes in the pore size and shape of the materials. Both MOFs also showed unusually high stability against atmospheric water, retaining over 80% of their accessible surface areas one week after full desolvation in ambient air. N-Arylation of these heterocycles gave four new chelating ligands L3–L6, which showed a range of coordination behaviours with zinc in the solid state, including discrete and polymeric species and an M2L5 assembly as a minor product. Investigation of their coordination chemistry in solution revealed that the 1H isomers L3 and L5 exhibit effectively equivalent coordination chemistry. However, the different steric demand in the 2H isomers promotes formation of both ML and ML2 complexes, and this in combination with the more favourable electronic configuration in L6 translated to an order of magnitude increase in association constant for this ligand compared to the three other isomers. For the development of functional metallosupramolecular materials, precise tuning of the combined sterics and electronics of increasingly densely functionalised organic ligands will be essential, and these studies have shown both the promise and potential complications offered by fused pyrazolopyridines towards this goal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Royal Society of Chemistry for provision of an Undergraduate Research Bursary to A. M. S. (U21-3181589040), and the School of Chemical and Physical Sciences, Keele University for financial support. We gratefully acknowledge the technical staff of the Lennard-Jones Laboratories and Central Science Laboratories at Keele University for their invaluable work in laboratory and instrument support.References
- S.-S. Zhao, J. Yang, Y.-Y. Liu and J.-F. Ma, Inorg. Chem., 2016, 55, 2261–2273 CrossRef CAS PubMed; E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC; K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119–4130 CrossRef PubMed.
- Y.-L. Huang, P.-L. Qiu, J.-P. Bai, D. Luo, W. Lu and D. Li, Inorg. Chem., 2019, 58, 7667–7671 CrossRef CAS PubMed; D. Sensharma, N. Zhu, S. Tandon, S. Vaesen, G. W. Watson and W. Schmitt, Inorg. Chem., 2019, 58, 9766–9772 CrossRef PubMed.
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS PubMed; X. Zhang, Z. Zhang, J. Boissonnault and S. M. Cohen, Chem. Commun., 2016, 52, 8585–8588 RSC; E. M. Miner, S. Gul, N. D. Ricke, E. Pastor, J. Yano, V. K. Yachandra, Y. Van Voorhis and M. Dincă, ACS Catal., 2017, 7, 7726–7731 CrossRef.
- C. Hua, P. W. Doheny, B. Ding, B. Chan, M. Yu, C. J. Kepert and D. D'Alessandro, J. Am. Chem. Soc., 2018, 140, 6622–6630 CrossRef CAS PubMed; H. C. Wentz, G. Skorupskii, A. B. Bonfim, J. L. Mancuso, C. H. Hendon, E. H. Oriel, G. T. Sazama and M. G. Campbell, Chem. Sci., 2020, 11, 1342–1346 RSC.
- D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef CAS PubMed.
- L. N. McHugh, M. J. McPherson, L. J. McCormick, S. A. Morris, P. S. Wheatley, S. J. Teat, D. McKay, D. M. Dawson, C. E. F. Sansome, S. E. Ashbrook, C. A. Stone, M. W. Smith and R. E. Morris, Nat. Chem., 2018, 10, 1096–1102 CrossRef CAS PubMed; A. Schneemann, V. Bon, I. Schedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC; A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef.
- M. I. Severino, E. Gkaniatsou, F. Nouar, M. L. Pinto and C. Serre, Faraday Discuss., 2021, 231, 326–341 RSC.
- R. E. Morris and L. Brammer, Chem. Soc. Rev., 2017, 46, 5444–5462 RSC; J. I. Feldblyum, M. Liu, D. W. Gidley and A. J. Matzger, J. Am. Chem. Soc., 2011, 133, 18257–18263 CrossRef CAS PubMed; Y. Wen, Q. Liu, S. Su, Y. Yang, X. Li, Q.-L. Zhu and X. Wu, Nanoscale, 2020, 12, 12767–12772 RSC.
- S. Trofimenko, Chem. Rev., 1972, 72, 497–509 CrossRef CAS; M. A. Halcrow, Dalton Trans., 2009, 2059–2073 RSC; C. Cuerva, J. A. Campo, P. Ovejero, M. R. Torres and M. Cano, Dalton Trans., 2014, 43, 8849–8860 RSC; J. S. Train, A. B. Wragg, A. J. Auty, A. J. Metherell, D. Chekulaev, C. G. P. Taylor, S. P. Argent, J. A. Weinstein and M. D. Ward, Inorg. Chem., 2019, 58, 2386–2396 CrossRef PubMed; I. Galadzhun, R. Kulmaczewski, N. Shahid, O. Cespedes, M. J. Howard and M. A. Halcrow, Chem. Commun., 2021, 57, 4039–4042 RSC.
- C. Pettinari, A. Tăbăcaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–13 CrossRef CAS; C. Heering, I. Boldog, V. Vasylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757–9768 RSC; H. S. Scott, S. Mukherjee, D. R. Turner, M. I. J. Polson, M. J. Zaworotko and P. E. Kruger, CrystEngComm, 2018, 20, 1193–1197 RSC; M. R. Bryant, A. D. Burrows, C. M. Fitchett, C. S. Hawes, S. O. Hunter, L. L. Keenan, D. J. Kelly, P. E. Kruger, M. F. Mahon and C. Richardson, Dalton Trans., 2015, 44, 9269–9280 RSC; J.-P. Zhang, S. Horike and S. Kitagawa, Angew. Chem., Int. Ed., 2007, 46, 889–892 CrossRef PubMed.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed; A. Nimmermark, L. Öhrström and J. Reedijk, Z. Kristallogr., 2013, 228, 311–317 Search PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS; H. Zhang, D. Liu, Y. Yao, B. Zhang and Y. S. Lin, J. Membr. Sci., 2015, 485, 103–111 CrossRef; S. Bhattacharyya, R. Han, W.-G. Kim, Y. Chiang, K. C. Jarachandrababu, J. T. Hungerford, M. R. Dutzer, C. Ma, K. S. Walton, D. S. Sholl and S. Nair, Chem. Mater., 2018, 30, 4089–4101 CrossRef; N. Novendra, J. M. Marrett, A. D. Katsenis, H. M. Titi, M. Arhangelskis, T. Friščić and A. Navrotsky, J. Am. Chem. Soc., 2020, 142, 21720–21729 CrossRef.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC; K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef CAS; C. R. Wade, T. Corrales-Sanchez, T. C. Narayan and M. Dincă, Energy Environ. Sci., 2013, 6, 2172–2177 RSC.
- C. S. Hawes, R. Babarao, M. R. Hill, K. F. White, B. F. Abrahams and P. E. Kruger, Chem. Commun., 2012, 48, 11558–11560 RSC; A. A. Garcia-Valdivia, M. Pérez-Mendoza, D. Choquesillo-Lazarte, J. Cepeda, B. Fernández, M. Souto, M. González-Tejero, J. A. García, G. M. Espallargas and A. Rodríguez-Diéguez, Cryst. Growth Des., 2020, 20, 4550–4560 CrossRef CAS; A. A. García-Valdivia, A. Zabala-Lekuona, G. B. Ramírez-Rodríguez, J. M. Delgado-López, B. Fernández, J. Cepeda and A. Rodríguez-Diéguez, CrystEngComm, 2020, 22, 5086–5095 RSC.
- A. A. Watson, D. A. House and P. J. Steel, Aust. J. Chem., 1995, 48, 1549–1572 CrossRef CAS; R. J. Weekes and C. S. Hawes, CrystEngComm, 2019, 21, 5152–5163 RSC; T. Godau, F. Platzmann, F. W. Heinemann and N. Burzlaff, Dalton Trans., 2009, 254–255 RSC.
- A. Schmidt, A. Beutler and B. Snovydovych, Eur. J. Org. Chem., 2008, 4073–4095 CrossRef CAS.
- N. Aljaar, M. Al-Noaimi, J. Conrad and U. Beifuss, J. Org. Chem., 2021, 86, 1408–1418 CrossRef CAS PubMed; X. Ding, J. Bai, H. Wang, B. Zhao, J. Li and F. Ren, Tetrahedron, 2017, 73, 172–178 CrossRef.
- C. S. Hawes and P. E. Kruger, Dalton Trans., 2014, 43, 16450–16458 RSC.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed; C. S. Hawes, Dalton Trans., 2021, 50, 6034–6049 RSC.
- M. E. A. Safy, M. Amin, R. R. Haikal, B. Elshazly, J. Wang, Y. Wang, C. Woll and M. H. Alkordi, Chem. – Eur. J., 2020, 26, 7109–7117 CrossRef CAS PubMed; P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef; J. B. DeCoste, G. W. Peterson, B. J. Schindler, K. L. Killops, M. A. Brow and J. J. Mahle, J. Mater. Chem. A, 2013, 1, 11922–11932 RSC; K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2012, 24, 3153–3167 CrossRef.
- Z. Zhang, J. Zhou, W. Xing, Q. Xue, Z. Yan, S. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523–2529 RSC; A. Kumar, C. Hua, D. G. Madden, D. O'Nolan, K.-J. Chen, L.-A. J. Keane, J. J. Perry IV and M. J. Zaworotko, Chem. Commun., 2017, 53, 5946–5949 RSC.
- P. Guo, D. Dutta, A. G. Wong-Foy, D. W. Gidley and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 2651–2657 CrossRef CAS PubMed.
- M. A. Halcrow, Coord. Chem. Rev., 2005, 249, 2880–2908 CrossRef CAS.
- C. M. Fitchett, C. Richardson and P. J. Steel, Org. Biomol. Chem., 2005, 3, 498–502 RSC.
- N. J. Williams, W. Gan, J. H. Reibenspies and R. D. Hancock, Inorg. Chem., 2009, 48, 1407–1415 CrossRef CAS PubMed; S. J. Butler, Chem. – Eur. J., 2014, 20, 15768–15774 CrossRef PubMed; V. Venkatesan, S. Kumar, S. K. Ashok Kumar and S. K. Sahoo, Inorg. Chem. Commun., 2019, 102, 171–179 CrossRef; Y. Mikata, Y. Sato, S. Takeuchi, Y. Kuroda, H. Konno and S. Iwatsuki, Dalton Trans., 2013, 42, 9688–9698 RSC.
- H. L. Dalton, C. S. Hawes and T. Gunnlaugsson, Cryst. Growth Des., 2017, 17, 4365–4376 CrossRef CAS.
- Z. Berkovitch-Yellin and L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 159–165 CrossRef CAS; L. Pedzisa and B. P. Hay, J. Org. Chem., 2009, 74, 2554–2560 CrossRef PubMed; G. R. Desiraju, Angew. Chem., Int. Ed., 2011, 50, 52–59 CrossRef PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. T. Baker, N. J. Ferguson, H. A. Goodwin and A. D. Rae, Aust. J. Chem., 1989, 42, 623–638 CrossRef CAS; J. W. Goodwin, P. E. Kruger and C. S. Hawes, J. Coord. Chem., 2021, 74, 341–360 CrossRef.
- P. King, M. Maeder and S. Clifford, ReactLab Equilibria, Jplus Consulting Ltd, Fremantle, Australia, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details and synthesis of HL1 and HL2, X-ray data tables, additional figures, thermogravimetric analysis data, additional gas adsorption data, X-ray powder diffraction data, UV-Visible absorption and fluorescence figures, NMR spectra. CCDC 2121405–2121415. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt04007c |
| This journal is © The Royal Society of Chemistry 2022 |