
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Use of group 13 aryloxides for the synthesis of green chemicals and oxide materials†
Rafał
Petrus
 *a,
Józef
Utko
b,
Joanna
Petrus
c,
Mohammad
Awashra
d and
Tadeusz
Lis
b
*a,
Józef
Utko
b,
Joanna
Petrus
c,
Mohammad
Awashra
d and
Tadeusz
Lis
b
aFaculty of Chemistry, Wrocław University of Science and Technology, 23 Smoluchowskiego, 50-370 Wrocław, Poland. E-mail: rafal.petrus@pwr.edu.pl
bFaculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland
cMaco Productions Polonia, 22 Szwajcarska, 54-405 Wrocław, Poland
dDepartment of Chemistry, Aix-Marseille University, Jardin du Pharo, 58 bd Charles Livon, 13284 Marseille Cedex 07, France
First published on 9th February 2022
Abstract
In this work, group 13 metal aryloxides [Al(MesalO)3] (1), [Me2Ga(MesalO)]2 (2), [AlLi3(MesalO)6] (3) and [Me2GaLi(MesalO)2(THF)] (4) were obtained by the reaction of methyl salicylate (MesalOH) with group-13 alkyls MMe3 (for M = Al, Ga) or their combination with BuLi in a THF/alcohol solution. The direct reaction of MMe3 (for M = Al, Ga) and MesalOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) led to compound 1 or 2, respectively. When the same reactions were carried out with additional BuLi, the heterometallic compound 3 or the mixture of 4 and [Li6(MesalO)6] (5) was obtained. Compounds 1–5 were used for the chemical conversion of glycerol to α-hydroxy acid glyceryl esters by alcoholysis of L-lactide (L-LA), glycolide (GA), and ε-caprolactone (ε-CL). Compounds 1–5 were also efficient initiators for the ring-opening polymerization (ROP) of L-LA, GA, and ε-CL using glycerol as a branching agent to synthesize 3-arm polyesters. Heterometallic compounds 3 and 4 were attractive molecular precursors for the preparation of group 13-lithium ceramics, i.e. γ-LiAlO2 and β-LiGaO2.
:3) led to compound 1 or 2, respectively. When the same reactions were carried out with additional BuLi, the heterometallic compound 3 or the mixture of 4 and [Li6(MesalO)6] (5) was obtained. Compounds 1–5 were used for the chemical conversion of glycerol to α-hydroxy acid glyceryl esters by alcoholysis of L-lactide (L-LA), glycolide (GA), and ε-caprolactone (ε-CL). Compounds 1–5 were also efficient initiators for the ring-opening polymerization (ROP) of L-LA, GA, and ε-CL using glycerol as a branching agent to synthesize 3-arm polyesters. Heterometallic compounds 3 and 4 were attractive molecular precursors for the preparation of group 13-lithium ceramics, i.e. γ-LiAlO2 and β-LiGaO2.
Introduction
Glycerol is an important raw material with extensive applications in the petrochemical, tobacco, food, cosmetic and pharmaceutical industries, i.e., as a food preservative and filler, sweetener, thickener, solvent, humectant, lubricant, demulcent, emollient, and antimicrobial and antiviral agents. It is also a major by-product of the conversion of vegetable oils to biodiesel. In the last ten years, increased biodiesel production has led to a glut of glycerol in the global market.1 Therefore, many routes or technologies have been implemented to transform crude glycerol into value-added chemicals or to use it for viable energy generation. The most common reactions are hydrogenolysis, esterification, etherification, selective oxidation or reduction, dehydration, pyrolysis, and gasification.2–4 Glycerol is usually converted into glyceryl esters, glyceric acid, 1,2-propanediol, dihydroxyacetone, mesoxalic acid, oxalic acid, tartronic acid, glycerol carbonate,5 polyglycerols, syngas, or hydrogen.6,7 Macromolecular glycerol derivatives are attracting increased attention due to the diversity of polymer compositions and architectures. Linear, hyperbranched, or dendritic glycerol-based polyethers, polycarbonates, and polyhydroxyacids were investigated mainly for the delivery of therapeutic agents. For example, poly(glycerol sebacate) was essential for cardiovascular, neurovascular, orthopedic, and soft tissue regeneration systems.8 Glycerol or polyglycerols were also applied as branching agents to synthesize aliphatic biodegradable polyesters for encapsulation purposes.9–12 Star-shaped glycerol-lactic acid oligomers were used for the surface modification of hygienic superabsorbent polymer hydrogels.13 Low molecular weight lactic acid glyceryl derivatives were investigated as dietary energy supplements,14 active ingredients in antiseptic and antimicrobial compositions,15 or promising plasticizers for poly(L-lactide) (PLLA). Plasticized PLLA had a lower glass transition temperature, 3.8 times greater elongation at break, and 1.7 times greater impact strength.16 Glyceryl glycolates were developed as anti-aging skin care ingredients in cosmetics. Glyceryl esters have high valorization potential and remarkable properties and can be applied as additives or precursors in many processes in the polymer field. Therefore, great interest has been focused on developing novel and inexpensive synthesis methods or new areas of their practical application.Based on the above, we chose glycerol as the external alcohol and branching agent to synthesize α-hydroxy acid glyceryl esters or star-shaped polyesters. We decided to explore the use of group 13 metal aryloxides because they are popular catalysts/initiators in organic synthesis17–20 and polymerization reactions.21–33 Recently, our interest has been focused on the synthesis of heterometallic compounds, in which the presence of two different Lewis centers can result in heterometallic cooperativity.34–38 For example, chiral lithium–aluminum binaphthoxide [AlLi(binol)2] is a highly effective asymmetric catalyst for the 1,4-addition of a Horner–Wadsworth–Emmons reagent to enones, Michael additions, and tandem Michael-aldol reactions.39,40 The heterobimetallic gallium–lithium catalyst [GaLi((binol-CH2)2O)] was used for the enantioselective meso-epoxide ring-opening reaction with a phenolic oxygen nucleophile to afford 1,2-diol monoethers in up to 94% yield and up to 96% ee.41 Although group 13-lithium aryloxides/alkoxides have been well studied in a range of organic transformations, the examples of heterometallic cooperativity in polymerization processes are limited. Within the selected group of compounds, only six aluminum and two indium derivatives have been investigated for the ROP of cyclic esters.42 Among them, sterically hindered anionic aluminum–lithium alkoxide complexes supported by diamido ether tridentate ligands [HAlLi(OBn)((RN-o-C6H4)2O)(THF)2] (R = C4H9, Cy) were inactive in the ROP of rac-LA; however, their bis-alkoxide analogs [AlLi(OBn)2((RN-o-C6H4)2O)(THF)2] polymerized 85% of the monomer in 16 h (rac-LA/Al = 230/1, 25 °C, CH2Cl2).43 For heterometallic aluminum aryloxides [Me2AlM(2,6-(MeO)2C6H3O)2]2 (M = Li, Na, K), a higher activity in the ROP of L-LA (L-LA/Al/BnOH = 100/1/1, 5 h, 125 °C, toluene) of the lithium analogue (78%) than those of sodium (48%) and potassium (20%) was observed.44 Lithium–indium complexes supported by dianionic fluorinated dialkoxy-diimino ligands {ONRNO}2− (R = C2H4, rac-1,2-cyclohexyl) converted 98–99% of rac-LA in 16 or 0.5 h (rac-LA/In/iPrOH = 100/1/1, 80 °C, toluene) but polymerization control was poor in both cases.45 The essential common feature of these compounds is that they are more active than their homometallic trivalent counterparts. Recently, the Williams group reported a series of heterodinuclear Al(III)/M(I) Schiff base catalysts (M = Na, K, Rb, Cs), which show exceptional activities for phthalic anhydride and cyclohexene oxide copolymerization. The best synergic heterometallic cooperation shows the Al(III)/K(I) catalyst (TOF = 1072 h−1, 0.25 mol% vs. anhydride, T = 100 °C) that combines high activity, quantitative alternating polyester selectivity, and excellent polymerization control at catalyst loadings below 1 mol%.46 Therefore, the development of new group 13-lithium complexes and their applications in the polymerization of cyclic esters are essential to understand the important catalyst features in heterometallic cooperativity.
Heterometallic group 13-lithium alkoxides are also attractive single-source molecular precursors to prepare LiMO2 (for M = Al, Ga) ceramics by a hydrothermal or sol–gel process.47,48 Among the materials of this group, special attention has been focused on γ-LiAlO2 and β-LiGaO2, which have unique physicochemical properties and a wide range of practical applications. γ-LiAlO2 is an essential material in battery technologies and is used to coat electrodes49,50 or used as an electrolyte matrix in molten carbonate fuel cells.51 Due to its chemical and thermal stability and good performance under high neutron and electron radiation, it is also considered a promising tritium breeder in nuclear fusion reactors.52 β-LiGaO2 doped with V3+ or Cr4+ ions is an attractive tunable room-temperature laser material. β-LiGaO2-ZnO alloys form semiconductors with a tunable direct bandgap between 3.3 and 5.6 eV and therefore were applied to fabricate laser diodes and photodetectors operating throughout much of the ultraviolet region.53,54 β-LiGaO2 and γ-LiAlO2 were also investigated as lattice-matched substrates for epitaxial growth of GaN,55,56 InN,57 and ZnO.58,59
Here, we report a new method of chemical conversion of glycerol to α-hydroxy acid glyceryl esters by alcoholysis of cyclic esters, i.e., L-lactide (L-LA), glycolide (GA), and ε-caprolactone (ε-CL), mediated by lithium and group 13 aryloxides. The preparation and characterization of six metal aryloxides, namely [Al(MesalO)3] (1), [Me2Ga(MesalO)]2 (2), [AlLi3(MesalO)6] (3), [Me2GaLi(MesalO)2(THF)] (4), [Li6(MesalO)6] (5) and [Me2GaLi(MesalO)2(H2O)] (6), MesalO = methyl salicylate ligand, are presented. Compounds 1–5 are effective initiators for the ROP of L-LA, GA, and ε-CL using glycerol as a branching agent to synthesize 3-arm polyesters. Compounds 3 and 4 were found to be attractive molecular precursors for the preparation of group 13-lithium ceramics, i.e. γ-LiAlO2 and β-LiGaO2.
Results and discussion
Synthesis and characterization of homometallic and heterometallic group 13 metal aryloxides
We focused on designing homo- and heterometallic compounds with a well-defined molecular structure obtained by the reaction of organometallic reagents of group 13 or lithium with a phenolic ligand. The selection of suitable ligands that would positively influence the solubilities and crystallization abilities of metal compounds was essential. We investigated methyl salicylate as a potential ligand because, after deprotonation, it can act as an O,O′-bidentate chelating agent for a number of metal ions, forming thermodynamically stable and neutral complexes.60–63 We also chose methyl salicylate for its commercial availability, low cost, and properties that allow its use in many industrial applications, including medicine, food, and cosmetics.64,65Our interest in synthesizing group 13 metal aryloxides arose from a simple reaction between AlMe3 and MesalOH (1:3) performed in a THF/MeOH solution (1:2). After several weeks of crystallization, colorless plate crystals of [Al(MesalO)3] (1, 65%) were obtained. Previously, Lewiński reported the synthesis of aluminum methyl salicylate compounds of [Me2Al(MesalO)]2 and [MeAl(MesalO)2] by the selective substitution of one or two Me groups in AlMe3 using the MesalOH proligand.66,67 We found that the introduction of MeOH into the synthesis procedure leads to the substitution of all alkyl groups in AlMe3. We then decided to use GaMe3 for comparison with AlMe3, but dimeric dimethyl-gallium aryloxide [Me2Ga(MesalO)]2 (2, 69%) was obtained as shown in Scheme 1.
 | ||
| Scheme 1 Synthesis of 1 and 2. | ||
It is well-established that the direct reaction of trialkylgallium reagents with phenols usually leads to the formation of dialkylgallium(aryloxides) [Me2Ga(OAr)]2. The lack of further reaction is probably due to electronic effects because the strong electron-donating aryloxide in [Me2Ga(OAr)]2 reduces the reactivity of Ga–Me bond towards protonolysis. When an elevated temperature (above 100 °C) was used, the synthesis of alkylgalliumbis(aryloxides) [MeGa(OAr)2], OAr = halido substituted 8-quinolinato, was also reported.68 There are no known examples of gallium trisaryloxide synthesized using organometallic reagents.69,70 In this work, all reactions involving the use of GaMe3 were carried out at room temperature, and the substitution of only one methyl group was observed.
Single-crystal X-ray diffraction (XRD), NMR spectroscopy, and Fourier-transform infrared attenuated total reflectance (FTIR-ATR) spectroscopy were used to determine the structures and physicochemical properties of the compounds obtained [ESI, Table S1 and Fig. S1–S6†]. The molecular structure of 1 presented in Fig. 1 revealed a distorted octahedral geometry around Al1 atoms with the O6 donor surrounding three MesalO ligands. It should also be noted that in coordination chemistry AlO6 octahedra are observed only in three mononuclear aluminum aryloxides [Al(OAr)3] for OAr = 1-(4-methoxy-2-oxyphenyl)ethanonato;71 3,6-di-t-butyl-1,8-diazatricyclo[6.2.2.02,7]dodeca-2,4,6-triene-4,5-diolato; 9-oxy-1H-phenalen-1-one;72 and 2-oxybenzaldehydato.73 The lengths of the Al–O(aryloxo) bonds, i.e., 1.817(3)–1.824(4) Å, are typical of octahedrally coordinated aluminum centers.74–76 The results of continuous-shape measure calculations showed an insignificant departure from an ideal octahedron for the coordination environment around Al1 with the S(Oh) parameter of 0.170.77
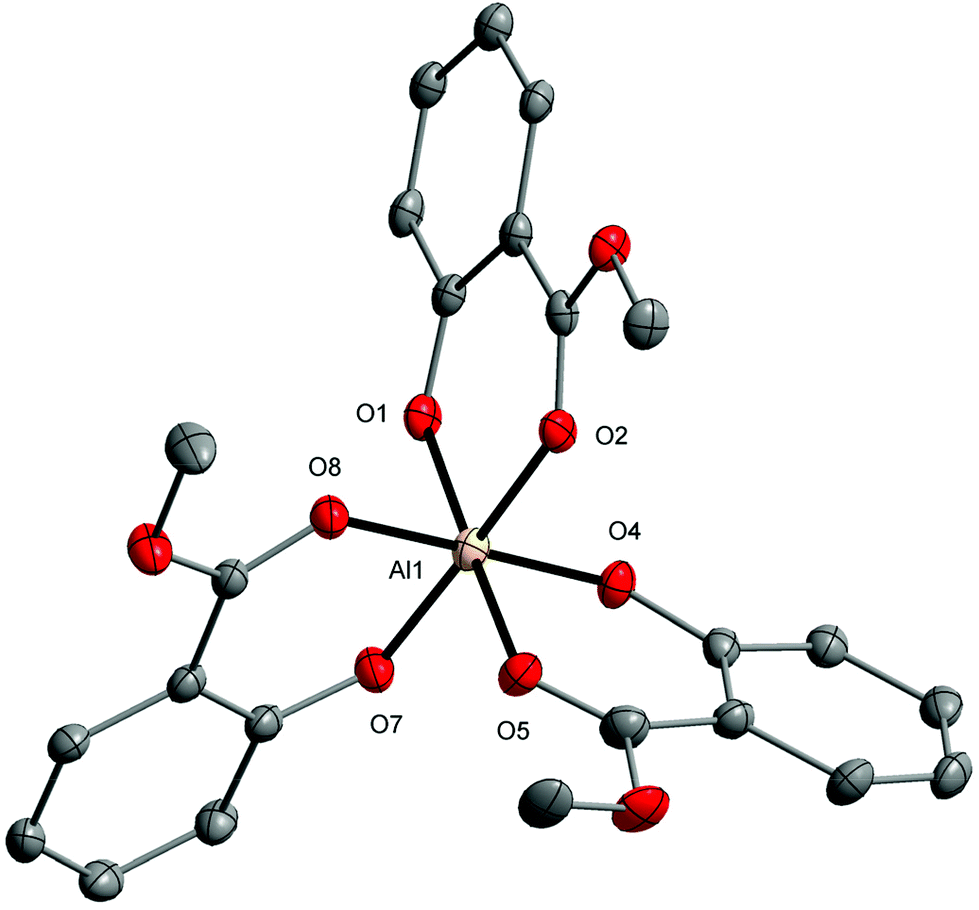 | ||
| Fig. 1 Molecular structure of [Al(MesalO)3] (1). Displacement ellipsoids are drawn at the 25% probability level. The second part of the disordered aryloxy ligands and hydrogen atoms are omitted for the sake of clarity. | ||
The mononuclear coordination aluminum compound [Al(EtsalO)3] (1a, 56%) (where EtsalO = ethyl salicylate ligand), was also synthesized using EtOH instead of MeOH (ESI, Table S1 and Fig. S7–S10†).
The structure of 2 was previously published together with the aluminum and indium isostructural analogs.67 However, the data deposited in the Cambridge Structural Database were obtained at room temperature, unlike the structure of 2 determined by us at 100 K. Therefore, we introduced a detailed description of the received molecular model in this work (Fig. 2). The lengths of Ga–O(aryloxo), i.e., 1.936(2)–2.275(2) Å and Ga-CH3 of 1.956(2)–1.958(2) Å are typical of alkoxo/aryloxo bridged dimeric gallium compounds.78–81 The comparison of Ga–O distance in the equatorial plane (1.936(2) Å) with those in the axial plane (2.161(2) and 2.275(2) Å) revealed the significant lengthening of axial Ga–O bonds, which results from the low Lewis acidity of the gallium(III) ions. The gallium atoms in 2 adopt a distorted trigonal bipyramidal geometry with S(TBPY-5) = 1.262. Dialkyl(aryloxides) of group 13 have been intensively investigated in the past as reagents in metal–organic chemical vapor deposition and various inorganic/organic transformations.82–86
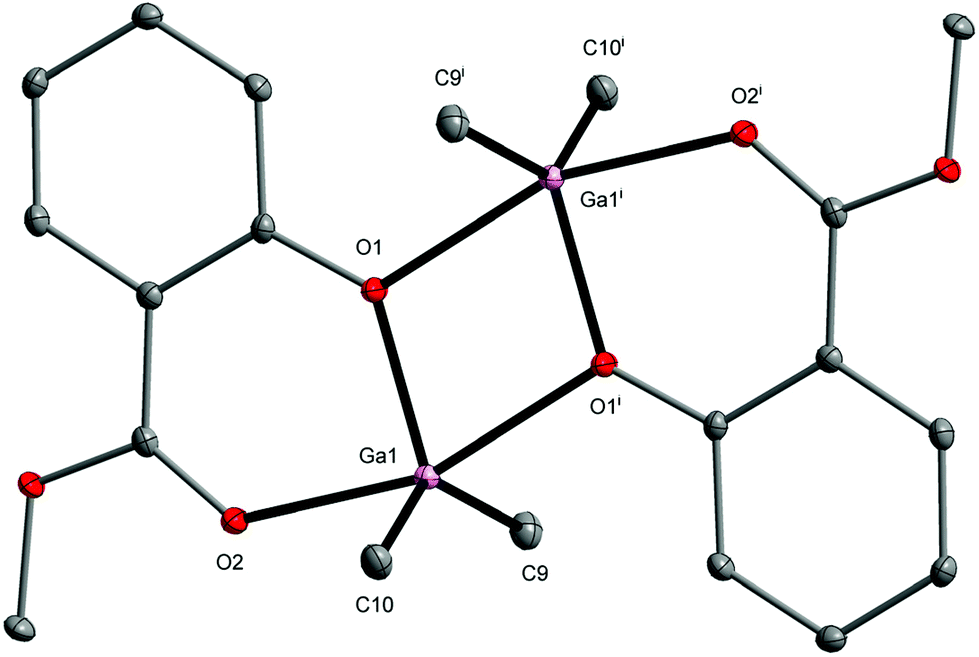 | ||
| Fig. 2 Molecular structure of [Me2Ga(MesalO)]2 (2). The displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for the sake of clarity [symmetry code: (i) −x + 1, −y + 1, −z + 1]. | ||
We investigate the synthesis of group 13-lithium aryloxides by the reaction of the ligand precursor with MMe3 (for M = Al, Ga) and BuLi, in a solution of THF/MeOH at stoichiometries of M/Li/MesalOH = 1/3/6, as shown in Scheme 2.
 | ||
| Scheme 2 Synthesis of 3–5. | ||
The XRD study of the received crystalline materials showed the formation of [AlLi3(MesalO)6] (3, 54%) or the mixture of [Me2GaLi(MesalO)2(THF)] (4, 25%) and [Li6(MesalO)6] (5, 64%). Compounds 3 and 4 were characterized by 1H, 13C, 1H-DOSY, and 7Li NMR, and FTIR-ATR spectroscopy (ESI, Fig. S11–S20†).
Aluminum and lithium ions in 3 held together by four μ- and two μ3-O(aryloxo) bridges form a double-opened dicubane core structure with two missing vertices. In this structure, the Li1–Li3 ions occupy two external vertices and one vertex of the common face of the tetranuclear unit. The second vertex of the common face is the Al1 ion (Fig. 3).
 | ||
| Fig. 3 Molecular structure of [AlLi3(MesalO)6] (3). The displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for the sake of clarity. | ||
In coordination chemistry, heterometallic aluminum–lithium aryloxides usually form dinuclear or tetranuclear compounds with an Al:Li ratio of 1:1, i.e., [Me2AlLi(OAr)2],87 and [MeAlLi(OAr)(OCMePhPh)(Et2O)]88 where OAr = 2,6-di-t-butyl-4-methylphenoxide; [AlLi(EDBP)2(THF)2] where EDBP = 2,2′-ethylidenebis(4,6-di-tert-butylphenolato);89 [AlLi(binol)(THF)2((CH2)5CO)];39 [AlLi(binol)(OtBu)2(PMDTA)] where PMDTA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine, [AlLi(binol)(OtBu)2(THF)2], [Al2Li2(binol)4(THF)4];90 [Me2AlLi(OAr)2]2 where OAr = 2,6-dimethoxyphenoxide,91 [Et2Al2Li2(OAr)4(DME)3]n where OAr = catecholato.92 For the Al:Li ratio of 2:1 or 1:3, only the structures of [R2Al2Li(OPh)6(THF)6] (for R = Et, nBu),93 or [AlLi3(binol)3(THF)6]90 have been reported. For heterometallic aluminum–lithium compounds, the double open dicubane unit was observed only in the structure of [Al2Li2(OR)8] for RO = hexafluoropropoxide.94 The Li1 and Li2 atoms in 3 adopt a trigonal bipyramidal geometry with the metric parameters S(TBPY-5) = 2.047 or 1.717, while the Li3 atom forms a vacant trigonal bipyramid (S(vTBPY-4) = 4.723). The six-fold coordinated Al1 atom with the O6 donor environment adopted an octahedral geometry (S(Oh) = 0.256). The 1H and 13C{1H} NMR spectra of 3 in THF-D8 revealed the presence of two sets of nonequivalent resonance signals with the 2:1 ratio corresponding to four μ-MesalO and two μ3-MesalO ligands (ESI, Fig. S11 and S12†). The 7Li NMR spectrum of 3 contains only one signal at 4.00 ppm (ESI, Fig. S13†). The DOSY 1H NMR spectra of 3 and 4 revealed the retention of solid-state structures in the THF-D8 solution (ESI, Fig. S14 and S19†). When for the synthesis of aluminum–lithium compound 3 EtOH was used instead of MeOH, the isostructural compound [AlLi3(EtsalO)6] (3a, 63%) was obtained (ESI, Table S1, Fig. S21–S25†).
The molecular structure of 4 formally consists of units {Me2Ga(MesalO)} and {Li(MesalO)(THF)} bridged by aryloxo oxygen atoms. The metal atoms in 4 adopt a distorted tetrahedral geometry (Ga1 with S(T-4) = 2.116) or a square pyramidal geometry (Li1 with S(SPY-5) = 0.643).95 The dinuclear gallium–lithium structure has previously been reported for [GaLi(MTB)2(THF)3] where MTB = 2-oxo-2′-mercapto-1,1′-binaphthyl,96 and [GaLi(OR)4(THF)2] where RO = 2-trifluoromethyl-2-propanolato.97 Heterometallic gallium–lithium alkoxides/aryloxides usually form tetranuclear clusters, i.e. [GaLi3(binol)3(THF)4], [GaLi3(binol)3(DME)3],98 or [Me2GaLi(OR)2]2 for RO = 2-methoxyethanolato.99 We also investigated the reactivity of alkyl groups in 4 by performing an NMR-scale reaction in THF-D8 solution with four molar equiv. of MeOH. These measurements revealed that Ga–Me bonds remain intact, and no evolution of methane was detected (ESI, Fig. S26†). When the crystals of compound 4 were exposed to air overnight, colorless needle-like crystals of [Me2GaLi(MesalO)2(H2O)] (6) were obtained (ESI, Fig. S27†). In 6, the water molecule coordinated to the lithium center replaces the THF molecule observed in 4 (Fig. 4).
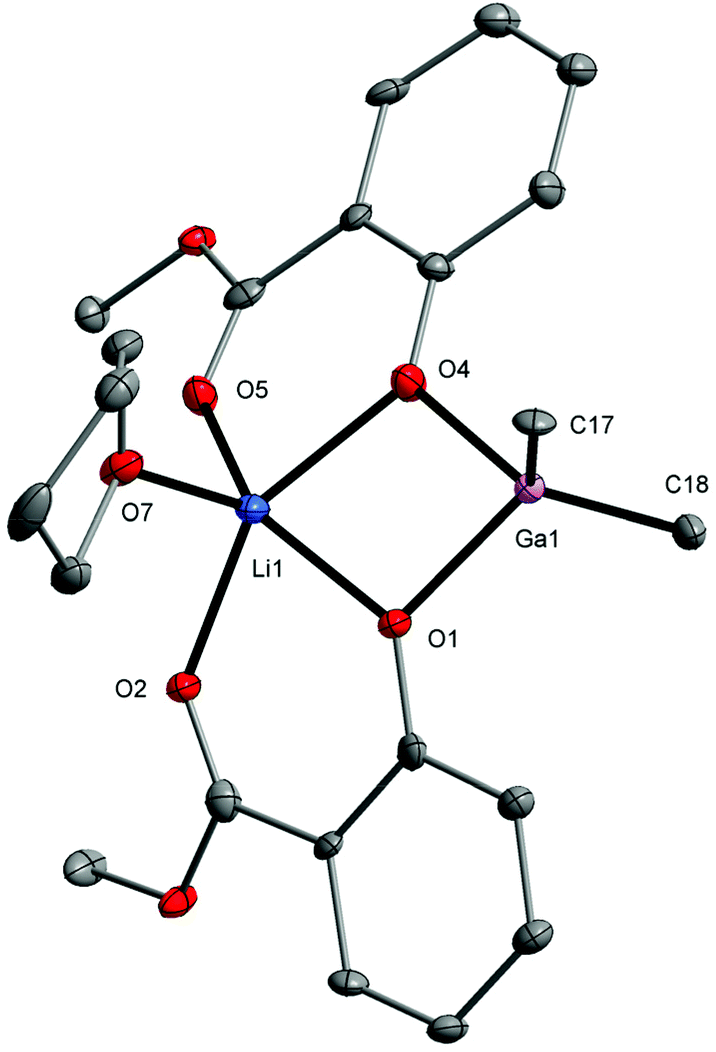 | ||
| Fig. 4 Molecular structure of [Me2GaLi(MesalO)2(THF)] (4). The displacement ellipsoids are drawn at the 30% probability level. | ||
The presence of two different metal elements in the structures of 3 and 4 makes them ideal candidates for use as molecular precursors to prepare group 13-lithium ceramics. Thermogravimetry and differential scanning calorimetry (TGA-DSC) were used to select the calcination temperatures required for the efficient removal of organic ligands and crystallization of heterometallic oxide phases. The thermal decompositions of compounds 3 and 4 from 25 to 1000 °C were performed with a heating rate of 10 °C min−1 under N2. The thermograms obtained presented in Fig. 5 indicate that compounds 3 and 4 underwent multistep thermal decomposition. The melting points of 3 and 4 determined by DSC were 200.5 °C and 124.4 °C, respectively. Compound 3 was stable up to 175 °C with a mass loss not exceeding 1%. For 4, the decomposition starts above 95 °C with the removal of the THF ligand. The aromatic ligands were decomposed and removed within 190 and 300 °C for 4 and 190 and 500 °C for 3. The overall mass loss of 84.8% for 3 corresponds well to the estimated value of 85.4% for the equimolar mixture of LiAlO2 and Li2CO3. The thermal decomposition of 4 with a mass loss of 76.9% compared to the theoretical value of 77.4% indicated the formation of LiGaO2. The final thermal decomposition temperatures were 850 °C for 3 and 751 °C for 4.
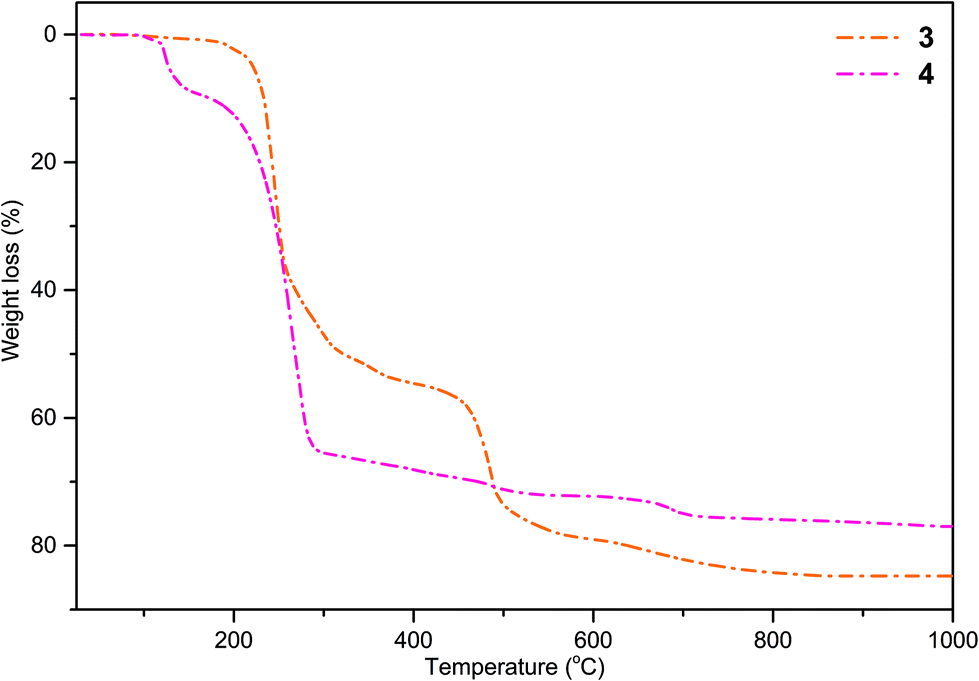 | ||
| Fig. 5 TGA curves for 3 and 4 measured at a heating rate of 10 °C min−1 under a nitrogen atmosphere over the temperature range 25–1000 °C. | ||
The PXRD patterns (Fig. 6) of oxide materials synthesized by calcination of 3 at 850 °C show the formation of γ-LiAlO2 as the main crystalline phase and small amounts of Li2CO3 (Fig. 6a). The resulting raw powder was then purified using the different water solubilities of both phases. The water-soluble Li2CO3 was removed by solvent extraction to give pure γ-LiAlO2 as shown in Fig. 6b. Thermolysis of 4 at 850 °C leads to the selective formation of β-LiGaO2 as shown in Fig. 7. The morphology of the resulting heterometallic oxide materials was investigated by transmission electron microscopy (TEM, Fig. 8, 9 and Fig. S28, S29 in the ESI†). The γ-LiAlO2 crystallites of diameter 36–45 nm are oval with a marked tendency for aggregation (Fig. 8). The β-LiGaO2 crystallites with a 35–400 nm size are well-dispersed and oval or spherical (Fig. 9).
 | ||
| Fig. 6 PXRD patterns of materials prepared by calcination of 3 at 850 °C (a), purified γ-LiAlO2 (b), γ-LiAlO2 [Crystallography Open Database (COD), 4002659, green] (c), Li2CO3 [COD, 9009642, purple] (d). | ||
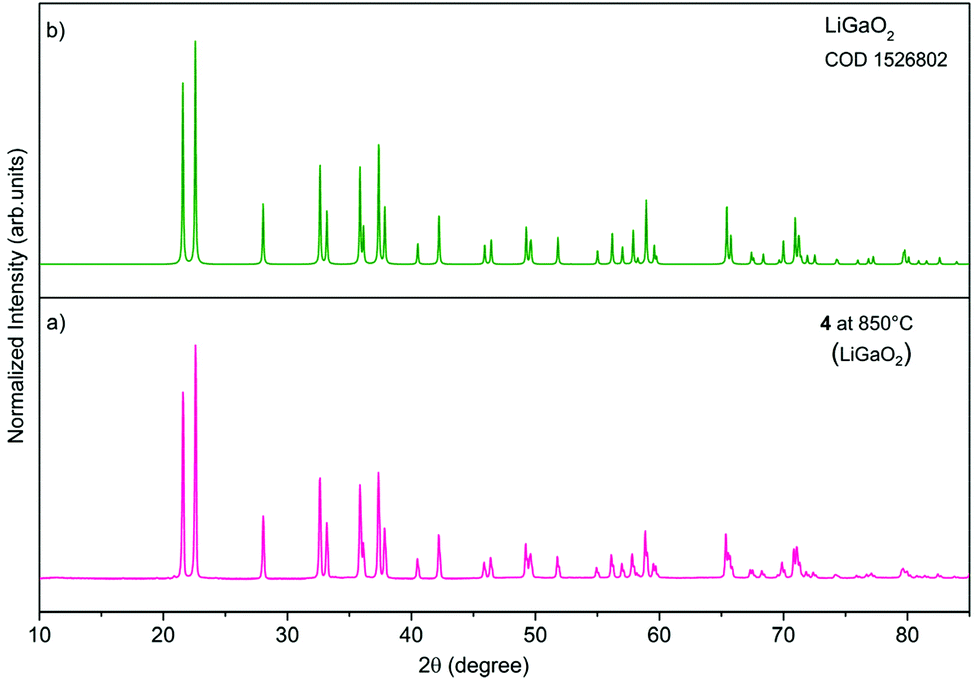 | ||
| Fig. 7 PXRD patterns of the material prepared by calcination of 4 at 850 °C (a), β-LiGaO2 [COD, 1526802, green] (b). | ||
 | ||
| Fig. 8 TEM micrographs of γ-LiAlO2 prepared by calcination of 3 at 850 °C (a–d). | ||
 | ||
| Fig. 9 TEM micrographs of β-LiGaO2 prepared by calcination of 4 at 850 °C (a–d). | ||
Alcoholysis of cyclic esters
We investigated the alcoholysis reactions of heterocyclic esters, namely L-LA, GA, and ε-CL, to synthesize α-hydroxy acid glyceryl esters (Scheme 3). Typical reactions were carried out in a THF solution at room temperature using 1 to 3 equivalents of glycerol (Gl) per lactone and 1–5 as catalysts. | ||
| Scheme 3 Catalyst-free alcoholysis of L-LA and GA. | ||
First, we conducted control experiments for the selected heterocyclic esters in the absence of a catalyst source. 1H NMR monitoring of alcoholysis reactions with a stoichiometry of X/Gl = 1/3 for X = L-LA, GA and ε-CL over time showed that after 168 h, 93% conversion of L-LA to glyceryl (S,S)-O-lactyllactate (GlL2) and 99% conversion of GA to the mixture of glyceryl glycolylglycolate (GlG2, 55%) and glyceryl tris(glycolyl)glycolate (GlG4, 44%) were obtained (ESI, Table S2, entry 27 and Table S3 entry 23, Fig. S30–S33†). Under these conditions, no reactivity of ε-CL was observed. When the same reactions were performed using only two molar excess of Gl, a significant decrease in the conversion of heterocyclic esters was observed, 70% for GlL2 and 40%/43% for GlG2/GlG4 (ESI, Table S2 entry 16 and Table S3† entry 15). All reactions were carried out using an excess of Gl, and therefore, monoglyceryl esters of hydroxy acids were predominantly formed. However, the formation of 1-monoglyceryl and 2-monoglyceryl esters of hydroxy acids was observed in the investigated reactions, as shown in Scheme 3. In this work, both isomers were treated as one kind of product. We cannot rule out the formation of di- and triglyceryl esters of hydroxy acids, but their presence was not observed in the amounts detectable by NMR spectroscopy.
When L-LA alcoholysis reactions were carried out with the stoichiometry of L-LA/Gl/M = 1/3/0.01, where M is a metal atom present in compounds 1–4, the results obtained were similar to those for catalyst-free reactions (ESI, Table S4,† entries 1–4). Under similar conditions, after 360 min, 5 led to the mixture of glyceryl lactate (GlL1) and GlL2 49/47% (ESI, Table S4,† entry 18). The catalyst concentration increased five times; compounds 3 and 4 converted 94–99% of L-LA in 2 minutes. After 30 min, the mixture of GlL1/GlL2 with individual conversion yields of 62%/37% for 3 and 8%/90% for 4 was achieved (ESI, Table S4, entries 28–35; Fig. S34 and S35†). Compounds 1 and 2 were inactive in this reaction, and 5 led to lactyl oligomers.
In GA alcoholysis reactions carried out with the GA/Gl/M = 1/3/0.01 stoichiometry using compounds 1–5, >98% substrate conversion was observed within the first 5 min. In the initial stage of these reactions, metal-aryloxide catalysts led to the formation of glycolic acid oligoesters (GlGn), which were then converted into glyceryl glycolate (GlG1) and GlG2 (ESI, Fig. S36–S38†). For example, after 30 min, the conversion of GA to GlG1/GlG2/GlGn was 4/56/40% for 1, 85/13/2% for 3, 64/24/11% for 4, and 63/26/11% for 5 (ESI, Table S5,† entries 5, 14, 21 and 33).
When the ε-CL alcoholysis reactions were carried out with the ε-CL/Gl/M = 1/2/0.08 stoichiometry after 300 min conversion of ε-CL into glyceryl 6-hydroxyhexanoate (GlCL1) was 60% for 3 and 77% for 4 (ESI, Table S6, entries 28 and 33, Fig. S39–S41†). Compounds 1 and 2 were inactive under the investigated conditions, and 5 in the reaction when using ε-CL/Gl/M = 1/2/0.02 after 96 min converts 97% of ε-CL (ESI, Table S6,† entry 22).
Summarizing the catalytic activity of 1–5 in the synthesis of α-hydroxy acid glyceryl esters, we found that 1 and 2 are inactive in the alcoholysis of L-LA and ε-CL. The most active in these two reactions was 5, leading to GlL1/GlL2 in 120 min or GlCL1 in 90 min with conversion yields of 73/24% or 97% in stoichiometry X/Gl/M = 1/2/0.02. When the catalyst concentration was increased 2.5 times in the L-LA alcoholysis reaction, the activity of 3 was almost eight times higher than that of 4. However, in the ε-CL alcoholysis, the catalytic activity of 4 was approximately 1.3 times better than those revealed by 3. We established that the most active one in the alcoholysis reaction of GA was 3, leading to GlG1 with a conversion of 20% higher than those obtained by 4 and 5 after 30 min. The presence of Al(III)/Ga(III) centers in 3 and 4 enhances their catalytic activity in GA alcoholysis compared to 5 (ESI, Table S4,† entries 14, 21 and 33). Compound 1 showed significantly greater reactivity than 2, but GA was mainly converted to the mixture of GlGn and GlG2.
The 1H NMR spectra of the reaction mixtures revealed the presence of glyceryl 1- and 2-lactate/glycolate/6-hydroxyhexanoate. Resonance signals from the glyceryl alkyl and hydroxyl groups were found in the same region for these compounds regardless of the units of hydroxy acids (Fig. 10 and Fig. S36, S39 in the ESI†). For example, the 1H NMR spectrum presented in Fig. 10 showed resonances of glyceryl CH2 at 4.09 and 3.94 ppm (diastereotopic protons) and 3.42 ppm; CH at 3.64 ppm, and OH at 4.89 and 4.64 ppm for glyceryl 1-lactate. The corresponding resonance signals of Gl protons in glyceryl 2-lactate were found at 4.74 ppm for CH, 3.50 for CH2, and 3.47–3.27 for the OH groups. Resonance signals from hydroxyl, methine, and methyl protons of the lactate unit were found at 5.34, 4.14, and 1.25 ppm. Electrospray ionization mass spectrometry (ESI-MS) studies confirmed the formation of α-hydroxy acid glyceryl esters (ESI, Fig. S42–S44†).
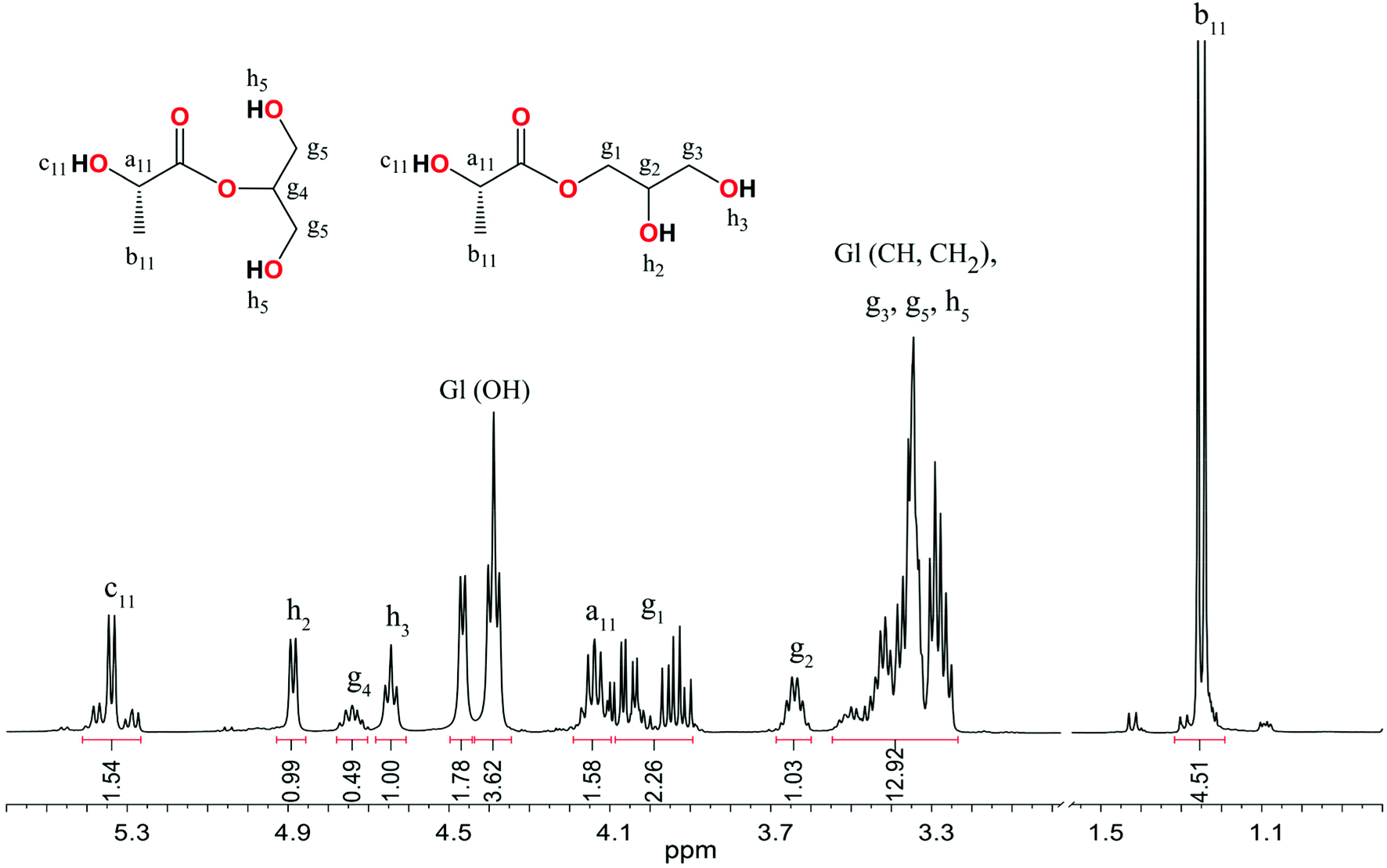 | ||
| Fig. 10 1H NMR spectrum in DMSO-D6 of GlL1 synthesized in the L-LA alcoholysis reaction in the presence of 3. | ||
Ring-opening polymerization of L-LA, GA, and ε-CL
The polymerization of L-LA, GA, and ε-CL was performed in THF solution or bulk using Gl as a co-initiator and compounds 1–5 as initiators to compare their catalytic activities. The amount of compound used in each reaction was calculated on the basis of the number of metal centers (M) in 1–5. Representative results for the L-LA, GA, and ε-CL polymerizations are summarized in Table 1. The number average molecular weights (Mn) and dispersity indexes (Đ) of the polymers were determined by size-exclusion chromatography (SEC) with a multiangle laser-light scattering (MALLS) or refractive index detector (RI).a
| Entry | Initiator | Monomer | [X]/[Gl]/[M] | t (min) | C (%) |
M
nc (kDa) |
Đ |
T
md (°C) |
T
maxd (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: [M]0 = 10 mM, 10 ml of THF as the solvent, temperature 25 °C, under an atmosphere of N2. b Obtained from 1H NMR analysis. c Obtained from SEC analysis with the RI (entries 1-8) or MALLS (entries 9-20) detector. d Obtained from TGA/DTA analysis; Tm – melting point; Tmax – temperature of the maximum degradation rate. e GA and L-LA polymerization performed in bulk at 110 °C. f polymerization performed in ε-CL at 70 °C. | |||||||||
| 1 | 1 | GA | 20/1/1 | 93 | 98 | 13.0 | 1.19 | 191.61 | 317.27 |
| 2 | 3 | GA | 20/1/1 | 4 | 99 | 14.5 | 1.23 | 200.18 | 325.72 |
| 3 | 4 | GA | 20/1/1 | 20 | 99 | 14.9 | 1.15 | 191.18 | 330.44 |
| 4 | 5 | GA | 20/1/1 | 1 | 99 | 13.4 | 1.16 | 221.04 | 327.96 |
| 5e | 1 | GA | 100/1/1 | 45 | 98 | 13.1 | 1.47 | 221.29 | 324.59 |
| 6e | 3 | GA | 100/1/1 | 8 | 97 | 10.9 | 1.27 | 219.23 | 331.61 |
| 7e | 4 | GA | 100/1/1 | 15 | 95 | 10.6 | 1.28 | 215.93 | 330.13 |
| 8e | 5 | GA | 100/1/1 | 4 | 98 | 11.0 | 1.29 | 219.19 | 330.32 |
| 9 | 1 | L-LA | 20/1/1 | 720 | 76 | 2.9 | 2.90 | — | 275.98 |
| 10 | 3 | L-LA | 20/1/1 | 38 | 92 | 3.9 | 2.88 | — | 265.37 |
| 11 | 4 | L-LA | 20/1/1 | 300 | 89 | 9.5 | 2.24 | — | 276.17 |
| 12 | 5 | L-LA | 20/1/1 | 45 | 98 | 4.6 | 2.84 | — | 274.40 |
| 13e | 1 | L-LA | 100/1/1 | 720 | 72 | 10.2 | 3.16 | 144.94 | 295.83 |
| 14e | 3 | L-LA | 100/1/1 | 120 | 90 | 5.4 | 1.83 | 131.82 | 294.65 |
| 15e | 4 | L-LA | 100/1/1 | 720 | 88 | 10.6 | 1.43 | 136.20 | 289.50 |
| 16e | 5 | L-LA | 100/1/1 | 120 | 94 | 4.5 | 1.62 | 133.20 | 282.34 |
| 17f | 1 | ε-CL | 200/1/1 | 240 | 82 | 7.0 | 1.80 | 61.31 | 233.72 |
| 18f | 3 | ε-CL | 200/1/1 | 240 | 90 | 9.5 | 2.92 | 62.05 | 248.73 |
| 19f | 4 | ε-CL | 200/1/1 | 240 | 87 | 9.4 | 2.67 | 61.24 | 246.63 |
| 20f | 5 | ε-CL | 200/1/1 | 240 | 97 | 6.9 | 2.05 | 61.06 | 224.24 |
In the ROP of GA performed with a GA/Gl/M stoichiometry of 20/1/1, polyglycolide (PGA) formation with Mn values ranging from 13.0 to 14.9 kDa and a Đ of 1.16 to 1.23 was observed. The received polymers were isolated as a crystalline precipitate after exceeding a certain molecular weight during these reactions. We suspect that the very poor solubility of the formed PGAs caused their similar Mn and Đ values. Initiator 5 was the most active in GA polymerization with a GA/M ratio of 20, resulting in a monomer conversion of 99% after 1 min. Compound 3 showed a similar conversion value after 4 minutes, compound 4 after 20 minutes, and 1 after 45 minutes (Table 1, entries 1–4). When GA polymerization was performed in bulk at a GA/Gl/M = 100/1/1, the resulting PGAs had Mn values of 10.6 to 13.1 kDa and Đ values within the range of 1.27 to 1.47.
Compounds 1–5 were also used as initiators for the bulk polymerization of ε-CL at 70 °C with the GA/Gl/M stoichiometry of 200/1/1. After 240 min of reaction carried out with 1, polycaprolactone (PCL) formation was observed with an Mn of 7.0 kDa and a Đ of 1.80. Under the same conditions, heterometallic compounds 3–4 led to polymers with Mn values of 9.4–9.5 and Đ values of 2.67–2.97 (Table 1, entries 18 and 19). The use of lithium aryloxide 5 resulted in a product with Mn = 7.2 and Đ = 2.69 (Table 1, entry 20).
In the L-LA polymerization reactions, the formation of PLLAs with Mn values of 2.90 to 10.6 kDa and Đ values of 1.43–3.16 was observed (Table 1, entries 9–16). When polymerization was performed with 1–5 in an L-LA/Gl/M molar ratio of 20/1/1, the Mn values of the PLLAs were 2.9 to 9.5 kDa and Đ was within the range of 2.24–2.90. The most active in the ROP of L-LA was 3, which converted 92% of the monomer in 38 min. To achieve similar results, 5 needs 45 min and 4 needs 300 min. When 1 was used for the ROP of L-LA after 720 min, 76% of the monomer conversion was achieved, and the resulting material had an Mn of 2.9 kDa and a Đ of 2.9. When the ratio of L-LA/Gl/M was 100/1/1, compounds 3–5 produced polymers with Mn values of 4.5 to 10.3 kDa and Đ of 1.43 to 1.83. High dispersity indices in the 1.5 to 2.9 range are typical of branched polyesters, and similar values were previously reported for 3-arm PLLAs.100,101
In the ROP of L-LA, the Mn and Đ values of the PLLAs obtained using 3 and 5 were comparable and were at least twice lower than those synthesized with 4. Furthermore, the SEC curves recorded for the PLLAs obtained using heterometallic 3 and 4 reveal the bimodal distribution of molecular weight. Representative SEC curves of bimodal PLLAs obtained in the presence of 3 and 4 in the reaction with L-LA/Gl/M stoichiometry of 100/1/1 are presented in Fig. 11. In these examples, the higher molecular weight fractions have estimated Mn values of 10.3 kDa with Đ = 1.30 for 3 and 12.5 kDa with Đ = 1.40 for 4. The lower molecular weight fractions have Mn values of 2.3 kDa with Đ = 2.13 for 3 and 8.3 kDa with Đ = 1.30 for 4. Conventionally, it is assumed that polymerization will be initiated from all functional groups using a bi- or trifunctional chain transfer agent; however, this assumption is not always substantiated.102 We investigated the structure of the resulting PLLAs using 1H-DOSY or 1H–13C HSQC measurements, but we did not find distinguishing signals characteristic of the formation of polymers with different architectures (ESI, Fig. S45–S48†). To exclude the possibility of the formation of different species in the solution, we performed NMR scale reactions of 3 and 4 with excess Gl, but no reaction between reagents was observed (ESI, Fig. S49 and S50†). Therefore, it is proposed that the steric hindrance and the nature of the –OH group in the chain transfer agent, the number of lithium centers in the initiator structure, and the prolonged reaction time influence the formation of the two molecular weight fractions. The sterically hindered Al(III) center in 3 is proposed to perform only a structural function, and thus only Li centers take an active part in the ROP of L-LA. Therefore, the catalytic activity of 3 is comparable to those observed for 5. In the presence of 4, the polymer was also proposed to be synthesized at the Li sites. However, the tetracoordinated Ga(III) center may participate in the formation of stable interactions between the carbonyl moiety of the polymer chain, and thus further activation of the L-LA monomer may be limited due to the decrease in the steric accessibility of the Li center.103 The prolonged reaction time and a temperature above 110 °C, favor the occurrence of transesterification side reactions and the formation of cyclic polymers, leading to a bimodal molecular weight distribution. These observations could also explain the reduced catalytic activity of 4 in L-LA alcoholysis, where mainly GlL2 was synthesized despite the high content of the active lithium site (Table S4,† entries 21 and 35).
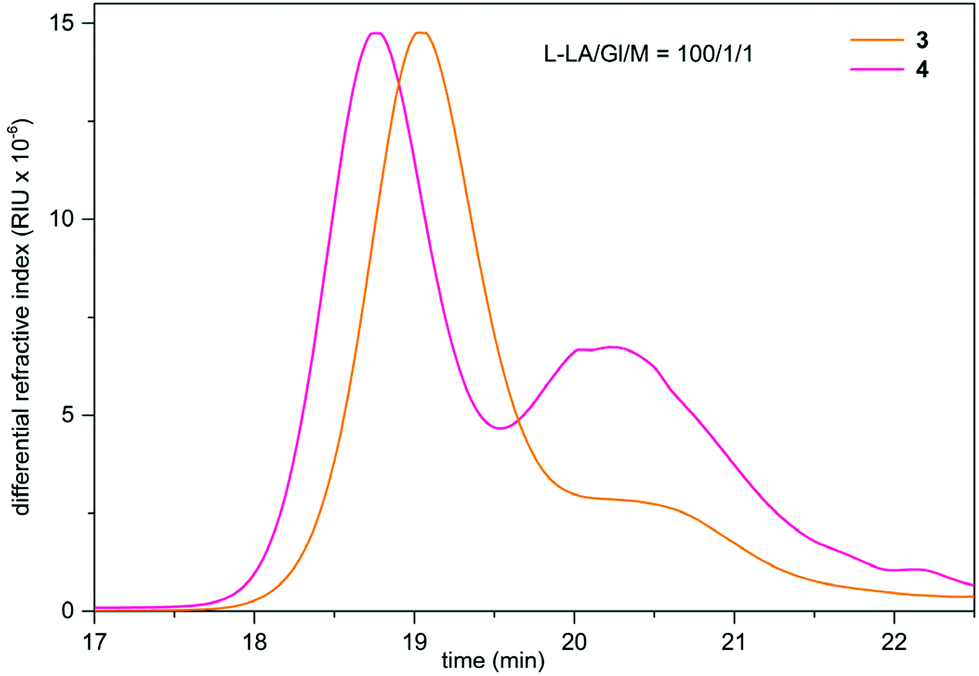 | ||
| Fig. 11 SEC chromatograms of bimodal PLLAs synthesized using heterometallic 3 and 4. | ||
However, PCLs synthesized using heterometallic 3 and 4 have similar Mn ∼9.4 kDa and Đ values of 2.7–2.9 despite the different lithium center content in the initiator structure. Furthermore, both polymers have higher Mn values and broader Đ than those obtained with homometallic 1 and 5. These results suggested that multiple factors are involved in the ROP of cyclic esters, including the type of monomers used, nature and number of metallic centers, the nature and steric hindrance of the ligands, the coordination number of metal ions, and the central core structure of the initiators.
The 1H NMR and ESI-MS analysis of the resulting macromolecular chain ends revealed the formation of star-shaped PGA, PLLA, or PCL with glyceryl branching points, indicating an activated monomer mechanism (Fig. 12, 13, and Fig. S51–S54 in the ESI†).104
For example, the 1H NMR spectrum of the resulting 3-arm PLLA in C6D6 showed resonance signals at 4.14, 2.64, and 1.12 ppm from the CH, OH, and CH3 groups of hydroxyl end units, the protons of the CH and CH3 groups of the main chain at 5.06 and 1.34 ppm, and resonances for the alkyl protons of the triglyceryl ester group at 3.93 and 3.80 ppm (Fig. 12). The ESI-MS spectrum presented in Fig. 13 confirmed the formation of branched PLLAs.
 | ||
| Fig. 12 1H NMR spectrum of 3-arm PLLA in C6D6. * – assigned ligand residues. | ||
 | ||
| Fig. 13 Mass spectrum of 3-arm PLLA. | ||
Conclusion
This study developed a new, simple, and efficient strategy for the preparation of various glycerol derivatives, i.e., α-hydroxy acid glyceryl esters and branched polyesters, by alcoholysis or polymerizations of L-LA, GA, and ε-CL. For these purposes, we use lithium and group 13 aryloxides [Al(MesalO)3] (1), [Me2Ga(MesalO)]2 (2), [AlLi3(MesalO)6] (3), [Me2GaLi(MesalO)2(THF)] (4), and [Li6(MesalO)6] (5) which can play dual roles as catalysts in ester synthesis and as polymerization initiators. This approach enabled the synthesis of very attractive green chemicals that, after the implementation of a purification procedure, have the potential to be used as dietary supplements, antiseptic and antimicrobial agents, polymer additives (plasticizers, surface modification agents), or polymeric encapsulation materials. The effect of the metal aryloxide catalyst on cyclic ester alcoholysis or polymerization reactions was investigated. We showed that the type of monomers used, the nature and number of metallic centers, and the central core structure of the used initiators strongly influence the polymerization of the cyclic esters and the physicochemical properties of the resulting polymers. The first heterometallic gallium-alkali metal initiator was developed for the alcoholysis and ROP of cyclic esters. In the field of heterometallic cooperativity, we established that the presence of Al(III)/Ga(III) centers in 3 and 4 enhances the catalytic activity of active Li sites in GA alcoholysis. In the alcoholysis and polymerization of L-LA, the presence of Ga(III) decreases the catalytic activity of 4 compared to the lithium analog 5.Metal aryloxides 1–6 were obtained by the reaction of the ligand precursor with group 13 alkyls MMe3 (for M = Al, Ga) or their combination with BuLi in a THF/alcohol solution. Direct reactions of MMe3 (for M = Al, Ga) and MesalOH (1:3) led to compound 1 or 2, respectively. When the same reactions were carried out with additional BuLi, heterometallic compound 3 or the mixture of 4 and [Li6(MesalO)6] (5) was obtained. In 3 and 4, the formed group 13 species retain the rearrangements that occurred in 1 or 2. In coordination chemistry, 3 is the second example of aryloxides with an Al:Li ratio of 1:3, while 4 and 6 are the fourth and fifth examples of gallium–lithium aryloxides.
Heterometallic aryloxides 3 and 4 are attractive molecular precursors for the preparation of group 13-lithium ceramics, i.e. γ-LiAlO2 and β-LiGaO2, by the thermal decomposition at 850 °C. These materials have various practical applications in coatings, battery and cell technologies, semiconductors, and laser diode fabrication.
Overall, we present new structurally authenticated group 13-lithium aryloxides, which could be helpful in the development of green chemicals, i.e., α-hydroxy acid glyceryl esters or 3-arm polyesters, as well as ceramic materials of industrial importance.
Experimental section
Materials and methods
All syntheses were performed in a dry N2 atmosphere using standard Schlenk techniques. Standard methods were used to purify the reagents: toluene, hexane, and THF were distilled over Na; CH2Cl2 was distilled over P2O5, and EtOH was distilled over Mg. All chemical reagents were purchased from commercial sources: methyl salicylate, metallic sodium, AlMe3 1.0 M in hexane, GaMe3 2.0 M in toluene, nBuLi solution 1.6 M in hexane, L-lactide, glycolide, ε-caprolactone (Sigma-Aldrich, St Louis, MO, USA); toluene, hexane, THF, CH2Cl2 and C2H5OH (POCH); CD2Cl2, DMSO-D6, and THF-D8 (Carl-Roth). L-Lactide and glycolide were recrystallized twice from toluene, sublimed, and kept over P2O5, andε-caprolactone was dried under vacuum and kept over molecular sieves. AlMe3 and GaMe3 are highly pyrophoric and should be handled and used only in an atmosphere of argon or nitrogen. The 1H, 7Li, and 13C{1H} NMR spectra were recorded at room temperature on a JEOL JNM-ECZ 400 MHz spectrometer. Chemical shifts were reported in parts per million and referenced to the residual protons in deuterated solvents. The 7Li spectra were referenced to a 0.1 M solution of LiNO3 in D2O. FTIR-ATR spectra were recorded on a Bruker Vertex 70 vacuum spectrometer. Elemental analyses were performed on a PerkinElmer 2400 CHN elemental analyzer. Metal ion concentrations were determined by ICP-OES using a Thermo Scientific iCAP 7400 Duo spectrometer. The thermal decomposition of the metal aryloxide precursors was performed using an NT 1313 furnace (Neotherm) equipped with a KXP4 thermostat under air. The resulting metal oxide materials were investigated by powder X-ray diffraction with an Empyrean (PANalytical) diffractometer. The samples were analyzed using the PDF-4+ and COD powder diffraction database. The morphologies of the oxide materials were examined using an FEI Tecnai G2 20 X-Twin TEM microscope equipped with a field emission gun (FEG) and an integrated energy dispersive spectrometer (EDAX). For the TEM observation, 200 mesh copper grids with lacey carbon films were used. The signals from copper and carbon (from the grids) were visible on the TEM EDX spectra. The SEC traces of PLLAs and PCLs were recorded at 30 °C with a system consisting of an Agilent 1100 Series isocratic pump, a degasser, an autosampler, a thermostatic box for columns, and a set of TSK Gel columns (2 × PLGel 5 μm MIXED-C). A Wyatt Optilab rEX interferometric refractometer and a MALLS DAWN EOS laser photometer (Wyatt Technology Corp., USA) were used as detectors. The eluent was CH2Cl2 with a flow rate of 0.8 ml min−1. The Mn and Đ values were calculated from the experimental traces using the Wyatt ASTRA v 4.90.07 program. PGA SEC traces were recorded at 40 °C with a system consisting of a SHIMADZU-LC-20AD pump, a degasser, an autosampler, a thermostatic box for columns, and a set of TSK Gel columns (2 × PLGel 5 μm MIXED-C). The RI-Optilab-T-rex-Wyatt interferometric refractometer (Wyatt Technology Corp., USA) was used as the detector. The eluent was DMF with a flow rate of 0.8 ml min−1. The Mn and Đ values were calculated from the experimental traces using the Wyatt ASTRA v 7.1.2 program. TGA/DSC was performed in a nitrogen atmosphere with a TGA/DSC 3+ system (Mettler-Toledo) at a heating rate of 10 °C min−1.Single-crystal X-ray diffraction studies
Single-crystal XRD data were collected using an Xcalibur Ruby, XtaLAB Synergy R, or Agilent SuperNova Dual Atlas diffractometer at 100 K for 1–4 and 6.105 Experimental details and crystal data are given in Table S1.† The structures were solved by direct methods and refined by the full-matrix least-squares method on F2, using the SHELXTL package.106 Nonhydrogen atoms were refined with anisotropic thermal parameters. All hydrogen atoms were positioned geometrically and added to the structure factor calculations but were not refined. Molecular graphics for the resulting structures were created using Diamond (version 3.1e).107 During the single-crystal XRD measurements of 4 a higher symmetry than the real one was assumed, and only 75% of the data completeness was achieved. CCDC 2076392–2076396, 2128614 and 2128703† contain the supplementary crystallographic data for this paper.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 170.7 (3C, C–O), 137.6 (3C, ArH), 130.2 (3C, ArH), 123.9 (3C, ArH), 114.9 (3C, ArH), 111.5 (3C, Ar), 111.3 (3C, ArH), 53.5 (3C, CH3). FTIR-ATR (cm−1): 3058 (vw), 3035 (vw), 2956 (w), 2865 (vw), 2712 (vw), 2605 (vw), 2324 (vw), 1927 (vw), 1814 (vw), 1740 (vw), 1629 (vs), 1602 (s), 1542 (s), 1508 (vw), 1461 (s), 1440 (s), 1407 (m), 1380 (w), 1353 (s), 1303 (vw), 1267 (m), 1244 (vs), 1198 (w), 1159 (m), 1137 (m), 1094 (m), 1028 (m), 956 (w), 880 (m), 858 (m), 839 (m), 797 (m), 759 (vs), 706 (m), 669 (m), 639 (s), 572 (m), 532 (w), 462 (m), 444 (m), 419 (w), 405 (w). For catalytic purposes, the crystals of 1 were recrystallized from ethanol and, as a result of transesterification, the formation of ethyl salicylate ligands was observed.
O), 170.8 (3C, C–O), 137.4 (3C, ArH), 130.2 (3C, ArH), 123.9 (3C, ArH), 114.7 (3C, ArH), 111.6 (3C, Ar), 63.4 (3C, CH2), 14.0 (3C, CH3). FTIR-ATR (cm−1): 3092 (vw), 3034 (vw), 3009 (vw), 2983 (w), 2804 (vw), 2701 (vw), 2605 (vw), 2493 (vw), 2324 (vw), 1928 (vw), 1911 (vw), 1817 (vw), 1627 (vs), 1601 (s), 1542 (s), 1509 (vw), 1463 (s), 1450 (s), 1404 (s), 1377 (m), 1346 (s), 1301 (vw), 1267 (m), 1239 (vs), 1179 (w), 1159 (m), 1137 (m), 1094 (m), 1028 (w), 992 (w), 893 (w), 873 (vw), 859 (w), 837 (m), 800 (w), 759 (vs), 707 (m), 669 (m), 637 (s), 572 (m), 534 (w), 479 (m), 455 (m), 436 (m), 420 (w).
O), 170.6 (2C, C–O), 137.6 (2C, ArH), 131.3 (2C, ArH), 124.2 (2C, ArH), 116.1 (2C, ArH), 112.1 (2C, Ar), 53.7 (2C, CH3), −6.7 (4C, GaCH3). FTIR-ATR (cm−1): 3188 (vw), 3072 (vw), 2956 (w), 2916 (vw), 1677 (s), 1616 (m), 1586 (m), 1554 (w), 1486 (m), 1440 (s), 1327 (m), 1304 (s), 1248 (s), 1217 (vs), 1158 (m), 1136 (m), 1091 (m), 1045 (vw), 1033 (w), 964 (w), 866 (w), 849 (w), 822 (vw), 800 (vw), 753 (s), 721 (vs), 698 (vs), 666 (m), 528 (vs), 438 (m).
O), 171.1 (4C, C–O), 168.2 (2C, C–O), 135.5 (2C, ArH), 134.9 (4C, ArH), 131.8 (2C, ArH), 130.6 (4C, ArH), 124.1 (4C, ArH), 123.8 (2C, ArH), 114.9 (4C, ArH), 114.7 (2C, ArH), 112.3 (6C, Ar), 52.2 (4C, CH3), 51.2 (2C, CH3). 7Li NMR (THF-D8, 155 MHz): δ 4.00. FTIR-ATR (cm−1): 3027 (vw), 2950 (w), 2844 (vw), 2319 (vw), 2234 (vw), 2162 (vw), 2141 (vw), 2109 (vw), 2081 (vw), 2049 (vw), 1979 (vw), 1925 (vw), 1703 (m), 1677 (m), 1641 (s), 1613 (vs), 1598 (vs), 1566 (w), 1546 (m), 1464 (vs), 1442 (s), 1387 (w), 1353 (m), 1325 (m), 1292 (w), 1254 (vs), 1223 (s), 1198 (m), 1153 (m), 1085 (m), 1035 (w), 954 (w), 898 (w), 861 (s), 817 (w), 798 (w), 764 (s), 755 (vs), 707 (s), 676 (w), 662 (w), 599 (m), 581(m), 558 (w), 533 (m), 493 (m), 472 (w), 451 (w), 431 (w).
O), 171.8 (4C, CO), 170.6 (4C, C–O), 168.3 (2C, C–O), 135.5 (2C, ArH), 134.7 (4C, ArH), 131.8 (2C, ArH), 130.6 (4C, ArH), 124.1 (6C, ArH), 115.0 (2C, ArH), 114.9 (4C, ArH), 112.0 (6C, Ar), 61.7 (4C, CH2), 60.2 (2C, CH2), 14.6 (2C, CH3), 14.3 (4C, CH3). 7Li NMR (THF-D8, 155 MHz): δ 4.01. FTIR-ATR (cm−1): 3072 (vw), 2982 (w), 2955 (vw), 2935 (vw), 2896 (vw), 2319 (vw), 2162 (vw), 2141 (vw), 1929 (vw), 1707 (m), 1676 (s), 1650 (s), 1620 (s), 1599 (s), 1564 (w), 1547 (m), 1467 (vs), 1445 (s), 1409 (m), 1379 (w), 1367 (w), 1342 (m), 1349 (m), 1319 (w), 1292 (m), 1255 (vs), 1230 (s), 1215 (vs), 1161 (m), 1149 (s), 1117 (vw), 1088 (s), 1076 (s), 1045 (vw), 1032 (m), 1012 (w), 952 (vw), 891 (m), 866 (m), 853 (m), 827 (m), 795 (w), 765 (vs), 758 (vs), 708 (s), 664 (m), 601 (s), 582 (m), 561(w), 534 (w), 494 (m), 476 (w), 458 (w), 425 (w), 405 (w).
O), 166.6 (2C, C–O), 135.4 (2C, ArH), 132.5 (2C, ArH), 121.1 (2C, ArH), 116.8 (2C, Ar), 116.1 (2C, ArH), 68.0 (2C, CH2), 51.6 (2C, CH3), 26.2 (2C, CH2), −6.4 (2C, GaCH3). 7Li NMR (THF-D8, 155 MHz): δ 3.76. FTIR-ATR (cm−1): 3363 (vw), 3059 (vw), 2952 (w), 2871 (w), 2651(vw), 2051 (vw), 1981 (vw), 1928 (vw), 1680 (vs), 1598 (m), 1561 (m), 1475 (s), 1445 (s), 1434 (s), 1317 (s), 1297 (m), 1264 (m), 1232 (vs), 1194 (m), 1160 (s), 1147 (m), 1087 (s), 1056 (m), 1044 (m), 969 (w), 953 (vw), 899 (w), 864 (m), 818 (w), 797 (w), 758 (s), 727 (m), 704 (s), 666 (m), 597 (s), 578 (s), 535 (m), 479 (m).
O), 170.7 (3C, C–O), 137.6 (3C, ArH), 130.2 (3C, ArH), 123.9 (3C, ArH), 114.9 (3C, ArH), 111.5 (3C, Ar), 111.3 (3C, ArH), 53.5 (3C, CH3). FTIR-ATR (cm−1): 3058 (vw), 3035 (vw), 2956 (w), 2865 (vw), 2712 (vw), 2605 (vw), 2324 (vw), 1927 (vw), 1814 (vw), 1740 (vw), 1629 (vs), 1602 (s), 1542 (s), 1508 (vw), 1461 (s), 1440 (s), 1407 (m), 1380 (w), 1353 (s), 1303 (vw), 1267 (m), 1244 (vs), 1198 (w), 1159 (m), 1137 (m), 1094 (m), 1028 (m), 956 (w), 880 (m), 858 (m), 839 (m), 797 (m), 759 (vs), 706 (m), 669 (m), 639 (s), 572 (m), 532 (w), 462 (m), 444 (m), 419 (w), 405 (w). For catalytic purposes, the crystals of 1 were recrystallized from ethanol and, as a result of transesterification, the formation of ethyl salicylate ligands was observed.
O), 170.8 (3C, C–O), 137.4 (3C, ArH), 130.2 (3C, ArH), 123.9 (3C, ArH), 114.7 (3C, ArH), 111.6 (3C, Ar), 63.4 (3C, CH2), 14.0 (3C, CH3). FTIR-ATR (cm−1): 3092 (vw), 3034 (vw), 3009 (vw), 2983 (w), 2804 (vw), 2701 (vw), 2605 (vw), 2493 (vw), 2324 (vw), 1928 (vw), 1911 (vw), 1817 (vw), 1627 (vs), 1601 (s), 1542 (s), 1509 (vw), 1463 (s), 1450 (s), 1404 (s), 1377 (m), 1346 (s), 1301 (vw), 1267 (m), 1239 (vs), 1179 (w), 1159 (m), 1137 (m), 1094 (m), 1028 (w), 992 (w), 893 (w), 873 (vw), 859 (w), 837 (m), 800 (w), 759 (vs), 707 (m), 669 (m), 637 (s), 572 (m), 534 (w), 479 (m), 455 (m), 436 (m), 420 (w).
O), 170.6 (2C, C–O), 137.6 (2C, ArH), 131.3 (2C, ArH), 124.2 (2C, ArH), 116.1 (2C, ArH), 112.1 (2C, Ar), 53.7 (2C, CH3), −6.7 (4C, GaCH3). FTIR-ATR (cm−1): 3188 (vw), 3072 (vw), 2956 (w), 2916 (vw), 1677 (s), 1616 (m), 1586 (m), 1554 (w), 1486 (m), 1440 (s), 1327 (m), 1304 (s), 1248 (s), 1217 (vs), 1158 (m), 1136 (m), 1091 (m), 1045 (vw), 1033 (w), 964 (w), 866 (w), 849 (w), 822 (vw), 800 (vw), 753 (s), 721 (vs), 698 (vs), 666 (m), 528 (vs), 438 (m).
O), 171.1 (4C, C–O), 168.2 (2C, C–O), 135.5 (2C, ArH), 134.9 (4C, ArH), 131.8 (2C, ArH), 130.6 (4C, ArH), 124.1 (4C, ArH), 123.8 (2C, ArH), 114.9 (4C, ArH), 114.7 (2C, ArH), 112.3 (6C, Ar), 52.2 (4C, CH3), 51.2 (2C, CH3). 7Li NMR (THF-D8, 155 MHz): δ 4.00. FTIR-ATR (cm−1): 3027 (vw), 2950 (w), 2844 (vw), 2319 (vw), 2234 (vw), 2162 (vw), 2141 (vw), 2109 (vw), 2081 (vw), 2049 (vw), 1979 (vw), 1925 (vw), 1703 (m), 1677 (m), 1641 (s), 1613 (vs), 1598 (vs), 1566 (w), 1546 (m), 1464 (vs), 1442 (s), 1387 (w), 1353 (m), 1325 (m), 1292 (w), 1254 (vs), 1223 (s), 1198 (m), 1153 (m), 1085 (m), 1035 (w), 954 (w), 898 (w), 861 (s), 817 (w), 798 (w), 764 (s), 755 (vs), 707 (s), 676 (w), 662 (w), 599 (m), 581(m), 558 (w), 533 (m), 493 (m), 472 (w), 451 (w), 431 (w).
O), 171.8 (4C, CO), 170.6 (4C, C–O), 168.3 (2C, C–O), 135.5 (2C, ArH), 134.7 (4C, ArH), 131.8 (2C, ArH), 130.6 (4C, ArH), 124.1 (6C, ArH), 115.0 (2C, ArH), 114.9 (4C, ArH), 112.0 (6C, Ar), 61.7 (4C, CH2), 60.2 (2C, CH2), 14.6 (2C, CH3), 14.3 (4C, CH3). 7Li NMR (THF-D8, 155 MHz): δ 4.01. FTIR-ATR (cm−1): 3072 (vw), 2982 (w), 2955 (vw), 2935 (vw), 2896 (vw), 2319 (vw), 2162 (vw), 2141 (vw), 1929 (vw), 1707 (m), 1676 (s), 1650 (s), 1620 (s), 1599 (s), 1564 (w), 1547 (m), 1467 (vs), 1445 (s), 1409 (m), 1379 (w), 1367 (w), 1342 (m), 1349 (m), 1319 (w), 1292 (m), 1255 (vs), 1230 (s), 1215 (vs), 1161 (m), 1149 (s), 1117 (vw), 1088 (s), 1076 (s), 1045 (vw), 1032 (m), 1012 (w), 952 (vw), 891 (m), 866 (m), 853 (m), 827 (m), 795 (w), 765 (vs), 758 (vs), 708 (s), 664 (m), 601 (s), 582 (m), 561(w), 534 (w), 494 (m), 476 (w), 458 (w), 425 (w), 405 (w).
O), 166.6 (2C, C–O), 135.4 (2C, ArH), 132.5 (2C, ArH), 121.1 (2C, ArH), 116.8 (2C, Ar), 116.1 (2C, ArH), 68.0 (2C, CH2), 51.6 (2C, CH3), 26.2 (2C, CH2), −6.4 (2C, GaCH3). 7Li NMR (THF-D8, 155 MHz): δ 3.76. FTIR-ATR (cm−1): 3363 (vw), 3059 (vw), 2952 (w), 2871 (w), 2651(vw), 2051 (vw), 1981 (vw), 1928 (vw), 1680 (vs), 1598 (m), 1561 (m), 1475 (s), 1445 (s), 1434 (s), 1317 (s), 1297 (m), 1264 (m), 1232 (vs), 1194 (m), 1160 (s), 1147 (m), 1087 (s), 1056 (m), 1044 (m), 969 (w), 953 (vw), 899 (w), 864 (m), 818 (w), 797 (w), 758 (s), 727 (m), 704 (s), 666 (m), 597 (s), 578 (s), 535 (m), 479 (m).
Glyceryl (S,S)-O-lactyllactate (GlL2)
O)), 5.05 (q, J = 7.0 Hz, 1H, CH(CO)), 4.92 (dd, J = 5.2, 3.4 Hz, 1H, HO(CH)), 4.65 (m, 1H, HO(CH2)), 4.20 (m, 1H, CH(CO)), 4.08, 3.95 (m, 2H, OCH2), 3.64 (m, 1H, CH(OH)), 3.42 (m, 2H, CH2(OH)), 1.42 (d, J = 7.0 Hz, 3H, CH3(CHO)), 1.30 (d, J = 7.0 Hz, 3H, CH3(CHOH)). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 174.1 (1C, CO), 170.3 (1C, CO), 72.5 (1C, CH2(OH)), 69.2 (1C, CH(OH)), 68.3 (1C, CH(CO)), 65.6 (1C, CH(CO)), 66.4 (1C, OCH2), 20.4 (1C, CH3(CHOH)), 16.7 (1C, CH3(CHO)).
O)), 5.05 (q, J = 7.0 Hz, 1H, CH(CO)), 4.74 (m, 1H, OCH), 4.20 (m, 1H, CH(CO)), 3.49 (m, 4H, CH2(OH)), 3.47–3.24 (s, 2H, HO(CH2)), 1.42 (d, J = 7.0 Hz, 3H, CH3(CHO)), 1.30 (d, J = 7.0 Hz, 3H, CH3(CHOH)). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 174.1 (1C, CO), 170.3 (1C, CO), 76.6 (1C, OCH), 68.3 (1C, CH(CO)), 65.6 (1C, CH(CO)), 59.5 (2C, CH2(OH)), 20.4 (1C, CH3(CHOH)), 16.7 (1C, CH3(CHO)).
Glyceryl glycolylglycolate (GlG2)
O)), 4.97 (d, J = 5.2 Hz, 1H, HO(CH)), 4.71 (s, 2H, CH2(CO)), 4.69 (t, J = 5.7 Hz, 1H, HO(CH2)), 4.14, 3.98 (m, 2H, OCH2), 4.11 (m, 2H, CH2(CO)), 3.66 (m, 1H, CH(OH)), 3.49 (m, 2H, CH2(OH)). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 172.3 (1C, CO), 167.8 (1C, CO), 72.3 (1C, CH2(OH)), 69.2 (1C, CH(OH)), 66.2 (1C, OCH2), 60.4 (1C, CH2(CO)), 59.4 (1C, CH2(CO)).
O)), 4.89 (m, 1H, OCH), 4.70 (s, 2H, CH2(CO)), 4.11 (m, 2H, CH2(CO), 3.54–3.45 (m, 4H, CH2(OH)), 3.45–3.24 (s, 2H, HO(CH2)).
Glyceryl tris(glycolyl)glycolate (GlG4)
O)), 4.97 (d, J = 5.2 Hz, 1H, HO(CH)), 4.88 (s, 2H, CH2(CO)), 4.83 (s, 2H, CH2(CO)), 4.78 (s, 2H, CH2(CO)), 4.69 (t, J = 5.7 Hz, 1H, HO(CH2)), 4.14, 3.98 (m, 2H, OCH2), 4.11 (m, 2H, CH2(CO)), 3.66 (m, 1H, CH(OH)), 3.49 (m, 2H, CH2(OH)).
O)), 4.89 (m, 1H, OCH), 4.88 (s, 2H, CH2(CO)), 4.83 (s, 2H, CH2(CO)), 4.78 (s, 2H, CH2(CO)), 4.11 (m, 2H, CH2(CO)), 3.54–3.45 (m, 4H, CH2(OH)), 3.45–3.24 (s, 2H, HO(CH2)).
Glyceryl (S)-lactate (GlL1)
O)), 4.89 (d, J = 5.4 Hz, 1H, HO(CH)), 4.64 (t, J = 5.5 Hz, 1H, HO(CH2)), 4.14 (m, 1H, CH(CO)), 4.09, 3.94 (m, 2H, OCH2), 3.64 (m, 1H, CH(OH)), 3.42 (m, 2H, CH2(OH)), 1.25 (d, J = 6.7 Hz, 3H, CH3). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 174.7 (1C, CO), 72.5 (1C, CH2(OH)), 69.3 (1C, CH(OH)), 66.0 (1C, CH(CO)), 65.7 (1C, OCH2), 20.5 (1C, CH3).
O)), 4.74 (m, 1H, OCH), 4.14 (m, 1H, CH(CO)), 3.50 (m, 4H, CH2(OH)), 3.47–3.24 (s, 2H, HO(CH2)), 1.25 (d, J = 6.7 Hz, 3H, CH3). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 174.5 (1C, CO), 75.8 (1C, OCH), 66.0 (1C, CH(CO)), 59.8 (2C, CH2(OH)), 20.6 (1C, CH3).
Glyceryl glycolate (GlG1)
O)), 4.89 (d, J = 5.5 Hz, 1H, HO(CH)), 4.64 (t, J = 5.5 Hz, 1H, HO(CH2)), 4.09, 3.94 (m, 2H, OCH2), 4.01 (m, 2H, CH2(CO)), 3.64 (m, 1H, CH(OH)), 3.42 (m, 2H, CH2(OH)). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 172.8 (1C, CO), 72.6 (1C, CH2(OH)), 69.3 (1C, CH(OH)), 65.7 (1C, OCH2), 59.7 (1C, CH2(CO)).
O)), 4.76 (m, 1H, OCH), 4.01 (m, 2H, CH2(CO), 3.49 (m, 4H, CH2(OH)), 3.45–3.24 (s, 2H, HO(CH2)). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 172.6 (1C, CO), 75.9 (1C, OCH), 59.8 (2C, CH2(OH)), 59.6 (1C, CH2(CO)).
Glyceryl 6-hydroxyhexanoate (GlCL1)
O), 2.29 (m, 2H, CH2(CO)), 1.52 (m, 2H, CH2(CH2CO)), 1.40 (m, 2H, CH2(CH2OH)), 1.28 (m, 2H, CH2(CH2CH2OH)). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 173.0 (1C, CO), 72.5 (1C, CH2(OH)), 69.3 (1C, CH(OH)), 65.5 (1C, OCH2), 59.8 (1C, CH2(CH2)4CO), 33.6 (1C, CH2(CO), 32.2 (1C, CH2(CH2OH)), 25.1 (1C, CH2(CH2CH2OH)), 24.4 (1C, CH2(CH2CO)).
O), 3.40–3.23 (s, 2H, HO(CH2)), 2.29 (m, 2H, CH2(CO)), 1.52 (m, 2H, CH2(CH2CO)), 1.40 (m, 2H, CH2(CH2OH)), 1.28 (m, 2H, CH2(CH2CH2OH)). 13C{1H} NMR (DMSO-D6, 101 MHz): δ 173.0 (1C, CO), 75.4 (1C, OCH), 60.5 (2C, CH2OH), 59.8 (1C, CH2(CH2)4CO), 33.6 (1C, CH2(CO), 32.2 (1C, CH2(CH2OH)), 25.1 (1C, CH2(CH2CH2OH)), 24.4 (1C, CH2(CH2CO)).
Author contributions
Rafał Petrus: involved in conceptualization, investigation, project administration, funding acquisition, and original draft writing and editing. Józef Utko: crystallization of 1, 3 and 3a. Joanna Petrus: IR investigations of 1–6 and writing – original draft. Mohammad Awashra: the study of L-LA and GA alcoholysis under catalyst-free conditions or using 5. Tadeusz Lis: single-crystal X-ray diffraction measurements of 1–3a and 6. All authors have read and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Polish National Science Center for financial support, grant number 2017/26/D/ST5/01123.Notes and references
- K. A. Wurzel, Glycerol, in Encyclopedia of Toxicology, ed. P. Wexler, Elsevier, 2nd edn, 2005, pp. 449–451 Search PubMed.
- H. W. Tan, A. A. A. Raman and M. K. Aroua, Glycerol Production and Its Applications as a Raw Material: A Review, Renewable Sustainable Energy Rev., 2013, 27, 118–127 CrossRef CAS.
- Y. Wang, J. Zhou and X. Guo, Catalytic Hydrogenolysis of Glycerol to Propanediols: A Review, RSC Adv., 2015, 5(91), 74611–74628 RSC.
- X.-L. Li, Q. Zhou, S.-X. Pan, Y. He and F. Chang, A Review of Catalytic Upgrading of Biodiesel Waste Glycerol to Valuable Products, Curr. Green Chem., 2020, 7(3), 259–266 CrossRef CAS.
- S. Sahani, S. N. Upadhyay and Y. C. Sharma, Critical Review on Production of Glycerol Carbonate from Byproduct Glycerol through Transesterification, Ind. Eng. Chem. Res., 2021, 60(1), 67–88 CrossRef CAS.
- A. G. Adeniyi and J. O. Ighalo, A Review of Steam Reforming of Glycerol, Chem. Pap., 2019, 73(11), 2619–2635 CrossRef CAS.
- S. Veluturla, N. Archna, D. S. Rao, N. Hezil, I. S. Indraja and S. Spoorthi, Catalytic Valorization of Raw Glycerol Derived from Biodiesel: A Review, Biofuels, 2018, 9(3), 305–314 CrossRef CAS.
- Y. Wang, S. Lu, P. Gabriele and J. J. Harris, Poly(Glycerol Sebacate) in Tissue Engineering and Regenerative Medicine, Mater. Matters, 2016, 11(3), 80–83 CAS.
- A. M. Fischer, C. Schüll and H. Frey, Hyperbranched Poly(Glycolide) Copolymers with Glycerol Branching Points via Ring-Opening Copolymerization, Polymer, 2015, 72, 436–446 CrossRef CAS.
- F. K. Wolf, A. M. Fischer and H. Frey, Poly(Glycolide) Multi-Arm Star Polymers: Improved Solubility via Limited Arm Length, Beilstein J. Org. Chem., 2010, 6, 67 Search PubMed.
- E. Göktürk, A. G. Pemba and S. A. Miller, Polyglycolic Acid from the Direct Polymerization of Renewable C1 Feedstocks, Polym. Chem., 2015, 6(21), 3918–3925 RSC.
- A. Michalski, M. Brzezinski, G. Lapienis and T. Biela, Star-Shaped and Branched Polylactides: Synthesis, Characterization, and Properties, Prog. Polym. Sci., 2019, 89, 159–212 CrossRef CAS.
- M. Ghasri, A. Jahandideh, K. Kabiri, H. Bouhendi, M. J. Zohuriaan-Mehr and N. Moini, Glycerol-Lactic Acid Star-Shaped Oligomers as Efficient Biobased Surface Modifiers for Improving Superabsorbent Polymer Hydrogels, Polym. Adv. Technol., 2019, 30(2), 390–399 CrossRef CAS.
- G. A. Brooks, Glycerol-Lactate Esters for Use as an Energy Supplement during Exercise and Recovery, US6743821B2, 2004. SciFinder Scholar 2008:23397.
- D. H. Bae and G. E. Lee, Antiseptic and Antibacterial Composition for External Application, Containing Glycerin Lactic Acid Ester as Active Ingredient, WO2018048042A11, 2018. SciFinder Scholar 2018:444780.
- Y. Yuan, Z. Hu, X. Fu, L. Jiang, Y. Xiao, K. Hu, P. Yan and J. Lei, Poly(Lactic Acid) Plasticized by Biodegradable Glyceryl Lactate, J. Appl. Polym. Sci., 2016, 133(21), 43460 CrossRef.
- H.-J. Jung, Y. Cho, D. Kim and P. Mehrkhodavandi, Cationic Aluminum, Gallium, and Indium Complexes in Catalysis, Catal. Sci. Technol., 2021, 11(1), 62–91 RSC.
- S. Saito and H. Yamamoto, Designer Lewis Acid Catalysts—Bulky Aluminium Reagents for Selective Organic Synthesis, Chem. Commun., 1997, 17, 1585–1592 RSC.
- S. Saito, Aluminum Trisphenoxide Polymer as a Lewis Acidic, Solid Catalyst, Synlett, 1999,(1), 57–58 CrossRef CAS.
- D. Tanaka, Y. Kadonaga, Y. Manabe, K. Fukase, S. Sasaya, H. Maruyama, S. Nishimura, M. Yanagihara, A. Konishi and M. Yasuda, Synthesis of Cage-Shaped Aluminum Aryloxides: Efficient Lewis Acid Catalyst for Stereoselective Glycosylation Driven by Flexible Shift of Four- to Five-Coordination, J. Am. Chem. Soc., 2019, 141(44), 17466–17471 CrossRef CAS PubMed.
- X.-X. Zheng and Z.-X. Wang, Synthesis of Aluminum Complexes Supported by 2-(1,10-Phenanthrolin-2-Yl)Phenolate Ligands and Their Catalysis in the Ring-Opening Polymerization of Cyclic Esters, RSC Adv., 2017, 7(44), 27177–27188 RSC.
- C. Zhang and Z.-X. Wang, Aluminum and Zinc Complexes Supported by Functionalized Phenolate Ligands: Synthesis, Characterization and Catalysis in the Ring-Opening Polymerization of ε-Caprolactone and Rac-Lactide, J. Organomet. Chem., 2008, 693(19), 3151–3158 CrossRef CAS.
- A. M. McCollum, A. M. Longo, A. E. Stahl, A. S. Butler, A. L. Rheingold, T. R. Cundari, D. B. Green, K. R. Brereton and J. M. Fritsch, Synthesis, Spectroscopy, and Crystallography of Mononuclear, Five-Coordinate Aluminum Complexes That Act as Cyclic Ester Polymerization Initiators, Polyhedron, 2021, 204(115233), 115233 CrossRef CAS.
- B. Gao, D. Li, Y. Li, Q. Duan, R. Duan and X. Pang, Ring-Opening Polymerization of Lactide Using Chiral Salen Aluminum Complexes as Initiators: High Productivity and Stereoselectivity, New J. Chem., 2015, 39(6), 4670–4675 RSC.
- Y. Chapurina, T. Roisnel, J.-F. Carpentier and E. Kirillov, Zinc, Aluminum and Group 3 Metal Complexes of Sterically Demanding Naphthoxy-Pyridine Ligands: Synthesis, Structure, and Use in ROP of Racemic Lactide and β-Butyrolactone, Inorg. Chim. Acta, 2015, 431, 161–175 CrossRef CAS.
- L. Qin, Y. Zhang, J. Chao, J. Cheng and X. Chen, Four- and Five-Coordinate Aluminum Complexes Supported by N,O-Bidentate β-Pyrazylenolate Ligands: Synthesis, Structure and Application in ROP of ε-Caprolactone and Lactide, Dalton Trans., 2019, 48(32), 12315–12325 RSC.
- C. Robert, T. E. Schmid, V. Richard, P. Haquette, S. K. Raman, M.-N. Rager, R. M. Gauvin, Y. Morin, X. Trivelli, V. Guérineau, I. del Rosal, L. Maron and C. M. Thomas, Mechanistic Aspects of the Polymerization of Lactide Using a Highly Efficient Aluminum(III) Catalytic System, J. Am. Chem. Soc., 2017, 139(17), 6217–6225 CrossRef CAS PubMed.
- K. Nie, C. Wang, X. Cheng, J. Li, Y. Han and Y. Yao, Unbridged bidentate aluminum complexes supported by diaroylhydrazone ligands: Synthesis, structure and catalysis in polymerization of ε-caprolactone and lactides, Appl. Organomet. Chem., 2020, 34, e5627 CrossRef CAS.
- E. Yue, F. Cao, J. Zhang, W. Zhang, Y. Jiang, T. Liang and W.-H. Sun, Bimetallic Aluminum Complexes Bearing Novel Spiro-Phenanthrene-Monoketone/OH Derivatives: Synthesis, Characterization and the Ring-Opening Polymerization of ε-Caprolactone, RSC Adv., 2021, 11(22), 13274–13281 RSC.
- F. Isnard, F. Santulli, M. Cozzolino, M. Lamberti, C. Pellecchia and M. Mazzeo, Tetracoordinate Aluminum Complexes Bearing Phenoxy-Based Ligands as Catalysts for Epoxide/Anhydride Copolymerization: Some Mechanistic Insights, Catal. Sci. Technol., 2019, 9(12), 3090–3098 RSC.
- N. Zhao, Q. Wang, G. Hou, H. Song and G. Zi, Synthesis, Structure, and Catalytic Activity of Aluminum Chloride Complexes with Chiral Biaryl Schiff-Base Ligands, Inorg. Chem. Commun., 2014, 44, 86–90 CrossRef CAS.
- D. Specklin, C. Fliedel, F. Hild, S. Mameri, L. Karmazin, C. Bailly and S. Dagorne, Mononuclear Salen-Gallium Complexes for Iso-Selective Ring-Opening Polymerization (ROP) of Rac-Lactide, Dalton Trans., 2017, 46(38), 12824–12834 RSC.
- M. T. Muñoz, M. Palenzuela, T. Cuenca and M. E. G. Mosquera, Aluminum Aryloxide Compounds as Very Active Catalysts for Glycidyl Methacrylate Selective Ring-Opening Polymerization, ChemCatChem, 2018, 10(5), 936–939 CrossRef.
- H. Sasai, T. Arai, Y. Satow, K. N. Houk and M. Shibasaki, The First Heterobimetallic Multifunctional Asymmetric Catalyst, J. Am. Chem. Soc., 1995, 117(23), 6194–6198 CrossRef CAS.
- W. Gruszka, A. Lykkeberg, G. S. Nichol, M. P. Shaver, A. Buchard and J. A. Garden, Combining Alkali Metals and Zinc to Harness Heterometallic Cooperativity in Cyclic Ester Ring-Opening Polymerisation, Chem. Sci., 2020, 11(43), 11785–11790 RSC.
- R. Petrus, J. Utko, R. Gniłka, M. G. Fleszar, T. Lis and P. Sobota, Solvothermal Alcoholysis Method for Recycling High-Consistency Silicone Rubber Waste, Macromolecules, 2021, 54(5), 2449–2465 CrossRef CAS.
- R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Deprotonative Metalation Using Ate Compounds: Synergy, Synthesis, and Structure Building, Angew. Chem., Int. Ed., 2007, 46, 3802–3824 CrossRef CAS PubMed.
- W. Gruszka, H. Sha, A. Buchard and J. A. Garden, Heterometallic cooperativity in divalent metal ProPhenol catalysts: combining zinc with magnesium or calcium for cyclic ester ringopening polymerisation, Catal. Sci. Technol., 2022 10.1039/d1cy01914g.
- T. Arai, H. Sasai, K.-I. Aoe, K. Okamura, T. Date and M. Shibasaki, A New Multifunctional Heterobimetallic Asymmetric Catalyst for Michael Additions and Tandem Michael–Aldol Reactions, Angew. Chem., Int. Ed. Engl., 1996, 35(1), 104–106 CrossRef CAS.
- T. Arai, H. Sasai, K. Yamaguchi and M. Shibasaki, Regioselective Catalytic Asymmetric Reaction of Horner-Wadsworth-Emmons Reagents with Enones: The Odyssey of Chiral Aluminum Catalysts, J. Am. Chem. Soc., 1998, 120, 441–442 CrossRef CAS.
- S. Matsunaga, J. Das, J. Roels, E. M. Vogl, N. Yamamoto, T. Iida, K. Yamaguchi and M. Shibasaki, Catalytic Enantioselective Meso-Epoxide Ring Opening Reaction with Phenolic Oxygen Nucleophile Promoted by Gallium Heterobimetallic Multifunctional Complexes, J. Am. Chem. Soc., 2000, 122(10), 2252–2260 CrossRef CAS.
- W. Gruszka and J. A. Garden, Advances in heterometallic ring-opening (co)polymerisation catalysis, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed.
- F. Hild, P. Haquette, L. Brelot and S. Dagorne, Synthesis and structural characterization of well-defined anionic aluminium alkoxide complexes supported by NON-type diamido ether tridentate ligands and their use for the controlled ROP of lactide, Dalton Trans., 2010, 39, 533–540 RSC.
- T. Muñoz, T. Cuenca and M. E. G. Mosquera, Heterometallic aluminates: alkali metals trapped by an aluminium aryloxide claw, Dalton Trans., 2014, 43, 14377–14385 RSC.
- M. Normand, E. Kirillov, T. Roisnel and J.-F. Carpentier, Indium Complexes of Fluorinated Dialkoxy-Diimino Salen-like Ligands for Ring Opening Polymerization of rac-Lactide: How Does Indium Compare to Aluminum?, Organometallics, 2012, 31, 1448–1457 CrossRef CAS.
- W. T. Diment, G. L. Gregory, R. W. F. Kerr, A. Phanopoulos, A. Buchard and C. K. Williams, Catalytic Synergy Using Al(III) and Group 1 Metals to Accelerate Epoxide and Anhydride Ring-Opening Copolymerizations, ACS Catal., 2021, 11, 12532–12542 CrossRef CAS.
- S. W. Kwon and S. B. Park, Effect of precursors on the morphology of lithium aluminate prepared by hydrothermal treatment, J. Mater. Sci., 2000, 35, 1973–1978 CrossRef CAS.
- O. Renoult, J.-P. Boilot and M. Boncoeur, Alkoxide-Hydroxide Route for the Preparation of γ-LiAlO2-Based Ceramics, J. Nucl. Mater., 1995, 26, 233–239 CrossRef.
- F. Bianchini, H. Fjellvåg and P. Vajeeston, A First Principle Comparative Study of the Ionic Diffusivity in LiAlO2 and NaAlO2 Polymorphs for Solid-State Battery Applications, Phys. Chem. Chem. Phys., 2018, 20(15), 9824–9832 RSC.
- E. Temeche, S. Indris and R. M. Laine, LiAlO2/LiAl5O8 Membranes Derived from Flame-Synthesized Nanopowders as a Potential Electrolyte and Coating Material for All-Solid-State Batteries, ACS Appl. Mater. Interfaces, 2020, 12(41), 46119–46131 CrossRef CAS PubMed.
- R. Baron, T. Wejrzanowski, J. Milewski, Ł. Szabłowski, A. Szczęśniak and K.-Z. Fung, Manufacturing of γ-LiAlO2 Matrix for Molten Carbonate Fuel Cell by High-Energy Milling, Int. J. Hydrogen Energy, 2018, 43(13), 6696–6700 CrossRef CAS.
- S. W. Kwon and S. B. Park, Effect of Precursors on the Preparation of Lithium Aluminate, J. Nucl. Mater., 1997, 246(2–3), 131–138 CrossRef CAS.
- C. Chen, C.-A. Li, S.-H. Yu and M. M. C. Chou, Growth and Characterization of β-LiGaO2 Single Crystal, J. Cryst. Growth, 2014, 402, 325–329 CrossRef CAS.
- L. Trinkler, A. Trukhin, B. Berzina, V. Korsaks, P. Ščajev, R. Nedzinskas, S. Tumėnas, M. M. C. Chou, L. Chang and C.-A. Li, Luminescence Properties of LiGaO2 Crystal, Opt. Mater., 2017, 69, 449–459 CrossRef CAS.
- E. S. Hellman, Z. Liliental-Weber and D. N. E. Buchanan, Epitaxial Growth and Orientation of GaN on (1 0 0) γ-LiAlO2, MRS Internet J. Nitride Semicond. Res., 1997, 2, 32 CrossRef.
- C. Chen, S. Sun, M. M. C. Chou and K. Xie, In Situ Inward Epitaxial Growth of Bulk Macroporous Single Crystals, Nat. Commun., 2017, 8, 2178 CrossRef PubMed.
- J. Wang, L. Pu, G. Tang and J. Zhang, Electronic and Structural Characterization of InN Heterostructures Grown on β-LiGaO2 (001) Substrates, Vacuum, 2015, 119, 106–111 CrossRef CAS.
- C. Chen, T. Yan, S.-H. Yu, C.-Y. Lee, C.-W. Chang and M. M. C. Chou, Microstructural and Optical Properties of High-Quality ZnO Epitaxially Grown on a LiGaO2 Substrate, RSC Adv., 2015, 5(45), 35405–35411 RSC.
- T. Yan, Y. Min, M. Lin, C. Chen, C. Lee, L. Zhao, N. Ye, M. M. C. Chou, H. Liu and W. Zhou, Intersected Nonpolar ZnO Nanosail Arrays Aligned Epitaxially on LiGaO2 Substrate towards Enhanced Photoelectrochemical Responses, Nano Sel., 2021, 2(6), 1233–1243 CrossRef CAS.
- J. Lewiński, J. Zachara and E. Grabska, Synthesis and Molecular Structure of (tBuOO)(tBuO)Al(μ-OtBu)2Al(mesal)2. The First Structurally Characterized (Alkylperoxo)aluminum Compound, J. Am. Chem. Soc., 1996, 118(28), 6794–6795 CrossRef.
- S. Dutta and V. Manivannan, A Mononuclear Manganese(III) Schiff Base Complex, [Mn(C14H10N2O2)(C8H7O3)(CH4O)], Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 813–815 CrossRef.
- F. E. Jacobsen and S. M. Cohen, Using Model Complexes To Augment and Advance Metalloproteinase Inhibitor Design, Inorg. Chem., 2004, 43, 3038–3047 CrossRef CAS PubMed.
- L. M. R. Hill, M. K. Taylor, V. W. L. Ng and C. G. Young, Toward Multifunctional Mo(VI–IV) Complexes: cis-Dioxomolybdenum(VI) Complexes Containing Hydrogen-Bond Acceptors or Donors, Inorg. Chem., 2008, 47, 1044–1052 CrossRef CAS PubMed.
- T. Ohta, T. Imagawa and S. Ito, Involvement of transient receptor potential vanilloid subtype 1 in analgesic action of methylsalicylate, Mol. Pharmacol., 2009, 75, 307–317 CrossRef CAS PubMed.
- A. Lapczynski, L. Jones, D. McGinty, S. P. Bhatia, C. S. Letizia and A. M. Api, Fragrance material review on methyl salicylate, Food Chem. Toxicol., 2007, 45, 428–452 CrossRef PubMed.
- J. Lewiński, J. Zachara, B. Mańk and S. Pasynkiewicz, Synthesis and Molecular Structure of AlMe[O·C(OMe)C6H4-o-O]2. The First Structurally Characterized Five-Coordinated Simple Alky, J. Organomet. Chem., 1993, 454(1–2), 5–7 CrossRef.
- J. Lewiński, J. Zachara and K. B. Starowieyski, Structure Investigation of Dimethyl-Aluminium, -Gallium and -Indium O,O′-Chelate Complexes in Solution and the Solid State. Molecular Structure of Five-Co-Ordinated [Me2M(μ-OC6H4CO2Me-2)]2 Adducts, J. Chem. Soc., Dalton Trans., 1997, 22, 4217–4222 RSC.
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, Alkyl gallium(III) quinolinolates: A new class of highly selective anti-leishmanial agents, Eur. J. Med. Chem., 2020, 186, 111895 CrossRef CAS PubMed.
- L. G. Bloor, C. J. Carmalt and D. Pugh, Single-source precursors to gallium and indium oxide thin films, Coord. Chem. Rev., 2011, 255, 1293–1318 CrossRef CAS.
- C. J. Carmalt and S. J. King, Gallium(III) and indium(III) alkoxides and aryloxides, Coord. Chem. Rev., 2006, 250, 682–709 CrossRef CAS.
- T. Li, R. Fang, B. Wang, Y. Shao, J. Liu, S. Zhang and Z. Yang, A Simple Coumarin as a Turn-on Fluorescence Sensor for Al(III) Ions, Dalton Trans., 2014, 43(7), 2741–2743 RSC.
- S. Müller, S. Steil, A. Droghetti, N. Großmann, V. Meded, A. Magri, B. Schäfer, O. Fuhr, S. Sanvito, M. Ruben, M. Cinchetti and M. Aeschlimann, Spin-Dependent Electronic Structure of the Co/Al(OP)3 Interface, New J. Phys., 2013, 15(11), 113054 CrossRef.
- A. R. Sanwaria, M. Nagar, R. Bohra, A. Chaudhary, S. M. Mobin, P. Mathur and B. L. Choudhary, Sol–Gel Synthesis of Highly Pure α-Al2O3 Nano-Rods from a New Class of Precursors of Salicylaldehyde-Modified Aluminum(Iii) Isopropoxide. Crystal and Molecular Structure of [Al(OC6H4CHO)3], RSC Adv., 2014, 4(57), 30081–30089 RSC.
- H. C. Aspinall, J. Bacsa, O. D. Beckingham, E. G. B. Eden, N. Greeves, M. D. Hobbs, F. Potjewyd, M. Schmidtmann and C. D. Thomas, Adding the Right (or Left) Twist to Tris-Chelate Complexes–Coordination Chemistry of Chiral Oxazolylphenolates with M3+ Ions (M = Al or Lanthanide), Dalton Trans., 2014, 43(3), 1434–1442 RSC.
- M. A. Bahili, E. C. Stokes, R. C. Amesbury, D. M. C. Ould, B. Christo, R. J. Horne, B. M. Kariuki, J. A. Stewart, R. L. Taylor, P. A. Williams, M. D. Jones, K. D. M. Harris and B. D. Ward, Aluminium-Catalysed Isocyanate Trimerization, Enhanced by Exploiting a Dynamic Coordination Sphere, Chem. Commun., 2019, 55(53), 7679–7682 RSC.
- J. E. Bollinger, J. T. Mague and D. M. Roundhill, Lipophilic Hexadentate Aluminum, Gallium, Indium, and Iron Complexes of a New Phenolate-Derivatized Cyclohexanetriamine Ligand, Inorg. Chem., 1994, 33(7), 1241–1242 CrossRef CAS.
- S. Alvarez, Distortion Pathways of Transition Metal Coordination Polyhedra Induced by Chelating Topology, Chem. Rev., 2015, 115, 13447–13483 CrossRef CAS PubMed.
- H. Schumann, M. Frick, B. Heymer and F. Girgsdies, Intramolecularly Stabilized Organoaluminium and Organogallium Compounds: Synthesis and X-Ray Crystal Structures of Some Dimethylaluminium and -Gallium Alkoxides Me2M–O–R–OR′ and Amides Me2M–NH–R–OR′, J. Organomet. Chem., 1996, 512(1–2), 117–126 CrossRef CAS.
- C. E. Knapp, L. Pemberton, C. J. Carmalt, D. Pugh, P. F. McMillan, S. A. Barnett and D. A. Tocher, Synthesis, AACVD and X-Ray Crystallographic Structures of Group 13 Monoalkoxometallanes, Main Group Chem., 2010, 9(1,2), 31–40 CAS.
- P. Horeglad, M. Cybularczyk, A. Litwińska, A. M. Dąbrowska, M. Dranka, G. Z. Żukowska, M. Urbańczyk and M. Michalak, Controlling the Stereoselectivity of Rac-LA Polymerization by Chiral Recognition Induced the Formation of Homochiral Dimeric Metal Alkoxides, Polym. Chem., 2016, 7(11), 2022–2036 RSC.
- D. G. Hendershot, M. Barber, R. Kumar and J. P. Oliver, Synthesis and Characterization of Alkylaluminum and -Gallium Derivatives of 2-(Methylthio)- and 2-Methoxyphenol. Crystal Structures of the Dimeric Complexes [R2M(μ-OC6H4-2-ECH3)]2 (M = Aluminum, E = S, R = Me, Iso-Bu; M = Al, E = O, R = Et, Iso-Bu; M = Gallium, E = O, R = Me) Featuring Five Coordinate Metal Centers, Organometallics, 1991, 10(9), 3302–3309 CrossRef CAS.
- C. E. Knapp and C. J. Carmalt, Solution Based CVD of Main Group Materials, Chem. Soc. Rev., 2016, 45(4), 1036–1064 RSC.
- L. G. Bloor, C. J. Carmalt and D. Pugh, Single-Source Precursors to Gallium and Indium Oxide Thin Films, Coord. Chem. Rev., 2011, 255(11–12), 1293–1318 CrossRef CAS.
- W. Ziemkowska, Group 13 Alkyl Compounds Incorporating Aliphatic and Aromatic Diolate Ligands, Coord. Chem. Rev., 2005, 249(21–22), 2176–2194 CrossRef CAS.
- J. A. Francis, S. G. Bott and A. R. Barron, Are intramolecularly stabilized compounds of aluminum suitable structural models of the SN2 transition state? Molecular structure of [(tBu)2Al(μ-OC6H4-2-OMe)]2, Polyhedron, 1999, 18, 2211–2218 CrossRef CAS.
- C. N. McMahon, S. G. Bott and A. R. Barron, Observation of an unusual amine oxidation reaction during the oxidation and hydrolysis of [(tBu)2Ga(o,-C6H4NMe2)]2: molecular structures of (tBu)2Ga(o-C6H4NMe2)(μ-OH)-Ga(tBu)[o-C6H4N(O)Me2](tBu)2Ga(o-C6H4NMe2)(O=PPh3), Polyhedron, 1997, 16, 3407–3413 CrossRef CAS.
- W. Clegg, E. Lamb, S. T. Liddle, R. Snaith and A. E. H. Wheatley, Towards an Understanding of the Conjugate Addition of Organolithium Reagents to α,β-Unsaturated Ketones: The Isolation and Solid-State Structure of a Monomeric Lithium Aluminate with Very Short Agostic Li⋯HC Interactions, J. Organomet. Chem., 1999, 573(1–2), 305–312 CrossRef CAS.
- M. B. Power, A. R. Barron, S. G. Bott and J. L. Atwood, .Pi.-Face Selectivity of Coordinated Ketones to Nucleophilic Additions: The Importance of Aluminum-Oxygen .Pi.-Bonding, J. Am. Chem. Soc., 1990, 112(9), 3446–3451 CrossRef CAS.
- X. Pan, A. Liu, L. Yao, L. Wang, J. Zhang, J. Wu, X. Zhao and C.-C. Lin, Synthesis, Characterization of Hetero-Bimetallic Complex and Application in the Polymerization of ε-Caprolactone, Inorg. Chem. Commun., 2011, 14(5), 763–766 CrossRef CAS.
- H. Nöth, A. Schlegel and M. Suter, A Study of Noyori's Reagent, J. Organomet. Chem., 2001, 621(1–2), 231–241 CrossRef.
- M. T. Muñoz, C. Urbaneja, M. Temprado, M. E. G. Mosquera and T. Cuenca, Lewis Acid Fragmentation of a Lithium Aryloxide Cage: Generation of New Heterometallic Aluminium-Lithium Species, Chem. Commun., 2011, 47(42), 11757–11759 RSC.
- H. Bock, R. Beck, Z. Havlas and H. Schödel, Tetralithium Bis(Ethylaluminum) Tetrakis(Catecholate) Pentakis(Dimethoxyethane): An Oxygen-Rich Cluster [(Al++2(Li+)4(O18)−8] in a C36H44 Hydrocarbon Shell, Eur. J. Inorg. Chem., 1998, 1998(12), 2075–2077 CrossRef.
- H. Bock, R. Beck, Z. Havlas and H. Schodel, Preparation, Structure, and Density Functional Calculation of the Solvent-Separated Ion Pair [(H5C2)Al(OC6H5)3−⋯Li+⋯−(H5C6O)3Al(C4H9)]−[Li+(DME)3], Inorg. Chem., 1998, 37, 5046–5049 CrossRef CAS.
- S. M. Ivanova, B. G. Nolan, Y. Kobayashi, S. M. Miller, O. P. Anderson and S. H. Strauss, Relative Lewis Basicities of Six Al(ORF)4− Superweak Anions and the Structures of LiAl{OCH(CF3)2}4 and [1-Et-3-Me-1,3-C3H3N2][Li{Al{OCH(CF3)2}4}2], Chem. – Eur. J., 2001, 7(2), 503–510 CrossRef CAS.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Shape maps and polyhedral interconversion paths in transition metal chemistry, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS.
- A. J. Blake, A. Cunningham, A. Ford, S. J. Teat and S. Woodward, Enantioselective Reduction of Prochiral Ketones by Catecholborane Catalysed by Chiral Group 13 Complexes, Chem. – Eur. J., 2000, 6(19), 3586–3594 CrossRef CAS.
- P. C. Andrews, C. M. Forsyth, P. C. Junk, I. Nuzhnaya and L. Spiccia, Synthesis and Structural Characterisation of Gallium and Indium Fluoroalkoxide ‘Ate’ Complexes, J. Organomet. Chem., 2009, 694(3), 373–381 CrossRef CAS.
- J. Pauls, S. Chitsaz and B. Neumüller, Chiral Gallium and Indium Alkoxometalates, Z. Anorg. Allg. Chem., 2000, 626(9), 2028–2034 CrossRef CAS.
- E. Jung, S. H. Yoo, T.-M. Chung, C. G. Kim, Y. Kim and D. Y. Jung, Heterobimetallic Lithium Organoaluminum and Organogallium Complexes: Potential Single Precursors for MOCVD of LiMO2 Thin Films, Inorg. Chem. Commun., 2002, 5(6), 439–441 CrossRef CAS.
- I. Arvanitoyannis, A. Nakayama, N. Kawasaki and N. Yamamoto, Novel Star-Shaped Polylactide with Glycerol Using Stannous Octoate or Tetraphenyl Tin as Catalyst: 1. Synthesis, Characterization and Study of Their Biodegradability, Polymer, 1995, 36(15), 2947–2956 CrossRef CAS.
- M. Bednarek, Branched Aliphatic Polyesters by Ring-Opening (Co)Polymerization, Prog. Polym. Sci., 2016, 58, 27–58 CrossRef CAS.
- Y. Zhu, C. Romain, V. Poirier and C. K. Williams, Influences of a Dizinc Catalyst and Bifunctional Chain Transfer Agents on the Polymer Architecture in the Ring-Opening Polymerization of ε-Caprolactone, Macromolecules, 2015, 48, 2407–2416 CrossRef CAS.
- A. B. Kremer, R. J. Andrews, M. J. Milner, X. R. Zhang, T. Ebrahimi, B. O. Patrick, P. L. Diaconescu and P. Mehrkhodavandi, A Comparison of Gallium and Indium Alkoxide Complexes as Catalysts for Ring-Opening Polymerization of Lactide, Inorg. Chem., 2017, 56(3), 1375–1385 CrossRef CAS PubMed.
- S.-C. Rosca, D.-A. Rosca, V. Dorcet, C. M. Kozak, F. M. Kerton, J.-F. Carpentier and Y. Sarazin, Alkali aminoether-phenolate complexes: synthesis, structural characterization and evidence for an activated monomer ROP mechanism, Dalton Trans., 2013, 42, 9361–9375 RSC.
- Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Brandenburg, DIAMOND, Crystal Impact GbR, Bonn, Germany, 2007 Search PubMed.
- R. Petrus, P. Fałat and P. Sobota, Use of Lithium Aryloxides as Promoters for Preparation of α-Hydroxy Acid Esters, Dalton Trans., 2020, 49(3), 866–876 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2076392–2076396, 2128614 and 2128703. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03777c |
| This journal is © The Royal Society of Chemistry 2022 |