
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of reinforcement learning in chemistry
Stephen
Gow
 *a,
Mahesan
Niranjan
b,
Samantha
Kanza
a and
Jeremy G
Frey
a
*a,
Mahesan
Niranjan
b,
Samantha
Kanza
a and
Jeremy G
Frey
a
aDepartment of Chemistry, University of Southampton, University Road, Southampton, Hants SO17 1BJ, UK. E-mail: s.r.gow@soton.ac.uk
bDepartment of Electronics and Computer Science, University of Southampton, University Road, Southampton, Hants SO17 1BJ, UK
First published on 30th August 2022
Abstract
The growth of machine learning as a tool for research in computational chemistry is well documented. For many years, this growth was heavily driven by the paradigms of supervised and unsupervised learning. Recently, however, there has been increased interest in the use of a third paradigm: reinforcement learning. This approach, in which an agent interacts with an environment to learn which actions it should take to maximise a long-term objective, is particularly suited to problems of planning or sequential decision making. In this review, we present an accessible summary of the theory behind reinforcement learning (and its common extension, deep reinforcement learning) tailored specifically to chemistry researchers. We also review the applications of reinforcement learning which already exist within the world of chemistry, and consider the future direction of research based on this promising technique.
1 Introduction
The subject of machine learning, drawing strengths from probabilistic inference, function approximation and optimization, theory of dynamical systems, and parallel and distributed computing, is the driver of all recently reported advances in artificial intelligence. Much of the applications of machine learning, including applications seen in chemistry, are centred around supervised and unsupervised learning formulations, solving regression, classification, density estimation and low rank matrix approximation problems. Advances in instrumentation and our collective ability to generate, archive and distribute vast quantities of data, along with advances in complex models and algorithmic tricks around their training have achieved step advances in several applications such as in silico screening of drugs.1Supervised and unsupervised learning paradigms essentially learn static or dynamic mappings in a space of features. Unsupervised learning aims to characterise the probability distributions of these features, or discover subspaces in which the derived data may live, while supervised learning uses labelled data to infer relationships between features. Such data is usually seen as arising from an underlying probabilistic generating mechanism, sampled in an identical and independently distributed IID manner.2
A machine learning paradigm that is distinct from the above two is that of reinforcement learning (RL),3 which addresses planning or sequential decision making problems. Reinforcement learning assumes a setting in which an agent interacts with an environment to acquire data and learn about the environment, and executes actions that will maximise a long term objective. The environment in this setting makes transitions between states whenever the agent executes an action. The resulting state the environment transitions into is either fully or partially observable by the agent, which is also given a short term reward resulting from the chosen action. A diagram of this process is shown in Fig. 1.
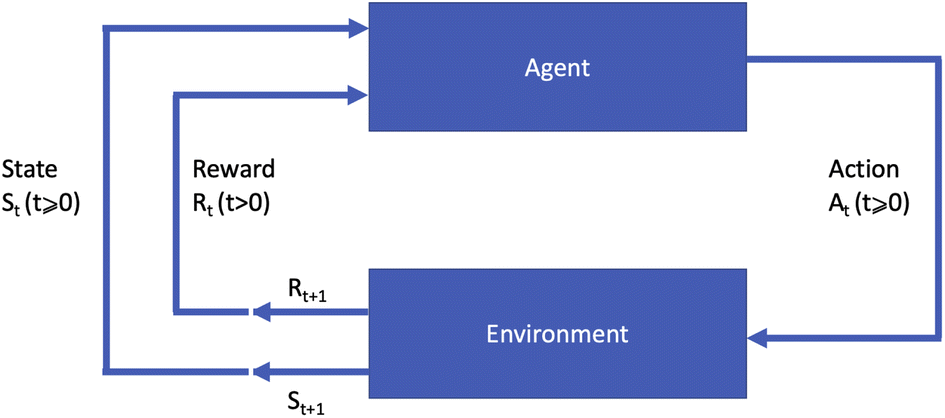 | ||
| Fig. 1 Diagram of the reinforcement learning process. The agent receives information from the environment via the state st and reward rt (t > 0) at time step t and chooses an action at which affects the environment at time step t + 1. | ||
The problem then is to combine the information contained in the observed state transitions and received immediate reward so as to act in a way that a long term objective is met. The challenge, of course, is that greedily accumulating the immediate rewards obtained need not be the optimal decision because such decisions could drive the agent along trajectories that may subsequently offer low rewards. The action an agent takes in any state is referred to as a policy, and discovering an optimal policy while characterising the uncertain behaviour of the environment is the learning challenge in this paradigm. The formulation is closely related to the subject of stochastic optimal control.4
Recent developments in the reinforcement learning have been heavily influenced by the use of powerful non-linear function approximation methods derived from neural network architectures, and the algorithmic developments around them.5 Such function approximations are used to model parametric forms of optimal policies or to approximate what we could loosely refer to as the usefulness of reaching any state along the trajectory towards the optimal one in the long term. It is this combination of learning paradigm and neural architectures that have led to several advances in artificial intelligence such as defeating the world's best human players of complex game of Go.6
Machine learning is now relatively widespread in chemistry research. Both supervised and unsupervised learning, driven by the rise of artificial neural networks, have seen use in virtually every important discipline within the field.7 Until recently, reinforcement learning was less widely utilised. In the last five years this has begun to change, and examples of RL can now be found in a wide variety of areas from drug discovery to reaction control. This review aims to act as a guide to the use of reinforcement learning within chemistry, setting out the theory required to use it, discussing the practical issues which may be encountered, and reviewing the applications that already exist.
The remainder of this paper is organised as follows. In Section two, we introduce the theoretical principles of reinforcement learning. In Section three, we present a brief review of deep neural network architectures. In Section four, we review the applications of RL to real problems within chemistry that have been conducted to date. Finally, we consider the future of RL as a tool in chemistry research.
2 Theory of reinforcement learning
Reinforcement learning is a vast subject with much prior work. This paper provides a high level summary; more detail on the considered concepts is provided by Sutton and Barto3 among others.A reinforcement learning agent is designed to select a particular action from a set of possible actions, given the current state of a system within a set of possible states. Both the state space and the action space may be continuous or discrete; the action space may differ between states. Actions are selected using a policy which gives the probability of selecting an action given the current state. Each action that may be selected both changes the state of the system at the next time step and leads to the agent receiving a different reward, which is defined by a reward function. The agent wishes to maximise the total reward received during the length of the simulation. In most applications, the simulation length is finite and reaching a specified number of time steps or a specified subset of the state space will cause the simulation to end, but infinite-step simulations are also possible. A key challenge in reinforcement learning is the exploit-explore problem of finding the correct balance between taking actions which are known to give high expected rewards and exploring actions which have not yet been investigated.
Reinforcement learning is often framed in the context of the Markov decision process,8 in which the current state is all that is needed to determine an action: knowing the previous history of the states and actions taken to reach the current state provides no additional information with which to select the next action. This allows the agent to learn efficiently, as the reward of taking the same action while in the same state is effectively constant. However, this is also a strong assumption, as there are real processes in which the most rewarding action may depend on the process history as well as the current state. These are referred to as hidden state problems, since the behaviour of the system would be Markovian if the state observed by the agent captured all of the information which defined the reward.9 They can be handled via the more general partially observable Markov decision process, allowing the agent to interact with an incomplete representation of the state space; a discussion of how different methods compare in this respect was provided by Jaakkola et al.10
The design of the reward function plays an important role in RL, as this determines what the agent is aiming to maximise through its policy learning. Reward functions can be discrete or continuous, and may include rewards at every time step of the simulation or at only the final time step. They may also include a discount value to decrease the importance of expected rewards as the number of time steps beyond the current one increases. The use of additional rewards not motivated directly by the underlying problem to help the agent to learn is called reward shaping, and has been the subject of significant research.11 Designing a reward function to ensure the agent performs well for the specified task is challenging and somewhat subjective, and most RL work in chemistry has approached this using subject-specific expert knowledge. In inverse reinforcement learning, the reward function is treated as unknown and a good reward function is learned by the agent based on a set of examples with properties which the agent wishes to replicate. Further details of this approach can be found in Hadfield-Menell et al.12
2.1 Types of reinforcement learning
In most RL settings the key aim of the agent is to learn a policy to maximise the expected reward as much as possible during the simulation. The two broad approaches for this are policy learning and value learning. In policy learning, the agent aims to directly learn an optimal policy to maximise the total expected reward. Value learning instead focuses on learning a value function which defines the expected reward of either every state or every pair of state and action, and uses this to select the most rewarding policy. Policy learning works directly with the quantity of interest and is very stable but can be highly sample inefficient, while value learning offers a greater sample efficiency at the cost of possible instability and the risk of diverging from the optimal policy.13Another important distinction for RL is whether an algorithm is model-based, in which a model for the dynamics of the environment is used during learning and action selection, or model-free, in which case a policy is learned and actions selected without the use of such a model. An algorithm may also be on-policy, if the states and actions used in training the agent are generated by the current policy, or off-policy if they are generated in some other way.
The scope of problems which can be addressed with traditional RL is limited by practical concerns around the size of the state space and action space. For example, in value learning, the expected value of every state–action pair must be recorded, which quickly becomes infeasible as the number of possible states increases. Recent advances in the field have been driven by deep reinforcement learning, in which unknown functions of the state and action space such as the policy or value function are approximated via a deep neural network. Since these functions can be approximated based on a small sample of observations, the dimensionality of the state space is no longer a limitation and more complex problems can be tackled.5 This approach does introduce additional complexities arising from the additional variance imposed by the approximation, and in the choice of neural network used; the types of neural network which may be considered for the purpose are discussed in Section 3.
2.2 Policy optimisation algorithms
Many different algorithms have been used for policy optimisation in chemistry. The most popular forms of value learning are based on Q-learning, a model-free method in which the agent learns a Q-function describing the expected rewards of state–action pairs. In its simplest form, Q-learning is a tabular method in which the agent learns by evaluating every state–action pair and updating Q values using value iteration. Watkins and Dayan14 proved that this method converges to the optimal policy if every action can be attempted multiple times at every state, but this is impractical if the state space is large. Deep Q-learning15 overcomes this obstacle by using a neural network called a deep Q-network to approximate the Q-function given a sample of state–action pairs. Using a non-linear approximation to the Q-function can cause instability in the learning process; this is addressed via a technique called experience replay, in which recent observations of the agent at each time step are stored in memory and Q value updates are made with respect to samples from the agent's experience instead of single values. A variant named double Q-learning16 uses two estimates of the Q-function trained on different experiences. This fixes an issue with standard Q-learning in which the expected value of certain actions can be over-estimated in stochastic environments.Most policy learning algorithms are based on two key concepts: policy gradient and actor-critic. Actor-critic methods consist of two modules: an “actor” model estimating the policy function, and a “critic” model estimating the value function. The parameters of the actor are updated based on the output of the critic given the current parameters to determine a policy which will lead to good expected rewards. Fig. 2 presents this form of RL agent diagrammatically. In contrast to Fig. 1, the agent is now split into the actor module choosing actions based on the current state and the value function estimate, and the critic module learning the value function based on the current state and reward.
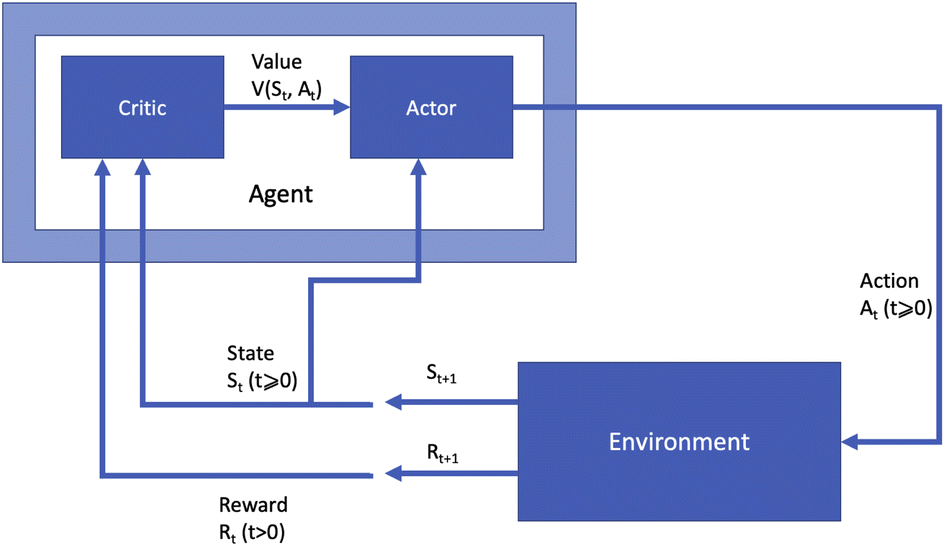 | ||
| Fig. 2 Diagram of a reinforcement learning agent using an actor-critic policy optimisation method. The agent is split into two modules, a critic which estimates the value of state–action pairs given the state st and reward rt, and an actor which chooses an action at given the state and the value. | ||
Actor-critic methods are frequently characterised by the nature of the critic module, such as the pioneering temporal difference actor-critic (TD-AC) algorithm of Barto et al.,17 and the advantage actor-critic (A2C) algorithm of Peters and Schaal;18 the latter was developed further by Mnih et al.19 into asynchronous advantage actor-critic (A3C). The actor may optimise its parameters using any method, but in recent work it is common for a version of policy gradient learning to be used.
In policy gradient methods, the parameters of the policy are optimised by updating them proportionately to the partial derivative of the performance of the policy with respect to the parameters (the gradient function). This is equivalent to optimising a stochastic policy via a function approximating the true policy. The optimisation step may be conducted using any gradient ascent method. Direct computation of the gradient is difficult as it depends on both the action selection under the current policy, and the distribution of states under an optimal policy; however, the policy gradient theorem states that it is possible to obtain an unbiased estimate of the gradient given a sample of experiences from a reasonable function approximation.20 The earliest policy gradient algorithm, named REINFORCE or ‘vanilla’ policy gradient, was introduced by Williams.21 This is a form of on-policy Monte Carlo approximation using the likelihood ratio trick, and remains popular due to its simplicity to implement. Other approaches to simple policy gradient learning include finite difference methods; a comparison of these early algorithms is given by Riedmiller et al.22 However, there are several disadvantages: they can be slow to converge and require many samples to learn a near-optimal policy, have very high variance and will often converge to a local optimum instead of a global one.
These weaknesses led to the development of more advanced methods incorporating actor-critic techniques. Schulman et al.23 developed Trust Region Policy Optimisation (TRPO), an algorithm based on a series of approximations to a theoretical result concerning policy improvement by minimising surrogate loss functions. A restriction on the divergence between the old and new policy is enforced to prevent overly large updates which may harm convergence, while still allowing large updates within a trust region where it is safe to do so. Proximal Policy Optimisation (PPO)24 follows a similar strategy but works with a clipped surrogate objective function, increasing the range of problems to which it can be applied and reducing the complexity of implementation while maintaining many of the benefits of TRPO.
Another way to improve the efficiency of actor-critic policy gradient algorithms is to utilise off-policy updates to reduce the amount of interaction required with the environment. Deep Deterministic Policy Gradient (DDPG)25 is an early example of this which can be viewed as an extension of deep Q-learning to a continuous action space: learning is conducted via gradient descent on the Q function and gradient ascent on the policy function, resulting in a model-free algorithm which is easy to apply and does not require discretisation of the action space. Fujimoto et al.26 combined DDPG with ideas from double Q-learning by using two critic networks, delaying policy updates until value learning is complete, and adding a regularisation step with the combined effect of reducing variance and potential overestimation bias; the resulting algorithm is named Twin Delayed Deep Deterministic Policy Gradient (TD3). DDPG and TD3 are extremely efficient, but can suffer from sensitivity to hyperparameter selection. An off-policy method that aims to address this is the soft actor-critic algorithm,27 in which the actor network aims to simultaneously maximise expected reward and entropy.
Finally, the popular decision-making algorithm Monte Carlo tree search (MCTS) can also be viewed as a form of reinforcement learning. MCTS was introduced by Coulom28 as a search algorithm for decision-making processes, drawing on a form of Monte Carlo simulation named rollout within a decision tree. More recent developments in MCTS have incorporated RL techniques, and several scholars have noted the links between the general MCTS formulation and reinforcement learning; a detailed study of the relationship between the two fields was undertaken by Vodopivec et al.29
The choice of which algorithm to use is a difficult one and somewhat subjective, although it will depend on the nature of the environment (for example if the state space is represented in a discrete or a continuous form). There have been several studies conducted to aid in this decision, including comparisons on the cart-pole problem by Nagendra et al.,30 a guide to statistical comparisons by Colas et al.,31 and a comparison across several benchmark problems by Jordan et al.32 It should be noted that the list of RL algorithms given here is not exhaustive, and that the development of algorithms is an active area of research – a recent example being the Invariant Decoupled Advantage Actor-critic method of Raileanu and Fergus,33 which aims to improve the ability of an agent to generalise to new environments.
3 Neural networks for deep reinforcement learning
The term neural network (NN) may in principle describe any computational network which aims to learn from data in a way which mimics the behaviour of a biological brain. Networks are composed of layers of nodes or neurons which are interconnected to allow the processing of information, and are closely related to the wider concept of machine learning.A neural network must contain an input layer and an output layer. Hidden layers between the input and output layers are required for all but the simplest computations to be achieved. Deep neural networks contain multiple hidden layers; while there is no general definition for the lowest number of layers required for a network to be considered deep instead of shallow, the most common threshold is that of any network with two or more hidden layers. Schmidhuber34 provides a more detailed account of the history of deep neural networks.
The number of layers in a neural network, and the number of nodes in each layer, can have a significant effect on the behaviour of the network. A network with more or larger layers is more powerful and can fit well to a wider range of underlying data generation processes, at the expense of increased complexity and a risk of overfitting to the training data. This is however rarely considered in reinforcement learning literature, where it is common to simply present the network used without consideration of alternatives. One approach adopted by some authors is to treat neural network architecture as an optimisation problem and search the space of possible architectures for the best model; this is discussed by Benardos and Vosniakos35 and Luo et al.36 among others.
3.1 Feedforward neural networks
The simplest and oldest form of neural network is a feedforward network, in which there are no cycles connecting the nodes of the network. In this structure, information only moves forwards from the input nodes to the output nodes.In principle, any directed acyclic graph may be used as the basis for a feedforward NN. The most common network structure is a fully connected network, in which every node in a layer has a connection to every node in the next layer. An example of a fully connected feedforward NN with two hidden layers is shown in Fig. 3. This type of network is often referred to as a multilayer perceptron (MLP), although this terminology is somewhat confusing as other authors restrict it to specific types of fully connected network. Feedforward neural network are usually trained using iterative optimisation methods such as gradient descent or stochastic gradient descent, with the backpropagation algorithm used to compute the gradient with respect to a loss function.
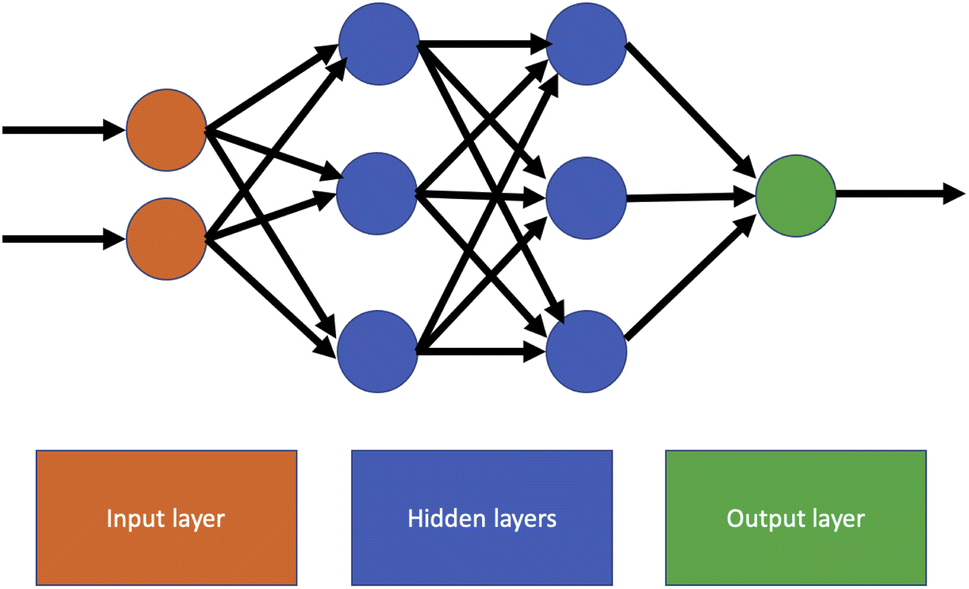 | ||
| Fig. 3 Example fully connected feedforward neural network with two inputs, two hidden layers of size three and one output. Information travels through the network strictly from left to right, and every node in one layer is connected to every node in the next layer. | ||
Feedforward neural networks have found widespread use in fields as diverse as facial recognition,37 dynamic systems control,38 geology and mining,39 and urban sustainability.40 In some application areas, however, the lack of ability for information to move backwards between layers can be a limitation.
3.2 Recurrent neural networks
A recurrent neural network (RNN) is a network which contains at least one cycle between nodes. The simplest possible example of an RNN is shown in Fig. 4. The structure of an RNN with practical uses will be more complicated in terms of its nodes and the connections between them, but the defining principle is the same: in contrast to an FNN, loops connecting nodes in the hidden layer(s) mean that information no longer travels strictly forwards. This allows information to be stored within the network via a form of internal memory RNNs are of particular use for sequential data and sequence modelling, and can handle inputs of variable lengths and time series data. RNNs have been used in numerous fields including musical composition41 and musical computation,42 water table modelling,43 and electrical load forecasting.44 An area of particular focus has been natural language processing45 and text classification and prediction;46 indeed, for language modelling, a review by Sundermeyer et al.47 demonstrated that RNNs offer significant advantages over comparable methods based on feedforward networks.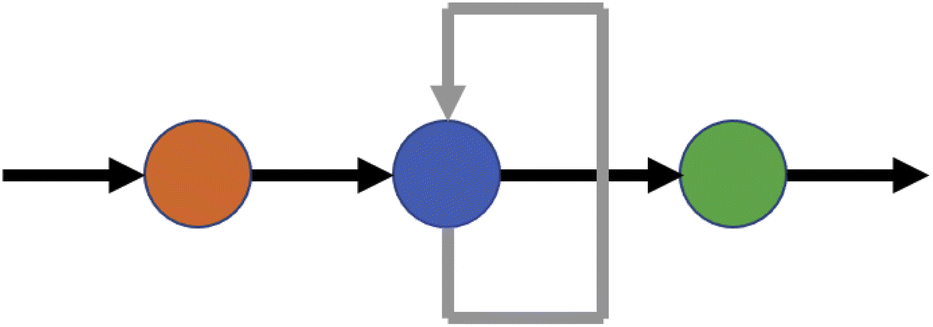 | ||
| Fig. 4 Simple recurrent neural network with one input, a single one-node hidden layer and one output. The grey arrow connecting the node in the hidden layer to itself is the source of recurrence in the network. | ||
The presence of cycles in RNNs affects the way in which the network can be trained, and methods based on backpropagation can be problematic due to repeated multiplication of gradients causing terms to vanish or explode. This was addressed by Hochreiter and Schmidhuber48 through the creation of Long-Short-Term Memory (LSTM), a system in which information is stored in a cell and the flow of information to and from the cell is regulated by a series of gates. The Gated Recurrent Unit (GRU) is a more recent development with a similar but simpler structure, and was shown by Chung et al.49 to offer comparable performance on a selected set of problems.
LSTM and GRU architectures can struggle with certain tasks due to limitations of their structure. Difficulties in sequence prediction with these methods motivated Joulin and Mikolov50 to investigate augmentation of RNNs with a neural stack to increase the memory capability of the network. Similar work on extending RNNs with structures analogous to stacks and queues was conducted by Grefenstette et al.51 to overcome challenges in learning natural language transductions. Stack-augmented RNNs (stack-RNN) extend the network with cells consisting of multiplicative gates defining a memory stack, on which a PUSH (insert) or POP (remove) operation may be conducted to change the makeup of the vectors stored within the network's memory. This allows the network to learn longer-term dependencies between features of the training data.
3.3 Generative adversarial networks
While traditional neural networks are extremely good at classification and regression problems, they are less well tuned to generative modelling. This is particularly important in chemistry problems such as drug design. The generative adversarial network (GAN) architecture was proposed by Goodfellow et al.52 as a framework designed specifically for generative modelling. It consists of a pair of networks working in competition with each other: a generator network G which aims to produce new data which could plausibly come from the training set, and a discriminator network D which is given a sample of data and aims to determine if this comes from the training set or the generator network. Based on the output of the discriminator, the generator can improve its generation process over time to generate more realistic samples. A diagram of this process is shown in Fig. 5.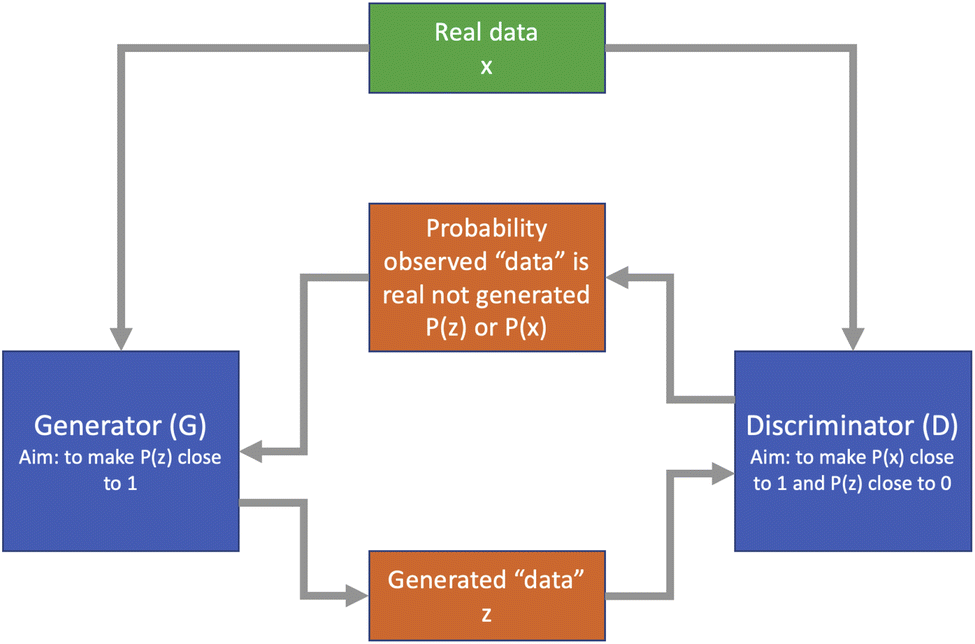 | ||
| Fig. 5 Diagram of a generative adversarial network architecture. | ||
The networks G and D may take several forms, but are typically FNNs or RNNs. GANs were extended to handle sequential data using reinforcement learning in the SeqGAN method of Yu et al.53 Application areas include text-to-image generation,54 astronomical image restoration55 and several problems in medical imaging.56 GANs are a developing area of machine learning, both in terms of structure and applications; a more detailed review of recent progress was provided by Alqahtani et al.57
3.4 Other networks
A convolutional neural network (CNN) is a network which includes hidden layers to perform convolution operations on the inputs to the layer. Compared to fully connected feedforward NNs, the convolutional layers act to regularise the network to reduce overfitting, and allow fewer neurons to be used. CNNs also commonly include pooling layers for dimension reduction. The input to a CNN is of tensor form, so they are particularly effective for spatial data: the most active field of application is image processing, for example by Krizhevsky et al.58 and Valueva et al.59 CNNS can work directly with graphical inputs, with applications in molecular chemistry.60 A more general CNN-based machine learning method for graphs named message passing neural network (MPNN) was developed by Gilmer et al.61 and applied to quantum chemistry problems.The variational autoencoder (VAE) was introduced by Kingma and Welling62 as an application of neural networks to variational Bayesian methods for approximate inference. It consists of two networks, an encoder and a decoder, which map inputs to and from a latent variable space which is otherwise intractable. The encoder and decoder must be selected from the stable of other neural network architectures: early work by Bowman et al.63 made use of RNN encoders and decoders for language modelling, and similar techniques have since been applied to automatic chemical design by Gómez-Bombarelli et al.64 and Griffiths and Hernández-Lobato.65
Another significant encoder–decoder structure is the transformer architecture of Vaswani et al.66 The encoder and decoder are built from a series of layers comprising FNNs and sub-layers based on attention mechanisms, a form of mapping which enhances the importance of some parts of the input data while reducing the importance of other parts. In contrast to RNNs, transformers process an entire input at once, even if the input is sequential in nature. Transformers have found success in natural language processing tasks, and have largely replaced RNNs as the method of choice in the field.67 Recent papers by Chen et al.68 and Janner et al.69 link the transformer method to reinforcement learning by framing RL as a sequence modelling problem.
4 Applications of reinforcement learning in chemistry
The range of chemistry problems to which RL has been applied is extremely varied, with at least some active areas of research in most sub disciplines of the field. Fig. 6 depicts these problems hierarchically by broad area of application. There is considerable variation in activity between fields: while most of the topics listed under “Other areas” have been the subject only one or two RL papers each, the field of structure design (both molecular and biochemical) has seen a vast array of relevant publications. This section is therefore divided into six headings: drug discovery and small molecule design, molecular geometry optimisation, biochemical sequence design, proteins, reaction management and process control, and other areas.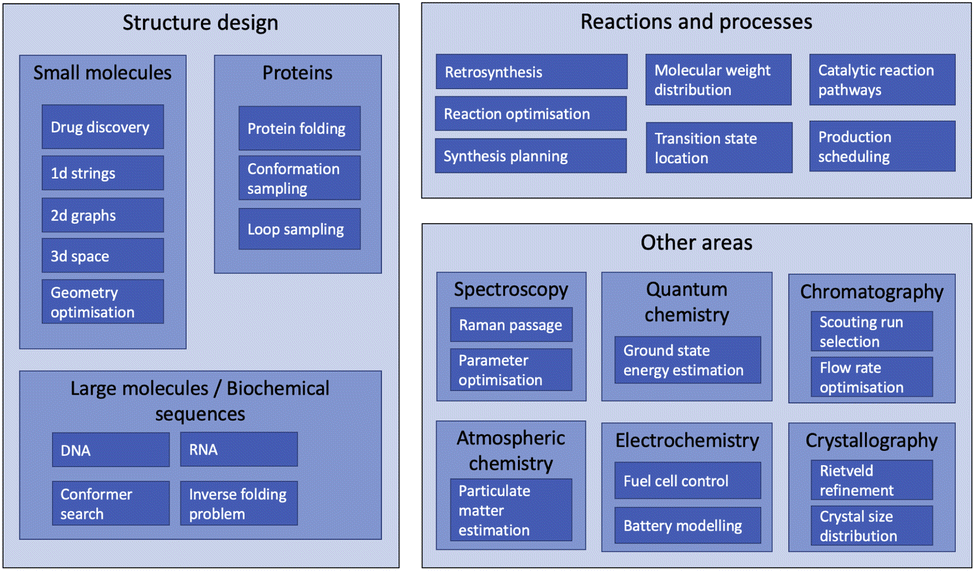 | ||
| Fig. 6 Hierarchical diagram of chemistry problems to which reinforcement learning has been applied. | ||
4.1 Drug discovery and small molecule design
The design and investigation of new molecules for use in pharmaceutical treatment is an extremely important problem, but there are many challenges associated with this process. As discussed by Torjesen,70 only around one in every 5000 possible new drugs will be successful, and even a successful design will require around 12 years of research and development. There has therefore been a great deal of interest in utilising machine learning to improve this.The area in which computational methods have shown the most promise is in the generation of candidate molecules with promising properties. In principle, a generative model can be used to propose molecules which are more likely to be successful for a given task. While this is not a new idea, the rise of neural networks in recent years has led to drug design becoming one of the most active areas of machine learning research in chemistry, and a wide variety of approaches have been taken. Authors such as Mandlik et al.,71 Schneider and Clark,72 Vamathevan et al.1 and Mouchlis et al.73 have provided detailed coverage of the topic. Most relevant to this review, however, is the large body of work considering molecular design as a sequential problem in which decisions are made to improve the properties of the molecule(s) being proposed. Reinforcement learning is an ideal tool to achieve this.
The use of reinforcement learning in drug discovery began with the work of Guimares et al.74 and Sánchez-Lengeling et al.75 The problem formulation is based on SMILES, a widely-used string representation of a molecule. The states of the RL agent are partially-completed SMILES strings, and the action space is the selection of the next character to be added to the string. Constructing a string in this fashion is a difficult problem, as SMILES syntax is notably fragile. The vast majority of combinations of characters are invalid, including those generated at intermediate time steps en route to a valid string. Among valid strings, very small changes may dramatically alter the chemical properties of the associated molecule, so it is difficult to construct a string which is likely to correspond to a desirable molecule. Non-RL methods to do this have been proposed with some success, notably that of Ikebata et al.,76 but even among these the proportion of valid, chemically desirable sequences is relatively low.
The method introduced by Sánchez-Lengeling et al.,75 named Objective-Reinforced Generative Adversarial Networks for Inverse-design Chemistry (ORGANIC), is based on the SeqGAN approach of Yu et al.53 It utilises a GAN neural network structure, with an LSTM RNN as the generator network and a CNN as the discriminator. The reward function is a linear combination of the discriminator and a quality metric based on the properties of the sequence; Monte Carlo rollout is used at intermediate time steps to estimate expected future rewards, with policy gradient optimisation used for policy learning. This work was later extended by Putin et al.77 using a differentiable neural computer in place of LSTM, allowing longer and more complex sequences to be learned and generated. The resulting Reinforced Adversarial Neural Computer (RANC) architecture was shown to generate a higher proportion of unique molecules with desirable properties.
Also in 2017, Olivecrona et al.78 proposed a different RL method for drug discovery. With the same state and action spaces, this method trains an RNN with three layers of 1024 GRUs each on a set of 1.5 million SMILES strings corresponding to existing molecules, and uses RL to augment the likelihood of the RNN so that molecules with desirable properties will be constructed. The authors state that a policy-based learning approach is more appropriate than a value-based approach for this problem, and make use of the REINFORCE algorithm to learn an optimal policy. The reward functions used are based solely on the desirability of the sequences created, for example to promote DRD2 activity. REINVENT, a direct extension of this approach incorporating a memory unit in the scoring function so that a more diverse range of molecules are proposed, was developed later by the same research group and published in a paper by Blaschke et al.79
There are many further examples of RL agents constructing SMILES strings one character at a time. Popova et al.80 used the REINFORCE algorithm and a reward function based only on molecular properties and no intermediate rewards while making use a generator-predictor structure: a memory augmented single layer stack-RNN was chosen to generate new SMILES strings directly without recourse to descriptor-based modelling, while an LSTM RNN was used for property prediction. This approach was shown to propose novel compounds to inhibit Janus kinase 2 (JAK2), a protein linked to several important cellular processes in the human body. Later, Yoshimori et al.81 maintained the same architecture as Olivecrona et al.,78 while replacing the reward function with the output of the LigandScout 3D pharmacophore model of Wolber and Langer82 to aid the discovery of molecules with the desired pharmacophores.
Neil et al.83 conducted a thorough exploration of several approaches to molecular design using RL. Focusing on multi-objective optimisation, the authors tested several different neural network architectures and policy optimisation methods against a set of 19 benchmarks for molecular design. Proximal policy optimisation with a single LSTM RNN was found to perform best of the RL approaches, outperforming both advantage actor-critic and vanilla policy gradient as well as GAN architectures (although as noted above, GANs have advanced significantly in the years since). The non-RL method Hillclimb-MLE, based on repeated maximum likelihood estimation, was also found to offer competitive performance levels.
Recent work by Pereira et al.84 used six-layer RNNs incorporating both LSTM and GRU structures within their layers to both generate SMILES strings and determine the reward to given to the generated molecule. Given a target receptor to inhibit, the IC50 is defined as the amount of a substance required to inhibit 50% of the receptor. The RL reward function is based on the negative predicted log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50 of the compound generated with respect to the target, with a penalty term if the molecule is lacking in novelty. This has the effect of biasing the generator model towards both desired chemical properties and increased molecular diversity. Another recent paper, Born et al.,85 directly incorporated information on the target disease (in this case cancer) into the molecular generation process. Molecules are generated using two VAEs running in conjunction, one generating a gene expression profile and the second generating SMILES strings for molecules based on the generated gene expression profile, with a reward function based on the IC50 as estimated by the PaccMann drug sensitivity model.
IC50 of the compound generated with respect to the target, with a penalty term if the molecule is lacking in novelty. This has the effect of biasing the generator model towards both desired chemical properties and increased molecular diversity. Another recent paper, Born et al.,85 directly incorporated information on the target disease (in this case cancer) into the molecular generation process. Molecules are generated using two VAEs running in conjunction, one generating a gene expression profile and the second generating SMILES strings for molecules based on the generated gene expression profile, with a reward function based on the IC50 as estimated by the PaccMann drug sensitivity model.
Another distinct approach to SMILES string generation for drug design is that introduced by Krishnan et al.86 This approach draws on transfer learning, in which policies learned by an agent in one problem may be applied to another related problem; it has previously seen use in both molecular library generation87 and RL research.88,89 A stack-augmented RNN with gated recurrent units is trained to generate strings, and is combined with a set of molecules known to inhibit proteins similar to the intended target using transfer learning to create a target-specific generative model. A property prediction model is then used to guide the generator towards more desirable molecules, with a reward function based on the predicted docking score of the generated molecule. The method was tested on the problem of inhibiting the JAK2 protein; it was able to both reproduce existing JAK2 inhibitors without similar molecules being included in the training set, and to design potential new inhibitors.
The wide variety of reward functions used in the work described so far highlights the importance of reward function construction in drug design problems. To bypass this challenge, Agyemang et al.90 treated drug design as an inverse RL problem and inferred the reward function for the agent from the SMILES strings of known molecules with desirable properties. Molecule generation is handled by a multiple layer stack-augmented RNN, with PPO for policy learning. The authors demonstrated this method on several examples, including JAK2 inhibition.
As previously mentioned, the challenging syntax of SMILES strings makes their construction difficult. For this reason, Thiede et al.91 worked with SELFIES strings, in which every combination of characters is valid and the substrings generated during the construction process can be directly interpreted. Optimisation is handled via PPO, while the reward function is defined by a combination of an extrinsic reward (based on the predicted properties of the molecule) and an intrinsic reward named curiosity to encourage increased exploration of the state space.
An alternative to string-based molecular representations are two-dimensional graphs, which offer increased robustness and interpretability, for example of partially-constructed graphs as molecular substructures. Fig. 7 shows a highly simplified example of how a molecular graph may be constructed using reinforcement learning. The action space consists of two distinct action types: addition of a new atom in the molecular graph and addition of a bond between existing atoms. A stopping criterion to determine when a molecule is considered “complete” is also required. The methods used in practical applications are significantly more complex than this, but are usually based on similar principles.
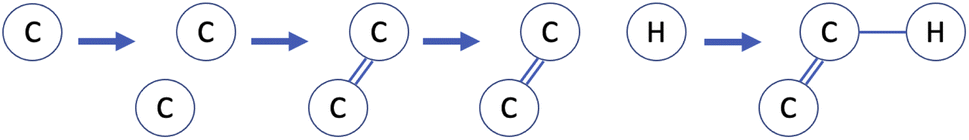 | ||
| Fig. 7 Simplified demonstration of the process of constructing a molecular graph using reinforcement learning. Starting from an empty canvas, the agent places atoms and adds bonds between them to create a molecule. | ||
The pioneering RL work for graphical molecular construction was that of You et al.92 Given a set of scaffold subgraphs corresponding to atoms or molecular substructures, the state space of the RL agent is defined as the set of graphs which can be constructed from these subgraphs, and the action space as the set of possible extensions to the existing graph by either connecting existing nodes in the graph or adding an additional scaffold subgraph. Two deep graph convolutional networks were used as a generator and a discriminator in an adversarial setting, with rewards based on molecular properties and adversarial loss. This method was tested on problems of molecular property optimisation and property targeting, and demonstrated to offer significant improvements over earlier approaches. Later, Khemchandani et al.93 integrated the work of You et al.92 with a neural network method for property prediction introduced by Yang et al.94 to create a new method for generation of molecules with desirable properties. A recent paper by Atance et al.95 uses a gated graph neural network in place of a convolutional network, with a memory-aware RL approach developed from that of Olivecrona et al.78 and a best agent reminder loss function. This method shows signs of strong performance on QED optimisation and DRD2 activity tasks.
Zhou et al.96 took a different approach, aiming to optimise existing molecules for drug discovery. By making only chemically valid changes – atom addition, bond addition or bond removal – to molecules, the authors ensure the final molecule is itself chemically valid, and avoid the need for pre-training on existing data to reduce the risk of bias in the model. A deep Q-network structure utilising a fully connected feedforward NN with four layers is used for value function learning, with a reward function (which includes intermediate rewards but weights the final reward most highly) defined by the molecular properties to be optimised.
In a similar vein, Ståhl et al.97 introduced DeepFMPO, a temporal difference actor-critic RL with two LSTM RNNs to discover novel molecules which optimise multiple objectives through molecular modification. First, a library of molecular fragments is created by fragmenting a set of initial molecules, and fragments are encoded in such a way that similar molecules have similar encodings. The agent alters one fragment in the molecule at each time step, with rewards given for molecular validity and improvement in the target properties. The reward function is updated over time as the agent discovers more molecules with desirable properties.
Two papers published in 2020 focus on ensuring that the proposed new molecules can be synthesised and that the synthesis route for the molecule can be determined. Gottipati et al.98 developed an approach named policy gradient for forward synthesis. The state space is the set of molecules to be used in a chemical reaction, and the action space is split into two parts: an intermediate action of selecting a reaction template, and a final action of selecting a reactant to combine with the current molecule to produce a new molecule with desirable chemical properties. A k-nearest neighbour algorithm is used to discretise the otherwise extremely vast reactant space, and the selection policy is optimised using twin delayed deep descent policy gradient (TD-DDPG). Three neural networks are used, one each for predicting the best reaction template, computing the action to be taken, and estimating the Q-value of the product molecule. Each network is a fully connected feedforward NN with four layers, although the number of neurons in the hidden layers varies between networks. Similarly, Horwood and Noutahi99 take a given set of initial molecules and reactants and define the transitions between states in terms of sequences of chemical reactions. Actions are treated hierarchically in terms of selecting a reaction template and then a reactant. Molecules are represented using Morgan fingerprints and reaction templates by SMARTS syntax. An advantage actor-critic algorithm is used for policy optimisation.
There has also been interest in using RL for three-dimensional molecular design, which can take advantage of spatial information not captured by string or graphical representations. Simm et al.100 developed an RL agent which designs molecules by placing atoms onto a canvas in 3D space. The reward function is based on fundamental physical properties of the constructed molecules, in effect encouraging the agent to learn the laws governing atomic interactions in three dimensions. The state space is the set of currently placed atoms and their positions plus a bag of atoms remaining to be placed (the initial set of atoms to be placed must be specified in advance), with the action space being the selection of the next atom to be placed and its location, and the placement action chosen based on the distance and dihedral angle with respect to an already-placed focal atom when the canvas is not empty. The same authors develop the method further in Simm et al.101 to exploit symmetry in the design process using rotationally covariant state–action representations and neural network architectures.
Another example of 3D molecular construction is the work of Bolcato and Boström,102 an extension of the DeepFMPO method of Ståhl et al.97 discussed above. The revised method uses conformer search on molecular fragments for 3D alignment so that electrostatic and shape similarities may be considered instead of simpler 2D similarity.
Recently, Meldgaard et al.103 proposed a method combining imitation learning and reinforcement learning to ensure that the generated molecules are stable. Imitation learning is a learning scheme in which an ML model may learn from demonstrations and has some history of use alongside RL.104 First, an imitation learning agent is trained on a molecular database to construct existing molecules and predict their stability using quantum chemistry and 3D information. A deep RL agent using a Q-learning algorithm then explores the molecular space to discover new stable molecules, while simultaneously updating the prediction model in areas of the space a long way from the training data.
4.2 Molecular geometry optimisation
A closely related problem to that of molecular design is the prediction and optimisation of molecular geometries. Instead of searching for a molecule, we now wish to determine the most likely geometry of a molecule given some information about it, and potentially also optimise that geometry with respect to some conditions. This is typically done using molecular dynamics simulations or density functional theory, but these methods can be computationally expensive. Two recent papers have attempted to address this using reinforcement learning.Cho et al.105 presented a method to predict the 3D structures of small molecules. Given the SMILES string of a target molecule and a randomised initial structure for its atoms, the agent takes actions corresponding to the movements of atoms within the molecule in 3D space, with rewards given by density functional theory energy calculations and deep deterministic policy gradient optimisation. The agent was tested on five simple hydrocarbon targets and was able to correctly locate the lowest-energy structure of the five molecules.
Ahuja et al.106 modified a conventional approach for optimisation of molecular geometries via minimisation of potential energy using the BFGS algorithm to additionally draw on RL. The state of the agent at each time step consists of the gradients of the potential energy surface and the updates made to the positions of the atoms in the molecule at each previous time step. The action to be taken is to propose a correction term to the BFGS calculation for the next atomic position update, with policy learning via PPO, and a neural network architecture consisting of several feedforward NNs linked by a self-attention layer for information sharing (the authors note the similarity of this structure to the transformer architecture discussed above). Since the aim is to reach an optimal geometry quickly, the reward function is fixed to −1 per time step. This approach is shown to reduce the number of time steps required to reach an optimum compared to non-RL methods on a set of test molecules.
4.3 Large molecule and biochemical sequence design
Molecular design is also a critical problem in biochemistry, where the molecules of interest are typically much larger. Design of biochemical sequence structures such as RNA, DNA and proteins is the focus of significant research in bio-engineering, as these structures play important roles in a wide range of medically significant processes such as signal transduction and transcription control. The nature of these sequences presents many challenges distinct from those found in small molecule design: the search space is very different in character, and substantially larger as each molecule contains more components. Biochemical sequence design is closely related to the inverse folding problem, in which the aim is to design a sequence that folds to a given structure. (Protein folding, an important sub-problem, is dealt with in Section 4.4 of this paper.)The earliest reinforcement learning work in this field was conducted by Eastman et al.107 in the shape of a deep RL agent for the RNA design or inverse folding problem. Given a sequence of RNA bases, the agent aims to find a new sequence which will fold to a target structure by repeated modification of the existing structure using a form of local search. Policy optimisation is handled by asynchronous advantage actor-critic algorithm, while the neural network used consists of several convolutional layers operating on either one or seven RNA bases at a time, plus a final fully connected layer for value function estimation and a softmax layer to output action probabilities. This approach is shown to improve on existing non-RL algorithms for RNA design on a test set of 100 target structures, although the authors note that further performance improvements should be possible.
Independently, Runge et al.108 introduced LEARNA, a deep RL algorithm to sequentially design an RNA sequence to fold to a target structure. Uniquely among chemistry literature, the neural network architecture is determined via architecture search: the network always includes an embedding layer and a fully-connected NN with at most 2 layers, and may also include a CNN with at most 2 layers and a LSTM RNN with at most 2 layers. PPO is used for policy learning, with the hyperparameters and environment parameters jointly optimised alongside the network architecture. The reward function is based on the Hamming distance between the observed structure from the folding process and the target structure, and incorporates a local improvement step. The authors also demonstrate an extension based on meta-learning, which concerns agents that can learn how to improve their own learning given relevant previous experience109 and has been used to improve the training of RL agents.110,111 Meta-LEARNA, which transfers knowledge learned from solving one RNA sequence problem to others, was found to improve on the other algorithms tested, solving a higher proportion of problems in a significantly shorter time across three sets of test problems.
The wider problem of general biological sequence design (for example of DNA or proteins) is tackled by Angermueller et al.112 Given a type of biological sequence to be designed, the state space is defined as the set of possible sequence prefixes, and the action space as the vocabulary of characters which can appear in the sequence (in the previously stated examples, DNA nucleotides and amino acids respectively). The reward function is treated as partially unknown: intermediate rewards are set to zero and the final reward penalises overly similar sequences, but other than this the reward function must be approximated during sequence construction. Several surrogate models of varying complexity are considered for the reward function, predominantly supervised regression models, including a Bayesian ensemble of neural networks. The policy is determined by a new algorithm named DyNA PPO, a form of model-based optimisation using PPO with additional simulated data from one or more of the candidate models for the reward function, with the effect of increasing the sample efficiency of the agent.
Conformer search – the prediction of stable 3D molecular geometries for flexible molecules – is also a relevant and significant challenge, analogous to geometry optimisation for small molecules. This is approached using RL by Gogineni et al.113 To represent the conformer search problem as a Markov decision process, the state of the RL agent is taken as the sequence of conformers observed during the search process so far. The action space is a discretised version of the torsional space, consisting of the choice of torsion angles for each rotatable bond in the current molecule. Node embedding is handled via a message passing graph convolutional neural network, a graph pooling operator and LSTM memory unit to convert the embeddings into a full representation with historical information incorporated, while torsion action selections are determined by a feedforward NN. The authors also demonstrate that the agent's performance may be enhanced via the concept of curriculum learning, a strategy in which the agent learns progressively over a series of increasingly complex related problems building up to the problem of interest.114
4.4 Proteins
Similar problems to those in the more general biochemical sequence field have been studied extensively in the specific case of proteins. In particular, the protein folding problem has attracted significant attention, including from a machine learning perspective. Much of the work conducted on protein folding using reinforcement learning concerns the 2D hydrophobic-polar model, a heavily simplified lattice model for folding introduced by Dill115 which was nonetheless shown by Berger and Leighton116 to be an NP-complete problem. Early work by Czibula et al.117 applies simple Q-learning to the HP model, with RL actions corresponding to four possible folding movements in 2D space and a final reward defined by the negative energy of the final protein structure; smaller intermediate rewards are offered for valid amino acid configurations.Li et al.118 took the first steps into deep RL for the 2D HP model by introducing FoldingZero, a protein self-folding architecture based on a deep convolutional network and upper confidence bound tree searching plus an actor-critic RL algorithm to iteratively improve both steps. In this formulation, the action space is of size three instead of four, as the folding moves conducted must form a self-avoiding walk. Rewards are provided for the amount of H–H contact in the final folded protein. Later, Jafari and Javidi119 utilised an RNN with three hidden LSTM layers for deep Q-learning RL via a vanilla policy gradient algorithm, with the action space defined identically to Czibula et al.117 but a novel reward function: the reward is −1 if amino acids are lattice neighbours and not consecutive in order and is a small positive value otherwise, and the agent attempts to maximise absolute value of the cumulative reward. Results demonstrated an improvement in both minimising the free energy in the final folded state and in the time required to reach a solution from several starting positions compared to previous bidimensional folding techniques.
Two recent attempts have been made to address protein folding in a more realistic framework. The masters thesis of Gao120 uses a graphical representation of the 3D structures of proteins: nodes in the graph contain information about residues in the conformation and edges contain information about relationships between them. The RL agent operates by changing the torsion angles of the conformation one at a time, so the action space consists of selecting torsion angle to be altered and the number of degrees by which to rotate it. Protein folding is viewed as an infinite-step process, with rewards given so that the agent aims to minimise the free energy of the conformation. The neural network architecture is very similar to that used by Gogineni et al.113 for the broader conformer search problem.
Panou and Reczko121 applied RL to the protein folding software game Foldit Standalone, introduced by Kleffner et al.122 as a development of the online game Foldit of Cooper et al.123 Foldit works by allowing human users to interact with an image of a protein in several ways, with a score awarded to each protein configuration based on the negative of its energy. In the work of Panou and Reczko,121 15 of the possible interactions with a protein form the action set for the RL agent, which works on pre-processed images of proteins taken from the game. Deep Q-learning with a CNN composed of several convolutional layers for feature extraction and two fully connected layers for prediction is used for policy optimisation, with the reward function based on the change in Foldit score resulting from an action. The agent was trained on 20 proteins and tested on 20 more; while performance is highly dependent on hyperparameter settings, an optimised agent is able to deliver consistent structural improvements within a reasonable time, although it does not exceed human performance at the same task.
Ideas derived from reinforcement learning have also been applied to more general protein conformation sampling. Shamsi et al.124 presented an extension to count-based adaptive sampling for exploration of protein conformation landscapes, in which reinforcement learning is used to choose the set of points in the protein structure at which molecular dynamics simulations should be run to identify low-energy states. The use of RL in this work is limited, however, as the policy is never optimised and must be supplied by a human; instead, the reward function is used to optimise a set of weight parameters corresponding to the directions in which sampling may move from an initial state. Barozet et al.125 developed two methods to sample the conformational space of protein loop portions, an important feature of many proteins which are often represented unrealistically by a single conformation. The more successful of the two methods in terms of sampling speed uses an RL-inspired heuristic for the selection of a tripeptide to be added during the loop generation process. Scoring is based on the probability of successfully closing a protein loop given previous loops generated using relevant tripeptides, although this is not a reward function in the traditional sense as there is no cumulative reward to be maximised.
4.5 Chemical reaction planning, optimisation and control
There are many open problems relating to the planning, management and optimisation of chemical reactions. Retrosynthetic planning, or retrosynthesis, concerns attempts to work backwards from a target molecule to determine a set of reactions which will lead to the generation of a reasonable quantity of the target at the lowest cost. The target may be a small chemical molecule or a larger biomolecular structure. Several synthesis routes may be possible, and the aim is to determine the most efficient route. An example of this process is shown in Fig. 8 for a target molecule A given some available initial reactants B, C, D, E, F, G. The two routes depicted also involve intermediate reaction products H, I, J, K, L, M; it is clear that the second route is to be preferred, as it requires only two reactions to be conducted instead of six. Retrosynthesis is commonly done with computer assistance, for example by Szymkuć et al.126 and Delépine et al.127 among many others, and has recently seen some use of RL as a strategy to make decisions about which reaction paths should be investigated.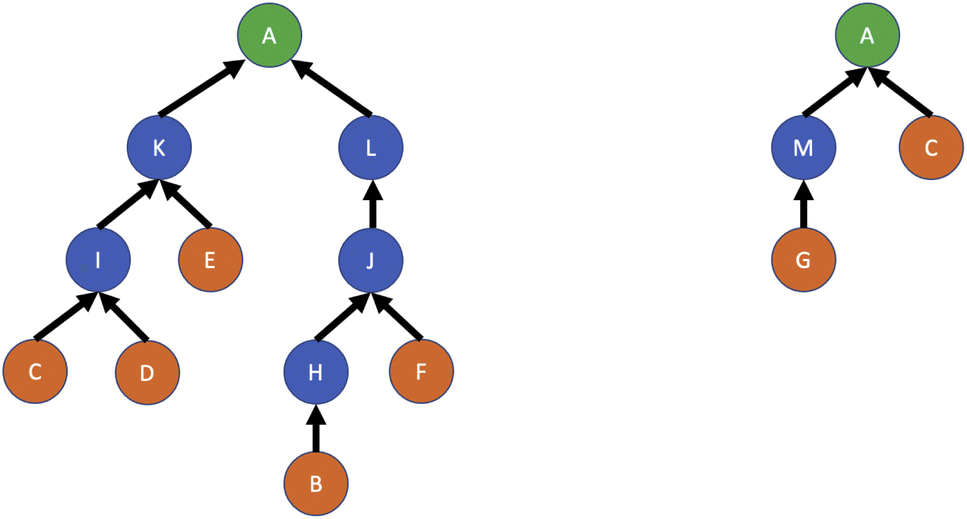 | ||
| Fig. 8 Example of retrosynthesis with a target molecule A. Circles represent molecules, and arrows depict relationships between the reactants and products of chemical reactions. Given the available initial reactants (orange), two different synthesis routes can be devised via different intermediate products (blue); the second route is preferable as it requires fewer reactions. | ||
For chemical retrosynthesis, Schreck et al.128 designed an RL agent which is given a set of possible reactions leading to a product (either the final product or an intermediate) and a set of commercially available molecules from which the target must be synthesised. The action space consists of selecting a reaction at each step in the synthesis chain, and the reward function is designed to minimise the cost of synthesising the target product, subject to the total number of reactions required being no greater than a specified threshold. A form of deep Q-learning is used for policy improvement. A different RL treatment of the problem was developed by Segler et al.129via a method to propose molecular transformations and determine their validity based on Monte Carlo tree search and three separate neural networks for expansion policy, rollout policy and prediction. Koch et al.130 later applied this approach to bioretrosynthesis, with extensions to handle biological compounds via reaction rules and combined biochemical scoring.
Having determined a reaction of interest, the next step is to choose the conditions under which the reaction will take place. This can significantly influence the volume of the target reaction product which can be produced, and there is substantial literature on optimising reaction conditions via various methods, for instance Gao et al.131 and Shields et al.132 The key work on RL for reaction optimisation is that of Zhou et al.133 The state space of the agent is defined as the set of all possible combinations of reaction conditions, and the action space by the set of changes that could be made to the current conditions. A policy gradient algorithm and a LSTM RNN with two hidden layers are used. When tested on four real reactions, the agent consistently finds the optimal reaction conditions faster than other non-RL algorithms.
In a related problem domain, Li et al.134 use asynchronous advantage actor-critic RL to control the molecular weight distribution in atom transfer radical polymerisation. Given the current reaction system, the agent may add a fixed amount of four possible reagents to the reaction system, provided the reagent budget has not been reached. A reward of 1 is given if the actual molecular weight distribution is very close to the target molecular weight distribution, and 0.1 given if it is further away but still sufficiently close to be worthy of further exploration in this approximate region. Two different neural networks are considered, with a CNN found to outperform a simple fully-connected NN.
Ma et al.135 treat control of the molecular weight distribution in nonlinear polymerisation reactions as a process control problem. The agent chooses the values to which the initiator and monomer flow rates in the polymerisation process are to be set, with a positive reward given if the difference between target and observed molecular weights is sufficiently small, and negative reward otherwise; the final molecular weights are given greater importance than intermediate values. DDPG and two feedforward NNs are used for policy optimisation. The training regime is adapted to incorporate historical measurement data as semi-batch experiments are non-Markovian. A different reaction control problem was also approached using DDPG by Alhazmi and Sarathy,136 focusing on the reaction temperature in a continuous stirred tank reactor (CSTR) network with complex dynamics and measurement uncertainty. The problem of temperature control of a CSTR system via reinforcement learning was earlier considered by Pandian and Noel,137 with the conclusion that using deep RL directly is a better approach than simply using RL to tune the parameters of a more traditional controller.
Also relevant is the recent work of Rajak et al.138 on the problem of optimal synthesis planning for inorganic materials. The authors use deep RL with policy gradient optimisation to generate time sequences of reaction conditions for quantum material synthesis via chemical vapour deposition.
Another approach to better understanding and explaining chemical reaction mechanisms is to locate the transition states in the reaction and use these to determine the factors that characterise the reaction. This is a difficult problem, as the set of possible factors is typically much larger than the subset which actually affect the reaction mechanism, but recent computational developments have led to renewed interest in attempting to learn reaction mechanisms automatically. RL was introduced to this effort by Zhang et al.139via a method to determine reaction dynamics and transition state locations using molecular dynamics simulation and RL with two fully connected feedforward NNs. The learning approach is named variational target optimisation and is a combination of two techniques closely related to actor-critic methods which were earlier described by the same research group.140
Reinforcement learning has recently begun to play a role in research into catalysis. Although the key problem of identifying new catalysts to improve reaction efficiency has yet to be attempted using RL, there has been promising work on related problems, including that of Yoon et al.141 on prediction of kinetic pathways and barriers in potential catalysts under reaction conditions. The state of the system consists of information about the energy surface structure, and the agent chooses one of four possible actions to modify the surface, with actor-critic TRPO for policy optimisation. The final reward is positive if kinetically feasible surface separation is observed and negative if the upper energy bound is exceeded, with positive intermediate rewards given when transition states are observed. The resulting CatGym method was tested on an Ni–Pd–Au alloy catalyst and was able to explore the surface efficiently and generate kinetic pathways to low-energy configurations. Similarly, Lan and An142 used a combination of deep RL and density functional theory to discover reaction pathways in the Haber–Bosch process with an Fe(111) catalyst, identifying a path with a lower free energy barrier than previously known pathways. The agent uses PPO with two fully connected NNs, with a state space defined by vectors of length 23 containing information about the reaction at different surface sites and the number of gas species along the reaction path, and rewards based on the free energy barrier connecting the previous and current state after an action is taken.
Finally, an unusual application of reinforcement learning to improve the yield of a chemical product was developed by Hubbs et al.143 in the form of an agent to determine optimal production schedules for a chemical manufacturing process. Here, the action space consists of constructing a schedule to determine which product is to be produced on each day of the simulation, and rewards are given based on the profit made from the production schedule. The RL implementation uses advantage actor-critic and a neural network with 12 hidden layers.
4.6 Other areas
However, this is perhaps an area of opportunity, as RL has seen some use in quantum physics which may have applications to chemistry fields. For example, both Niu et al.148 writing on an application of RL to quantum control and Bolens and Heyl149 on RL for digital quantum simulation highlight their potential uses in quantum chemistry but do not attempt this themselves. Similarly, the work of Nguyen et al.150 on measurement of double quantum dot devices may also be relevant to quantum chemistry research in future.
RL has recently seen use in battery modelling. Unagar et al.156 calibrate a model for lithium-ion battery discharge by using an RL agent to select the degradation parameters of the model. The agent uses the Lyapunov variation of the actor-critic algorithm and two fully connected NNs, and is shown to generate better calibrated models than a Kalman filter method. Li et al.157 use deep Q-learning to construct an energy management strategy for battery electric vehicles, with the reward function constructed so that the agent will minimise the energy loss of the system while ensuring its safety.
5 Conclusion
Reinforcement learning is a technique which is growing in popularity across many academic fields, and chemistry is no exception. Recent advances in deep reinforcement learning, the development of more efficient algorithms and improved neural network structures have driven a significant increase in the range of potential applications within chemistry. In this paper, we have reviewed the work conducted so far in several broad areas and specific problems, considering both the theoretical approaches chosen by different authors and the practical challenges posed by different problem settings.The growth of RL within chemistry has been notably non-uniform, with much more focus on some areas than others. In particular, molecular design for drug discovery has seen an extremely large volume of work, with many different variants of the wider design problem being attempted independently by different research groups using several varieties of RL. Biochemistry problems of sequence design and protein analysis have also been a key area of RL research in recent years. In contrast, the use of RL in fields such as chromatography, crystallography and quantum chemistry has so far been extremely limited. It is possible that there is untapped potential for RL to aid researchers in some of these areas, as planning and sequential decision making problems to which it is well suited are present in these fields.
One area of concern arising from this review is the relatively limited impact of previous RL research on practical chemistry. For example, despite the many promising computational results obtained in drug discovery problems, it is somewhat concerning to note that few (if any) of the molecules discovered through these methods have yet been synthesised. Similarly, despite the encouraging performance of RL in reaction optimisation tasks, there is little evidence that this has influenced the choice of reaction conditions in real problems. There are many reasons why this may be the case, but it does highlight the importance of ensuring that practical chemists – and not just those already engaged in computational work – are in a position to take advantage of new developments.
In practical terms, reinforcement learning has become significantly more accessible in recent years. As interest in the topic has increased, so too has the volume of easily available literature. In addition, open-source code libraries are available in a variety of programming languages, removing a significant hurdle to easy implementation of RL. This, combined with its increasingly widespread use across different disciplines, suggests that the importance of RL as a practical technique within chemistry will only continue to grow.
Author contributions
SG undertook the main research for this article. MN, SK and JGF organised the project and reviewed the material. SG and MN wrote the bulk of the manuscript and all authors reviewed and approved the manuscript before submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the AI for Scientific Discovery Network, funded by UKRI EPSRC under grant no: EP/S000356/1. The authors would like to thank Colin Bird for his work in proof-reading this manuscript and providing valuable feedback on its domain-specific content.Notes and references
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah and M. Spitzer, et al. , Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS PubMed.
- S. Guo and Z. Qu, Edge Learning for Distributed Big Data Analytics: Theory, Algorithms, and System Design, Cambridge University Press, 2022 Search PubMed.
- R. S. Sutton and A. G. Barto, Reinforcement Learning: An Introduction, MIT Press, 2018 Search PubMed.
- D. Bertsekas, Reinforcement Learning and Optimal Control, Athena Scientific, 2019 Search PubMed.
- K. Arulkumaran, M. P. Deisenroth, M. Brundage and A. A. Bharath, IEEE Signal Process. Mag., 2017, 34, 26–38 Search PubMed.
- D. Silver, A. Huang, C. J. Maddison, A. Guez, L. Sifre, G. Van Den Driessche, J. Schrittwieser, I. Antonoglou, V. Panneershelvam and M. Lanctot, et al. , Nature, 2016, 529, 484–489 CrossRef CAS PubMed.
- A. C. Mater and M. L. Coote, J. Chem. Inf. Model., 2019, 59, 2545–2559 CrossRef CAS PubMed.
- M. v. Otterlo and M. Wiering, in Reinforcement Learning, Springer, 2012, pp. 3–42 Search PubMed.
- S. D. Whitehead and L.-J. Lin, Artif. Intell., 1995, 73, 271–306 CrossRef.
- T. Jaakkola, S. Singh and M. Jordan, Adv. Neural Inf. Process. Syst., 1994, 7, 345–352 Search PubMed.
- A. Y. Ng, D. Harada and S. Russell, ICML, 1999, 278–287 Search PubMed.
- D. Hadfield-Menell, S. Milli, P. Abbeel, S. Russell and A. Dragan, Adv. Neural Inf. Process. Syst., 2017, 30, 6768–6777 Search PubMed.
- O. Nachum, M. Norouzi, K. Xu and D. Schuurmans, Adv. Neural Inf. Process. Syst., 2017, 30, 2272–2282 Search PubMed.
- C. J. Watkins and P. Dayan, Mach. Learn., 1992, 8, 279–292 Search PubMed.
- V. Mnih, K. Kavukcuoglu, D. Silver, A. A. Rusu, J. Veness, M. G. Bellemare, A. Graves, M. Riedmiller, A. K. Fidjeland and G. Ostrovski, et al. , Nature, 2015, 518, 529–533 CrossRef CAS PubMed.
- H. Hasselt, Adv. Neural Inf. Process. Syst., 2010, 23, 2613–2621 Search PubMed.
- A. G. Barto, R. S. Sutton and C. W. Anderson, IEEE Trans. Syst. Man Cybern., 1983, 13, 834–846 Search PubMed.
- J. Peters and S. Schaal, Neurocomputing, 2008, 71, 1180–1190 CrossRef.
- V. Mnih, A. P. Badia, M. Mirza, A. Graves, T. Lillicrap, T. Harley, D. Silver and K. Kavukcuoglu, International Conference on Machine Learning, 2016, pp. 1928–1937 Search PubMed.
- R. S. Sutton, D. A. McAllester, S. P. Singh and Y. Mansour, Adv. Neural Inf. Process. Syst., 2000, 1057–1063 Search PubMed.
- R. J. Williams, Mach. Learn., 1992, 8, 229–256 Search PubMed.
- M. Riedmiller, J. Peters and S. Schaal, 2007 IEEE International Symposium on Approximate Dynamic Programming and Reinforcement Learning, 2007, pp. 254–261 Search PubMed.
- J. Schulman, S. Levine, P. Abbeel, M. Jordan and P. Moritz, International Conference on Machine Learning, 2015, pp. 1889–1897 Search PubMed.
- J. Schulman, F. Wolski, P. Dhariwal, A. Radford and O. Klimov, Proximal policy optimization algorithms, arXiv preprint arXiv:1707.06347, 2017.
- T. P. Lillicrap, J. J. Hunt, A. Pritzel, N. Heess, T. Erez, Y. Tassa, D. Silver and D. Wierstra, Continuous control with deep reinforcement learning, arXiv preprint arXiv:1509.02971, 2015.
- S. Fujimoto, H. Hoof and D. Meger, International Conference on Machine Learning, 2018, pp. 1587–1596 Search PubMed.
- T. Haarnoja, A. Zhou, P. Abbeel and S. Levine, International Conference on Machine Learning, 2018, pp. 1861–1870 Search PubMed.
- R. Coulom, International Conference on Computers and Games, 2006, pp. 72–83 Search PubMed.
- T. Vodopivec, S. Samothrakis and B. Ster, J. Artif. Intell. Res., 2017, 60, 881–936 CrossRef.
- S. Nagendra, N. Podila, R. Ugarakhod and K. George, 2017 International Conference on Advances in Computing, Communications and Informatics (ICACCI), 2017, pp. 26–32 Search PubMed.
- C. Colas, O. Sigaud and P.-Y. Oudeyer, A Hitchhiker's Guide to Statistical Comparisons of Reinforcement Learning Algorithms, arXiv preprint arXiv:1904.06979, 2019.
- S. Jordan, Y. Chandak, D. Cohen, M. Zhang and P. Thomas, International Conference on Machine Learning, 2020, pp. 4962–4973 Search PubMed.
- R. Raileanu and R. Fergus, International Conference on Machine Learning, 2021, pp. 8787–8798 Search PubMed.
- J. Schmidhuber, Neural Netw., 2015, 61, 85–117 CrossRef PubMed.
- P. Benardos and G.-C. Vosniakos, Eng. Appl. Artif. Intell., 2007, 20, 365–382 CrossRef.
- R. Luo, F. Tian, T. Qin, E. Chen and T.-Y. Liu, Adv. Neural inf. Process. Syst., 2018, 31, 7816–7827 Search PubMed.
- A. Eleyan and H. Demirel, Computational Intelligence and Bioinspired Systems, 2005 Search PubMed.
- D. H. Nguyen and B. Widrow, IEEE Control Systems Magazine, 1990, 10, pp. 18–23 Search PubMed.
- P. Tahmasebi and A. Hezarkhani, Nat. Resour. Res., 2011, 20, 25–32 CrossRef CAS.
- A. Haldorai and A. Ramu, Neural Process. Lett., 2021, 53, 2385–2401 CrossRef.
- D. Eck and J. Schmidhuber, A first look at music composition using LSTM recurrent neural networks, Istituto Dalle Molle Di Studi Sull Intelligenza Artificiale, 2002, vol. 103, p. 48 Search PubMed.
- J. A. Franklin, INFORMS J. Comput., 2006, 18, 321–338 CrossRef.
- P. Coulibaly, F. Anctil, R. Aravena and B. Bobée, Water Resour. Res., 2001, 37, 885–896 CrossRef.
- J. Zheng, C. Xu, Z. Zhang and X. Li, 2017 51st Annual Conference on Information Sciences and Systems (CISS), 2017, pp. 1–6 Search PubMed.
- T. Mikolov, M. Karafiát, L. Burget, J. Cernocký and S. Khudanpur, Interspeech, 2010, 1045–1048 Search PubMed.
- A. Khalifa, G. A. Barros and J. Togelius, Deeptingle, arXiv preprint arXiv:1705.03557, 2017.
- M. Sundermeyer, H. Ney and R. Schlüter, IEEE/ACM Trans. Audio, Speech, Language Process., 2015, 23, 517–529 Search PubMed.
- S. Hochreiter and J. Schmidhuber, Neural Comput., 1997, 9, 1735–1780 CrossRef CAS PubMed.
- J. Chung, C. Gulcehre, K. Cho and Y. Bengio, Empirical evaluation of gated recurrent neural networks on sequence modeling, arXiv preprint arXiv:1412.3555, 2014.
- A. Joulin and T. Mikolov, Adv. Neural Inf. Process. Syst., 2015, 28, 190–198 Search PubMed.
- E. Grefenstette, K. M. Hermann, M. Suleyman and P. Blunsom, Adv. Neural Inf. Process. Syst., 2015, 28, 1828–1836 Search PubMed.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Adv. Neural Inf. Process. Syst., 2014, 27, 2672–2680 Search PubMed.
- L. Yu, W. Zhang, J. Wang and Y. Yu, Proceedings of the AAAI Conference on Artificial Intelligence, 2017 Search PubMed.
- T. Xu, P. Zhang, Q. Huang, H. Zhang, Z. Gan, X. Huang and X. He, Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, 2018, pp. 1316–1324 Search PubMed.
- K. Schawinski, C. Zhang, H. Zhang, L. Fowler and G. K. Santhanam, Mon. Not. R. Astron. Soc.: Lett., 2017, 467, L110–L114 Search PubMed.
- X. Yi, E. Walia and P. Babyn, Med. Image Anal., 2019, 58, 101552 CrossRef PubMed.
- H. Alqahtani, M. Kavakli-Thorne and G. Kumar, Arch. Comput. Methods Eng., 2021, 28, 525–552 CrossRef.
- A. Krizhevsky, I. Sutskever and G. E. Hinton, Adv. Neural Inf. Process. Syst., 2012, 25, 1097–1105 Search PubMed.
- M. V. Valueva, N. Nagornov, P. A. Lyakhov, G. V. Valuev and N. I. Chervyakov, Math. Comput. Simul., 2020, 177, 232–243 CrossRef.
- D. Duvenaud, D. Maclaurin, J. Aguilera-Iparraguirre, R. Gómez-Bombarelli, T. Hirzel, A. Aspuru-Guzik and R. P. Adams, Adv. Neural Inf. Process. Syst., 2015, 28, 2224–2232 Search PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, International Conference on Machine Learning, 2017, pp. 1263–1272 Search PubMed.
- D. P. Kingma and M. Welling, Auto-encoding Variational Bayes, arXiv preprint arXiv:1312.6114, 2013.
- S. R. Bowman, L. Vilnis, O. Vinyals, A. M. Dai, R. Jozefowicz and S. Bengio, Generating sentences from a continuous space, arXiv preprint arXiv:1511.06349, 2015.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef.
- R.-R. Griffiths and J. M. Hernández-Lobato, Chem. Sci., 2020, 11, 577–586 RSC.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser and I. Polosukhin, Adv. Neural Inf. Process. Syst., 2017, 30, 6000–6010 Search PubMed.
- T. Wolf, L. Debut, V. Sanh, J. Chaumond, C. Delangue, A. Moi, P. Cistac, T. Rault, R. Louf and M. Funtowicz, et al., Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations, 2020, pp. 38–45 Search PubMed.
- L. Chen, K. Lu, A. Rajeswaran, K. Lee, A. Grover, M. Laskin, P. Abbeel, A. Srinivas and I. Mordatch, Adv. Neural Inf. Process. Syst., 2021, 34, 15084–15097 Search PubMed.
- M. Janner, Q. Li and S. Levine, Adv. Neural Inf. Process. Syst., 2021, 34, 1273–1286 Search PubMed.
- I. Torjesen, Pharm. J., 2015, Online, URI: 20068196 Search PubMed.
- V. Mandlik, P. R. Bejugam and S. Singh, in Artificial Neural Network for Drug Design, Delivery and Disposition, Elsevier, 2016, pp. 123–139 Search PubMed.
- G. Schneider and D. E. Clark, Angew. Chem., Int. Ed., 2019, 58, 10792–10803 CrossRef CAS PubMed.
- V. D. Mouchlis, A. Afantitis, A. Serra, M. Fratello, A. G. Papadiamantis, V. Aidinis, I. Lynch, D. Greco and G. Melagraki, Int. J. Mol. Sci., 2021, 22, 1676 CrossRef PubMed.
- G. L. Guimares, B. Sánchez-Lengeling, P. L. C. Farias and A. Aspuru-Guzik, Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models, arXiv preprint arXiv:1705.10843, 2017.
- B. Sánchez-Lengeling, C. Outeiral, G. Guimaraes and A. Aspuru-Guzik, Optimizing Distributions Over Molecular Space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC), 2017, https://chemrxiv.org/articles/ORGANIC_1_pdf/5309668 Search PubMed.
- H. Ikebata, K. Hongo, T. Isomura, R. Maezono and R. Yoshida, J. Comput. Aided Mol. Des., 2017, 31, 379–391 CrossRef CAS PubMed.
- E. Putin, A. Asadulaev, Y. Ivanenkov, V. Aladinskiy, B. Sanchez-Lengeling, A. Aspuru-Guzik and A. Zhavoronkov, J. Chem. Inf. Model., 2018, 58, 1194–1204 CrossRef CAS.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, J. Cheminformatics, 2017, 9, 1–14 CrossRef PubMed.
- T. Blaschke, O. Engkvist, J. Bajorath and H. Chen, J. Cheminformatics, 2020, 12, 1–17 CrossRef.
- M. Popova, O. Isayev and A. Tropsha, Sci. Adv., 2018, 4, eaap7885 CrossRef CAS PubMed.
- A. Yoshimori, E. Kawasaki, C. Kanai and T. Tasaka, Chem. Pharm. Bull., 2020, 68, 227–233 CrossRef CAS PubMed.
- G. Wolber and T. Langer, J. Chem. Inf. Model., 2005, 45, 160–169 CrossRef CAS PubMed.
- D. Neil, M. H. S. Segler, L. Guasch, M. Ahmed, D. Plumbley, M. Sellwood and N. Brown, ICLR, 2018 Search PubMed.
- T. Pereira, M. Abbasi, B. Ribeiro and J. P. Arrais, J. Cheminformatics, 2021, 13, 1–17 CrossRef PubMed.
- J. Born, M. Manica, A. Oskooei, J. Cadow, G. Markert and M. R. Martínez, iScience, 2021, 24, 102269 CrossRef CAS PubMed.
- S. R. Krishnan, N. Bung, G. Bulusu and A. Roy, J. Chem. Inf. Model., 2021, 61, 621–630 CrossRef CAS PubMed.
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed.
- T. G. Karimpanal and R. Bouffanais, Adapt. Behav., 2019, 27, 111–126 CrossRef.
- S. Gamrian and Y. Goldberg, International Conference on Machine Learning, 2019, pp. 2063–2072 Search PubMed.
- B. Agyemang, W.-P. Wu, D. Addo, M. Y. Kpiebaareh, E. Nanor and C. Roland Haruna, Brief. Bioinform., 2021, 22, bbaa364 CrossRef PubMed.
- L. A. Thiede, M. Krenn, A. Nigam and A. Aspuru-Guzik, Curiosity in exploring chemical space: intrinsic rewards for deep molecular reinforcement learning, arXiv preprint arXiv:2012.11293, 2020.
- J. You, B. Liu, Z. Ying, V. S. Pande and J. Leskovec, NeurIPS, 2018 Search PubMed.
- Y. Khemchandani, S. O'Hagan, S. Samanta, N. Swainston, T. J. Roberts, D. Bollegala and D. B. Kell, J. Cheminformatics, 2020, 12, 1–17 CrossRef.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef CAS.
- S. R. Atance, J. V. Diez, O. Engkvist, S. Olsson and R. Mercado, De novo drug design using reinforcement learning with graph-based deep generative models, ChemRxiv preprint, 2021.
- Z. Zhou, S. Kearnes, L. Li, R. N. Zare and P. Riley, Sci. Rep., 2019, 9, 1–10 CrossRef PubMed.
- N. Ståhl, G. Falkman, A. Karlsson, G. Mathiason and J. Boström, J. Chem. Inf. Model., 2019, 59, 3166–3176 CrossRef PubMed.
- S. K. Gottipati, B. Sattarov, S. Niu, Y. Pathak, H. Wei, S. Liu, S. Blackburn, K. Thomas, C. Coley and J. Tang, et al., International Conference on Machine Learning, 2020, pp. 3668–3679 Search PubMed.
- J. Horwood and E. Noutahi, ACS Omega, 2020, 5, 32984–32994 CrossRef CAS.
- G. Simm, R. Pinsler and J. M. Hernández-Lobato, International Conference on Machine Learning, 2020, pp. 8959–8969 Search PubMed.
- G. Simm, R. Pinsler, G. Csányi and J. M. Hernández-Lobato, International Conference on Learning Representations, 2020 Search PubMed.
- G. Bolcato and J. Boström, On the value of using 3D-shape and electrostatic similarities in deep generative methods, ChemRxiv preprint, 2021.
- S. A. Meldgaard, J. Köhler, H. L. Mortensen, M.-P. V. Christiansen, F. Noé and B. Hammer, Generating stable molecules using imitation and reinforcement learning, arXiv preprint arXiv:2107.05007, 2021.
- T. Hester, M. Vecerik, O. Pietquin, M. Lanctot, T. Schaul, B. Piot, D. Horgan, J. Quan, A. Sendonaris and I. Osband, et al., Thirty-second AAAI Conference on Artificial Intelligence, 2018 Search PubMed.
- Y. Cho, S. Kim, P. P. Li, M. P. Surh, T. Y.-J. Han and J. Choo, Workshop at the 33rd Conference on Neural Information Processing Systems (NeurIPS), 2019 Search PubMed.
- K. Ahuja, W. H. Green and Y.-P. Li, J. Chem. Theory Comput., 2021, 17, 818–825 CrossRef CAS.
- P. Eastman, J. Shi, B. Ramsundar and V. S. Pande, PLoS Comput. Biol., 2018, 14, e1006176 CrossRef PubMed.
- F. Runge, D. Stoll, S. Falkner and F. Hutter, International Conference on Learning Representations, 2019 Search PubMed.
- C. Finn and S. Levine, Meta-learning and universality: Deep representations and gradient descent can approximate any learning algorithm, arXiv preprint arXiv:1710.11622, 2017.
- J. X. Wang, Z. Kurth-Nelson, D. Tirumala, H. Soyer, J. Z. Leibo, R. Munos, C. Blundell, D. Kumaran and M. Botvinick, Learning to reinforcement learn, arXiv preprint arXiv:1611.05763, 2016 Search PubMed.
- A. Gupta, B. Eysenbach, C. Finn and S. Levine, Unsupervised meta-learning for reinforcement learning, arXiv preprint arXiv:1806.04640, 2018.
- C. Angermueller, D. Dohan, D. Belanger, R. Deshpande, K. Murphy and L. Colwell, Proceedings of the International Conference on Learning Representations, 2020 Search PubMed.
- T. Gogineni, Z. Xu, E. Punzalan, R. Jiang, J. Kammeraad, A. Tewari and P. Zimmerman, Adv. Neural Inf. Process. Syst., 2020, 33, 20142–20153 Search PubMed.
- S. Narvekar, B. Peng, M. Leonetti, J. Sinapov, M. E. Taylor and P. Stone, Curriculum learning for reinforcement learning domains: a framework and survey, 2020, arXiv preprint arXiv:2003.04960.
- K. Dill, Biochemistry, 1985, 24, 1501–1509 CrossRef CAS PubMed.
- B. Berger and T. Leighton, J. Comput. Biol., 1998, 5, 27–40 CrossRef CAS PubMed.
- G. Czibula, I. Bocicor and I. Czibula, International Journal of Computer Technology and Applications, 2011, 2, 171–182 Search PubMed.
- Y. Li, H. Kang, K. Ye, S. Yin and X. Li, Workshop on Deep Reinforcement Learning at NeurIPS, 2018 Search PubMed.
- R. Jafari and M. M. Javidi, SN Appl. Sci., 2020, 2, 259 CrossRef CAS.
- W. Gao, M.Sc. thesis, The Johns Hopkins University, 2020.
- D. Panou and M. Reczko, DeepFoldit – A Deep Reinforcement Learning Neural Network Folding Proteins, arXiv preprint arXiv:2011.03442, 2020.
- R. Kleffner, J. Flatten, A. Leaver-Fay, D. Baker, J. B. Siegel, F. Khatib and S. Cooper, Bioinformatics, 2017, 33, 2765–2767 CrossRef CAS PubMed.
- S. Cooper, F. Khatib, A. Treuille, J. Barbero, J. Lee, M. Beenen, A. Leaver-Fay, D. Baker, Z. Popovic and D. Eccles, Nature, 2010, 466, 756–760 CrossRef CAS PubMed.
- Z. Shamsi, K. J. Cheng and D. Shukla, J. Phys. Chem. B, 2018, 122, 8386–8395 CrossRef CAS PubMed.
- A. Barozet, K. Molloy, M. Vaisset, T. Siméon and J. Cortés, Bioinformatics, 2020, 36, 1099–1106 CAS.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef.
- B. Delépine, T. Duigou, P. Carbonell and J.-L. Faulon, Metab. Eng., 2018, 45, 158–170 CrossRef.
- J. S. Schreck, C. W. Coley and K. J. Bishop, ACS Cent. Sci., 2019, 5, 970–981 CrossRef CAS.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS.
- M. Koch, T. Duigou and J.-L. Faulon, ACS Synth. Biol., 2020, 9, 157–168 CrossRef CAS PubMed.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- Z. Zhou, X. Li and R. N. Zare, ACS Cent. Sci., 2017, 3, 1337–1344 CrossRef CAS PubMed.
- H. Li, C. R. Collins, T. G. Ribelli, K. Matyjaszewski, G. J. Gordon, T. Kowalewski and D. J. Yaron, Mol. Syst. Des. Eng., 2018, 3, 496–508 RSC.
- Y. Ma, W. Zhu, M. G. Benton and J. Romagnoli, J. Process Control, 2019, 75, 40–47 CrossRef CAS.
- K. Alhazmi and S. M. Sarathy, 2020 European Control Conference (ECC), 2020, pp. 1066–1068 Search PubMed.
- B. J. Pandian and M. M. Noel, Chem. Prod. Process Model., 2018, 13, 20170040 CAS.
- P. Rajak, A. Krishnamoorthy, A. Mishra, R. Kalia, A. Nakano and P. Vashishta, npj Comput. Mater., 2021, 7, 1–9 CrossRef.
- J. Zhang, Y.-K. Lei, Z. Zhang, X. Han, M. Li, L. Yang, Y. I. Yang and Y. Q. Gao, Phys. Chem. Chem. Phys., 2021, 23, 6888–6895 RSC.
- J. Zhang, Y. I. Yang and F. Noé, J. Phys. Chem. Lett., 2019, 10, 5791–5797 CrossRef CAS PubMed.
- J. Yoon, Z. Cao, R. K. Raju, Y. Wang, R. Burnley, A. J. Gellman, A. B. Farimani and Z. W. Ulissi, Mach. Learn.: Sci. Technol., 2021, 2, 045018 Search PubMed.
- T. Lan and Q. An, J. Am. Chem. Soc., 2021, 143, 16804–16812 CrossRef CAS PubMed.
- C. D. Hubbs, C. Li, N. V. Sahinidis, I. E. Grossmann and J. M. Wassick, Comput. Chem. Eng., 2020, 141, 106982 CrossRef CAS.
- I. Paparelle, L. Moro and E. Prait, Phys. Lett. A, 2020, 384, 126266 CrossRef CAS.
- C. Monea, 13th International Conference on Electronics, Computers and Artificial Intelligence (ECAI), 2021 Search PubMed.
- J. Westermayr and P. Marquetand, Chem. Rev., 2020, 121, 9873–9926 CrossRef.
- M. Ostaszewski, L. Trenkwalder, W. Masarczyk, E. Scerri and V. Dunjko, Adv. Neural Inf. Process. Syst., 2021, 34, 18182–18194 Search PubMed.
- M. Y. Niu, S. Boixo, V. N. Smelyanskiy and H. Neven, npj Quantum Inf., 2019, 5, 33 CrossRef.
- A. Bolens and M. Heyl, Phys. Rev. Lett., 2021, 127, 110502 CrossRef CAS PubMed.
- V. Nguyen, S. Orbell, D. T. Lennon, H. Moon, F. Vigneau, L. C. Camenzind, L. Yu, D. M. Zumbühl, G. A. D. Briggs and M. A. Osborne, et al. , npj Quantum Inf., 2021, 7, 1–9 CrossRef.
- A. Kensert, G. Collaerts, K. Efthymiadis, G. Desmet and D. Cabooter, J. Chromatogr. A, 2021, 1638, 461900 CrossRef CAS PubMed.
- S. Nikita, A. Tiwari, D. Sonawat, H. Kodamana and A. S. Rathore, Chem. Eng. Sci., 2021, 230, 116171 CrossRef CAS.
- S.-W. Chang, C.-L. Chang, L.-T. Li and S.-W. Liao, IEEE Access, 2020, 8, 9864–9874 Search PubMed.
- M. Karimi, M. Imanzadeh, P. Farhadi and N. Ghadimi, Int. J. Inf. Electron. Eng., 2012, 2, 752–756 Search PubMed.
- J. Li, T. Yu and B. Yang, IEEE Access, 2021, 9, 6063–6075 Search PubMed.
- A. Unagar, Y. Tian, M. A. Chao and O. Fink, Energies, 2021, 14, 1361 CrossRef CAS.
- W. Li, H. Cui, T. Nemeth, J. Jansen, C. Ünlübayir, Z. Wei, L. Zhang, Z. Wang, J. Ruan, H. Dai, X. Wei and D. U. Sauer, J. Energy Storage, 2021, 36, 102355 CrossRef.
- M. Vollmar and G. Evans, Crystallogr. Rev., 2021, 1–48 Search PubMed.
- Z. Feng, Q. Hou, Y. Zheng, W. Ren, J.-Y. Ge, T. Li, C. Cheng, W. Lu, S. Cao, J. Zhang and T. Zhang, Comput. Mater. Sci., 2019, 156, 310–314 CrossRef.
- V. Manee, R. Baratti and J. A. Romagnoli, Chem. Eng. Res. Des., 2022, 178, 111–123 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |