
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Consideration of predicted small-molecule metabolites in computational toxicology†
Marina
Garcia de Lomana
 ab,
Fredrik
Svensson
c,
Andrea
Volkamer
d,
Miriam
Mathea
*a and
Johannes
Kirchmair
*b
ab,
Fredrik
Svensson
c,
Andrea
Volkamer
d,
Miriam
Mathea
*a and
Johannes
Kirchmair
*b
aBASF SE, 67063 Ludwigshafen am Rhein, Germany. E-mail: miriam.mathea@basf.com; Tel: +49-621-60-29054
bDepartment of Pharmaceutical Sciences, Faculty of Life Sciences, University of Vienna, 1090 Vienna, Austria. E-mail: johannes.kirchmair@univie.ac.at; Tel: +43-1-4277-55104
cAlzheimer's Research UK UCL Drug Discovery Institute, University College London, London WC1E 6BT, UK
dIn Silico Toxicology and Structural Bioinformatics, Institute of Physiology, Charité Universitätsmedizin Berlin, 10117 Berlin, Germany
First published on 23rd February 2022
Abstract
Xenobiotic metabolism has evolved as a key protective system of organisms against potentially harmful chemicals or compounds typically not present in a particular organism. The system's primary purpose is to chemically transform xenobiotics into metabolites that can be excreted via renal or biliary routes. However, in a minority of cases, the metabolites formed are toxic, sometimes even more toxic than the parent compound. Therefore, the consideration of xenobiotic metabolism clearly is of importance to the understanding of the toxicity of a compound. Nevertheless, most of the existing computational approaches for toxicity prediction do not explicitly take metabolism into account and it is currently not known to what extent the consideration of (predicted) metabolites could lead to an improvement of toxicity prediction. In order to study how predictive metabolism could help to enhance toxicity prediction, we explored a number of different strategies to integrate predictions from a state-of-the-art metabolite structure predictor and from modern machine learning approaches for toxicity prediction. We tested the integrated models on five toxicological endpoints and assays, including in vitro and in vivo genotoxicity assays (AMES and MNT), two organ toxicity endpoints (DILI and DICC) and a skin sensitization assay (LLNA). Overall, the improvements in model performance achieved by including metabolism data were minor (up to +0.04 in the F1 scores and up to +0.06 in MCCs). In general, the best performance was obtained by averaging the probability of toxicity predicted for the parent compound and the maximum probability of toxicity predicted for any metabolite. Moreover, including metabolite structures as further input molecules for model training slightly improved the toxicity predictions obtained by this averaging approach. However, the high complexity of the metabolic system and associated uncertainty about the likely metabolites apparently limits the benefit of considering predicted metabolites in toxicity prediction.
Introduction
The metabolic system has evolved as the primary defense system against xenobiotic, potentially toxic substances. Its protective function is based on the biotransformation of xenobiotics into more hydrophilic and, hence, more rapidly excretable compounds (metabolites). However, a minority of metabolites produced by the metabolic system are more active than their parent compound (which is exploited by the prodrug concept) or even toxic.1The important role of metabolism in the toxicity of small organic molecules highlights the need for the consideration of metabolic pathways also in the computational prediction of toxicity. However, so far only a few in silico models for toxicity prediction have integrated metabolism information. For example, Dmitriev et al.2 built linear models for the prediction of rat acute toxicity using self-consistent regression, thereby considering parent compounds and measured metabolites. More specifically, they trained a model on about 3000 parent compounds and used it to predict the LD50 value of 37 test parent compounds and their measured metabolites (around 200 known metabolites). To calculate the final LD50 value, different strategies for averaging the LD50 values predicted for the parent compounds and their metabolites were investigated. However, only minor improvements in the overall performance of the model were achieved compared to using only the predicted probability of the parent compounds (R2 increased from 0.78 to 0.81 and RMSE remained at 0.49). In a more recent study from the same research group,3 classification models based on a Bayesian approach were trained on parent compounds with annotated bioactivity data for a variety of endpoints. The bioactivity of a compound was then calculated as the maximum probability predicted among the parent compound and its measured metabolites. For the 28 endpoints in the “toxic and adverse effects” category (with data sets ranging from 15 to 5583 toxic and non-toxic compounds), an increase of up to 0.14 in the precision and 0.16 in the recall during leave-one-out cross-validation (CV) was obtained on average (compared to taking the predicted probability of the parent compound only). These results show that the consideration of metabolism in prediction models can substantially improve the identification of potentially toxic compounds.
Data on measured metabolites can be valuable for estimating the toxicity of compounds but such approaches rely on the availability of experimental data. For this reason, in silico approaches to predict the likely metabolites of substances based on molecular structures are in high demand. Several predictors of this kind are available today, including BioTransformer,4 CyProduct,5 GloryX,6 Meteor Nexus,7 SyGMA,8 TIssue MEtabolic Simulator (TIMES)9 and XenoSite.10
In previous works, researchers from the Laboratory of Mathematical Chemistry (LMC) have combined in silico models for toxicity prediction with their TIMES metabolite predictor. The first model from LMC taking into account the parent compound and its metabolites (predicted with the S-9 metabolism simulator of TIMES) was developed for the prediction of in vitro mutagenicity (i.e. outcomes of the AMES assay).11,12 This AMES model was based on decision trees trained on the reactivity profile of compounds and labeled a compound as toxic if any of its predicted metabolites were predicted as toxic. The evaluation of the model on the training data showed that the metabolism-aware approach resulted in lower sensitivity (0.77) and specificity (0.74) compared to the performance of the model considering only the parent compound (sensitivity 0.82; specificity 0.94). The lower sensitivity obtained by this approach may be related to the fact that compounds without any predicted metabolites were automatically classified as inactive. Another drawback of this approach is the decrease in specificity due to false positive predictions derived from non-mutagenic parents with metabolites predicted as mutagenic. In addition to the training data, the model was evaluated on a test set of 36 mutagenic compounds, obtaining a sensitivity of 0.58 (corresponding to 21 correctly classified compounds). Despite the overall drop in performance, the metabolism-aware approach correctly identified compounds of which their mutagenicity is related to the metabolites formed.
Two further decision tree models from LMC targeting skin and respiratory sensitization, respectively,13,14 also included the evaluation of several properties of predicted metabolites (e.g. reactivity profile or ability to cross-link proteins) to classify the parent compounds as non-sensitizers or sensitizers (further distinguished between strong or weak sensitizers in the case of the skin). The evaluation of this skin sensitization model on the training data yielded 80% correct predictions for strong sensitizers, 34% for weak sensitizers and 72% for non-sensitizers, while the respiratory sensitization model obtained a sensitivity of 0.89 and a specificity of 0.52.
A further model of this kind from LMC was reported for the in vivo micronucleus test (MNT).15 By comparing the assay outcomes of the (in vitro) AMES assay with a liver genotoxicity and an MNT in vivo assays, bioactivated compounds and “bioexhausted” compounds (i.e. highly reactive compounds interacting with off-targets before reaching the target) were analyzed to establish in vitro–in vivo relationships. Based on this analysis, an in vivo rat liver metabolism predictor reproducing phase II conjugation reactions and detoxification pathways was developed. The toxicity prediction model of MNT applied on the predicted metabolites (derived with the in vivo metabolite predictor) reached a sensitivity of 0.82 and a specificity of 0.61 on the training data.
The performance of this MNT model, as well as the skin and respiratory sensitization models, was not compared to the performance of models not considering predicted metabolites. Therefore it is not possible to conclude on the benefits or drawbacks of these metabolism-aware models compared to models considering only parent compounds.
Overall, these recent reports on efforts to enhance toxicity prediction of small organic molecules by the consideration of their biotransformation provide valuable insights and starting points for the further development of methods for computational toxicology. Although metabolism is key to understanding the pharmacokinetics and toxicity of compounds, the inherent uncertainty of the complex metabolic data could also hinder the improvement of models integrating this information. So far, the existing works on this topic are either based on only a few parent compounds and their measured metabolites, or focused on a single endpoint, making it therefore difficult to derive more general conclusions.
With this work, we aim to provide a systematic study on how, and to what extent, the consideration of metabolism can help the in silico prediction of toxicity. In order to cover a wide chemical space and make models applicable to new, untested compounds, we referred to the use of predicted metabolites. Five relevant toxicological endpoints and assays were selected for investigation: the in vitro AMES assay (considering metabolic activation with S-9 liver extract), the in vivo micronucleus test (MNT), a skin sensitization assay (the murine local lymph node assay, LLNA), and the drug-induced liver injury (DILI) and cardiological complications (DICC) endpoints.16–18 All selected endpoints and assays have in common that their outcome is known to be related, to some extent, to the biological activity of metabolites. Positive outcomes of the genotoxicity assays (AMES and MNT) and the skin sensitization assay (LLNA) can be produced by reactive metabolites that bind to DNA or skin proteins. The in vitro AMES assay (considering metabolic activation) was specifically chosen to evaluate the impact of adding metabolism information to a less complex endpoint (that is less dependent on pharmacokinetic variables than other in vivo endpoints). Moreover, reactive metabolites are also known to be a recurrent trigger of idiosyncratic adverse effects of drugs (i.e. unpredictable and infrequent adverse reactions often unrelated to dose).16 The role of metabolites in the two organ toxicity endpoints (DILI and DICC), often triggered by idiosyncratic adverse reactions, was hence also investigated.17,18
Materials and methods
Data sets
More specifically, the CCRIS database (stored in XML file format) was queried for mutagenicity studies based on the AMES assay, resulting in 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 907 study results (i.e. experimental assay outcomes on a set of compounds). For extracting these studies, the word “ames” was queried in the test system field (“mstu/tsstm”) of the XML file. The retrieved AMES data were further filtered for experiments that test for metabolic activation, by querying the data for the words “liver”, “hepatocytes”, “s9” and “s-9” in the “matvm” field. The resulting data (38267 study results) were further curated by removing any inconclusive or potentially ambiguous results. This was achieved by removing studies with results labeled as “weak” or as both “positive” and “negative” (e.g. “positive (retest was negative)”). Also inconclusive results caused by precipitating compounds were removed from the data set by querying the labels “negative” and “precipitation” (e.g. “negative, precipitation at 3 highest doses.”).
907 study results (i.e. experimental assay outcomes on a set of compounds). For extracting these studies, the word “ames” was queried in the test system field (“mstu/tsstm”) of the XML file. The retrieved AMES data were further filtered for experiments that test for metabolic activation, by querying the data for the words “liver”, “hepatocytes”, “s9” and “s-9” in the “matvm” field. The resulting data (38267 study results) were further curated by removing any inconclusive or potentially ambiguous results. This was achieved by removing studies with results labeled as “weak” or as both “positive” and “negative” (e.g. “positive (retest was negative)”). Also inconclusive results caused by precipitating compounds were removed from the data set by querying the labels “negative” and “precipitation” (e.g. “negative, precipitation at 3 highest doses.”).
The remaining data (38200 study results) were labeled as “toxic” if the results field matched the word “positive”, or “non-toxic” if the results field matched the word “negative”. To obtain only one result per compound, the data were deduplicated based on the CAS number and any compounds with conflicting class labels were removed from the data set. This resulted in 4721 compounds with AMES data.
The GENE-TOX database was obtained from PubChem.22 The different genotoxicity study types contained in this database were queried to select only those studies belonging to the AMES assay (i.e. matching the “Histidine reverse gene mutation, Ames assay” assay type). From the 1057 compounds with AMES data only the 238 results considering metabolic activation (i.e. matching “with metabolic activation” in the “activation” field) were conserved. The activity labels were used as is.
The NTP AMES data set contains 64246 study results. Results from assay setups without S-9 activation and from assays with microsome-activating conditions of less than 5% were removed from the data set. Results without an activity label reported in the study conclusion and results labeled as “equivocal” were removed from the data set. These filtering steps resulted in 40859 study results. Study outcomes with a “positive” or “weakly positive” study conclusion label were annotated as “toxic”, and study outcomes with the “negative” conclusion label as “non-toxic”. Compounds were deduplicated based on the CAS number, and duplicate compounds with conflicting labels were removed from the data set. In contrast to the above data sets, the NTP set did not include SMILES strings for the tested compounds. The SMILES strings were obtained by querying PubChem via the PUG REST interface23 using the CAS numbers provided with the NTP data set. This resulted in 1959 compounds annotated with AMES results.
The data from the three databases were merged based on the canonical SMILES (see section Structure preparation for details). Compounds with identical canonical SMILES but differing AMES activity labels (72 compounds) were removed from the data set. This resulted in a total of 5061 compounds (1908 toxic and 3153 non-toxic compounds; Table 1).
| Endpoint | Number of | Ratio | |
|---|---|---|---|
| Toxic compounds | Non-toxic compounds | ||
| AMES | 1908 | 3153 | 1:2 |
| MNT | 315 | 1460 | 1:5 |
| DILI | 435 | 226 | 2:1 |
| DICC | 965 | 2243 | 1:2 |
| LLNA | 521 | 749 | 1:1 |
Structure preparation
The standardization of the molecular structures followed the same procedure as described by Garcia de Lomana et al.24 (with one exception, indicated below). Briefly, the SMILES strings were standardized with the ChemAxon Standardizer31 node in KNIME32 to remove solvents and salts, annotate aromaticity, neutralize charges and mesomerize structures (i.e. returning the canonical resonant form of the molecule). Moreover, compounds containing any element other than H, B, C, N, O, F, Si, P, S, Cl, Se, Br and I as well as multi-component compounds were removed from the data set. Lastly, compounds with fewer than four heavy atoms or with molecular weight greater than 1000 Da (this criterion has been introduced for the current work only) were filtered out from the respective data set.For the remaining standardized structures, canonical SMILES were derived with RDKit33 in KNIME. These canonical SMILES were used for the deduplication of compounds in each data set. Compounds with identical canonical SMILES but conflicting labels for an endpoint were removed from the respective endpoint data set.
Descriptor calculation
Molecular structures were encoded with count-based Morgan fingerprints with a radius of 2 bonds and a length of 2048 bytes (computed with the “RDKit Count-Based Fingerprint” node in KNIME) plus 119 1D and 2D physicochemical property descriptors (computed with the “RDKit descriptor calculation” node in KNIME). These RDKit physicochemical property descriptors capture properties such as the number of occurrences of a specific atom type, bond or ring, as well as global molecular properties such as polarity and solubility. Moreover, up to two acidic and two basic pKa values were calculated for each molecule with the “pKa” KNIME node from ChemAxon.34 For molecules with fewer than two acidic or basic groups, the remaining pKa feature values were filled with the mean value of the respective data set.Model development and evaluation
Prior to model development, a variance filter was applied to all input features to remove those with a variance of less than 0.001. The remaining features were scaled with the StandardScaler class of scikit-learn35 by subtracting the mean and scaling to unit variance. Both variance filtering and scaling were performed individually for each data set.The models were evaluated within a 5-fold cross-validation (CV) framework by splitting the data into 80% training and 20% test set with the StratifiedShuffleSplit class of scikit-learn. To account for data imbalance, oversampling with SMOTENC (an extension of SMOTE that handles categorical features)36 was performed on the training set (with a ratio of samples in the minority class with respect to the majority class of 0.8). All molecular fingerprints and discrete RDKit descriptor features (e.g. number of hydrogen bond donors or ring count) were specified as categorical features in SMOTENC.
For each training set, random forest (RF) models were trained with the RandomForestClassifier of scikit-learn, with default parameters, except for num_trees = 1000, min_samples_leaf = 3 and class_weights = “balanced”.
For evaluating the performance of the models, the precision, recall, F1 score and Matthews Correlation Coefficient (MCC) were calculated on the respective test set of the CV. The precision measures the proportion of true positive predictions out of all positive predictions, while the recall measures the proportion of correctly predicted positive samples. The F1 score is the harmonic mean of precision and recall. The MCC takes into consideration all four classes of predictions (true positive, true negative, false positive and false negative predictions) and ranges between −1 and +1 (being +1 the perfect prediction). Both the F1 score and the MCC are robust against data imbalance.
Differences in the performance between models were evaluated with the nonparametric Mann–Whitney U test.37 For comparing a pair of models, the values for a given performance metric obtained in the different CV runs were used as input for the “mannwhitneyu” function implemented in SciPy.38 The p-value threshold of 0.05 was applied to consider a difference as significant. Due to the negligible number of significant results, a correction of the p-value accounting for the number of comparisons performed was deemed to be not necessary.
Metabolite prediction with Meteor Nexus
The metabolites were predicted with Meteor Nexus,7,39 a leading software package for metabolism prediction that is widely applied in the industries. Meteor Nexus covers a broad range of approximately 500 manually curated biotransformations gathered from several public sources and proprietary data sets from member organizations of Lhasa Limited.In this study, starting from the prepared molecular structures (canonical SMILES), four generations of metabolites were predicted and subsequently scored with the “Site of Metabolism (SOM) Scoring” method,40 which is the default scoring method of Meteor Nexus. Other processing options were retained at their default setting. The score given to each metabolite is based on experimental data for compounds that are chemically related to the query compound around the site of metabolism. The molecular structures of the predicted metabolites were prepared and standardized following the same procedure described for the parent compounds (starting from the SMILES string output by Meteor Nexus).
Predicted metabolite information as input descriptors for parent compounds
Two different approaches for including metabolite information as input features in machine learning were explored (Fig. 1A). In the first approach, the above-mentioned molecular fingerprints and physicochemical properties for each parent compound were concatenated with chemical descriptors calculated for the top-5 predicted metabolites of that parent compound (if available; metabolite scoring with Meteor). The chemical descriptors of the metabolites comprise count-based Morgan fingerprints (radius of 2; length of 1024 bytes) and all of the 200 physicochemical property descriptors of RDKit listed under “rdkit.Chem.Descriptors._descLis”. For parent compounds with fewer than five predicted metabolites, the empty values of the Morgan fingerprint vectors from the remaining metabolites were filled with zeros (indicating the absence of the structural feature) and the features corresponding to RDKit descriptors were filled with the mean value of the whole data set for that feature. Models were trained combining the molecular descriptors of the parent compounds with (a) Morgan fingerprints of the metabolites, (b) RDKit physicochemical property descriptors of the metabolites or (c) a combination of both. | ||
| Fig. 1 Overview of the different strategies explored to integrate predicted metabolite information into the in silico models. | ||
In the second approach, the above-mentioned fingerprints and physicochemical properties for each parent compound were concatenated with a count-based “biotransformation fingerprint”. The biotransformation fingerprint encodes the number of occurrences of a particular biotransformation (as labeled by Meteor Nexus) in the predicted metabolic tree. For each endpoint data set only those biotransformation predicted for at least one parent compound were included in the fingerprint. The feature length of the fingerprint ranges from 238 for the LLNA data set to 330 for the AMES data set. In addition to models based on the complete descriptor vector, models were also built on subsets of features selected prior to model building (in an attempt to reduce noise related to the sparsity of the biotransformation fingerprints). The feature selection was conducted on all descriptors (including fingerprints and physicochemical descriptors) and using the LassoCV implementation from scikit-learn within a 5-fold CV. Any feature with an output coefficient of zero was removed from the data prior to the training of the RF models.
Combination of the probabilities of toxicity predicted for a parent compound and its predicted metabolites
Two types of models were used for predicting the probability of toxicity:
(i) Baseline model: without the consideration of metabolites (i.e. trained only on the parent compounds).
(ii) Metabolism-aware model: with the consideration of metabolites (i.e. trained on the parent compounds and labeled metabolites).
The molecular descriptors defined in the “Descriptor calculation” section were used as input features for the parent compounds and metabolites in both types of models. For the metabolism-aware model the labels of the metabolites were assigned according to the workflow described in “Assignment of toxicity labels to metabolites”. The predicted probabilities for the parent compounds (with the baseline model) were used as a baseline result to analyze whether model performance improves when considering metabolites for the prediction of toxicity.
In an attempt to obtain the most accurate predicted probability for the parent compounds and metabolites, two approaches combining the baseline model and metabolism-aware model were investigated:
(a) Baseline-approach: baseline model for the prediction of both parent compounds and metabolites.
(b) Hybrid-approach: baseline model for the prediction of parent compounds plus metabolism-aware model for the prediction of metabolites.
To obtain the overall probability of toxicity of a compound (i.e. with the consideration of its metabolites), the selected model was applied to calculate the probability of toxicity of the parent compound and that of the predicted metabolites (up to four generations; Fig. 2). In addition, a number of different strategies for filtering predicted metabolites according to their relevance to toxicity were explored by a grid search. These filters are based on calculated logP, the Meteor score and/or predicted phase II metabolism, and are intended to remove any non-toxic (since readily excretable or unlikely) metabolites. The investigated threshold values, below which metabolites were removed, are 0 and 3 for logP, and 100, 200 and 300 for the Meteor score. When the phase II metabolism filter was applied, metabolites formed by phase II reactions, as well as those metabolites further transformed by phase II reactions, were filtered out. A grid search over the 23 possible combinations of filters (always including the possibility of not filtering for one or more properties) was performed.
 | ||
| Fig. 2 Workflow for calculating the overall probability of toxicity. The baseline model or the metabolism-aware model are used to predict the probability of toxicity of parent compounds and predicted metabolites independently. The predictions for a compound and its predicted metabolites are then combined into an overall probability to obtain the toxicity label. | ||
The predicted probabilities of toxicity calculated for the selected metabolites were then combined with the predicted probability for the respective parent compound. For the combination of the predicted probabilities of toxicity, four strategies were explored (Fig. 1B):
(1) Strategy 1: mean predicted probability over all compounds (i.e. the parent compound and all predicted metabolites).
(2) Strategy 2: median predicted probability over all compounds (i.e. the parent compound and all predicted metabolites).
(3) Strategy 3: maximum predicted probability among the parent compound and its predicted metabolites.
(4) Strategy 4: mean between the predicted probability of the parent compound and the maximum probability among all predicted metabolites.
If the overall probability was above 0.5, the compound was predicted as toxic and otherwise as non-toxic.
(1) All metabolites with identical canonical SMILES as a parent compound were assigned the toxicity label of the parent compound.
(2) All metabolites not covered by step 1 and originating from non-toxic parent compounds were labeled as “non-toxic”.
(3) All metabolites not covered by step 1 and originating from toxic parent compounds were compared with the already labeled metabolites. If an identical metabolite (based on the canonical SMILES) was labeled in one of the previous steps (as toxic or non-toxic), the same label was assigned.
(4) The remaining unlabeled metabolites from toxic parent compounds were labeled as “toxic” (Table 2).
| Endpoint | Number of metabolites | Percentage of metabolites | |||
|---|---|---|---|---|---|
| With the same molecular structure as a parent compound (step 1) (%) | Originating from non-toxic parent compounds (step 2) (%) | Originating from toxic parent compounds already labeled as toxic (step 3) (%) | Labeled as toxic as part of step 4 (%) | ||
| AMES | 86629 |
5.19 | 59.03 | 3.43 | 32.34 |
| MNT | 27105 |
2.11 | 81.53 | 2.22 | 14.14 |
| DILI | 10730 |
0.40 | 32.25 | 4.60 | 62.75 |
| DICC | 46881 |
2.21 | 67.43 | 4.82 | 25.54 |
| LLNA | 16842 |
3.46 | 51.62 | 5.66 | 39.26 |
To ensure that no data leak occurred in the metabolism-aware model due to the presence of identical metabolites in the training and test sets, the following procedure was conducted on each split:
(1) Stratified shuffle split was applied on the parent compounds (see Model development for details).
(2) The metabolites from the parent compounds in the test and training set were collected independently.
(3) The metabolites in the training set, which were also present in the test set (as parent or metabolite), were removed from the training set.
(4) The compounds of the training set were deduplicated based on the canonical SMILES (duplicates may appear due to repeated metabolites or metabolites identical to parent compounds).
Machine learning methods for further modeling optimization
RF, gradient boosted trees and k-nearest neighbors models with optimized hyperparameters were also trained in the hybrid-approach. The scikit-learn implementations ‘GradientBoostingClassifier’ and ‘KNeighborsClassifier’ were used for training the gradient boosted trees and k-nearest neighbor models, respectively. The hyperparameter optimization was conducted on the training set within a grid search evaluated on an inner 5-fold CV over the hyperparameters shown in Table 3.| Method | Hyperparameter | Values |
|---|---|---|
| Random forest | n_estimators | 400, 700, 1000 |
| Min_samples_leaf | 1, 2, 3 | |
| Class_weight | ‘Balanced’ | |
| Gradient boosted trees | n_estimators | 200, 400, 600 |
| Min_samples_leaf | 1, 2, 3 | |
| Learning_rate | 0.1, 0.01 | |
| K-nearest neighbors | n_neighbors | 3, 5, 8 |
| Weights | ‘Uniform’, ‘distance’ |
A further set of molecular descriptors, the Continuous and Data-Driven molecular Descriptors (CDDD),41 was employed as input for RF models. These descriptors are derived from a neural network trained to translate between two syntactically different molecular representations. In order to make the translation, the model first learns to compress meaningful information for the representation of molecules into a vector. This vector can hence be used as a data-driven molecular descriptor, offering a conceptually different method to represent molecules, compared to the fixed Morgan fingerprints and RDKit physicochemical descriptors.
Results and discussion
Analysis of the chemical space of the parent compounds and their predicted metabolites
To understand the nature and composition of the metabolites predicted for the parent compounds in each data set, several characteristics of the predicted metabolites were analyzed.The predicted metabolites result from phase I or phase II reactions (considering up to four generations of metabolites). The number of unique metabolites for the individual parent compounds (after removing duplicate metabolites from the respective metabolic tree) varied greatly (from 0 to 828). However, the median number of predicted metabolites among all parent compounds of an endpoint-specific data set was between 8 and 12 in all cases (Table 4).
| Endpoint | Mean number of metabolites per compound | Median number of metabolites per compound | Percentage of parent compounds without any predicted metabolite | Percentage of parent compounds with fewer than five predicted metabolites |
|---|---|---|---|---|
| AMES | 17.34 | 10 | 1.28 | 19.67 |
| MNT | 15.52 | 9 | 1.66 | 20.90 |
| DILI | 16.28 | 12 | 0.30 | 11.53 |
| DICC | 14.74 | 10 | 0.88 | 15.94 |
| LLNA | 13.38 | 8 | 0.87 | 23.75 |
By comparing the molecular properties of the parent compounds and their predicted metabolites (Fig. 3 reports on the AMES and MNT data sets; the graphs for the other endpoints are provided in Fig. S1†) we found the latter to have, averaged over all endpoints, a higher molecular weight (+43.9 Da) as well as a larger polar surface area (+44.4 Å2). The predicted metabolites also tended to have a lower logP value than the parent compounds (−1.5; averaged over all endpoints). These shifts are primarily a result of the addition of polar groups to the parent compounds, which make them more water soluble and therefore easier to excrete. This observation is in concordance with the higher number of hydrogen bond donors and acceptors observed in metabolites compared to parent compounds (1.8 more hydrogen bond donors and acceptors on average; Fig. 3). Overall, the shifts in the physicochemical property space between the parent compounds and the predicted metabolites are consistent with those observed for parent compounds and experimentally detected metabolites,42 a fact that supports the relevance of the predicted metabolites.
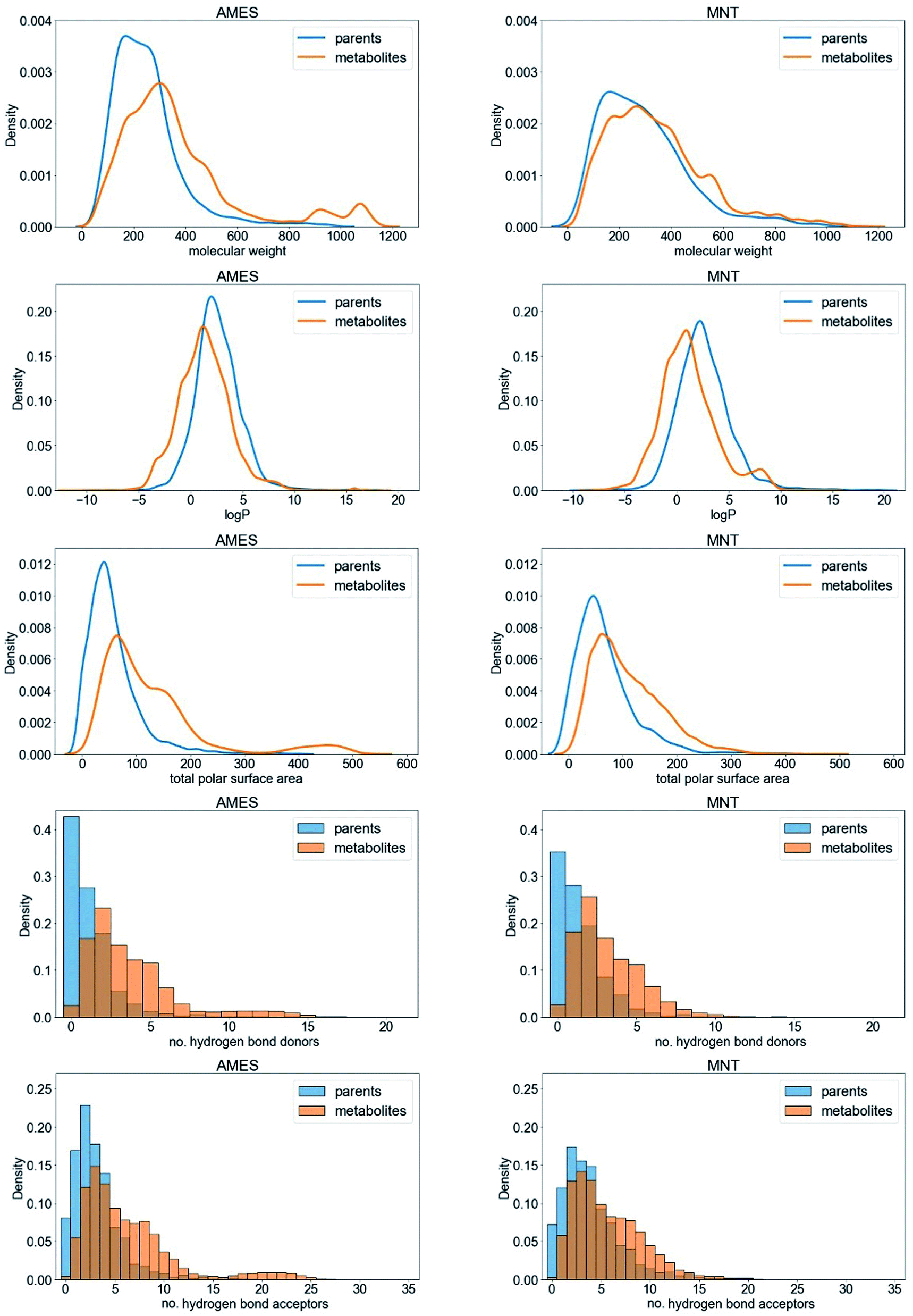 | ||
| Fig. 3 Comparison of the physicochemical properties of the parent compounds (blue) and predicted metabolites (orange) represented in the AMES and MNT data sets. | ||
Analysis of metabolites originating from toxic and non-toxic parent compounds
The toxicity observed for a compound may be a direct result of the parent compound or of one or several of its metabolites. Understanding the differences in the metabolites formed by toxic and non-toxic compounds may therefore help in their discrimination. However, when comparing the physicochemical properties of the (predicted) metabolites originating from toxic and from non-toxic parent compounds, we did not detect any substantial, systematic differences. This is not surprising because toxic effects may be related to a single metabolite, which is difficult to detect.Most notable was a minor shift in the logP distribution (see Fig. S2† for an example of the logP distributions of AMES and MNT): the logP of metabolites originating from non-toxic compounds was generally lower (logP of 0.8; averaged over all metabolites of all endpoints) than for metabolites from toxic compounds (logP of 1.2; averaged over all metabolites of all endpoints). The higher logP of metabolites originating from toxic parent compounds could be related to the observed toxicity, as these metabolites are more likely to evade excretion and to cross membranes.
Another aspect that could differ from toxic to non-toxic compounds are the types of biotransformations that they are undergoing. Testa et al.43 observed that some reactions are more prone to generate reactive or toxic metabolites than others. They showed that toxic metabolites are mainly formed by redox reactions, followed by conjugation reactions and, lastly, hydrolysis. Hence, the type of biotransformation that a compound undergoes may be an indicator of the compound's toxicity. To investigate whether the types of biotransformations in the metabolic trees of toxic and non-toxic compounds differ, the percentage of parent compounds of each toxicity class undergoing each biotransformation (as labeled by Meteor Nexus) was calculated for all endpoints.
We observed that some biotransformations occur more frequently in toxic parent compounds than in non-toxic ones (and vice versa). However, there was no single biotransformation observed to be related to the same toxicity class for all endpoints (see Fig. S3† for the examples of AMES and MNT). For instance, “aromatic reductive dehalogenation” is predicted more frequently for toxic compounds in the MNT assay (than for non-toxic compounds in this assay) while it is more often observed for non-toxic compounds in the AMES assay (than for toxic compounds in this assay).
In an analogous way, the enzymes catalyzing biotransformations in the metabolic tree of toxic and non-toxic compounds were also investigated. Similar results as for the biotransformations were observed, but, in this case, the differences between classes were smaller (i.e. there were few enzymes metabolizing a higher percentage of toxic or non-toxic compounds).
Baseline performance of the models
To enable the (later) quantification of the added value of metabolism prediction in toxicity prediction we generated baseline models trained exclusively on physicochemical properties of the parent compounds (encoded by count-based Morgan fingerprints and RDKit physicochemical property descriptors; see Materials and methods section for details).The mean F1 score obtained by the baseline models within 5-fold CV ranged from 0.64 (for MNT) to 0.82 (for AMES; Table 5). The superior performance of the AMES baseline model (F1 score at least 0.09 higher than for any other baseline model) is attributed to the larger size of the data set (it is the biggest data set considered in this study with at least 1853 compounds more than any other data set) as well as the nature of the endpoint: the AMES test is an in vitro assay carried out on bacteria, hence representing a more simple problem than the in vivo endpoints based on living mammals and considered in this work. Among the in vivo endpoints, the model for the LLNA assay, a skin sensitization assay measuring cellular proliferation in the draining lymph nodes of mice, obtained the highest mean F1 score (0.73). The lowest F1 score (0.64) was obtained by the MNT baseline model. The precision and recall yielded by each endpoint-specific model were on a similar level in all cases, indicating a balanced ratio of false positive and false negative predictions.
| Endpoint | F1 score | MCC | Precision | Recall |
|---|---|---|---|---|
| a Numbers reported in parentheses are the standard deviations. | ||||
| AMES | 0.82 (±0.01) | 0.65 (±0.03) | 0.83 (±0.01) | 0.82 (±0.01) |
| MNT | 0.64 (±0.03) | 0.29 (±0.05) | 0.67 (±0.02) | 0.62 (±0.03) |
| DILI | 0.68 (±0.04) | 0.37 (±0.08) | 0.69 (±0.04) | 0.68 (±0.04) |
| DICC | 0.69 (±0.02) | 0.39 (±0.04) | 0.71 (±0.02) | 0.69 (±0.03) |
| LLNA | 0.73 (±0.02) | 0.47 (±0.04) | 0.74 (±0.02) | 0.73 (±0.02) |
Metabolite information as input descriptors for parent compounds
When comparing the performance of these models containing metabolite information with that of the baseline models, no improvements of performance were observed (Table S2†). The few minor gains in performance did not exceed a value of +0.04 among all evaluated metrics and were not significant (at a p-value of 0.05; Table S3†). In several cases the addition of descriptors for the predicted metabolites led to small decreases in performance (up to a value of −0.09 among all metrics).
Within the 5-fold CV framework, the performance of these models was comparable to the baseline performance of each endpoint. For all evaluated metrics the difference from the baseline performance did not exceed ±0.01 (Tables S4 and S5†). The lack of an improvement in performance may be related to the sparsity of the biotransformation fingerprint: most of the biotransformations were not predicted to take place on more than 10% of the compounds. This low coverage of compounds may not be sufficient to enhance toxicity prediction. In order to remove possible noise caused by the sparse fingerprints, feature selection with a lasso model was applied to all input features (in order to discard irrelevant features prior to the training of the RF model). However, no relevant improvement in the performance compared to the baseline models was observed when feature selection was included prior to model training (F1 score deviations ranged from −0.05 to +0.01 among all endpoints).
Combination of predicted probabilities for parent compounds and metabolites
Another approach for considering metabolite information in toxicity prediction is the calculation of an “Overall predicted probability of toxicity” by combining the probabilities predicted for the parent compounds and their metabolites. A related approach (although based on distinct modeling methods and utilizing measured metabolites; explored for different endpoints) was applied, with some success, by Dmitriev et al.2 and Filimonov et al.3 (see the Introduction section for details).In this work, we explored four strategies to combine prediction probabilities:
Strategy 1: mean of the probabilities of the parent compound and all predicted metabolites.
Strategy 2: median probability of the parent compound and all predicted metabolites.
Strategy 3: maximum probability among the parent compound and all predicted metabolites.
Strategy 4: mean between the predicted parent compound probability and the maximum probability among all metabolites (i.e. the probability of the metabolite that the model deems most likely to be toxic, among all predicted metabolites).
To evaluate model performance, the obtained “Overall probability of toxicity” (derived by the different strategies) was compared to the experimental toxicity label of each parent compound (within 5-fold CV; see the “Data splitting” section for details). Note that all predicted metabolites (not only the five best-scored metabolites) were considered here.
The four strategies were applied to two approaches that differ in the underlying models used for calculating the predicted probabilities (Fig. 4ii). In the baseline-approach, we applied the baseline models on the test parent compounds and their metabolites and combined them with each of the above-mentioned strategies. With strategy 1, strategy 2 and strategy 3, a drop in F1 score and MCC was observed for all investigated endpoints. Strategies 1 and 2 especially showed a decrease in recall (up to −0.17), which was sometimes compensated, to some extent, by an increased precision (up to +0.04), while the opposite effect was observed for strategy 3 (Table S6†).
 | ||
| Fig. 4 Overview of the steps (i–iv) of the workflow for combining the predicted probability of parent compounds and predicted metabolites, showing the variations investigated at each stage. A grid search among all combinations of parameters at the different stages was conducted to identify the optimum solution. | ||
Out of the four strategies, the best classification performance was obtained, in general, with strategy 4. However, the gain in F1 scores compared to the respective baseline models was 0.02 or less (and hence not significant, according to the Mann–Whitney U test; see Table S7† for details). Compared to strategies 1 and 2, strategy 4 may provide a well-balanced compromise between an improved capacity to detect toxicity related to metabolism and noise introduced by the predicted metabolites. A similar result was also observed in the study by Dmitriev et al.,2 where several strategies to combine the predicted LD50 value (for acute rat toxicity) for parent compounds and their measured metabolites were investigated (mean of the predicted LD50 of all metabolites; mean of the predicted LD50 of the parent compound and all metabolites; maximum predicted LD50 among all metabolites; mean of the predicted LD50 of the parent compound and the most toxic metabolite). In agreement with our observations, Dimitriev et al. obtained their best results for the prediction of acute rat toxicity when taking the mean of the predicted LD50 for the parent compound and that of the most toxic metabolite. Also the increase in model performance (compared to taking the prediction of the parent compound only) in their case was minor (+0.03 in R2 and no differences in RMSE).
In the hybrid-approach, the predicted probabilities of the metabolites to be toxic were calculated with a dedicated model. We addressed the possibility that the absence of relevant improvement by the four above-mentioned strategies was due to a deficient coverage of the chemical space of the metabolites by the baseline model. The differences observed in the chemical space of parent compounds and metabolites (see the section “Analysis of the chemical space of the parent compounds and predicted metabolites” for details) could indicate that some metabolites fall outside the applicability domain of the models trained only on parent compounds (baseline models).
To expand the chemical space coverage of the models and try to improve the toxicity predictions for the metabolites, models including metabolites as input data (i.e. with their molecular descriptors as input features and the assigned toxicity as class label) were also developed (metabolism-aware models). The toxicity label of the metabolites for these models was assigned following the workflow described in the section “Assignment of toxicity labels to metabolites”. Instead of applying this straightforward labeling approach, the toxicity labels of the metabolites could have also been predicted with the baseline model. However, we did not investigate this option further as it would increase the complexity of the workflow and does not fit the purpose of this study. By labeling the metabolites we pretend to analyze whether the reason for the small model performance improvement is due to poor quality of the predicted probabilities of toxicity of the metabolites. Hence, predicting the toxicity label of the metabolites would suffer from the same limitation. We acknowledge that any manual or automatic metabolite labeling approach is a limitation of this study. The only way to overcome this limitation is the use of a large dataset of metabolites with measured toxicities. However, to our best knowledge no such dataset is in existence in the public domain.
With the hybrid-approach we aim to obtain the best predictions for each compound by predicting the probability of the parent compound to be toxic with the baseline model, and the probability to be toxic of the individual metabolites with the metabolism-aware model. Note that we also investigated the possibility to predict both the toxicity of the parent compound and the metabolites with the metabolism-aware model, but we did not see a relevant improvement compared to the baseline- or hybrid-approaches in this case and therefore did not further investigate this direction.
Compared to the baseline-approach, the hybrid-approach yielded better results in toxicity prediction. However, with improvements in the F1 scores and MCCs not exceeding 0.03 and 0.05, respectively, these results are not significantly better (based on the Mann–Whitney U test) than those obtained with the baseline model (Table 6). Few significant improvements were recorded for precision or recall for the MNT and DICC models (Table S8†).
| Endpoint | Combinationa | F1 score | MCC | Precision | Recall |
|---|---|---|---|---|---|
| a The baseline performance corresponds to models considering only parent compounds. Strategies 1, 2 and 3 correspond to taking the mean, median and maximum predicted probability among the parent compound and its metabolites, respectively. Strategy 4 corresponds to the mean between the predicted probability for the parent compound and the highest probability predicted for any of its metabolites. | |||||
| AMES | Baseline performance | 0.82 (±0.01) | 0.65 (±0.03) | 0.83 (±0.01) | 0.82 (±0.01) |
| Strategy 1 | 0.80 (±0.01) | 0.61 (±0.02) | 0.82 (±0.01) | 0.80 (±0.01) | |
| Strategy 2 | 0.80 (±0.01) | 0.61 (±0.02) | 0.81 (±0.01) | 0.79 (±0.01) | |
| Strategy 3 | 0.79 (±0.02) | 0.60 (±0.03) | 0.79 (±0.01) | 0.81 (±0.01) | |
| Strategy 4 | 0.83 (±0.02) | 0.65 (±0.03) | 0.82 (±0.02) | 0.83 (±0.02) | |
| MNT | Baseline performance | 0.64 (±0.03) | 0.29 (±0.05) | 0.67 (±0.02) | 0.62 (±0.03) |
| Strategy 1 | 0.61 (±0.03) | 0.31 (±0.05) | 0.75 (±0.02) | 0.59 (±0.03) | |
| Strategy 2 | 0.61 (±0.04) | 0.29 (±0.06) | 0.74 (±0.03) | 0.59 (±0.03) | |
| Strategy 3 | 0.65 (±0.02) | 0.31 (±0.03) | 0.64 (±0.02) | 0.67 (±0.02) | |
| Strategy 4 | 0.66 (±0.03) | 0.33 (±0.06) | 0.69 (±0.04) | 0.65 (±0.03) | |
| DILI | Baseline performance | 0.68 (±0.04) | 0.37 (±0.08) | 0.69 (±0.04) | 0.68 (±0.04) |
| Strategy 1 | 0.66 (±0.03) | 0.33 (±0.06) | 0.67 (±0.03) | 0.66 (±0.03) | |
| Strategy 2 | 0.66 (±0.03) | 0.32 (±0.06) | 0.67 (±0.03) | 0.65 (±0.03) | |
| Strategy 3 | 0.59 (±0.05) | 0.31 (±0.07) | 0.73 (±0.03) | 0.60 (±0.03) | |
| Strategy 4 | 0.66 (±0.03) | 0.37 (±0.05) | 0.73 (±0.02) | 0.65 (±0.03) | |
| DICC | Baseline performance | 0.69 (±0.02) | 0.39 (±0.04) | 0.71 (±0.02) | 0.69 (±0.03) |
| Strategy 1 | 0.68 (±0.02) | 0.40 (±0.03) | 0.75 (±0.02) | 0.66 (±0.01) | |
| Strategy 2 | 0.68 (±0.02) | 0.39 (±0.04) | 0.73 (±0.02) | 0.66 (±0.02) | |
| Strategy 3 | 0.68 (±0.01) | 0.38 (±0.01) | 0.67 (±0.00) | 0.70 (±0.00) | |
| Strategy 4 | 0.72 (±0.02) | 0.44 (±0.03) | 0.72 (±0.01) | 0.72 (±0.02) | |
| LLNA | Baseline performance | 0.73 (±0.02) | 0.47 (±0.04) | 0.74 (±0.02) | 0.73 (±0.02) |
| Strategy 1 | 0.70 (±0.02) | 0.42 (±0.04) | 0.73 (±0.02) | 0.70 (±0.02) | |
| Strategy 2 | 0.71 (±0.03) | 0.44 (±0.05) | 0.73 (±0.02) | 0.71 (±0.03) | |
| Strategy 3 | 0.69 (±0.01) | 0.42 (±0.03) | 0.71 (±0.02) | 0.71 (±0.01) | |
| Strategy 4 | 0.74 (±0.02) | 0.48 (±0.05) | 0.74 (±0.02) | 0.74 (±0.03) | |
The decrease in performance with strategies 1 and 2 (considering the predictions of all metabolites) in combination with the hybrid-approach was in general not as drastic as with the baseline-approach. This may indicate that the predicted probabilities for the metabolites were more accurate and did not include as much noise in the overall prediction. Again in this case, the best performance was observed with strategy 4 (averaging the probability of the parent compound and the most toxic metabolite), with only minor improvements in the F1 score of up to +0.03. Only for the DILI endpoint the F1 score decreased (by −0.02) with this strategy.
In addition, we analyzed whether the improvements in model performance may be limited due to the consideration of metabolites that are irrelevant to the observed toxic effect. In order to reduce the noise in the prediction caused by these metabolites, we applied several metabolite filters removing predicted metabolites that (a) have a low Meteor Nexus prediction score, (b) have a low calculated logP, or (c) are predicted to be further metabolized by conjugating enzymes (Fig. 4i).
Metabolites predicted with a low score by Meteor Nexus may be less likely to be observed in vivo and hence irrelevant to toxicity prediction. Metabolic reactions often lead to compounds with low logP values, making them more water soluble and therefore easier to excrete. These metabolites are also unlikely to cross membranes and they are less likely to induce toxic effects. Along the same lines, phase II metabolism facilitates the conjugation of compounds with polar moieties, making them more water soluble. It has already been observed that only few conjugation reactions lead to toxic metabolites.43 Following this reasoning, several thresholds for removing metabolites based on their Meteor Nexus score as well as calculated logP values were investigated. Also strategies to remove metabolites formed by phase II reactions, or remove metabolites which are further transformed by phase II reactions were explored. A grid search over all filtering possibilities (and all above-mentioned approaches and strategies) was conducted on each data set to obtain the most favorable combinations.
In most cases, reducing the number of metabolites considered for the prediction based on these parameters did not yield better models. Among the five top-ranked models (based on the F1 score) of the grid search, only in a few cases, minor improvements of up to +0.06 among all metrics and endpoints were observed (Table S9†). However, these performance improvements were not significant for any endpoint compared to the baseline performance (Table S10†).
Exploration of further modeling approaches with the hybrid-approach
To evaluate whether the predictions may be improved by optimizing the modeling approach, different machine learning modeling methods with optimized hyperparameters (within a grid search; see Materials and methods section for details) and a further, distinct set of descriptors (CDDD descriptors)41 were investigated at the example of the best performing approach, namely the hybrid-approach.The F1 score obtained for the following machine learning setup combinations is shown in Table S11:† RF, gradient boosted trees and k-nearest neighbors, each with and without the use of oversampling with SMOTENC (based on Morgan fingerprint and RDKit physicochemical descriptors as input descriptors). Moreover, the performance of RF models trained on CDDD descriptors (including oversampling with SMOTE) are also provided.
The results obtained with these new models do not deviate from those obtained with the models generated with the initial modeling setup (i.e. RF with fixed hyperparameters; combination of Morgan fingerprints and RDKit physicochemical descriptors; oversampling with SMOTENC; results reported in Table 6): the largest observed improvement in F1 scores yielded by the new models was of just +0.01. The conclusions derived in the ‘Combination of predicted probabilities for parent compounds and metabolites’ section remain consistent with the new results. The explicit incorporation of predicted metabolite information in toxicity prediction models did not significantly improve the toxicity predictions of these models either. Although there was often no benefit compared to the baseline models (or the benefit was small), the best strategy for combining the predicted probabilities of parent compounds and metabolites was, also in this case, strategy 4 (taking the mean between the predicted probability of the parent compound and the maximum probability among all predicted metabolites).
Conclusions
In this work we systematically investigated a variety of strategies to enhance toxicity prediction by taking into account xenobiotic metabolism. Our results show that none of these strategies produces models that consistently outperform others. The best results were obtained by averaging the probability of toxicity predicted for the parent compound and the maximum probability of toxicity predicted for any metabolite. This approach yielded models with F1 scores up to +0.03 higher than the baseline models disregarding metabolism.We observed that models trained exclusively on the parent compounds often produce poor predictions for the metabolites as their chemistry often differs. Including labeled metabolites in the training set of the models slightly improved the predictions of toxicity for the metabolites and hence the overall result of averaging the probabilities of toxicity for parent compounds and their metabolites. In some cases, discarding unlikely or water-soluble metabolites slightly improved the predictions (F1 score up to +0.04 higher than for the baseline models).
While metabolites can be key to detecting and understanding toxicity, they also add a new layer of complexity. The metabolites formed, their concentrations in the organism, and their excretion kinetics are often unknown. Therefore, including metabolism data in toxicity prediction poses veritable challenges. The fragile balance between added signal and added noise, when working with predicted metabolites in machine learning, may explain the small differences in performance of the models including metabolism information for toxicity prediction compared to the baseline models. It is clear from these results that there is still a long way to go in the development of sufficiently accurate models for metabolism prediction which, in turn, can boost toxicity prediction.
Abbreviations
| CCRIS | Chemical carcinogenesis research information system |
| CV | Cross-validation |
| DICC | Drug-induced cardiological complications |
| DILI | Drug-induced liver injury |
| ECHA | European chemicals agency |
| EFSA | European food safety authority |
| FDA | U.S. food and drug administration |
| GENE-TOX | Genetic toxicology data bank |
| LLNA | Murine local lymph node assay |
| LMC | Laboratory of mathematical chemistry |
| MCC | Matthews correlation coefficient |
| MNT | Micronucleus test |
| NTP | National toxicology program |
| RF | Random forest |
| TIMES | Tissue metabolic simulator |
Data availability
All data sets used in this study are publicly available. Due to licensing reasons, the original data and the predicted metabolites cannot be provided with this publication. However, a detailed protocol for the reproducible collection and preprocessing of the data utilized in this work is provided in the Materials and methods section. Moreover, Table S1† contains links for downloading the original data and complementary information about the data sets. Also detailed KNIME workflows used for preprocessing each data set and calculating the chemical descriptors of the parent compounds are provided in the ESI.† The workflows and parameters used for developing the models and necessary for reproducing the results are described in detail in the Materials and methods section. The code used for model training and evaluation can be accessed at https://github.com/marinaglr/metabio.Conflicts of interest
MGL and MM are employed at BASF SE. AV served as consultant for BASF SE.Acknowledgements
We thank Robert Landsiedel and Roland Buesen from BASF SE for their valuable comments and suggestions based on their expertise in toxicology. A. V. is supported by the Federal Ministry of Education and Research (BMBF), Germany (grant 031A262C).References
- M. Pirmohamed, N. R. Kitteringham and B. Kevin Park, The Role of Active Metabolites in Drug Toxicity, Drug Saf., 1994, 11, 114–144 CrossRef CAS PubMed.
- A. Dmitriev, A. Rudik, D. Filimonov, A. Lagunin, P. Pogodin, V. Dubovskaja, V. Bezhentsev, S. Ivanov, D. Druzhilovsky, O. Tarasova and V. Poroikov, Integral Estimation of Xenobiotics' Toxicity With Regard to Their Metabolism in Human Organism, Pure Appl. Chem., 2017, 89, 1449–1458 CAS.
- D. A. Filimonov, A. V. Rudik, A. V. Dmitriev and V. V. Poroikov, Computer-Aided Estimation of Biological Activity Profiles of Drug-Like Compounds Taking into Account Their Metabolism in Human Body, Int. J. Mol. Sci., 2020, 21, 7492 CrossRef CAS PubMed.
- Y. Djoumbou-Feunang, J. Fiamoncini, A. Gil-de-la-Fuente, R. Greiner, C. Manach and D. S. Wishart, BioTransformer: A Comprehensive Computational Tool for Small Molecule Metabolism Prediction and Metabolite Identification, J. Cheminf., 2019, 11, 2 Search PubMed.
- S. Tian, X. Cao, R. Greiner, C. Li, A. Guo and D. S. Wishart, CyProduct: A Software Tool for Accurately Predicting the Byproducts of Human Cytochrome P450 Metabolism, J. Chem. Inf. Model., 2021, 61, 3128–3140 CrossRef CAS PubMed.
- C. de Bruyn Kops, M. Šícho, A. Mazzolari and J. Kirchmair, GLORYx: Prediction of the Metabolites Resulting from Phase 1 and Phase 2 Biotransformations of Xenobiotics, Chem. Res. Toxicol., 2021, 34, 286–299 Search PubMed.
- C. A. Marchant, K. A. Briggs and A. Long, In Silico Tools for Sharing Data and Knowledge on Toxicity and Metabolism: Derek for Windows, Meteor, and Vitic, Toxicol. Mech. Methods, 2008, 18, 177–187 CrossRef CAS PubMed.
- L. Ridder and M. Wagener, SyGMa: Combining Expert Knowledge and Empirical Scoring in the Prediction of Metabolites, ChemMedChem, 2008, 3, 821–832 CrossRef CAS PubMed.
- O. Mekenyan, S. Dimitrov, T. Pavlov, G. Dimitrova, M. Todorov, P. Petkov and S. Kotov, Simulation of Chemical Metabolism for Fate and Hazard Assessment. V. Mammalian Hazard Assessment, SAR QSAR Environ. Res., 2012, 23, 553–606 CrossRef CAS PubMed.
- J. Zaretzki, M. Matlock and S. J. Swamidass, XenoSite: Accurately Predicting CYP-Mediated Sites of Metabolism with Neural Networks, J. Chem. Inf. Model., 2013, 53, 3373–3383 CrossRef CAS PubMed.
- O. Mekenyan, S. Dimitrov, R. Serafimova, E. Thompson, S. Kotov, N. Dimitrova and J. D. Walker, Identification of the Structural Requirements for Mutagenicity by Incorporating Molecular Flexibility and Metabolic Activation of Chemicals I: TA100 Model, Chem. Res. Toxicol., 2004, 17, 753–766 Search PubMed.
- G. M. Ovanes, D. D. Sabcho, S. P. Todor and D. V. Gilman, A Systematic Approach to Simulating Metabolism in Computational Toxicology. I. The TIMES Heuristic Modelling Framework, Curr. Pharm. Des., 2004, 10, 1273–1293 CrossRef PubMed.
- S. D. Dimitrov, L. K. Low, G. Y. Patlewicz, P. S. Kern, G. D. Dimitrova, M. H. I. Comber, R. D. Phillips, J. Niemela, P. T. Bailey and O. G. Mekenyan, Skin Sensitization: Modeling Based on Skin Metabolism Simulation and Formation of Protein Conjugates, Int. J. Toxicol., 2005, 24, 189–204 CrossRef CAS PubMed.
- O. Mekenyan, G. Patlewicz, C. Kuseva, I. Popova, A. Mehmed, S. Kotov, T. Zhechev, T. Pavlov, S. Temelkov and D. W. Roberts, A Mechanistic Approach to Modeling Respiratory Sensitization, Chem. Res. Toxicol., 2014, 27, 219–239 Search PubMed.
- O. G. Mekenyan, P. I. Petkov, S. V. Kotov, S. Stoeva, V. B. Kamenska, S. D. Dimitrov, M. Honma, M. Hayashi, R. Benigni, E. M. Donner and G. Patlewicz, Investigating the Relationship between in Vitro–in Vivo Genotoxicity: Derivation of Mechanistic QSAR Models for in Vivo Liver Genotoxicity and in Vivo Bone Marrow Micronucleus Formation Which Encompass Metabolism, Chem. Res. Toxicol., 2012, 25, 277–296 Search PubMed.
- T. Cho and J. Uetrecht, How Reactive Metabolites Induce an Immune Response That Sometimes Leads to an Idiosyncratic Drug Reaction, Chem. Res. Toxicol., 2017, 30, 295–314 Search PubMed.
- N. P. Chalasani, P. H. Hayashi, H. L. Bonkovsky, V. J. Navarro, W. M. Lee and R. J. Fontana, ACG Clinical Guideline: The Diagnosis and Management of Idiosyncratic Drug-Induced Liver Injury, Am. J. Gastroenterol., 2014, 109, 950–966 CrossRef PubMed ; quiz 967..
- I. Hopper, Cardiac Effects of Non-Cardiac Drugs, Aust. Prescr., 2011, 34, 52–54 CrossRef.
- National Institutes of Health, Chemical Carcinogenesis Research Information System (CCRIS), accessed February 19, 2021, https://ftp.nlm.nih.gov/projects/ccrislease/.
- National Institutes of Health, GENE-TOX, accessed February 19, 2021, https://www.nlm.nih.gov/databases/download/genetox.html.
- U.S. Department of Health and Human Services, National Toxicology Program, accessed February 19, 2021, https://cebs.niehs.nih.gov/datasets/search/ames.
- NCBI, PubChem Bioassay Record for AID 1259408, GENE-TOX Mutagenicity Studies, Source: Genetic Toxicology Data Bank (GENE-TOX), accessed February 19, 2021, https://pubchem.ncbi.nlm.nih.gov/bioassay/1259408.
- S. Kim, P. A. Thiessen, T. Cheng, B. Yu and E. E. Bolton, An Update on PUG-REST: RESTful Interface for Programmatic Access to PubChem., Nucleic Acids Res., 2018, 46, W563–w570 CrossRef CAS PubMed.
- M. Garcia de Lomana, A. Morger, U. Norinder, R. Buesen, R. Landsiedel, A. Volkamer, J. Kirchmair and M. Mathea, ChemBioSim: Enhancing Conformal Prediction of In Vivo Toxicity by Use of Predicted Bioactivities, J. Chem. Inf. Model., 2021, 61, 3255–3272 CrossRef CAS PubMed.
- eChemPortal, accessed August 6, 2020, https://www.echemportal.org/echemportal/.
- R. Benigni, C. Laura Battistelli, C. Bossa, A. Giuliani, E. Fioravanzo, A. Bassan, M. Fuart Gatnik, J. Rathman, C. Yang and O. Tcheremenskaia, Evaluation of the Applicability of Existing (Q)SAR Models for Predicting the Genotoxicity of Pesticides and Similarity Analysis Related With Genotoxicity of Pesticides for Facilitating of Grouping and Read Across, EFSA Support. Publ., 2019, 1598E Search PubMed.
- J. W. Yoo, N. L. Kruhlak, C. Landry, K. P. Cross, A. Sedykh and L. Stavitskaya, Development of Improved QSAR Models for Predicting the Outcome of the in Vivo Micronucleus Genetic Toxicity Assay, Regul. Toxicol. Pharmacol., 2020, 113, 104620 CrossRef CAS PubMed.
- M. Chen, A. Suzuki, S. Thakkar, K. Yu, C. Hu and W. Tong, DILIrank: The Largest Reference Drug List Ranked by the Risk for Developing Drug-Induced Liver Injury in Humans, Drug Discovery Today, 2016, 21, 648–653 CrossRef CAS PubMed.
- C. Cai, J. Fang, P. Guo, Q. Wang, H. Hong, J. Moslehi and F. Cheng, In Silico Pharmacoepidemiologic Evaluation of Drug-Induced Cardiovascular Complications Using Combined Classifiers, J. Chem. Inf. Model., 2018, 58, 943–956 CrossRef CAS PubMed.
- A. Wilm, U. Norinder, M. I. Agea, C. de Bruyn Kops, C. Stork, J. Kühnl and J. Kirchmair, Skin Doctor CP: Conformal Prediction of the Skin Sensitization Potential of Small Organic Molecules, Chem. Res. Toxicol., 2021, 34, 330–344 Search PubMed.
- Standardizer was used for structure canonicalization and transformation, JChem 3.5.0, ChemAxon, http://www.chemaxon.com Search PubMed.
- M. R. Berthold, N. Cebron, F. Dill, T. R. Gabriel, T. Kötter, T. Meinl, P. Ohl, C. Sieb, K. Thiel and B. Wiswedel, KNIME: The Konstanz Information Miner, in Studies in Classification, Data Analysis, and Knowledge Organization (GfKL 2007), Version 4.3.3., Springer, 2007 Search PubMed.
- G. Landrum, RDKit: Open-Source Cheminformatics Software, Version 4.2.0., 2016 Search PubMed.
- The pKa Plugin was used for the calculation of the pKa constant value of molecules, JChem 3.5.0, ChemAxon, http://www.chemaxon.com Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, Scikit-learn: Machine Learning in Python, version 0.22.1, 2011, vol. 12, pp. 2825–2830 Search PubMed.
- N. V. Chawla, K. Bowyer, L. O. Hall and P. O. Kegelmeyer, SMOTE: Synthetic Minority Over-Sampling Technique, 2002, 16, 321–357 Search PubMed.
- H. B. Mann and D. R. Whitney, On a Test of Whether one of Two Random Variables is Stochastically Larger Than the Other, Ann. Math. Stat., 1947, 18, 50–60 CrossRef.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt, A. Vijaykumar, A. P. Bardelli, A. Rothberg, A. Hilboll, A. Kloeckner, A. Scopatz, A. Lee, A. Rokem, C. N. Woods, C. Fulton, C. Masson, C. Häggström, C. Fitzgerald, D. A. Nicholson, D. R. Hagen, D. V. Pasechnik, E. Olivetti, E. Martin, E. Wieser, F. Silva, F. Lenders, F. Wilhelm, G. Young, G. A. Price, G.-L. Ingold, G. E. Allen, G. R. Lee, H. Audren, I. Probst, J. P. Dietrich, J. Silterra, J. T. Webber, J. Slavič, J. Nothman, J. Buchner, J. Kulick, J. L. Schönberger, J. V. de Miranda Cardoso, J. Reimer, J. Harrington, J. L. C. Rodríguez, J. Nunez-Iglesias, J. Kuczynski, K. Tritz, M. Thoma, M. Newville, M. Kümmerer, M. Bolingbroke, M. Tartre, M. Pak, N. J. Smith, N. Nowaczyk, N. Shebanov, O. Pavlyk, P. A. Brodtkorb, P. Lee, R. T. McGibbon, R. Feldbauer, S. Lewis, S. Tygier, S. Sievert, S. Vigna, S. Peterson, S. More, T. Pudlik, T. Oshima, T. J. Pingel, T. P. Robitaille, T. Spura, T. R. Jones, T. Cera, T. Leslie, T. Zito, T. Krauss, U. Upadhyay, Y. O. Halchenko, Y. Vázquez-Baeza and C. SciPy, SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python (Version 1.4.1.), Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- Meteor Nexus v3.1.0, Lhasa Limited Search PubMed.
- C. A. Marchant, E. M. Rosser and J. D. Vessey, A k-Nearest Neighbours Approach Using Metabolism-related Fingerprints to Improve In Silico Metabolite Ranking, Mol. Inf., 2017, 36, 1600105 CrossRef PubMed.
- R. Winter, F. Montanari, F. Noé and D. A. Clevert, Learning Continuous and Data-Driven Molecular Descriptors by Translating Equivalent Chemical Representations, Chem. Sci., 2019, 10, 1692–1701 RSC.
- J. Kirchmair, A. Howlett, J. E. Peironcely, D. S. Murrell, M. J. Williamson, S. E. Adams, T. Hankemeier, L. van Buren, G. Duchateau, W. Klaffke and R. C. Glen, How Do Metabolites Differ from Their Parent Molecules and How Are They Excreted?, J. Chem. Inf. Model., 2013, 53, 354–367 CrossRef CAS PubMed.
- B. Testa, A. Pedretti and G. Vistoli, Reactions and Enzymes in the Metabolism of Drugs and Other Xenobiotics. Drug Discov, Today, 2012, 17, 549–560 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dd00018g |
| This journal is © The Royal Society of Chemistry 2022 |